

Abstract
Gene duplication and loss contribute to gene content differences as well as phenotypic divergence across species. However, the extent to which gene content varies among closely related plant species and the factors responsible for such variation remain unclear. Here, using the Solanaceae family as a model and Pfam domain families as a proxy for gene families, we investigated variation in gene family sizes across species and the likely factors contributing to the variation. We found that genes in highly variable families have high turnover rates and tend to be involved in processes that have diverged between Solanaceae species, whereas genes in low-variability families tend to have housekeeping roles. In addition, genes in high- and low-variability gene families tend to be duplicated by tandem and whole genome duplication, respectively. This finding together with the observation that genes duplicated by different mechanisms experience different selection pressures suggest that duplication mechanism impacts gene family turnover. We explored using pseudogene number as a proxy for gene loss but discovered that a substantial number of pseudogenes are actually products of pseudogene duplication, contrary to the expectation that most plant pseudogenes are remnants of once-functional duplicates. Our findings reveal complex relationships between variation in gene family size, gene functions, duplication mechanism, and evolutionary rate. The patterns of lineage-specific gene family expansion within the Solanaceae provide the foundation for a better understanding of the genetic basis underlying phenotypic diversity in this economically important family.
Keywords: Solanaceae, domain family, whole genome duplication, tandem duplication, pseudogenes, evolutionary fate
Introduction
Biological diversity can be attributed to the influence of the environment as well as genetic differences. One prominent source of genetic variation within and between species is gene copy number. Due to differential gene gains and losses, there can be substantial variation in the number of gene copies in a gene family, with some families exhibiting high turnover rates and others having similar sizes across species. In some cases genes in families with high turnover rates are involved in divergent biological processes (Tatusov et al. 1997; Rubin 2000; Hahn et al. 2007; Guo 2013). Thus, this high degree of turnover in gene family membership is expected to contribute significantly to divergence in cellular and developmental processes across species. Consequently, differences in gene family content can be shaped by natural selection (Pál et al. 2006; Schrider and Hahn 2010; Albalat and Canestro 2016) and are central to the evolutionary diversification and ecological adaptation of species (Demuth and Hahn 2009; Żmieńko et al. 2014; Carretero-Paulet et al. 2015). Thus, comparative studies of the patterns of gene family turnover are fundamental for understanding and assessing the functional, evolutionary, and ecological significance of duplicate genes.
In eukaryotes, gene duplication is the primary source of new genes, which serve as the raw material for the evolution of novel functions (Ohno 1970; Zhang 2003). Duplicate genes can be generated through several mechanisms, such as whole genome duplication (WGD), segmental duplication, tandem duplication, and transposon-induced duplication (Panchy et al. 2016), each of which can have different impacts on duplicate gene functions and evolutionary fates and genomic architecture. WGD, for example, simultaneously doubles the number of all genes, and the requirement to maintain dosage balance leads to the preferential retention of genes encoding components of macromolecular complexes (Edger and Pires 2009; Birchler and Veitia 2014; Tasdighian et al. 2017). Segmental duplications in metazoans, where genomic segments that are hundreds to millions of base pairs long are duplicated in unlinked locations, can lead to chromosomal instability (Samonte and Eichler 2002). Tandem duplication can lead to new gene structures through the formation of chimeric genes (Rogers et al. 2017) and contributes to preferential retention of genes involved in stress response (Hanada et al. 2008). Transposon-induced duplication generates duplicates such as retrogenes that are mostly dead on arrival (Brosius 1991) but may modify gene expression (Flagel and Wendel 2009).
Although duplicates can be preserved through acquisition of novel functions (neofunctionalization; Zhang 2003), partitioning of ancestral functions among duplicates (subfunctionalization; Force et al. 1999), and/or other mechanisms (Lehti-Shiu et al. 2017), the majority of duplicate genes experience a brief period of relaxed selection and become pseudogenes within a few million years (Myr) (Lynch and Conery 2000). Because of differential gains, mostly due to differences in rates of gene duplication and loss through pseudogenization, gene family sizes and duplicate gene turnover rates are highly variable across species, including yeast (Hahn et al. 2005), fruit flies (Hahn et al. 2007), mammals (Demuth et al. 2006), and plants (Guo 2013). The existing studies of gene family turnover in plants have focused on highly divergent taxa, ranging from green algae to flowering plants (Guo 2013) or across the core eudicots (Carretero-Paulet et al. 2015). Thus, the extent of gene family size variation and the factors, particularly gene duplication mechanisms and pseudogenization, that contribute to this variation among closely related plant species remain unclear.
Here, we used the Solanaceae family as a model to investigate gene family variation among closely related species because a number of economically important species/cultivars in this family have been sequenced recently, including tomato (Tomato Genome Consortium 2012), potato (Potato Genome Sequencing Consortium 2011), eggplant (Hirakawa et al. 2014), pepper (Kim et al. 2014; Qin et al. 2014), tobacco (Sierro et al. 2013), and petunia (Bombarely et al. 2016). Fruits, tubers, leaves, and flowers of these species/cultivars have been used by humans as food, medicine, stimulants and decoration. In addition, Solanaceae species are important models for functional characterization of plant genes (Vanden Bossche et al. 2013; Fan et al. 2016) and for evolutionary and ecological studies (Hu and Saedler 2007; Nakazato et al. 2010; Särkinen et al. 2013). Furthermore, the genome sizes vary widely across Solanaceae species, ranging from 900 Mb in tomato (Tomato Genome Consortium 2012) to 4.5 Gb in tobacco (Sierro et al. 2013). We used the presence of a domain to define a family because sequences with the same protein domain are most likely homologous, and a gene may contain multiple protein domains that have divergent evolutionary histories and origins. Through a comparative genomics analysis of 12 Solanaceae and 3 outgroup species, we first determined the number of domain family gains and losses in each lineage. Next we assessed the extent of variation in domain family size across species and the domain family turnover rate for each branch in the Solanaceae species phylogeny. Finally, we determined how gene duplication mechanisms and pseudogenization contribute to domain family size variability.
Materials and Methods
Genome Annotation and Domain Family Designation
The genome sequences and annotations for each Solanaceae species and three outgroup species were downloaded from National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/genome/) or Solanaceae Genomics Network (SGN, https://solgenomics.net/): Solanum lycopersicum V2.5 (NCBI), S. pennellii SPENNV200 (NCBI), S. tuberosum V3.4 (SGN), S. melongena r2.5.1 (SGN), Capsicum annuum L. zunla-1 V2.0 (SGN), C. annuum_var. glabriusculum V2.0 (SGN), Nicotiana tabacum TN90 NGS (SGN), N. tomentosiformis V01 (NCBI), N. sylvestris GCF_000393655.1 (NCBI), N. benthamiana V1.0.1 (SGN), Petunia axillaris V1.6.2 (SGN), P. inflata V1.0.1 (SGN), Ipomoea trifida V1.0 (NCBI), Sesamum indicum V1.0 (NCBI), and Coffea canephora Vx (SGN). For two species with no annotation GFF files (S. melongena and I. trifida), CDS sequences were obtained from NCBI and used as queries in searches against the respective genome sequences using the BLAST-like alignment tool (BLAT) (Kent 2002) to find the location of each coding region in the genome. The criteria used for mapping were a threshold of 100% identity and no gap tolerated if located within an aligned block. GFF files were then generated based on the BLAT output with blat2gff.pf (Kent 2002).
Pfam domain Hidden Markov Models (HMMs, Version.3.0) were downloaded from the Pfam database (Finn et al. 2014), and transposase domains or domains found in proteins with transposase domains were excluded from downstream analyses. Protein sequences of genes in each Solanaceae species were used as queries in searches against the Pfam HMMs using HMMER3 (Finn et al. 2011) with the trusted cutoff. If >1 domains overlapped, the overlapping region was annotated with the Pfam domain with the smallest E-value. All protein sequences containing the same Pfam domain were considered to be in the same domain family. Because a gene may contain multiple protein domains that have divergent evolutionary histories and origins, genes with >1 types of protein domain were counted as being members of each domain family. Thus, a single gene can belong to multiple domain families. Using this definition, 26% genes in 12 Solanaceae species belonged to ≥2 domain families. For comparison, the sizes of domain families in eukaryotic species from representative phyla were downloaded from the Pfam database (Finn et al. 2014). To avoid the confounding effects of WGD in coefficient of variation (CoV) calculations, domain family sizes from N. tabacum and N. benthamiana, which have experienced recent WGDs (Bombarely et al. 2012; Sierro et al. 2013), were excluded.
Species Tree and Ancestral Presence/Absence State Inference
To build the species tree, genes in domain families with only a single copy in each species, or one randomly chosen copy if there were >1 copies in N. tabacum or N. benthamiana, which have experienced recent WGD (Bombarely et al. 2012; Sierro et al. 2013), were used. For each domain family, amino acid sequences were aligned using MUSCLE (Edgar 2004), and poorly aligned regions were removed using trimal (Capella-Gutiérrez et al. 2009) with a gt cutoff value of 0.8 (i.e., columns with gaps in more than 20% of the sequences are removed). The alignments were then combined and used to build a phylogenetic tree using RAxML/8.0.6 (Stamatakis 2014) with the following parameters: −f a −x12345 −p 12345 −# 1000 −m GTRGAMMA, with sequences from Co. canephora set as outgroups.
