

Abstract
Adenosine‐to‐inosine (A‐to‐I) microRNA editing is associated with tumor phenotypes in various cancer types. Recent analyses of The Cancer Genome Atlas (TCGA) dataset have shown several microRNAs that undergo A‐to‐I editing in human cancers, some of which have been reported to be associated with prognosis. Herein, we examined published small RNA deep sequencing data of 74 cases of lung adenocarcinoma (AD) and the corresponding normal counterpart (NC) specimen in silico in order to identify A‐to‐I microRNA editing events. Editing levels of miR‐379‐5p, miR‐99a‐5p, and miR‐497‐5p were lower in AD than in NC and, in a large number of cases, the editing level of miR‐200b‐3p was higher in AD than in NC. Difference in the editing level between AD and NC was largest for miR‐99a‐5p. Then, we examined the editing level of miR‐99a‐5p in 50 surgically resected lung adenocarcinoma cases at our institution by a conventional sequence‐based method, and its association with clinical outcomes. The editing level of miR‐99a‐5p was significantly lower in 19 cases of AD (38%) than in corresponding NC. These cases showed a shorter overall survival as assessed using the log‐rank test (P = .047). This trend was consistent with previous analyses of TCGA dataset. The altered editing level of microRNAs in lung adenocarcinoma could serve as a potential biomarker.
Keywords: biomarker, computational biology, lung adenocarcinoma, microRNA, RNA editing
1. INTRODUCTION
Lung cancer is the leading cause of cancer‐related death worldwide.1 Adenocarcinoma, which is a category of non‐small‐cell lung cancer (NSCLC), is the most common histological type among all lung cancer cases.2 Treatment outcome of unresectable or recurrent NSCLC has been improving with the introduction of novel molecular‐targeted drugs3, 4 and immune checkpoint inhibitors;5, 6, 7 nevertheless, these epoch‐making drugs do not cure NSCLC. In the American Society of Clinical Oncology (ASCO) guidelines for completely resected NSCLC, adjuvant chemotherapy is recommended depending on the pathological stage alone.8 To pursue individualized treatments, a novel biomarker for predicting the clinical outcome after surgery is desired.
Adenosine‐to‐inosine (A‐to‐I) RNA editing is a posttranscriptional modification of specific RNAs, catalyzed by proteins of the adenosine deaminases acting on the RNA (ADAR) family, ADAR1, and ADAR2.9 As inosine base‐pairs with cytosine in double strand nucleotides, A‐to‐I editing is functionally equivalent to an A‐to‐G mutation. The editing level largely varies among organs: The most frequently edited is brain mRNA, whereas lung mRNA is the second most frequently edited.10, 11 Since 2004, when primary microRNAs, precursors of microRNAs were first demonstrated to undergo A‐to‐I editing,12 some studies have shown the role of microRNA editing in normal organs13, 14 and various types of cancers.15, 16, 17 MicroRNAs are single‐stranded RNA molecules that are approximately 22 nucleotides in length and function as posttranscriptional gene regulators. They bind to the 3′ UTR of mRNAs which contain a sequence complementary to the seed sequence (positions 2‐8) on the microRNAs, resulting in degradation or downregulation of the target mRNAs.18 As a consequence of microRNA editing, altered base complementation of a primary microRNA molecule affects the cleavage activity of microRNA processing proteins, DROSHA and DICER.19, 20 Moreover, when the seed sequence of a microRNA undergoes RNA editing, the edited microRNA targets another set of mRNAs. Therefore, the alteration in the editing levels of microRNAs affects the phenotype of a cancer through the alterations of the gene expression profile.13 Recently, in silico analyses of small RNA deep sequencing data of The Cancer Genome Atlas (TCGA) dataset showed the comprehensive microRNA editing profile of 32 types of cancer.21, 22 In the cases of lung adenocarcinoma, significant differences in the editing levels of 5 microRNAs (miR‐200b‐3p, miR‐379‐5p, miR‐411‐5p, miR‐497‐5p, and miR‐99a‐5p) were found between adenocarcinoma (AD) samples and the corresponding normal counterpart (NC) samples, and correlations were found between the editing levels of 4 of these microRNAs (miR‐376c‐3p, miR‐379‐5p, miR‐381‐3p, and miR‐99a‐5p) in AD and the survival of the patients.
In the present study, we analyzed the published small RNA deep sequencing data of 74 cases of lung adenocarcinoma, including the data of both AD and NC in silico, and detected 3 microRNAs showing loss of editing in AD (the editing level was significantly lower in AD than in NC) and 1 microRNA showing gain of editing in AD (the editing level was significantly higher in AD than in NC). Regarding miR‐99a‐5p, which showed the largest difference of the editing level between AD and NC, we set up a conventional sequence‐based method to quantify the editing levels. We examined surgically resected specimens from 50 cases of lung adenocarcinoma at our institution to detect differences of the editing level of miR‐99a‐5p between AD and NC. In our dataset, loss of editing of miR‐99a‐5p was significantly associated with shorter overall survival (OS), which was consistent with the analyses of TCGA dataset.
2. MATERIALS AND METHODS
2.1. In silico analysis of deep sequencing data
We searched the Sequence Read Archives (SRA) at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/sra) using a search phrase, “miRNA lung adenocarcinoma.” We included paired data of lung adenocarcinomas and the corresponding normal tissues published before March 2015.
The downloaded SRA files were analyzed using perl scripts as previously described.23, 24 In short, 5′ and 3′ adaptors were removed by Process_reads.pl. The reads were mapped to the human reference genome (hg38) using Bowtie.25, 26 Here, we trimmed 2 bases at the 3′ end that undergo massive modifications.27 We allowed a maximum of 1 mismatch per read with a quality score of at least 30, best unique alignment, and no cross‐mapping. Then, the reads were remapped to the pre‐microRNA sequence database by Analyze_mutation.pl, to count the read depth and the number of reads with mismatch. The original script used release 18 from miRBase (http://www.mirbase.org) as reference. Instead, we used the latest version, release 21. The data format differs slightly according to versions; revised scripts are shown in Docs S1 and S2, and reference files are shown in Docs [Link], [Link], [Link]. Finally, sequencing errors were removed for each position of each pre‐microRNA by Binomial_analysis.pl. Output data included name of the pre‐microRNA, mismatch position, mismatch type (such as C‐to‐U), number of mismatched reads, total number of reads in the position, and P‐value with Benjamini‐Hochberg correction with a false discovery rate (FDR) of 0.05. Known single‐nucleotide polymorphisms (SNP) were discarded by reference to miRNASNP Release 2.0 (http://bioinfo.life.hust.edu.cn/miRNASNP2/).
2.2. Patient population and collection of samples
The study protocol was approved by the Institutional Ethical Review Board at the University of Tokyo Hospital (Tokyo, Japan), and written informed consent was obtained from each patient. Samples were obtained from patients who underwent lung resection for primary lung adenocarcinoma at the University of Tokyo Hospital between August 2007 to September 2011. We included all 27 patients with pathological stage IIA to IIIA disease and consecutive 23 patients with pathological stage IB disease. Pathological stage was defined according to the seventh edition of the TNM classification by the UICC. Recurrence‐free survival (RFS) was defined as the period from the date of resection for the lung cancer to the date of diagnosis of disease recurrence or death from any cause. OS was defined as the period from the date of resection to the date of death from any cause. Characteristics of the 50 patients are listed in Table 1.
Table 1.
Characteristics of 50 patients from University of Tokyo hospital enrolled in the present study
| Total | Recurrence | Death | |||
|---|---|---|---|---|---|
| n = 50 | n = 22 | P‐value | n = 11 | P‐value | |
| Age, y, mean (range) | 68.6 (48‐83) | 66.9 | 0.161 | 67.8 | 0.728 |
| Gender | |||||
| Male | 28 | 11 | 0.569 | 6 | 1 |
| Female | 22 | 11 | 5 | ||
| Smoking | |||||
| (+) | 23 | 10 | 1 | 5 | 1 |
| (−) | 27 | 12 | 6 | ||
| Stage | |||||
| IB | 23 | 6 | 0.024 | 4 | 0.515 |
| IIA | 7 | 3 | 2 | ||
| IIB | 9 | 4 | 1 | ||
| IIIA | 11 | 9 | 4 | ||
| EGFR mutation | |||||
| (+) | 13 | 8 | 0.51 | 3 | 1 |
| (−) | 30 | 14 | 6 | ||
| Adjuvant chemotherapy | |||||
| (+) | 28 | 14 | 0.398 | 5 | 1 |
| (−) | 22 | 8 | 4 | ||
EGFR, epidermal growth factor receptor.
2.3. Total RNA extraction from tissue
Fresh tissue specimens were immersed into 500 μL RNAlater solution (Ambion, Austin, TX, USA) overnight at 4°C. After removing the RNAlater solution, tissue specimens were frozen and stored at −20°C. Finely divided tissue pieces were placed in the ceramic beads tube of the Precellys Lysing Kit (Bertin Technologies, Saint‐Quentin en Yveline, France) containing 1000 μL RNAiso plus (TaKaRa, Shiga, Japan), and homogenized twice using Precellys24 (Bertin Technologies) at the oscillation frequency of 6500 per minute for 23 seconds each. The homogenized solution was centrifuged at 20 000 × g for 5 minutes and total RNA in the supernatant was extracted with chloroform, precipitated with isopropanol and eluted with 20 μL DEPC (diethylpyrocarbonate)‐treated water (Wako Pure Chemical Industries, Osaka, Japan).
2.4. Quantification of the editing level of miR‐99a‐5p and validation of the method
An overview of this original method is shown in Figure 1. First‐strand cDNA was synthesized from 5 ng total RNA using the TaqMan Advanced miRNA cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA), in accordance with the manufacturer's instructions.28 Then, 2 μL cDNA was amplified using 50 μL AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific) and primers at a final concentration of 400 nmol/L (F, 5′‐GAT CGA ATT CCA TCT CAT CCC TGC GTG TCT‐3′; R, 5′‐GAT CGA ATT CTT TTT CAC AAG ATC GGA TCT AC‐3′). Thermal cycling conditions consisted of preheating at 95°C for 10 minutes, followed by 37‐40 cycles (optimized for each sample) at 95°C for 15 seconds, and 60°C for 60 seconds. The PCR product was purified using Wizard SV Gel and PCR Clean‐Up System (Promega, Madison, WI, USA), in accordance with the manufacturer's instructions. The eluate was digested with EcoRI‐HF (New England Biolabs, Ipswich, MA, USA) at 37°C for 120 minutes, followed by heat inactivation at 65°C for 20 minutes. Then, T4 DNA ligase (New England Biolabs) was added, followed by incubation at room temperature for 30 minutes to concatenate the amplicons.29 Formation of a ladder with intervals of 73 base pairs was confirmed by electrophoresis. Taq DNA Polymerase (New England Biolabs), dATP, and dTTP were added, followed by incubation at 70°C for 20 minutes. The solution was loaded onto 1.2% Low Melting Agar (PH Japan, Hiroshima, Japan) with 1% Gel Indicator (BioDynamics Laboratory, Tokyo, Japan), and electrophoresed for 30 minutes. The ladder between approximately 300‐700 base pairs was cut out, and melted at 65°C. β‐Agarase (New England Biolabs) was added, followed by incubation at 42°C for 60 minutes. The solution was salted out, precipitated with ethanol, and eluted with nuclease‐free water. The concatenated amplicons were ligated into the pGEM‐T Easy Vector System (Promega), and clones with inserts of the expected length were sequenced using BigDye Terminator v3.1 (Thermo Fisher Scientific) with a primer on the vector, 5′‐GCC AAG CTA TTT AGG TGA C‐3′. We repeated the process of cloning and sequencing until we obtained in excess of 100 amplicons for each sample. The editing level in each sample was calculated as the number of edited reads divided by total reads.