To estimate the species divergence times, 4-fold degenerate transversion rates (4DTv) were used for the Molecular Clock Test in MEGA (Tamura et al. 2013), using the General Time Reversible model and Gamma Distributed (G) rates among sites. The evolutionary rate was set as 6 × 10−9 per site per year (Wolfe et al. 1989). The estimated species divergence times based on the molecular clock are consistent with those in an earlier study (Särkinen et al. 2013). The ancestral presence/absence states of domain families were first inferred using the parsimony method (supplementary table S1, Supplementary Material online) in Mesquite (Maddison and Maddison 2017). For nodes with ambiguous states, the maximum likelihood method was used to choose the more likely states (supplementary table S2, Supplementary Material online).
Expression Data Sources and Processing
S. lycopersicum RNA-sequencing data for five hormone treatments (Gupta et al. 2013; Shi et al. 2013; Wang, Tao, et al. 2013; Livne et al. 2015; Capua and Eshed 2017) and 13 stress treatments (Chen et al. 2013; Rosli et al. 2013; Pombo et al. 2014; Alkan et al. 2015; Chen et al. 2015; Du et al. 2015; Loraine et al. 2015; Yang et al. 2015; Fragkostefanakis et al. 2016; Worley et al. 2016; Pombo et al. 2017; Sarkar et al. 2017; Zheng et al. 2017) were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/) and DNA Data Bank of Japan (DDBJ, http://trace.ddbj.nig.ac.jp/). Reads were trimmed using Trimmomatic with the parameters, LEADING: 3 TRAILING: 3 SLIDINGWINDOW: 4:20, and seed mismatches = 2, palindrome clip threshold = 30, simple clip threshold = 10 (Bolger et al. 2014), based on the sequence quality in FastQC reports (Andrews 2010). Reads were mapped to the S. lycopersicum V2.5 genome using TopHat2 with –min-intron-length = 50, –max-intron-length = 5,000, and –max-multihits = 20 (Kim et al. 2013), and samples with an overall read mapping rate ≥80% were kept for calculating RPKM using cufflinks with max-intron-length = 5,000 (Trapnell et al. 2010). Fold changes (FC) in gene expression levels between hormone/stress treated and control samples were calculated using the Bioconductor package edgeR (Robinson et al. 2010). Genes with |log2(FC)| >1 were considered differentially expressed between samples.
Functional Annotation
Protein sequences were used as queries in BLASTP searches against the NCBI nr protein database with an E-value cut-off of 1e-5, and gene ontology (GO) annotations were inferred using blast2go (Conesa and Götz 2008) with default parameters. Some genes may be annotated with GO terms related to nonplant activities, for example, GO: 0001568 (blood vessel development); therefore, GO terms from Arabidopsis thaliana (http://www.arabidopsis.org/), Oryza sativa V7.0 (http://genome.jgi.doe.gov/), and S. lycopersicum ITAG2.4 (ftp://ftp.solgenomics.net/) annotations were used as reference lists to filter out nonplant GO terms. Plant GO Slim terms (http://www.geneontology.org/) were used to obtain a broad overview of functional annotation. Gene set enrichment analysis was performed using Fisher’s exact test, and the P-value was adjusted to account for multiple testing (Benjamini and Hochberg 1995). GO terms with adjusted P-values (q) smaller than 1e-5 were considered significantly over/under-represented.
Sequence Evolutionary Rate Calculations and WGD Inference
To calculate synonymous (Ks) and nonsynonymous (Ka) substitution rates for a gene pair, protein sequences were first aligned using Clustalw-2.1 (Larkin et al. 2007), and by comparing the amino acid sequences to the coding nucleotide sequences, the corresponding CDS alignments were generated and used as input in the yn00 program in PAML version 4.4.5 (Yang 2007) with default parameters. The Ks values were used to determine the relative timing of WGD and speciation events among Solanaceae lineages based on peak values of Ks distributions of reciprocal best matches from all-against-all BLASTp searches within species (pairs of paralogs) and between species (pairs of putative orthologs), respectively (supplementary fig. S1, Supplementary Material online). Consistent with earlier studies (Leitch et al. 2008; Potato Genome Sequencing Consortium 2011; Tomato Genome Consortium 2012; Hoshino et al. 2016), these Ks distributions (supplementary fig. S1, Supplementary Material online) indicated that the Solanaceae species experienced the γ triplication (γ WGD) shared by stem lineages of core eudicots (Vekemans et al. 2012), and the Solanaceae-specific triplication (Sol WGD). N.tabacum and N. benthamiana independently became polyploids after the Solanaceae-specific triplication (Bombarely et al. 2012; Sierro et al. 2013). I.trifida and Se. indicum also have lineage-specific WGD events (Wang et al. 2014; Hoshino et al. 2016).
Domain Family Turnover Rate
We used the likelihood-based method implemented in BadiRate v1.35 (Librado et al. 2012) to estimate the rate of gene gains and losses (turnover rate λ). Three different branch models including Free Rates (FR, each branch has its own turnover rate), Global Rates (GR, all branches have the same turnover rate), and Branch-specific Rates (BR, particular branches have specific turnover rates), were used to estimate λ values. To take into account potential differences in gene turnover rates due to overall domain family expansion and rapid gene loss after lineage-specific WGDs (Sankoff et al. 2010; Inoue et al. 2015), in the BR model, all the branches leading to lineages with WGDs were assigned branch-specific turnover rates, whereas other branches were assumed to have the same turnover rate. For large domain families, where no results were obtained after a runtime of >100 h, Markov Clustering (Enright et al. 2002) was used to divide each domain family into smaller subfamilies with the parameter −I = 3. The best turnover rate model for each domain family was chosen based on likelihood ratio tests (Peers 1971). To evaluate the robustness of the inferred numbers of gene gain/loss event for each of 8,651 families/subfamilies, 100 gene gain/loss events estimates were generated based on replicated BadiRate runs using the best model and 100 random seed values. Runs for all but the 24 largest families/subfamilies finished within two months. Because it was not feasible to conduct 100 replicates, only 5 replicate analyses were run for these 24 families/subfamilies. Branches leading to species that have experienced WGD events tended to have more gene gains/losses and larger standard deviations (supplementary fig. S2, Supplementary Material online).
Classification of Genes Based on Duplication Mechanisms
To classify duplicate genes into different categories according to duplication mechanism, the software MCScanX-transposed (Wang, Li, et al. 2013) was used. First, based on intra- and interspecies all-against-all BLASTp results, the five sequences with the best matches to each query sequence with E-value ≤1e-10 were assumed to be homologs and retained. Then the chromosomal locations of these homologous genes were compared using MCScanX-transposed. For a given species, all other species in the same genus, one representative species from each of the other Solanaceae genera, and two non-Solanaceae species were used as reference species. For example, when gene duplication mechanisms were assessed for S. lycopersicum genes, gene sequences and chromosomal locations from S. pennellii, S. tuberosum, S. melongena, C. annuum_var. glabriusculum, N. tomentosiformis, P. axillaris, I. trifida, and Co. canephora were used in MCScanX-transposed.
Duplicate genes were classified into four categories: 1) syntenic duplicates—paralogs are located in corresponding collinear blocks within a species; 2) dispersed duplicates—one paralog and its ortholog are both located in corresponding interspecies collinear blocks, whereas the other paralog and its ortholog are not; 3) tandem duplicates—paralogs are immediately adjacent to each other; 4) proximal duplicates—paralogs are adjacent each other but separated by ≤10 nonhomologous genes. Duplicates that did not belong to any of the above categories were removed from our analysis as their mechanism of duplication is ambiguous.
Note that instead of using the category names in MCScanX, we renamed the “segmental duplicates” as “syntenic duplicates”, because genes in alignable blocks within each genome could have been duplicated by either WGD or segmental duplication (Cannon et al. 2004; Singh et al. 2015). We also renamed the “transposed duplicates” as “dispersed duplicates” because these genes could have been duplicated through transposition, WGD and subsequent rearrangement of one of the copies, recombination between repeat sequences in unlinked regions, or nonhomologous end-joining of double-stranded breaks (Woodhouse et al. 2010). Because the Ks distributions of both syntenic and dispersed duplicates showed two similar peaks (supplementary fig. S3, Supplementary Material online) corresponding to the Sol and γ WGD events (supplementary fig. S1, Supplementary Material online), we further assigned duplicates to these two WGDs by estimating the Ks value distribution as a mixture of Gaussian distributions:
where a, b, and c are fitted constants obtained using nonlinear (weighted) least-squares estimation (nls) of the Ks distribution in the R environment (Nash 2014). After values of a, b, and c were fitted for both WGD duplicate categories, distributions of simulated Ks were obtained. Cutoff values of Ks used to define the boundaries of these two WGD events were chosen based on two criteria: 1) To maximize the difference in the area under the curve between the two distributions (i.e., choosing cutoff values yielding the largest difference in area under the two fitted distributions); 2) for any given Ks value, the number of gene pairs from the distribution corresponding to the Sol WGD is more than twice the number with Ks values corresponding to the γ WGD distribution. Because the Sol and γ WGDs were experienced by all Solanaceae species and the synonymous substitution rate is often assumed to be neutral, we used the Ks cutoff values from S. lycopersicum, which has the genome assembly with the highest N50, to define WGD duplicate gene pairs in other species.