Figure 1.
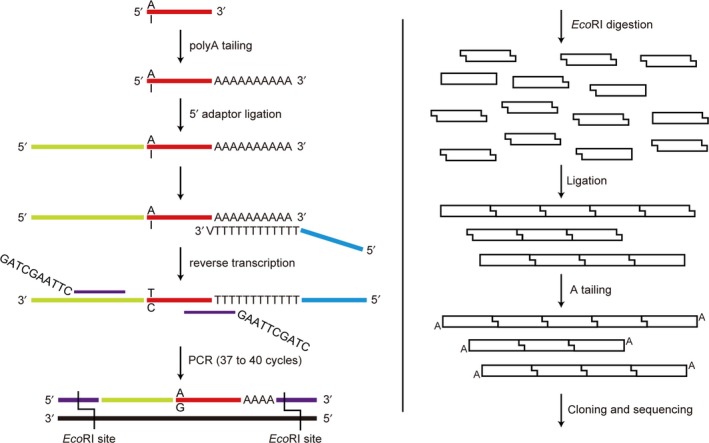
Schema of the method for quantifying the editing level of miR‐99a‐5p. Position 1 of miR‐99a‐5p is “A” in the absence of editing, and “I” with editing. First‐strand cDNA carries “T” and “C”, respectively. The proportion of edited miR‐99a‐5p is assumed to be constant before and after PCR and ligation. “GAATTC” in the PCR primer is the EcoRI site
To confirm that the proportion of edited reads reflected the editing level of miR‐99a‐5p, the unedited amplicon which consists of the 5′ adaptor, unedited miR‐99a‐5p and polyA was ligated into the pGEM‐T Easy Vector System (Promega). The edited amplicon was ligated similarly. Edited and unedited plasmids were mixed, so that the proportion of the copy number of the edited plasmids was 5%, 15%, and 25%. These 3 templates were amplified, concatenated, and sequenced by the same method described above until amplicons in excess of 100 were obtained for each template.
2.5. Quantitative RT‐PCR
For mRNA analysis, 1 μg total RNA treated with DNase I (Invitrogen, Carlsbad, CA, USA) was reverse transcribed with Superscript III (Invitrogen) using random hexamer primers, in accordance with the manufacturer's instructions. The cDNA was amplified with 10 μL Thunderbird SYBR qPCR Mix (Toyobo, Tokyo, Japan) and primers at a final concentration of 400 nmol/L, using the 7500 Fast Real‐Time PCR System (Applied Biosystems, Carlsbad, CA, USA). Thermal cycling conditions consisted of preheating of 95°C for 60 seconds, followed by 40 cycles at 95°C for 15 seconds and 60°C for 60 seconds. Specificity of the PCR was confirmed by analyzing the melting curves. PCR was carried out in triplicate, and mean Ct values of 3 samples were used for the analysis. Relative expression level of each gene was determined after normalization to the expression level of GAPDH. For survival analysis, the cutoff values for high or low expression level were determined using receiver operating characteristic (ROC) curves.30 The following primer sets were used: ADAR1 (F, 5′‐ACC GGT GCT TCA ACA CTC TGA CTA‐3′; R, 5′‐CGG GAG ATT TCT GCA TGG‐3′), ADAR2 (F, 5′‐TGT CAA CTG GAC GGT AGG CGA CT‐3′; R, 5′‐TGC CGC CAG CTT GGA CTC AT‐3′), and GAPDH (F, 5′‐CAC CAC CAA TGC TTA GCA C‐3′; R, 5′‐TGG CAG GTT TTT CTA GAC GG‐3′).
2.6. Statistical analysis
Student's t test (age) and Fisher's exact test (the others) were used to compare patient characteristics between the two groups. Statistical significance of the difference in the editing level between AD and NC was evaluated using the chi‐squared test with Benjamini‐Hochberg correction with a FDR of 0.05 (in silico analysis) and Fisher's exact test (conventional sequence‐based analysis). Survival analyses were carried out using the Kaplan‐Meier method, and survival curves were compared using the log‐rank test and a Cox proportional‐hazards model. Correlation between the editing level of each sample and the relative expression level of each gene (−ΔCt value) was evaluated using Spearman's rank‐order correlation test. All statistical analyses outside the perl scripts were carried out using IBM SPSS Statistics version 24 (IBM SPSS, Chicago, IL, USA). All the tests in this study were two sided, and the statistical significance level was set at P < .05.
3. RESULTS
3.1. In silico analysis of deep sequencing data
We downloaded the SRA files of 94 cases (188 samples) as candidates for analysis. They were from a single study which examined the relationship between smoking history and alteration of microRNA expression level in lung adenocarcinoma.31 Twenty samples had an average spot length of 30 bases, and further analysis showed that these samples had extremely low alignment proportions of approximately 1% (mapped reads divided by total reads). Therefore, these 20 samples and corresponding tissue samples were excluded, and the remaining 148 samples were analyzed. Profiles of the 74 patients are shown in Table 2.
Table 2.