Pseudogene Identification
To identify pseudogenes, protein sequences from A. thaliana, O. sativa, and S. lycopersicum were used as queries in TBLASTN (Altschul et al. 1990) searches against the unannotated genomic regions of target species. The alignments between the unannotated genomic regions with significant similarity to protein-coding genes and their protein-coding gene homologs were further processed with the pipeline from Campbell et al. (2014) to identity those that had premature stops/frameshifts and/or were truncated (<30% of functional paralogs) (Zou et al. 2009). To evaluate the Ka and Ks values between gene–pseudogene pairs, or between pseudogene–pseudogene pairs, nucleotide sequences of stop codons and frameshift positions in the pseudogenes were removed from the pairwise CDS alignments.
Results and Discussion
Domain Family Presence/Absence Variation
Variation in gene family size among taxa, which contributes to evolutionary divergence, is due to differential gain and loss of duplicates. Prior to assessing variation in gene family size, we evaluated the extent to which gene families were shared among species by examining the presence/absence distribution of gene families (using Pfam domains as a proxy, see Materials and Methods) across 12 Solanaceae species. In total, 4,313 families had ≥1 member in ≥1 species and were analyzed further. Of these domain families, 87.6% (3,775) were present in ≥10 species (fig. 1A), suggesting they were present in the Solanaceae common ancestor, 2.9% of domain families (126) were present in 2–6 species, and 4.0% of domain families (174) were species-specific. To rule out the possibility that these lineage-/species-specific domain families are false negatives, we investigated three technical sources of error including: 1) missing annotations, 2) contamination during sequencing, and 3) genome assembly coverage and quality.
Fig. 1.

—Distribution of domain families and domain family gains/losses in Solanaceae species. Frequency of domain families present in (A) the annotated genomic regions and (B) the annotated plus intergenic regions of different numbers of Solanaceae species. (C) The number of N. sylvestris-specific domain families (defined based on searches of annotated regions) present in intergenic regions of different numbers of Solanaceae species. (D) Inferred numbers of domain family gain/loss events across Solanaceae lineages. Blue/red numbers: the number of domain family gains and losses, respectively. Green bars: standard errors for divergence time estimates.
To determine if a domain family in a species was absent because it was not annotated, we used the seed sequences of each domain family to search against the intergenic sequences of that species. Out of 1,281 domain families that were absent in ≥1 species, sequences for 702 (54.8%) could be recovered in the intergenic regions of ≥1 other species, leading to an increased proportion (90.5%) of domain families present in ≥10 species and a reduction in species-specific domain families (3.1%) (fig. 1B). This indicates that annotation significantly impacts the number of domain families identified in a species. On the other hand, consistent with the contamination hypothesis, 72.4% of the species-specific domain families (126 of 174) were present only in N. sylvestris. Of the 126 N. sylvestris-specific domains, only 32 could be found in the intergenic regions of other species (fig. 1C), further supporting the notion that some of these domain families may have been encoded by contaminating DNA introduced during sample collection or DNA extraction. Therefore, we excluded all N. sylvestris domain families and 41 other species-specific domain families that were not identified in the intergenic regions of any other species, leaving a total of 4,146 domain families.
Considering that the genomes we analyzed are of draft quality, we next determined the correlation between the scaffold N50 and the number of domain families absent in each species. We found no significant correlation (Pearson’s correlation coefficient [PCC] =-0.23, P = 0.49), suggesting that even though incomplete genome assembly is expected to impact domain family discovery, the effect of this impact is not large enough to detect. Taken together, most domain families are present in nearly all Solanaceae species, indicating common ancestry. The existence of a subset of lineage/species-specific domain families, is largely explained by missing annotations, and 167 of these families appear to be derived from contamination.
Inference of Ancestral Domain Presence/Absence States
With potential false negative cases identified and potential contaminating sequences removed, we next assessed the contribution of differential gains and losses to the lineage-specific distribution of domain families by inferring the ancestral presence/absence states of domain families in the 11 Solanaceae species (fig. 1D). Of 757 domain families absent in ≥1 Solanaceae species, 660 and 71 were inferred to have been present and absent in the Solanaceae common ancestor, respectively, and 26 had ambiguous ancestral states (supplementary tables S1–S3, Supplementary Material online). To further assess the ancestral states of domain families in Solanaceae, we also analyzed the absence/presence distribution of domain families in other land plant/algae species (fig. 2A and B; supplementary table S3, Supplementary Material online). We found that 75% (73 of 97) of the domain families inferred to be absent or ambiguous based on analysis of Solanaceae species are present in multiple (>3) other plants/algae, indicating that these 73 families may also have been present in the Solanaceae common ancestor but had a higher loss rate compared with other domains. In addition to differential loss, the lineage-specific distribution of these families could also be due to high evolutionary rates where homologous domains are no longer recognized as belonging to the same domain family. To assess this possibility, we compared the Ka/Ks ratios, which are used as a proxy of the selective pressures acting on gene pairs, of reciprocal best match gene pairs from S. lycopersicum and S. pennellii for domain families present in 2–11 species, regardless of ancestral state inference, and found no significant difference (Wilcoxon rank sum test, supplementary fig. S4, Supplementary Material online). This suggests that the lineage-specific distribution of domain families may not be significantly influenced by high evolutionary rates. Therefore, among the lineage-specific domain families inferred to exist in the common ancestor of Solanaceae species, most have likely been lost independently in ≥1 species.
Fig. 2.

—Distribution of domain families that are absent in ≥1 Solanaceae species. The species examined include 11 Solanaceae species, 36 other plant/algal species, and 50 representative prokaryotic and eukaryotic phyla. Heat maps showing the presence/absence in each species for (A) 660 domain families inferred to be present in the Solanaceae common ancestor, (B) 73 domain families inferred to be absent in the Solanaceae common ancestor or that have ambiguous presence/absence ancestral states but are present in >3 other plant/algal species, and (C) 24 remaining domain families that are present in ≤3 other plant/algal species. Cyan: present; grey: absent. Color scale: the number of Solanaceae species with a given domain family. The phylogenic tree shows the same phylogenic relationships as figure 1D. Red and black branches indicate Solanaceae and outgroup species, respectively. Streptophyta and Chlorophyta species names and fungal, metazoan and prokaryotic phyla are shown in supplementary tables S3 and S4, Supplementary Material online, respectively.
The remaining 24 families absent in ≥1 Solanaceae species and in most of the examined algal/plant species may have been: 1) present in the Solanaceae common ancestor but not identified in the Pfam hmmscan analysis based on the parameters we used (see Materials and Methods); 2) acquired due to de novo emergence of novel domains; 3) acquired through horizontal gene transfer (HGT); or 4) contamination. Based on our Pfam hmmscan analysis in other plants and algal species, case (1) cannot be completely ruled out but is unlikely. We extended our analysis to examine the distributions of these 24 lineage-specific domains in 50 other representative prokaryotic and eukaryotic phyla (fig. 2C). We found two families that are only present in a monophyletic group of closely related Solanaceae species but not in any other organisms examined. These include the Sar8_2 family, which is present only in non-Petunia species and has members involved in response to microbial infection (Alexander 1992; Verberne et al. 2000), and the Prosystemin family, which is present only in S. lycopersicum, S. pennellii, and S. tuberosum, and has members involved in wound response (Constabel et al. 1998). Although this may suggest the de novo origin of these two families, prosystemin represents a clear case of rapid divergence as structural homologs are also present in Nicotiana (Ryan and Pearce 2003). We also found that seven families present in monophyletic groups of Solanaceae species are also present in nonplant organisms (fig. 2C; supplementary table S4, Supplementary Material online). Some of these domain families may have arisen through HGT, and this requires further analysis. In summary, the independent losses and gene annotation issues noted in the previous section are the two primary contributors to the limited distribution of some domain families, whereas other factors, such as de novo gains, rapid divergence, HGT, and contamination, likely only account for the limited distribution of a very small number of families.
Variation in Domain Family Size among Species and Its Relationship to Gene Functions
After examining the distribution of domain families among Solanaceae species, we next assessed how the sizes of these families vary across species by measuring the CoV (standard deviation in domain family size divided by the mean size) of each domain family (supplementary table S5, Supplementary Material online). Because the mean domain family size is the denominator in CoV, similar degrees of changes in size have a greater effect for smaller families. To minimize this impact, we first binned the domain families based on their average sizes across species, determined the 95th and 5th percentile values of the CoV distribution for each bin, and fitted 95th and 5th percentile values across bins (fig. 3A). Domain families above the 95th and below the 5th percentile trend lines were defined as having significantly higher and lower size variability compared with the genome-wide average, respectively. In total, there were 228 high-variability families and 410 low-variability families (supplementary table S5, Supplementary Material online).
Fig. 3.

—Relationship between domain family size (number of genes) and size variability. (A) Distribution of the CoV of domain family size among average domain family size bins. Domain families were assigned to bins based on the average number of genes in a domain family across species. Purple shade: region between the 5th (blue) and 95th (red) percentile trend lines. (B) GO Slim enrichment of genes in high- and low-variability domain families. Color scale: -log10(q). Red and blue: Over- and under-representation, respectively. (C) Enrichment of signal transduction child terms for genes in high- and low-variability domain families. Over-represented: -log10(q)>5. Under-represented terms not shown. Blue and red: Child terms over-represented for genes in low- and high-variability families, respectively. (D) Enrichment of signal transduction genes in domain families with ≥1 annotated signal transduction category genes. Blue and red: Low- and high-variability families enriched for signal transduction genes, respectively. (E) Proportion of genes up- and/or down-regulated upon hormone (H) and stress (S) treatments in high- and low-variability domain families. *P of Wilcoxon signed-rank test < 0.05; **P < 0.01. Only genes with >2-fold change in expression levels between treatment and control were considered.