Characteristics of the 74 patients in the published database
| n = 74 | Proportion, % | |
|---|---|---|
| Gender | ||
| Male | 20 | 27 |
| Female | 54 | 73 |
| Smoking | ||
| Current | 38 | 51 |
| Former | 20 | 27 |
| Never | 16 | 22 |
| Stage | ||
| I | 45 | 61 |
| II | 19 | 26 |
| III | 7 | 9 |
| IV | 3 | 4 |
The perl scripts output, including all mismatch types, included a total of 4518 significant mismatch events in 148 samples. The events were detected in 186 mismatch spots. We detected 64 mismatch hotspots, in which 1‐base mismatch events were detected in a common mismatch spot in at least 10 samples. However, most of the non‐A‐to‐G mismatch hotspots had extremely low mismatch frequencies (the number of reads with a mismatch divided by the number of total mapped reads) and were detected in fewer samples than the A‐to‐G mismatch hotspots (Figure 2). Therefore, we regarded the non‐A‐to‐G mismatch hotspots as noises, random sequencing errors which could not be eliminated by statistical procedure in the perl scripts. In regard to the 10 A‐to‐G mismatch hotspots, all of them were also detected in the previous study of TCGA dataset.22 These mismatch hotspots can be regarded as true editing sites, because they were firmly reproducible; we call them “editing hotspots” from here.
Figure 2.
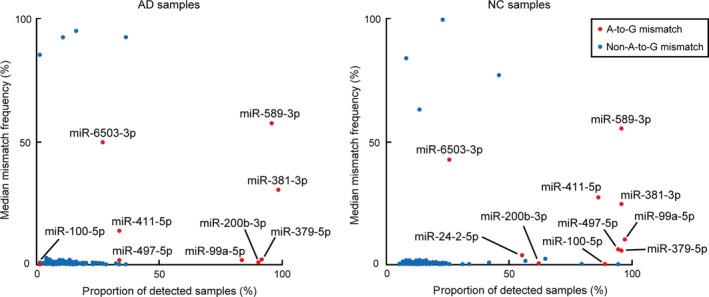
Distribution plot of the mismatch hotspots in AD (left panel) and NC (right panel). Each dot represents the proportion of the number of samples (out of 74) in which statistically significant mismatch events were observed (horizontal axis) and the median editing level among the cases (vertical axis). AD, adenocarcinoma; NC, normal counterpart
For the 10 A‐to‐I editing hotspots, we compared the editing levels of 8 microRNAs (paired editing hotspots) between AD and NC samples; for the remaining 2 microRNAs (miR‐100‐5p, miR‐24‐2‐5p), only 1 editing event was detected in 74 AD samples. Distribution of the editing levels of the 8 microRNAs is shown in Figure 3. Loss of editing was detected in position 5 of miR‐379‐5p (52 cases out of the 65 cases in which RNA editing was detected in both AD and NC; 52/65), position 1 of miR‐99a‐5p (60/60), and position 2 of miR‐497‐5p (20/23) (Figure 3A). Gain of editing was detected in position 5 of miR‐200b‐3p (19/41) (Figure 3B). Among these 4 microRNAs, median value of the editing level difference between AD and NC was the largest for miR‐99a‐5p. Most cases showed no significant difference of editing level at position 4 of miR‐381‐3p (53/70), position 6 of miR‐589‐3p (67/68), position 5 of miR‐411‐5p (14/22), or position 7 of miR‐6503‐3p (7/7) (Figure 3C).
Figure 3.
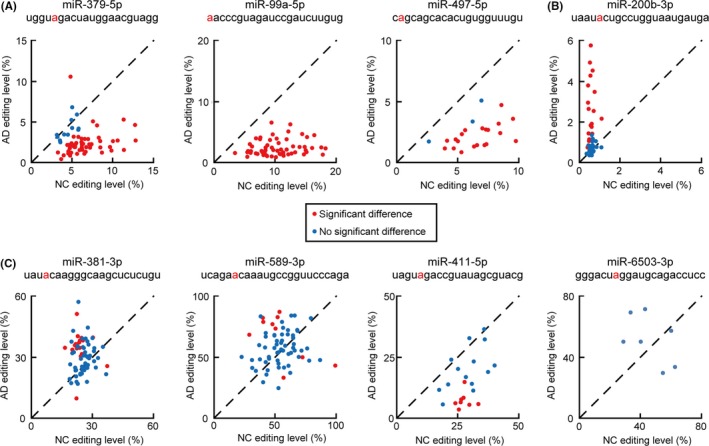
Distribution of A‐to‐I microRNA editing levels by in silico analysis. These 8 microRNAs showed significant A‐to‐I editing in more than 5 cases each of both AD and NC. Editing sites are shown in red letters. Each dot represents the editing level of AD and NC of each case showing (red) or not showing (blue) a statistically significant difference of the editing level. A, miR‐379‐5p (n = 65), miR‐99a‐5p (n = 60), and miR‐497‐5p (n = 23): loss of editing was seen in most cases. B, miR‐200b‐3p (n = 41): nearly half of the cases showed gain of editing. C, miR‐381‐3p (n = 71), miR‐589‐3p (n = 68), miR‐411‐5p (n = 22), and miR‐6503‐3p (n = 7): no significant differences of the editing level were observed in most cases. AD, adenocarcinoma; NC, normal counterpart
3.2. Quantification of the editing level of miR‐99a‐5p
Our improved method to quantify the editing level of miR‐99a‐5p shown in Figure 1 is based on two assumptions: (i) PCR efficiency of edited and unedited miR‐99a‐5p is equivalent; and (ii) the proportion of edited and unedited amplicons in concatemers is equivalent to that before ligation. To confirm that the proportion of edited reads reflected the editing level of the sample, we examined the standard template which contains 5%, 15%, and 25% edited amplicons (5′ adaptor, miR‐99a‐5p, and polyA). As shown in Table 3, each template showed results consistent with its mixture fractions.