To assess the functions that genes in high/low-variability domain families tend to have, we conducted a gene set enrichment analysis (see Materials and Methods). We found that genes in high-variability families tend to have functions related to, for example, secondary metabolic process, pollen-pistil interaction, cell death, abscission, and fruit ripening (fig. 3B;supplementary table S6, Supplementary Material online). An example of a high-variability domain family is 2OG-Fe(II) oxygenase (CoV = 0.26, 84.6th percentile), and diversification of 2OG-FeII-Oxy domain-containing genes is a key factor contributing to the diversity and complexity of specialized metabolites in land plants (Farrow and Facchini 2014; Kawai et al. 2014). Another example is the NB-ARC domain (CoV = 0.42, 100th percentile), which is enriched in genes involved in cell death (De Oliveira et al. 2016). The eukaryotic protein serine/threonine/tyrosine kinase domain family (Pkinase, CoV = 0.15, 66.7th percentile) is also highly variable, mostly due to receptor-like kinases involved in self/nonself-recognition (Lehti-Shiu and Shiu 2012). The high CoV values observed for these families likely reflect rapid changes in response to the environment, particularly biotic factors.
In contrast, genes in domain families with low-variability tend to be involved in central metabolism processes and housekeeping functions, including cell differentiation and growth, and lipid, protein and DNA metabolism (fig. 3B). This indicates that negative selection contributes to low gene family variability. Surprisingly, genes in low-variability domain families tend to have functions in the response to stress, suggesting that some stress response processes may be consistently maintained across Solanaceae species. This may also suggest that stress-related genes turnover quickly, as shown in previous studies (Guo 2013; Wu et al. 2015), but that their turnover rates are remarkably similar across lineages. Genes from 4 to 38 domain families were annotated to each of the above GO categories, revealing how genes from different domain families interact to influence the underlying processes. For example, domain families enriched in genes related to tropism include PHY (Phytochrome) and two PHY-associated domains (HisKA and HATPase_c), as well as AUX_IAA. Phytochromes function as photoreceptors, whereas Aux/IAA genes regulate auxin-induced gene expression and also mediate light responses (Reed 2001). Our observations are consistent with previous studies showing the connection between light sensing and auxin signaling (Colon-Carmona et al. 2000; Halliday et al. 2009; Pedmale et al. 2010).
Interestingly, three processes were enriched for genes in both high- and low-variability families (fig. 3B), including postembryonic development, signal transduction and photosynthesis. When we examined the signal transduction category further as an example, we found that two child terms, intracellular signal transduction and immune response-regulating signaling pathway, were enriched in high-variability family genes, whereas six child terms were enriched in low-variability family genes, including light signaling, organelle-nucleus signaling, and defense response signaling pathways (fig. 3C;supplementary table S7, Supplementary Material online). Second, we asked to what extent the variabilities of domain families enriched in signal transduction genes (q < 1e-5, Fisher’s Exact Test) differed. Among 154 signaling gene-enriched families, the variability (percentiles of CoV) ranged from 0.004 to 0.992. These families included 15 and 6 families with low (e.g., the PHY domain found in Phytochrome; Pedmale et al. 2010) and high (e.g., the WAK domain, involved in receptor kinase signaling in cell expansion and defense response, Wagner and Kohorn 2001; Delteil et al. 2016) variability, respectively (fig. 3D;supplementary table S8, Supplementary Material online). These results highlight both conserved signaling processes such as light signaling conserved among major plant lineages, as well as species-specific signaling pathways that may be important for phenotypic and adaptive divergence across species.
Given the connection between family size variability and signaling and environmental response, we further explored potential functional bias of genes in high- and low-variability domain families by analyzing transcriptome data sets for cultivated tomato treated with five hormones and 13 stresses (see Materials and Methods). We found that a significantly higher proportion of genes in high-variability families were responsive (either up or down-regulated) to the hormone treatments compared with that in low-variability families (fig. 3E). Thus, there are significant differences in hormone-mediated processes across species that contribute to divergence between species. Similarly, a significantly higher proportion of genes in high-variability families were down-regulated in response to stress treatments. In contrast, there was no significant difference in the proportion of up-regulated genes (fig. 3E). Considering that plant stress down-regulated genes tend to be involved in plant growth and development (Zeller et al. 2009), the correlation between down regulation and high family size variability may again reflect differences in developmental processes across species.
Taken together, there are considerable differences in domain family size variation across species. The families with high size variability tend to be those that function in plant–environment interactions, particularly biotic interactions, where the high variability is likely a consequence of an evolutionary arms-race. In contrast, low-variability families tend to have housekeeping roles where strong negative selection has likely contributed to the maintenance of consistent family sizes across species.
Gene Gain and Loss Patterns among Domain Families
Domain family sizes can vary across species due to differences in domain family expansion or contraction rates in different lineages. To evaluate how gene gain and loss events have contributed to the size variation of each domain family, we used a likelihood-based method (BadiRate, see Materials and Methods) to estimate the numbers of gene gain and loss events for internal and external branches in the Solanaceae species tree (fig. 4A;supplementary fig. S2, Supplementary Material online). The estimated average gene turnover (gain or loss) rate (λ) is 3.5e-2 events per gene per Myr, which is ∼25-fold higher than an earlier estimate of λ across Viridiplantae (1.4e-3, including species from green algae to core eudicots spanning ∼725 MY of evolution) (Guo 2013). Because the Solanaceae species we included in our analysis span only ∼26 MY of evolution, one possibility is that the shorter divergence time scale allowed us to better detect fluctuations in λ values that were masked across longer time scales (Demuth and Hahn 2009). However, the Solanaceae λ is also ∼17- to 30-fold higher than the turnover rates among yeast species (λ = 2.0e-3, ∼32 MY) (Hahn et al. 2005), Drosophila species (λ = 1.2e-3, ∼60 MY) (Hahn et al. 2007), and mammals (λ = 1.6e-3, ∼93 MY) (Demuth et al. 2006).
Fig. 4.

—Gene gain and loss events and duplication mechanisms. (A) The median number of gene gain/loss events across all families for each internal and external branch of five gain/loss inference replicate runs (see Materials and Methods) is shown. Results from the five and 100 replicate runs are shown in supplementary figure S4, Supplementary Material online. The numbers are colored based on inferred turnover rate (λ) as shown in the legend at the top-left corner. Green bars: Standard errors for divergence time estimates. (B) Ks distribution of S. lycopersicum duplicates derived from four different duplication mechanisms. Due to the high proportion of recent tandem duplicate genes and possible saturation of Ks, only duplicate genes with Ks values between 0.005 and 3.0 are shown. Gray dashed lines show the fitted distributions of Sol and γ WGD duplicates. Means/standard deviations (μ/σ) of the Sol and γ WGD distributions were 0.66/0.14 and 1.56/0.68, respectively. The ranges of Ks for the Sol and γ WGDs are indicated by green shading, and the cutoff values of Ks (μ ± 1.5σ and μ ± 0.9σ, respectively) are shown. (C) Enrichment of GO terms for S. lycopersicum genes duplicated by WGD, tandem and proximal duplications. Color scale: −log10 (q). Red and blue: Over- and under-representation, respectively.
Because divergence of these groups of species occurred on a similar time-scale as the Solanaceae species, another possibility is that the high Solanaceae λ is the consequence of recent large-scale duplication events. To assess this possibility, we more closely examined two branches with λ values larger than the average value. The first is the branch leading to N. tabacum (λ = 6.2e-1), which is derived from a recent allopolyploidy event, the hybridization of N. tomentosiformis and N. sylvestris (Sierro et al. 2013), and N. tabacum and N. tomentosiformis only diverged ∼0.7 MYA (fig. 4A). The second largest λ (4.0e-2) is on the branch leading to C. annuum var. glabriusculum, which was reported to have rapid amplification of transposable elements (Park et al. 2012; Qin et al. 2014). It is possible that the elevated transposable element activity may have led to more transposable element-mediated gene duplication events (Feschotte and Pritham 2007; Freeling 2009), resulting in a higher positive turnover rate. If these two highest λ values are removed, the average λ is 1.5e-3, similar to the gene turnover rate in Viridiplantae and other eukaryotes. Therefore, recent WGD and, to a lesser extent, transposon-mediated duplication, likely contributed to the significantly higher gene turnover rate among Solanaceae species.
The average λ varied not only between different branches, but also between different domain families. We hypothesized that high-variability domain families would have higher turnover rates. Consistent with this hypothesis, when the two branches leading to N. tabacum and C. annuum var. glabriusculum were excluded, the average λ for each domain family was significantly and positively correlated with the CoV percentile value (ρ = 0.43, P < 2.2e-16). The average λ for high-variability domain families (1.9e-3) is multiple orders of magnitude higher than that for low-variability domain families (3.4e−8). This is also true if the N. tabacum and C. annum species are included (λ=.5e-2 and 1.3e-5 for high- and low-variability families, respectively). These findings indicate that, as expected, higher variability is the result of higher gene turnover.