Table 3.
Results of the validation test for quantifying the editing level of miR‐99a‐5p. Mixture fraction indicates the proportion of the template with edited amplicon
| Mixture fraction | 5% | 15% | 25% |
|---|---|---|---|
| Total amplicon | 103 | 105 | 110 |
| Edited amplicon | 8 | 12 | 23 |
| Observed proportion | 7.8% | 11.4% | 20.9% |
| 95% CI | 3.4%‐14.7% | 6.0%‐19.1% | 13.7%‐29.7% |
| P‐value (binomial test) | 0.144 | 0.189 | 0.19 |
Next, we extracted total RNA from 100 resected tissue specimens obtained from a cohort of 50 cases of lung adenocarcinoma and examined the editing level of miR‐99a‐5p in these specimens. In 43 cases (86%), the editing level in AD was lower than that in NC. After statistical analysis, 19 cases (38%) showed significant loss of editing and 1 case (2%) showed significant gain of editing (Figure 4A). Loss of editing was not significantly associated with any unique clinical features (age, P = 0.750; gender, P = 0.560; smoking history, P = 1; stage, P = 0.247; epidermal growth factor receptor (EGFR) mutation, P = 0.736; adjuvant chemotherapy, P = 0.389). Kaplan‐Meier curves stratified by loss of editing are shown in Figure 4B‐D. Cases with loss of editing showed significantly shorter OS (P = 0.047; hazard ratio [HR], 3.25; 95% confidence interval [CI], 0.95‐11.13) and a tendency toward shorter RFS (P = 0.103; HR 1.91; 95% CI, 0.87‐4.19). In the subgroup analysis, loss of editing was significantly associated with shorter RFS in the subgroup of pathological stage IB disease (n = 23; P = 0.028; HR, 5.08; 95% CI, 1.01‐25.58).
Figure 4.
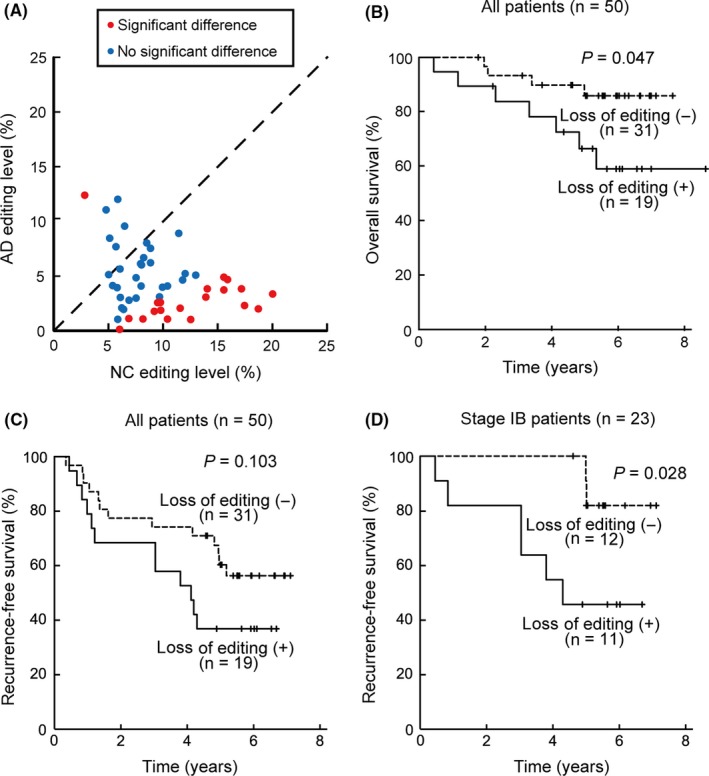
A, Editing levels of miR‐99a‐5p in the 50 cases at our institution. The format of the scatter plot is the same as that in Figure 2. B‐D, Kaplan‐Meier curves of the overall survival and the recurrence‐free survival, stratified by loss of editing of miR‐99a‐5p. D, represents the subgroup of pathological stage IB disease (n = 23)
Then, we compared the editing level of the 8 microRNAs with paired editing hotspots among our hospital dataset, downloaded SRA dataset, and previously reported TCGA dataset by Pinto et al22 (Table 4). For miR‐99a‐5p, the editing level ranges largely overlapped between our hospital dataset and the SRA dataset. Overall editing level (total number of edited reads of all cases divided by the total number of mapped reads of all cases) of each microRNA in TCGA dataset was within the corresponding range in the SRA dataset, except for miR‐497‐5p. For miR‐497‐5p, the overall editing level in AD from TCGA dataset reported by Wang et al21 (approximately 2.8% for AD; n = 483, raw data not available) was somehow within the corresponding range in the SRA dataset.
Table 4.