Influence of Duplication Mechanism on Gene Gains
Gene duplication and pseudogenization are two major factors leading to gene gains and losses, respectively. Because genes duplicated by different mechanisms are retained at different rates, we next assessed the extent to which different duplication mechanisms contributed to gene gains among Solanaceae domain families. To evaluate whether gene duplication mechanisms impact domain family size variation and gene turnover rate, we classified duplicate genes into four categories: 1) Syntenic—duplicates in collinear blocks within a genome, which are likely derived from WGD or segmental duplication, 2) dispersed—duplicates located in unlinked locations but not in collinear blocks, 3) tandem—duplicates immediately adjacent one another, and 4) proximal—duplicates in close proximity but with intervening nonhomologous gene(s) (see Materials and Methods). To determine when these duplication events took place, the synonymous substitution rate (Ks) between duplicates was used as a proxy for duplicate divergence time. In tomato for example, most tandem and proximal duplicate pairs have smaller Ks values than syntenic and dispersed duplicates, indicating they had a relatively more recent origin (fig. 4B). This is also true for the other Solanaceae species (supplementary fig. S3, Supplementary Material online). The syntenic and dispersed duplicates were further divided into two bins, each corresponding to one of two rounds of WGD (Solanaceae-specific [Sol] and γ, see Materials and Methods).
Because gene retention is also influenced by gene functions (Zhang 2003; Hanada et al. 2008; Edger and Pires 2009), we next asked whether genes duplicated through different mechanisms tend to have different functions. For this analysis, we used S. lycopersicum as a representative species because it is the most extensively annotated among our target species. We found that genes duplicated by WGD tend to be involved in translation processes and in nonsense-mediated decay (fig. 4C; supplementary table S9, Supplementary Material online), consistent with findings from earlier studies (Papp et al. 2003; Wu et al. 2008). In contrast, genes duplicated by tandem/proximal duplication tend to function in stress responses and secondary metabolic processes, which is also consistent with analyses of tandem duplicates in other plant species (Rizzon et al. 2006; Hanada et al. 2008).
The relative proportions of duplicates derived from different mechanisms and the patterns of Ks distribution vary greatly across species (supplementary fig. S3, Supplementary Material online). In particular, some species have either very few (e.g., S. melongena) or no syntenic duplicates (e.g., N. tomentosiformis). We found that assembly quality significantly influenced the discovery of syntenic duplicate genes, as genomes with smaller N50s tended to have fewer syntenic duplicates (ρ = 0.817, P = 0.002). With this caveat in mind, we found that genes in 87.7% (2,440), 7.1% (197), and 3.1% (85) of domain families were predominantly duplicated by WGD (includes syntenic and dispersed pairs that have Ks values corresponding to the Sol and γ WGDs; fig. 4B), tandem, and proximal mechanisms, respectively (fig. 5A;supplementary fig. S5 and table S10, Supplementary Material online). Interestingly, 1,254, 113, and 72 families were exclusively duplicated by WGD (e.g., Ribosomal_L5e involved in rRNA binding, Michael and Dreyfuss 1996), tandem (e.g., Sar8_2 involved in the development of systemic acquired resistance, Alexander 1992) and proximal duplication (e.g., Dehydrin involved in response to abiotic stresses, Puhakainen et al. 2004; Saavedra et al. 2006), respectively.
Fig. 5.

—Contribution of duplication mechanism to domain family size variation in 11 Solanaceae species. (A) Proportion of duplicate pairs in each domain family (x-axis) that were predominantly duplicated by WGD, tandem, or proximal mechanisms. (B) Enrichment of members of DNA-binding transcription factor (orange) and enzyme (black) domain families that were duplicated via different mechanisms (the full list of domains and their associated statistics are available in supplementary table S11, Supplementary Material online). Left panel: Domain families that tend to be enriched in WGD duplicates. Right panel: Families that tend to be enriched in tandem/proximal duplicates. (C) Correlation between domain family size variability (represented by CoV percentile) and the proportion of genes duplicated by different duplication mechanisms. The insert shows the enrichment of genes in high- and low-variability domain families duplicated via different mechanisms, tested using Fisher’s exact test. Color scale: −log10 (q). Red and blue: Over- and under-representation, respectively.
We next determined whether members of a family were significantly more likely to be duplicated via a particular mechanism (supplementary table S11, Supplementary Material online). We found that 11 out of 47 plant DNA-binding transcription factor (TF) domain families (as defined in Lehti-Shiu et al. 2017) tended to be duplicated by WGD (all P < 5.1e-06), whereas only 3 TF families (SRF-TF, B3, and AP2) tended to be duplicated by tandem/proximal duplications (all P < 1.2e-06) (orange text, fig. 5B). Additionally, out of 867 domains found in tomato metabolic genes (see Materials and Methods), 18 predominantly primary metabolic enzyme domain families tended to be duplicated by WGD (all P < 5.2e-06), whereas 31 mostly specialized metabolic enzyme families (e.g., UDPGT and 2OG-FeII_Oxy) tended to be duplicated by tandem/proximal duplications (all P < 8.8e-06; fig. 5B;supplementary table S11, Supplementary Material online). These results are consistent with studies postulating that TFs and primary metabolism gene duplicates are likely retained due to dosage balance requirements, whereas secondary metabolism genes have likely expanded lineage-specifically (Rizzon et al. 2006; Birchler and Veitia 2007; Hanada et al. 2008; Freeling 2009; Chae et al. 2014).
We next assessed the contribution of different duplication mechanisms to variation in domain family size. Because tandem/proximal duplications are more likely to be lineage-specific compared with WGDs, we expected and found that genes in high-variability domain families tended to be duplicated by tandem/proximal duplications (cyan and blue lines, fig. 5C). In contrast, the proportion of WGD duplicates is anticorrelated with CoV percentile (red line, fig. 5C). Consistent with these observations, genes in high-variability domain families (above the 95th percentile trend line, fig. 3A) tended to be duplicated by tandem and proximal duplication, whereas genes in low-variability domain families (below the 5th percentile trend line) tended to be duplicated by WGDs (insert, fig. 5C). These patterns highlight the fact that different duplication mechanisms contribute differently to gene gains/losses among Solanaceae domain families. In particular, tandem/proximal duplications are the main contributor to lineage-specific differences. In contrast, the finding that WGD duplicates tend to be enriched in low-variability domain families suggests that these duplicates are consistently either retained or lost postduplication among different lineages.
Relationship between the Timing and Mechanism of Duplication and Selective Pressure
As described in previous sections, variability in gene family size is strongly correlated with duplication mechanism and gene function. We next asked if Solanaceae genes duplicated by different mechanisms have significantly different evolutionary rates based on the ratio of nonsynonymous substitution rate (Ka) to Ks of each duplicate pair. To capture duplication events more thoroughly, the duplicates examined in the 11 Solanaceae species included both annotated genes and pseudogenes, the latter defined based on the presence of premature stops, frameshifts or truncations (see Materials and Methods). Thus, we focused on three types of duplicate pairs: 1) GG: gene–gene, 2) GP: gene–pseudogene, and 3) PP: pseudogene–pseudogene pairs. These pairs were further classified based on the potential duplication mechanism (supplementary fig. S6, Supplementary Material online; fig. 6).
Fig. 6.

—Evolutionary rates of different duplicates in representative species. (A) Gene–gene (GG), gene–pseudogene (GP) and pseudogene–pseudogene (PP) pairs duplicated by different mechanisms (syntenic, dispersed, tandem and proximal) in S. lycopersicum (S.ly). Three high density regions in the S.ly syntenic GG Ka–Ks plot correspond to the Sol, γ and pre-angiosperm WGDs, respectively. (B) Syntenic GG pairs in C. annuum L. zunla-1 (C.zu), C. annuum_var. glabriusculum (C.gl), N. tabacum (N.ta), and N. benthamiana (N.be). Each point in a Ka–Ks dot plot represents a single pair of duplicate sequences, and darker blue denotes a higher density of points. Red lines indicate the expectation under neutral selection, and blue lines connect the median Ka value of each log10 (Ks) bin as shown in supplementary figure S7B and C, Supplementary Material online. The vertical purple dashed line shows the lower boundary (Ks=0.44) for defining duplicates derived from the Sol WGD.
In S. lycopersicum for example, there were significantly fewer syntenic (43.7%) and tandem (43.3%) GP/PP duplicate pairs than GG duplicate pairs (Fisher’s exact tests comparing the numbers of GG and GP/PP pairs, all P < 2.2e-16). In contrast, there were significantly more dispersed (81.6%) and proximal (84%) GP/PP duplicate pairs than GG duplicate pairs (all P < 2.2e-16), which may indicate that dispersed and proximal duplicates are more likely to become pseudogenes, and thus, may be evolving faster. Consistent with this interpretation, syntenic GG pairs had the lowest Ka/Ks values, followed by dispersed, tandem, and proximal GG pairs (Wilcoxon signed-rank test, all P < 6.1e-5, fig. 6A;supplementary fig. S7A, Supplementary Material online). One potential reason why tandem GG pairs had higher Ka/Ks values than dispersed GG pairs is that most tandem GG pairs were duplicated more recently (figs. 4B and 6A), and younger duplicates tend to experience more relaxed selection (Lynch and Conery 2000). For each duplication mechanism, the Ka/Ks values tended to be the lowest for GG pairs, followed by GP and PP pairs. This is expected given that pseudogenes, by definition, were once functional and later became nonfunctional. Thus, after duplication, the pseudogene branch would experience a period of negative selection followed by a period of presumably neutral evolution. As a result, pseudogenes, particularly those that became pseudogenized recently, could have Ka/Ks values similar to those of functional genes. Therefore, the PP pairs with high Ks values but low Ka/Ks ratios (the third column, fig. 6A), likely underwent pseudogenization relatively recently because the signature of past selection remains. Thus, these PP pairs are examples of duplicate pairs that persisted for a long period of time (tens of millions of years) but eventually became pseudogenes.
As expected, a high proportion of syntenic GG pairs are likely derived from WGD (referred to as WGD pairs), and three high density regions in a plot of GG Ka versus Ks values correspond to the Sol, γ and preangiosperm WGDs (fig. 6A). Only 1.4% of syntenic GG pairs had Ks < 0.44 (lower bound for defining the Sol WGD) and are likely derived from recent segmental duplication (referred to as segmental pairs). We also found that the Ka/Ks values of WGD GG pairs (0.17 on average) were significantly lower than those of more recently duplicated, segmental GG pairs (0.47 on average) (Wilcoxon signed-rank test, P = 3.76e-13, fig. 6A;supplementary fig. S7B, Supplementary Material online). These observations indicate that recent segmental GG pairs may evolve faster than WGD GG pairs and tend to become pseudogenes quickly. To rule out the impact of divergence time on the evolutionary rate of duplicate genes (Lynch and Conery 2000), we compared syntenic duplicates in two C. annuum cultivars with a large number of recent, segmental GG pairs (Ks < 0.44) against those of two Nicotiana species (N. tabacum and N. benthamiana) that experienced a recent WGD (fig. 6B;supplementary fig. S7C, Supplementary Material online). We found that C. annuum segmental duplicates had significantly higher Ka/Ks values than Nicotiana WGD duplicates (Wilcoxon signed-rank test, all P < 2.2e-16). Therefore, recent segmental GG pairs have higher Ka/Ks even when divergence time is taken into account. Taken together, these results suggest that genes duplicated by different mechanisms experience distinct selection pressures, consistent with a previous study (Yang and Gaut 2011), which eventually results in different rates of gene retention and loss.
Contribution of Pseudogenization to Variation in Domain Family Size
On average, 20.9% and 52.0% of duplicate pairs in S. lycopersicum are GP and PP pairs, respectively (supplementary fig. S6, Supplementary Material online). This indicates that gene loss occurred frequently. Therefore, we expect that gene loss significantly contributes to variability in domain family sizes among Solanaceae species. Although variability in domain family size is significantly correlated with pseudogene number, the correlation is weak (Spearman’s rank correlation coefficient, ρ = 0.15, P < 2.2e-16, fig. 7A). We also found that species with more genes do not necessarily have more pseudogenes (ρ = 0.1, P = 0.78, fig. 7B). For example, although N. tabacum and N. benthamiana experienced the most recent WGD and have the largest number of protein-coding genes, they do not have the largest number of pseudogenes (fig. 7B). Instead, there is a significant positive correlation between pseudogene number and genome size (ρ = 0.81, P = 0.003, fig. 7C), consistent with the hypothesis that a larger genome size is likely the consequence of less efficient removal and/or more frequent expansion of “nonfunctional” sequences (Lefebure et al. 2017). We also found that larger domain families tend to have more pseudogenes (fig. 7D), indicating that these domain families tend to experience both more frequent gene birth and death events.
Fig. 7.
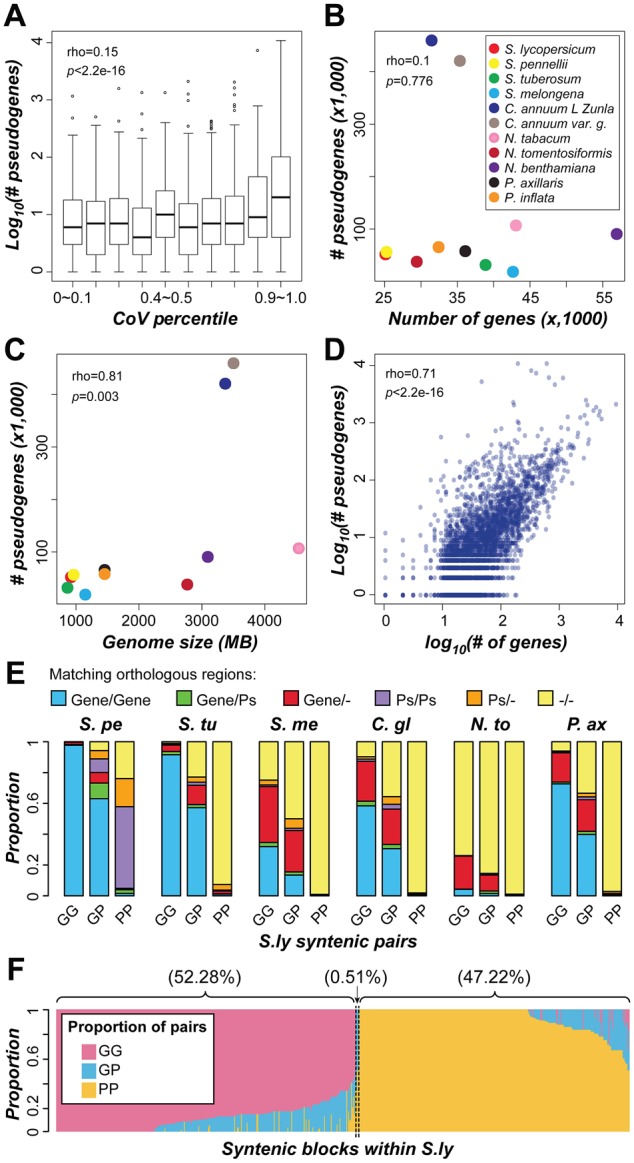
—Contribution of pseudogenization to domain family size variation. (A) Relationship between domain family size variability (CoV percentile) and logarithmic number of pseudogenes. (B) Relationship between the number of genes and pseudogenes among Solanaceae species. (C) Correlation between genome size and number of pseudogenes. (D) Correlation between the logarithmic number of genes and pseudogenes in a domain family. Each dot indicates a domain family. The ρ and P value for Spearman’s rank correlation are shown. (E) Proportion of different S. lycopersicum (S.ly) syntenic duplicate pairs that have orthologous sequences in collinear regions in other species. The orthologous sequences were defined giving priority to protein-coding genes over pseudogenes. If orthologous protein-coding genes were not identified for a given gene, then orthologous pseudogenes were searched for. S.pe, S. pennellii; S.tu, S. tuberosum; S.me, S. melongena; C.gl, C. annuum_var. glabriusculum; N.to, N. tomentosiformis; P.ax, P. axillaris; Ps, pseudogene; “–”, no orthologous sequence was found. (F) Proportion of duplicate pairs in S. lycopersicum collinear blocks that are predominantly (>50%) GG, GP, or PP pairs. Each column represents a pair of collinear blocks within S. lycopersicum. GG, gene–gene pair; GP, gene–pseudogene pair; PP, pseudogene–pseudogene pair.
In the previous section, we discussed PP pairs that are likely derived from WGD events but became pseudogenes independently (PP column, fig. 6A). We also noted the presence of a substantial number of PP pairs derived from more recent duplication events (Ks < 0.44, fig. 6A). These recent PP pairs may be derived from independent pseudogenization of originally functional duplicates or may be duplicates of pseudogenes. To distinguish between these possibilities, we searched for orthologs of S. lycopersicum pseudogenes in six other Solanaceae species. We found that, out of 2,011 recent segmental PP pairs with Ks < 0.44, 1, 885 (93.7%) (fig. 7E) have either pseudogenes as orthologs or no apparent orthologous sequence in the syntenic regions of any Solanaceae species analyzed. This proportion (93.7%) is significantly higher than that observed for GG (0.4%) and GP (13.3%) pairs (Fisher’s exact test, both P < 2.2e-16) and is inconsistent with the expectation that, if both sequences in a PP pair were pseudogenized independently after duplication, the corresponding functional homologs should be found in ≥1 other Solanaceae species. Thus, most recent segmental PP pairs are likely derived from pseudogene duplication, rather than pseudogenization after duplication of functional genes.
The origin of recent segmental PP pairs by duplication is also supported by the high proportion of pseudogenes in collinear, duplicated blocks within species. Among 593 pairs of collinear blocks in S. lycopersicum, 310 (52.28%) and 280 (47.22%) have predominantly GG and PP pairs, respectively (fig. 7F), which is significantly deviated from the random expectation (z-scores 7.4 and 63.4, respectively; P values <6.8e-14). Thus, collinear blocks tend to contain either GG or PP pairs. This pattern supports the notion that pseudogenes in these blocks are products of pseudogene duplication because, to explain this pattern based on independent pseudogenization of functional ancestral genes, a large number of additional, independent loss events would be required. The observation that recent segmental PP pairs tend to be located in regions with a high density of repeats (supplementary fig. S8, Supplementary Material online; ρ =0.14, P = 1.54e-08) suggests that these duplicates may be derived from repeat-mediated duplication mechanisms.
Conclusions
Genomes of an increasing number of plant species have been sequenced, facilitating comparative studies aimed at evaluating genome and gene content evolution among related plant species and, specifically in this study, identifying factors contributing to gene family size variation. Here, we used domain family as a proxy for gene family, and an unintended consequence of this practice was that genes with more protein domains contributed to more data points in the analysis. With this caveat in mind, we show that the distribution of domain families across Solanaceae species varies due to lineage-specific gains or losses and that different duplication mechanisms have contributed to domain family size variation. Genes in domain families with higher variability are more likely to have been duplicated by tandem duplication. Most of the observed tandem duplicates were duplicated recently and tend to be involved in processes that are highly diverse among Solanaceae species, for example, secondary metabolism (Chowański et al. 2016) and fruit ripening (Knapp 2002). Genes duplicated though different mechanisms also have different evolutionary rates. For example, tandem and recent segmental duplicate genes experience more relaxed selection than WGD duplicate genes. Taken together, these findings suggest that lineage-specific gene family expansion through tandem duplication plays an important role in the evolution of organisms and diversification among closely related species. Comparative evolutionary and functional analysis (e.g., of gene structures and expression patterns) of new tandem or segmental duplicate genes and ancestral genes, may help to uncover genetic changes underlying lineage-specific innovations.
The abundance of pseudogenes is often used to estimate the extent to which gene loss has impacted gene family size (Demuth and Hahn 2009). However, we found that pseudogenes are also frequently duplicated and remain readily detectable just like functional genes, indicating that the number of pseudogenes is not an accurate proxy for gene loss. Pseudogene duplication may happen randomly, producing duplicates that are not under selection. Alternatively, some pseudogene duplicates may be retained due to their effects on, for example, regulation of their protein-coding relatives (Pink et al. 2011). Further studies will be necessary to distinguish between these possibilities. We found that recent segmental PP pairs are closely associated with repeat sequences. It remains to be determined whether these recent segmental PP duplications in Solanaceae were produced by a recombination-like transposable element-mediated mechanism, as in humans (Zhou and Mishra 2005), or by another yet to be discovered mechanism.
Supplementary Material
Acknowledgments
We thank John P. Lloyd and Christina B. Azodi for helpful discussions. This work was supported by National Science Foundation IOS-1546617 and DEB-1655386 to S.-H.S.
Literature Cited
- Albalat R, Canestro C. 2016. Evolution by gene loss. Nat Rev Genet. 17:379–391. [DOI] [PubMed] [Google Scholar]
- Alexander D. 1992. A new multigene family inducible by tobacco mosaic virus or salicylic acid in tobacco. Mol Plant Microbe Interact. 5(6):513–515. [DOI] [PubMed] [Google Scholar]
- Alkan N, et al. 2015. Simultaneous transcriptome analysis of Colletotrichum gloeosporioides and tomato fruit pathosystem reveals novel fungal pathogenicity and fruit defense strategies. New Phytol. 205(2):801–815. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Andrews S. 2010. Fast QC. A quality control tool for high throughput sequence data In: Reference Source. Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300. [Google Scholar]
- Birchler JA, Veitia RA.. 2007. The gene balance hypothesis: from classical genetics to modern genomics. Plant Cell 19(2):395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA.. 2014. The gene balance hypothesis: dosage effects in plants. Methods Mol Biol. 1112:25–32. [DOI] [PubMed] [Google Scholar]
- Bombarely A, et al. 2016. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat Plants 2(6):16074. [DOI] [PubMed] [Google Scholar]
- Bombarely A, et al. 2012. A draft genome sequence of Nicotiana benthamiana to enhance molecular plant–microbe biology research. Mol Plant Microbe Interact. 25(12):1523–1530. [DOI] [PubMed] [Google Scholar]
- Bolger AM, et al. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius J. 1991. Retroposons–seeds of evolution. Science 251(4995):753.. [DOI] [PubMed] [Google Scholar]
- Campbell MS, et al. 2014. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164(2):513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SB, et al. 2004. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S, et al. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25(15):1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capua Y, Eshed Y.. 2017. Coordination of auxin-triggered leaf initiation by tomato LEAFLESS. Proc Natl Acad Sci U S A. 114(12):3246–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero-Paulet L, et al. 2015. High gene family turnover rates and gene space adaptation in the compact genome of the carnivorous plant Utricularia gibba. Mol Biol Evol. 32(5):1284–1295. [DOI] [PubMed] [Google Scholar]
- Chae L, et al. 2014. Genomic signatures of specialized metabolism in plants. Science 344(6183):510–513. [DOI] [PubMed] [Google Scholar]
- Chen T, et al. 2013. Comparative transcriptome profiling of a resistant vs. susceptible tomato (Solanum lycopersicum) cultivar in response to infection by tomato yellow leaf curl virus. PLoS One 8(11):e80816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, et al. 2015. A comparison of the low temperature transcriptomes of two tomato genotypes that differ in freezing tolerance: Solanum lycopersicum and Solanum habrochaites. BMC Plant Biol. 15:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowański S, et al. 2016. A review of bioinsecticidal activity of Solanaceae alkaloids. Toxins 8(3):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Carmona A, et al. 2000. Aux/IAA proteins are phosphorylated by phytochrome in vitro. Plant Physiol. 124(4):1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S.. 2008. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008:619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constabel CP, et al. 1998. Prosystemin from potato, black nightshade, and bell pepper: primary structure and biological activity of predicted systemin polypeptides. Plant Mol Biol. 36(1):55–62. [DOI] [PubMed] [Google Scholar]
- De Oliveira AS, et al. 2016. Cell death triggering and effector recognition by Sw-5 SD-CNL proteins from resistant and susceptible tomato isolines to tomato spotted wilt virus. Mol Plant Pathol. 17(9):1442–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delteil A, et al. 2016. Several wall-associated kinases participate positively and negatively in basal defense against rice blast fungus. BMC Plant Biol. 16:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuth JP, Hahn MW.. 2009. The life and death of gene families. BioEssays 31(1):29–39. [DOI] [PubMed] [Google Scholar]
- Demuth JP, et al. 2006. The evolution of mammalian gene families. PLoS One 1:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, et al. 2015. Comparative transcriptome analysis of resistant and susceptible tomato lines in response to infection by Xanthomonas perforans race T3. Front Plant Sci. 6:1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger PP, Pires JC.. 2009. Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 17(5):699–717. [DOI] [PubMed] [Google Scholar]
- Enright AJ, et al. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30(7):1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, et al. 2016. In vitro reconstruction and analysis of evolutionary variation of the tomato acylsucrose metabolic network. Proc Natl Acad Sci U S A. 113(2):E239–E248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow SC, Facchini PJ.. 2014. Functional diversity of 2-oxoglutarate/Fe(II)-dependent dioxygenases in plant metabolism. Front Plant Sci. 5:524.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ.. 2007. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 41:331–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39(Web Server issue):W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. 2014. Pfam: the protein families database. Nucleic Acids Res. 42(Database issue):D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagel LE, Wendel JF.. 2009. Gene duplication and evolutionary novelty in plants. New Phytol. 183(3):557–564. [DOI] [PubMed] [Google Scholar]
- Force A, et al. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151(4):1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkostefanakis S, et al. 2016. HsfA2 controls the activity of developmentally and stress-regulated heat stress protection mechanisms in tomato male reproductive tissues. Plant Physiol. 170(4):2461–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M. 2009. Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol. 60:433–453. [DOI] [PubMed] [Google Scholar]
- Guo YL. 2013. Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant J. 73(6):941–951. [DOI] [PubMed] [Google Scholar]
- Gupta S, et al. 2013. Transcriptome profiling of cytokinin and auxin regulation in tomato root. J Exp Bot. 64(2):695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, et al. 2005. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 15(8):1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, et al. 2007. Gene family evolution across 12 Drosophila genomes. PLoS Genet. 3(11):e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday KJ, et al. 2009. Integration of light and auxin signaling. Cold Spring Harb Perspect Biol. 1(6):a001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, et al. 2008. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol. 148(2):993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakawa H, et al. 2014. Draft genome sequence of eggplant (Solanum melongena L.): the representative Solanum species indigenous to the old world. DNA Res. 21(6):649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino A, et al. 2016. Genome sequence and analysis of the Japanese morning glory Ipomoea nil. Nat Commun. 7:13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JY, Saedler H.. 2007. Evolution of the inflated calyx syndrome in Solanaceae. Mol Biol Evol. 24(11):2443–2453. [DOI] [PubMed] [Google Scholar]
- Inoue J, et al. 2015. Rapid genome reshaping by multiple-gene loss after whole-genome duplication in teleost fish suggested by mathematical modeling. Proc Natl Acad Sci U S A. 112(48):14918–14923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai Y, et al. 2014. Evolution and diversity of the 2-oxoglutarate-dependent dioxygenase superfamily in plants. Plant J. 78(2):328–343. [DOI] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT–the BLAST-like alignment tool. Genome Res. 12(4):656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, et al. 2014. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet. 46(3):270–278. [DOI] [PubMed] [Google Scholar]
- Knapp S. 2002. Tobacco to tomatoes: a phylogenetic perspective on fruit diversity in the Solanaceae. J Exp Bot. 53(377):2001–2022. [DOI] [PubMed] [Google Scholar]