Comparison of the editing levels of the 8 microRNAs among the 3 datasets: the University of Tokyo Hospital, SRA, and TCGA
| Unit: (%) | Our dataset | SRA | TCGA | ||||
|---|---|---|---|---|---|---|---|
| n = 50 | n = 74 | n = 36 | |||||
| AD | NC | AD | NC | AD | NC | ||
| miR‐99a‐5p | Range | 0‐12.3 | 2.8‐20.0 | 0.79‐6.49 | 2.12‐18.24 | ||
| Overall | 4.37 | 9.50 | 2.34 | 10.21 | 3.57 | 12.37 | |
| miR‐379‐5p | Range | 0.35‐10.53 | 3.09‐12.78 | ||||
| Overall | 1.40 | 5.76 | 1.54 | 4.50 | |||
| miR‐497‐5p | Range | 0.80‐5.05 | 2.60‐10.18 | ||||
| Overall | 2.10 | 6.40 | 5.56 | 17.43 | |||
| miR‐200b‐3p | Range | 0.30‐5.95 | 0.33‐1.07 | ||||
| Overall | 1.30 | 0.52 | 1.41 | 0.66 | |||
| miR‐381‐3p | Range | 9.48‐57.14 | 16.31‐37.14 | ||||
| Overall | 25.75 | 24.92 | 36.33 | 28.46 | |||
| miR‐589‐3p | Range | 24.32‐87.02 | 23.08‐100 | ||||
| Overall | 60.9 | 55.57 | 70.73 | 67.98 | |||
| miR‐411‐5p | Range | 3.30‐36.36 | 9.38‐60.00 | ||||
| Overall | 8.67 | 28.27 | 19.17 | 35.32 | |||
| miR‐6503‐3p | Range | 23.08‐76.92 | 22.22‐80.00 | ||||
| Overall | 47.72 | 42.33 | 39.26 | 32.00 | |||
AD, adenocarcinoma; NC, normal counterpart; SRA, Sequence Read Archives; TCGA, The Cancer Genome Atlas.
3.3. Analysis of ADAR1 and ADAR2 expressions
Finally, we carried out quantitative RT‐PCR for expression of ADAR1 and ADAR2, with normalization to the expression of GAPDH. Amplification efficiencies of ADAR1, ADAR2, and GAPDH calculated from the slope of the standard curves from standard samples were 91.4%, 95.5%, and 93.0%, respectively. Expression levels of both ADAR1 and ADAR2 were downregulated in AD as compared to NC (median fold change (2−ΔΔCt) of ADAR1, 0.52; ADAR2, 0.16). There was a moderate positive correlation between the editing level in each sample and the expression level of ADAR2 (r s = 0.258, P = 0.009 for ADAR1, and r s = 0.424, P < 0.001 for ADAR2) (Figure 5A), consistent with a previous study in which miR‐99a‐5p was reported to be edited by ADAR2.21
Figure 5.
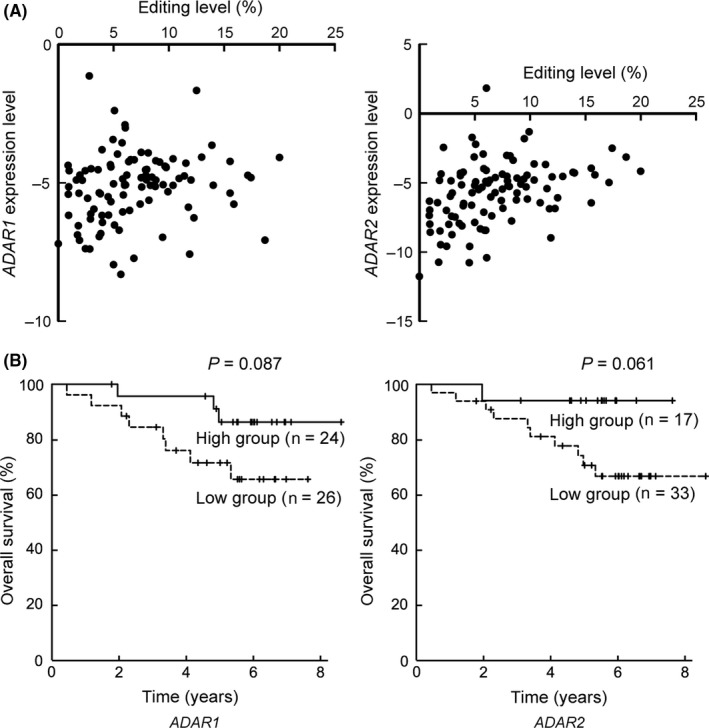
A, Scatter plots of the editing level of miR‐99a‐5p and the relative expression level (−ΔCt value) of ADAR1 and ADAR2 in each sample (n = 100). B, Kaplan‐Meier curves of the overall survival, grouped by the relative expression level of ADAR1 and ADAR2 in adenocarcinoma specimen (−ΔΔCt value) in each case (n = 50)
Then, the patients were divided into 2 groups according to the expression level (ΔΔCt value) of ADAR1 (high group, n = 24; low group, n = 26) and ADAR2 (high group; n = 17, low group; n = 33). Kaplan‐Meier curves are shown in Figure 5B. The ADAR1‐high group (P = 0.087; HR, 0.331; 95% CI, 0.087‐1.251) and the ADAR2‐high group (P = 0.061; HR, 0.176; 95% CI, 0.023‐1.376) showed a tendency toward a longer OS.
4. DISCUSSION
In the present study, in silico analysis identified 8 microRNAs with paired A‐to‐I editing hotspots. Furthermore, we demonstrated an improved method to quantify the editing level of miR‐99a‐5p. In our patient cohort, loss of editing was associated with poor prognosis after complete resection of lung adenocarcinoma. This tendency was consistent with the results of previous studies of TCGA dataset;21, 22 reproducibility was confirmed by a well‐validated conventional RT‐PCR method. Along with these studies, we believe that the alteration of microRNA editing in lung adenocarcinoma could serve as a potential biomarker for predicting patient outcomes.