- Larkin MA, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948. [DOI] [PubMed] [Google Scholar]
- Lefebure T, et al. 2017. Less effective selection leads to larger genomes. Genome Res. 27(6):1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehti-Shiu MD, Shiu SH.. 2012. Diversity, classification and function of the plant protein kinase superfamily. Philos Trans R Soc Lond B Biol Sci. 367(1602):2619–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehti-Shiu MD, et al. 2017. Diversity, expansion, and evolutionary novelty of plant DNA-binding transcription factor families. Biochim Biophys Acta 1860(1):3–20. [DOI] [PubMed] [Google Scholar]
- Leitch IJ, et al. 2008. The ups and downs of genome size evolution in polyploid species of Nicotiana (Solanaceae). Ann Bot. 101(6):805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, et al. 2012. BadiRate: estimating family turnover rates by likelihood-based methods. Bioinformatics 28(2):279–281. [DOI] [PubMed] [Google Scholar]
- Livne S, et al. 2015. Uncovering DELLA-independent gibberellin responses by characterizing new tomato procera mutants. Plant Cell 27(6):1579–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loraine AE, et al. 2015. Analysis and visualization of RNA-seq expression data using RStudio, Bioconductor, and Integrated Genome Browser. Methods Mol Biol. 1284:481–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS.. 2000. The evolutionary fate and consequences of duplicate genes. Science 290(5494):1151–1155. [DOI] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR.. 2017. Mesquite: a modular system for evolutionary analysis. Version 3.2. Available from: http://mesquiteproject.org
- Michael WM, Dreyfuss G.. 1996. Distinct domains in ribosomal protein L5 mediate 5 S rRNA binding and nucleolar localization. J Biol Chem. 271(19):11571–11574. [DOI] [PubMed] [Google Scholar]
- Nakazato T, et al. 2010. Ecological and geographic modes of species divergence in wild tomatoes. Am J Bot. 97(4):680–693. [DOI] [PubMed] [Google Scholar]
- Nash JC. 2014. Nonlinear parameter optimization using R tools. Chichester (United Kingdom: ): John Wiley & Sons Ltd. [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. New York, USA.: Springer-Verlag. [Google Scholar]
- Pál C, et al. 2006. Chance and necessity in the evolution of minimal metabolic networks. Nature 440(7084):667–670. [DOI] [PubMed] [Google Scholar]
- Panchy N, et al. 2016. Evolution of gene duplication in plants. Plant Physiol. 171(4):2294–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, et al. 2003. Dosage sensitivity and the evolution of gene families in yeast. Nature 424(6945):194–197. [DOI] [PubMed] [Google Scholar]
- Park M, et al. 2012. Evolution of the large genome in Capsicum annuum occurred through accumulation of single-type long terminal repeat retrotransposons and their derivatives. Plant J. 69(6):1018–1029. [DOI] [PubMed] [Google Scholar]
- Pedmale UV, et al. 2010. Phototropism: mechanism and outcomes. Arabidopsis Book 8:e0125.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers HW. 1971. Likelihood ratio and associated test criteria. Biometrika 58(3):577. [Google Scholar]
- Pink RC, et al. 2011. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA 17(5):792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo MA, et al. 2014. Transcriptomic analysis reveals tomato genes whose expression is induced specifically during effector-triggered immunity and identifies the Epk1 protein kinase which is required for the host response to three bacterial effector proteins. Genome Biol. 15(10):492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo MA, et al. 2017. Use of RNA-seq data to identify and validate RT-qPCR reference genes for studying the tomato-Pseudomonas pathosystem. Sci Rep. 7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potato Genome Sequencing Consortium, et al. 2011. Genome sequence and analysis of the tuber crop potato. Nature 475:189–195. [DOI] [PubMed] [Google Scholar]
- Puhakainen T, et al. 2004. Overexpression of multiple dehydrin genes enhances tolerance to freezing stress in Arabidopsis. Plant Mol Biol. 54(5):743–753. [DOI] [PubMed] [Google Scholar]
- Qin C, et al. 2014. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc Natl Acad Sci U S A. 111(14):5135–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JW. 2001. Roles and activities of Aux/IAA proteins in Arabidopsis. Trends Plant Sci. 6(9):420–425. [DOI] [PubMed] [Google Scholar]
- Rizzon C, et al. 2006. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput Biol. 2(9):e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, et al. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, et al. 2017. Tandem duplications lead to novel expression patterns through exon shuffling in Drosophila yakuba. PLoS Genet. 13(5):e1006795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosli HG, et al. 2013. Transcriptomics-based screen for genes induced by flagellin and repressed by pathogen effectors identifies a cell wall-associated kinase involved in plant immunity. Genome Biol. 14(12):R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, et al. 2000. Comparative genomics of the eukaryotes. Science 287(5461):2204–2215., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CA, Pearce G.. 2003. Systemins: a functionally defined family of peptide signal that regulate defensive genes in Solanaceae species. Proc Natl Acad Sci U S A. 100(Suppl 2):14577–14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra L, et al. 2006. A dehydrin gene in Physcomitrella patens is required for salt and osmotic stress tolerance. Plant J. 45(2):237–249. [DOI] [PubMed] [Google Scholar]
- Samonte RV, Eichler EE.. 2002. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 3(1):65–72. [DOI] [PubMed] [Google Scholar]
- Sankoff D, et al. 2010. The collapse of gene complement following whole genome duplication. BMC Genomics 11:313.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, et al. 2017. Integrated miRNA and mRNA expression profiling reveals the response regulators of a susceptible tomato cultivar to early blight disease. DNA Res. 24(3):235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Särkinen T, et al. 2013. A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol Biol. 13:214.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Hahn MW.. 2010. Gene copy-number polymorphism in nature. Proc R Soc B 277(1698):3213–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, et al. 2013. Transcriptome analysis of cytokinin response in tomato leaves. PLoS One 8(1):e55090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro N, et al. 2013. Reference genomes and transcriptomes of Nicotiana sylvestris and Nicotiana tomentosiformis. Genome Biol. 14(6):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PP, et al. 2015. Identification of ohnolog genes originating from whole genome duplication in early vertebrates, based on synteny comparison across multiple genomes. PLoS Comput Biol. 11(7):e1004394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, et al. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdighian S, et al. 2017. Reciprocally retained genes in the angiosperm lineage show the hallmarks of dosage balance sensitivity. Plant Cell 29(11):2766–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ.. 1997. A genomic perspective on protein families. Science 278(5338):631–637. [DOI] [PubMed] [Google Scholar]
- Tomato Genome Consortium 2012. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28(5):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Bossche R, et al. 2013. Transient expression assays in tobacco protoplasts. Methods Mol Biol. 1011:227–239. [DOI] [PubMed] [Google Scholar]
- Vekemans D, et al. 2012. Gamma paleohexaploidy in the stem lineage of core eudicots: significance for MADS-box gene and species diversification. Mol Biol Evol. 29(12):3793–3806. [DOI] [PubMed] [Google Scholar]
- Verberne MC, et al. 2000. Overproduction of salicylic acid in plants by bacterial transgenes enhances pathogen resistance. Nat Biotechnol. 18(7):779–783. [DOI] [PubMed] [Google Scholar]
- Wagner TA, Kohorn BD.. 2001. Wall-associated kinases are expressed throughout plant development and are required for cell expansion. Plant Cell 13(2):303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li J, Paterson AH. 2013. MCScanX-transposed: detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics 29(11):1458–1460. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tao X, et al. 2013. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genomics 14(1):841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, et al. 2014. Genome sequencing of the high oil crop sesame provides insight into oil biosynthesis. Genome Biol. 15(2):R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, et al. 1989. Rates of synonymous substitution in plant nuclear genes. J Mol Evol. 29(3):208–211. [Google Scholar]
- Woodhouse MR, et al. 2010. Transposed genes in Arabidopsis are often associated with flanking repeats. PLoS Genet. 6(5):e1000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley JN, et al. 2016. A novel method of transcriptome interpretation reveals a quantitative suppressive effect on tomato immune signaling by two domains in a single pathogen effector protein. BMC Genomics 17(1):229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, et al. 2015. Retained duplicate genes in green alga Chlamydomonas reinhardtii tend to be stress responsive and experience frequent response gains. BMC Genomics 16:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, et al. 2008. The preferential retention of starch synthesis genes reveals the impact of whole-genome duplication on grass evolution. Mol Biol Evol. 25(6):1003–1006. [DOI] [PubMed] [Google Scholar]
- Yang L, Gaut BS.. 2011. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol Biol Evol. 28(8):2359–2369. [DOI] [PubMed] [Google Scholar]
- Yang Y-X, et al. 2015. RNA-seq analysis reveals the role of red light in resistance against Pseudomonas syringae pv. tomato DC3000 in tomato plants. BMC Genomics 16:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
- Zeller G, et al. 2009. Stress-induced changes in the Arabidopsis thaliana transcriptome analyzed using whole-genome tiling arrays. Plant J. 58(6):1068–1082. [DOI] [PubMed] [Google Scholar]
- Zhang JZ. 2003. Evolution by gene duplication: an update. Trends Ecol Evol. 18(6):292–298. [Google Scholar]
- Zheng Y, et al. 2017. Comprehensive transcriptome analyses reveal that potato spindle tuber viroid triggers genome-wide changes in alternative splicing, inducible trans-acting activity of phased secondary small interfering RNAs, and immune responses. J Virol. 91(11):e00247–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Mishra B.. 2005. Quantifying the mechanisms for segmental duplications in mammalian genomes by statistical analysis and modeling. Proc Natl Acad Sci U S A. 102(11):4051–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Żmieńko A, et al. 2014. Copy number polymorphism in plant genomes. Theor Appl Genet. 127(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, et al. 2009. Evolutionary and expression signatures of pseudogenes in Arabidopsis and rice. Plant Physiol. 151(1):3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.