Before this biomarker can be applied practically in clinical medicine, the method for quantifying the editing level of microRNA needs to be further refined. In the first study to report microRNA editing, the authors amplified the precursor of miR‐22, pri‐miR‐22, cloned the PCR product and sequenced at least 100 amplicons.12 However, because editing of a primary microRNA affects the processing efficiency of the microRNA catalyzed by the double‐strand RNase enzymes, DROSHA and DICER, the editing level of primary microRNA is not always equal to the editing level of mature microRNA, which finally functions as the posttranscriptional regulator.32, 33 A recent comprehensive analysis showed that the difference in the editing level between a primary microRNA and mature microRNA is >10% in approximately half of the microRNAs.34 An improved method to amplify mature microRNA was published in 2012.28 In this study, mature microRNA was 3′ polyadenylated and 5′ adaptor ligated, reverse‐transcribed with oligo‐dT primer, specifically amplified, and cloned. At least 100 amplicons were sequenced. Use of the TaqMan Advanced miRNA cDNA Synthesis Kit (Thermo Fisher Scientific) reduced the operation hours of this method. Furthermore, to reduce cost entailed by the vast amount of sequencing, we made use of the concept of serial analysis of gene expression (SAGE)29 and established a protocol to concatenate the amplicons, as described above. Our improved method takes approximately 6 extra hours to concatenate the amplicon, while the number of sequenced samples was almost one‐half (5322 samples used to sequence 11 110 amplicons). Moreover, our validation experiment showed the reliability of this method.
Another candidate method, which would be less time‐consuming and less expensive, is to make use of TaqMan MGB (minor groove binder) Probe with 2 colors of fluorescent dye. After synthesizing first‐strand cDNA with the 5′ adaptor and polyA using TaqMan Advanced miRNA cDNA Synthesis Kit, the expression level of each microRNA can be quantified with a specific primer set and TaqMan MGB Probe, which are contained in TaqMan Advanced miRNA Assays (Thermo Fisher Scientific). Moreover, a method involving the use of the TaqMan Probe with 2 colors of fluorescent dye, FAM and VIC, to quantify the editing level of mRNA was established in a previous study.35 Combining these methods, both the expression and editing levels of microRNAs can be measured. A significant difficulty is that while securing the specificity of amplifying the microRNA, there is little space left for the TaqMan Probe to anneal on the editing spot, even with an MGB.
Previous studies have shown that miR‐99a‐5p itself functions as a tumor‐suppressing microRNA through repressing invasion, migration,36 and stemness.37 However, the functional impact of editing miR‐99a‐5p is not clear, because it undergoes RNA editing at position 1. The seed sequence of the microRNAs, spanning positions 2‐8, must be completely complementary to the 3′ UTR part of the target mRNA.18 A previous study reported that miR‐17‐5p and miR‐106a‐5p, whose sequences differ only in position 1, are regulated and function in parallel.38 Although the previous study of TCGA dataset showed that the editing level of miR‐99a‐5p was correlated with the mutation frequency of 14 important genes, including TP53, HRAS, NRAS and PIK3CA in 6 cancer types,21 this cannot be explained by the known function of microRNA editing. Therefore, we assume that the difference in the editing level of miR‐99a‐5p being the largest among the paired editing hotspots caused a “passenger change” in lung adenocarcinoma, rather than alteration of a specific function of miR‐99a‐5p.
In a previous study of TCGA dataset, expression level of ADAR1 in lung adenocarcinoma was reported to be upregulated as compared with that in the corresponding normal tissue.39 Moreover, amplification of the ADAR1 region40 and higher expression level of ADAR1 in cancerous tissue21 were associated with a tendency toward a poorer prognosis, although the association was not statistically significant for either. Our dataset showed an inverse tendency. We did not identify any clinical features (age, gender, smoking history, stage, EGFR mutation status) that were significantly associated with the expression levels of ADAR1 in our dataset. The largest difference between our cohort and TCGA cohort was in the ethnicity of the patients (100% East Asian vs 67.2% White and 1.4% Asian), which may have affected tumor biogenesis. We consider that study of more cases is needed to verify the function of ADAR1 in lung adenocarcinoma. Regarding ADAR2, our findings were consistent with previous studies; ADAR2 was downregulated in lung adenocarcinoma tissues,39 and a lower ADAR2 expression level was significantly associated with a poorer prognosis.41
In summary, we analyzed the alterations of microRNA editing in lung adenocarcinoma both in silico and by a conventional RT‐PCR method. Loss of editing of miR‐99a‐5p in cancerous tissue was associated with poor survival in surgically resected lung adenocarcinoma specimens. Further investigations are required to elucidate the cause and effect of this association and to refine the method for quantification of the editing level.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This study was supported by a Grant‐in‐Aid for Young Scientists (B) (JSPS KAKENHI No. 15K20917 and 15K18440), a Grant‐in‐Aid for Scientific Research (C) (JSPS KAKENHI No. 16K09574), and a Grant‐in‐Aid for Scientific Research (A) (JSPS KAKENHI No. 16H02653) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Maemura K, Watanabe K, Ando T, et al. Altered editing level of microRNAs is a potential biomarker in lung adenocarcinoma. Cancer Sci. 2018;109:3326–3335. 10.1111/cas.13742
Funding information
JSPS KAKENHI (15K18440, 15K20917, 16H02653, 16K09574), Ministry of Education, Culture, Sports, Science and Technology of Japan
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;5:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 2. Walters S, Maringe C, Coleman MP, et al. Lung cancer survival and stage at diagnosis in Australia, Canada, Denmark, Norway, Sweden and the UK: a population‐based study, 2004‐2007. Thorax. 2013;6:551‐564. [DOI] [PubMed] [Google Scholar]
- 3. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum‐pemetrexed in EGFR T790M‐positive lung cancer. N Engl J Med. 2017;7:629‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK‐positive non‐small‐cell lung cancer. N Engl J Med. 2017;9:829‐838. [DOI] [PubMed] [Google Scholar]
- 5. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med. 2015;17:1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med. 2015;2:123‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reck M, Rodriguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;19:1823‐1833. [DOI] [PubMed] [Google Scholar]
- 8. Kris MG, Gaspar LE, Chaft JE, et al. Adjuvant systemic therapy and adjuvant radiation therapy for stage I to IIIA completely resected non‐small‐cell lung cancers: American Society of Clinical Oncology/Cancer Care Ontario Clinical Practice Guideline Update. J Clin Oncol. 2017;25:2960‐2974. [DOI] [PubMed] [Google Scholar]
- 9. Watanabe K, Takai D. Disruption of the expression and function of microRNAs in lung cancer as a result of epigenetic changes. Front Genet. 2013;4:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paul MS, Bass BL. Inosine exists in mRNA at tissue‐specific levels and is most abundant in brain mRNA. EMBO J. 1998;4:1120‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. Profiling RNA editing in human tissues: towards the inosinome atlas. Sci Rep. 2015;5:14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA. 2004;8:1174‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine‐to‐inosine editing of miRNAs. Science. 2007;5815:1137‐1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng Y, Li T, Ren R, Shi D, Wang S. Revealing editing and SNPs of microRNAs in colon tissues by analyzing high‐throughput sequencing profiles of small RNAs. BMC Genom. 2014;15(Suppl 9):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choudhury Y, Tay FC, Lam DH, et al. Attenuated adenosine‐to‐inosine editing of microRNA‐376a* promotes invasiveness of glioblastoma cells. J Clin Invest. 2012;11:4059‐4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shoshan E, Mobley AK, Braeuer RR, et al. Reduced adenosine‐to‐inosine miR‐455‐5p editing promotes melanoma growth and metastasis. Nat Cell Biol. 2015;3:311‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tomaselli S, Galeano F, Alon S, et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin‐14 by lin‐4 mediates temporal pattern formation in C. elegans . Cell. 1993;5:855‐862. [DOI] [PubMed] [Google Scholar]
- 19. Yang W, Chendrimada TP, Wang Q, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;1:13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA‐151 precursor blocks cleavage by the Dicer‐TRBP complex. EMBO Rep. 2007;8:763‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Xu X, Yu S, et al. Systematic characterization of A‐to‐I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017;7:1112‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pinto Y, Buchumenski I, Levanon EY, Eisenberg E. Human cancer tissues exhibit reduced A‐to‐I editing of miRNAs coupled with elevated editing of their targets. Nucleic Acids Res. 2018;1:71‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alon S, Mor E, Vigneault F, et al. Systematic identification of edited microRNAs in the human brain. Genome Res. 2012;8:1533‐1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alon S, Eisenberg E. Identifying RNA editing sites in miRNAs by deep sequencing. Methods Mol Biol. 2013;1038:159‐170. [DOI] [PubMed] [Google Scholar]
- 25. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;3:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010; Chapter 11:Unit 11‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Burroughs AM, Ando Y, Hoon MJ, et al. A comprehensive survey of 3′ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res. 2010;10:1398‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawahara Y. Quantification of adenosine‐to‐inosine editing of microRNAs using a conventional method. Nat Protoc. 2012;7:1426‐1437. [DOI] [PubMed] [Google Scholar]
- 29. Yamamoto M, Wakatsuki T, Hada A, Ryo A. Use of serial analysis of gene expression (SAGE) technology. J Immunol Methods. 2001;1‐2:45‐66. [DOI] [PubMed] [Google Scholar]
- 30. Fluss R, Faraggi D, Reiser B. Estimation of the Youden Index and its associated cutoff point. Biom J. 2005;4:458‐472. [DOI] [PubMed] [Google Scholar]
- 31. Vucic EA, Thu KL, Pikor LA, et al. Smoking status impacts microRNA mediated prognosis and lung adenocarcinoma biology. BMC Cancer. 2014;14:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kawahara Y, Megraw M, Kreider E, et al. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;16:5270‐5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slezak‐Prochazka I, Durmus S, Kroesen BJ, van den Berg A. MicroRNAs, macrocontrol: regulation of miRNA processing. RNA. 2010;6:1087‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li L, Song Y, Shi X, et al. The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res. 2018;1:132‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong K, Lyddon R, Dracheva S. TaqMan‐based, real‐time quantitative polymerase chain reaction method for RNA editing analysis. Anal Biochem. 2009;2:173‐180. [DOI] [PubMed] [Google Scholar]
- 36. Sun M, Hong S, Li W, et al. MiR‐99a regulates ROS‐mediated invasion and migration of lung adenocarcinoma cells by targeting NOX4. Oncol Rep. 2016;5:2755‐2766. [DOI] [PubMed] [Google Scholar]
- 37. Feliciano A, Garcia‐Mayea Y, Jubierre L, et al. miR‐99a reveals two novel oncogenic proteins E2F2 and EMR2 and represses stemness in lung cancer. Cell Death Dis. 2017;10:e3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dhar S, Kumar A, Rimando AM, Zhang X, Levenson AS. Resveratrol and pterostilbene epigenetically restore PTEN expression by targeting oncomiRs of the miR‐17 family in prostate cancer. Oncotarget. 2015;29:27214‐27226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paz‐Yaacov N, Bazak L, Buchumenski I, et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015;2:267‐276. [DOI] [PubMed] [Google Scholar]
- 40. Anadon C, Guil S, Simo‐Riudalbas L, et al. Gene amplification‐associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene. 2016;33:4407‐4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valles I, Pajares MJ, Segura V, et al. Identification of novel deregulated RNA metabolism‐related genes in non‐small cell lung cancer. PLoS ONE. 2012;8:e42086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials