

Abstract
The molecular roles of the dually targeted ElaC domain protein 2 (ELAC2) during nuclear and mitochondrial RNA processing in vivo have not been distinguished. We generated conditional knockout mice of ELAC2 to identify that it is essential for life and its activity is non‐redundant. Heart and skeletal muscle‐specific loss of ELAC2 causes dilated cardiomyopathy and premature death at 4 weeks. Transcriptome‐wide analyses of total RNAs, small RNAs, mitochondrial RNAs, and miRNAs identified the molecular targets of ELAC2 in vivo. We show that ELAC2 is required for processing of tRNAs and for the balanced maintenance of C/D box snoRNAs, miRNAs, and a new class of tRNA fragments. We identify that correct biogenesis of regulatory non‐coding RNAs is essential for both cytoplasmic and mitochondrial protein synthesis and the assembly of mitochondrial ribosomes and cytoplasmic polysomes. We show that nuclear tRNA processing is required for the balanced production of snoRNAs and miRNAs for gene expression and that 3′ tRNA processing is an essential step in the production of all mature mitochondrial RNAs and the majority of nuclear tRNAs.
Keywords: cardiomyopathy, non‐coding RNAs, RNA processing, RNA‐Seq, snoRNAs
Subject Categories: Protein Biosynthesis & Quality Control, RNA Biology
Introduction
Most biological processes require non‐coding RNAs (ncRNAs): rRNAs and tRNAs are essential for protein synthesis and thereby life, small nuclear RNAs (snRNAs) catalyze pre‐mRNA splicing, telomerase RNA prevents loss of genetic information from the ends of chromosomes, and a catalytic ncRNA is required for 5′ tRNA processing as part of the RNase P RNA–protein complex 1. There are many different species of ncRNAs in addition to the best‐known rRNAs and tRNAs, including long ncRNAs, miRNAs, small RNAs, and piwi RNAs, that are important regulators of gene expression, and their impaired expression has been implicated in development and disease 1. Consequently, the regulation and production of ncRNAs are of particular interest and there is a growing body of knowledge about the different classes and regulators of ncRNAs; however, mechanistic and genetic models that validate their molecular roles and origins in vivo are still rare.
Eukaryotic nuclear tRNAs are transcribed as precursor transcripts by RNA polymerase III that contain 5′ leader and 3′ trailer sequences 2. The 5′ leader of nuclear tRNAs is processed by the RNase P complex 3, and the 3′ trailer sequences are cleaved by an RNase Z enzyme in the nucleus 4. Nuclear tRNA processing by RNase P is well understood in different systems; however, 3′ RNA processing by RNase Z, also known as ELAC2, is poorly understood in mammals 5. This is particularly crucial because tRNA and rRNA levels are compromised when 3′ RNA processing is impaired in yeast 5 but little is known about the consequences of RNase Z loss in mammals. There is evidence that 3′ RNA processing is important for the stability of precursor transcripts encoding tRNAs and generation of other regulatory non‐coding RNAs, including MALAT1 (also known as Neat2) and sRNAs 6.
In contrast to nuclear tRNAs, mitochondrial tRNAs (mt‐tRNAs) in animals are transcribed as long polycistronic transcripts that span the entire heavy or light stand of the genome 7 but do not contain 5′ leader and 3′ trailer sequences 8. Instead, mt‐tRNAs are flanked by mRNAs or rRNAs and the respective mitochondrial RNase P and RNase Z enzymes are responsible for their cleavage and hierarchical processing from the polycistronic transcripts 9, 10, 11, 12, 13. We have shown that impaired processing of mitochondrial tRNAs at the 5′ end has profound effects on mitochondrial gene expression, the assembly of the protein synthesis machinery, and the overall level of translation 10, but the mechanisms of 3′ end tRNA processing and consequences of its loss in vivo are still unclear. Understanding the role of RNA processing is essential because all mitochondrial RNAs are produced from large precursor transcripts and many diseases are caused by mutations in regulatory mtDNA‐encoded genes such as tRNAs and in nuclear‐encoded mitochondrial RNA‐binding proteins that regulate mitochondrial RNA metabolism [reviewed in 14].
ELAC2 shares homology with ElaC enzymes that have RNase Z activity. The ELAC2 mRNA has two different start codons that can produce a longer protein containing a mitochondrial targeting sequence and a shorter protein that lacks the targeting sequence and is localized to the nucleus 15. In cells, ELAC2 has been shown to process the 3′ ends of mitochondrial tRNAs 11, 12; however, its role and targets in the nucleus are not known. To understand the role of 3′ RNA processing in the nucleus and mitochondria in vivo, we generated conditional Elac2 knockout mice and show that ELAC2 is essential for the balanced production of tRNAs, miRNAs, and the C/D box snoRNAs that are required for protein synthesis.
Results
Loss of ELAC2 causes dilated cardiomyopathy and premature death by 4 weeks
A conditional knockout allele of the mouse Elac2 gene was generated by flanking exon 8 with loxP sites in embryonic stem (ES) cells (Fig 1A). Mice containing the germline transmission of this allele (Elac2 +/loxP‐neo) were crossed with transgenic mice expressing the Flp recombinase to excise the neomycin cassette (Fig 1A). The resulting Elac2 +/loxP mice were crossed to mice expressing Cre recombinase under the control of the β‐actin promoter to generate heterozygous Elac2 knockout mice (Elac2 +/−). We intercrossed the Elac2 +/− mice to produce Elac2 +/− and Elac2 +/+ mice in Mendelian proportions; however, the homozygous knockout mice (Elac2 −/−) were not observed. Proteins involved in mitochondrial gene expression often exhibit embryonic lethality at E8.5 10, 16, 17, 18, 19, 20; therefore, we analyzed the embryos at this stage to identify that Elac2 −/− embryos were not viable (Fig EV1A), indicating that ELAC2 is required for embryo growth and survival.
Figure 1. Heart‐specific knockout of ELAC2 causes early‐onset cardiomyopathy.
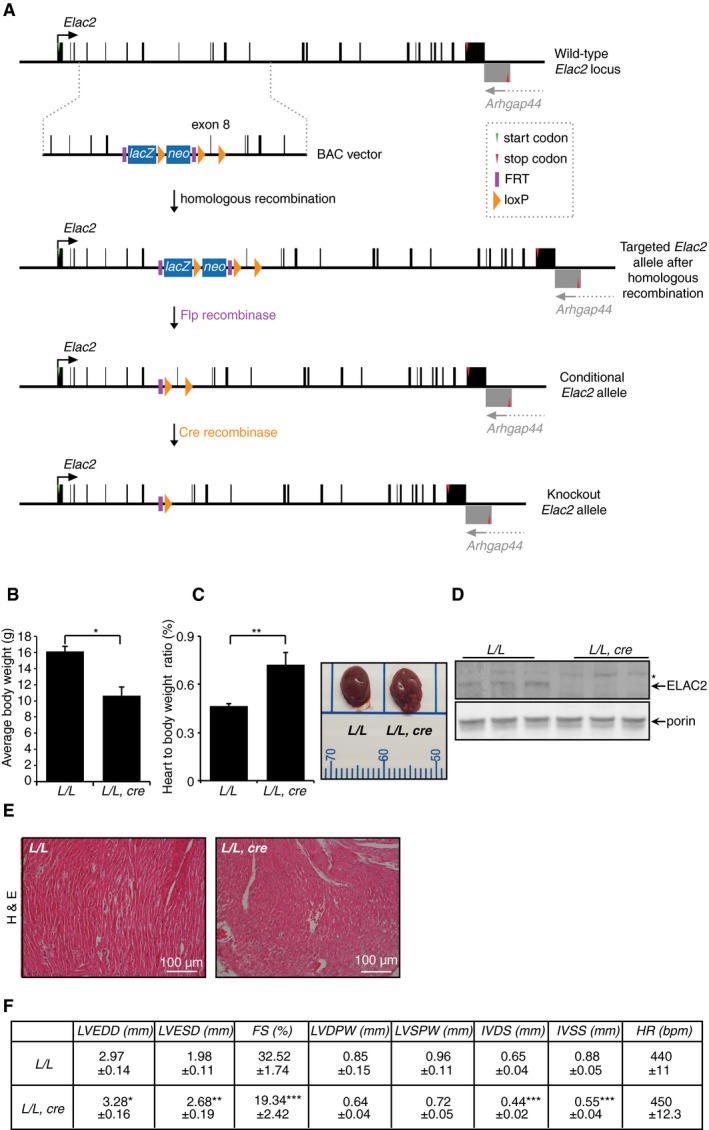
- Schematic showing the disruption of the Elac2 gene. LoxP sites flanking exon 8 of the Elac2 gene were inserted in the mouse genome by homologous recombination.
- Weight differences between control (L/L) and knockout mice (L/L, cre) at 4 weeks of age; L/L n = 19, L/L, cre n = 19. Error bars indicate SEM; *P < 0.05, Student's t‐test.
- Heart weight‐to‐body weight ratio in control (L/L) and knockout mice (L/L, cre) at 4 weeks; L/L n = 15, L/L, cre n = 15; photographic representation of size difference between L/L and L/L, cre hearts at 4 weeks of age. Error bars indicate SEM; **P < 0.01, Student's t‐test.
- Immunoblot showing the loss of ELAC2 in the L/L, cre mice compared to L/L control mice. Porin was used as a loading control. An asterisk indicates a non‐specific protein detected by the antibody.
- Hematoxylin and eosin staining of hearts from control (L/L) and knockout mice (L/L, cre). Scale bar is 100 μm.
- Echocardiographic parameters for Elac2 L/L (n = 5) and Elac2 L/L,cre (n = 5) 4‐week‐old mice. LVEDD, left ventricular end diastolic diameter; LVESD, left ventricular end systolic diameter; FS, fractional shortening; LVDPW, left ventricular posterior wall in diastole; LVSPW, left ventricular posterior wall in systole; IVDS, intraventricular septum in diastole; IVSS, intraventricular septum in systole; HR, heart rate. Values are means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with Elac2 L/L, Student's t‐test.
Figure EV1. Related to Fig 1: The effects of ELAC2 loss on survival and skeletal muscle.
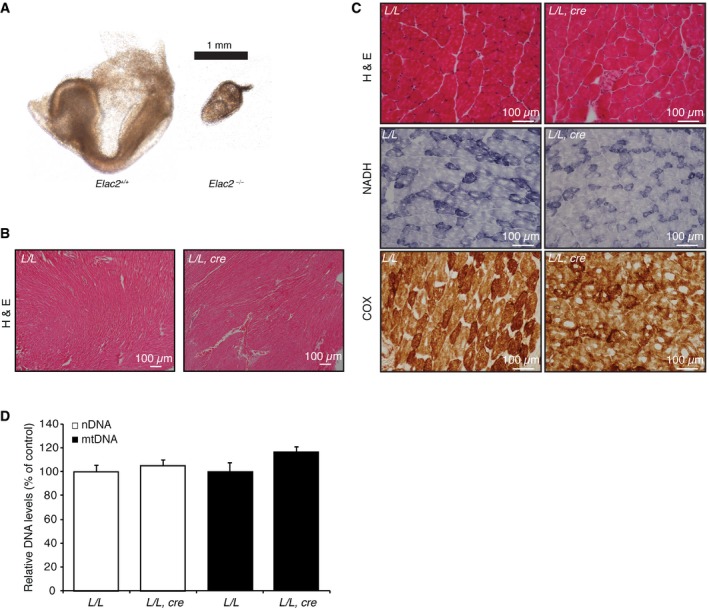
- Morphology of the Elac2 +/+ and Elac2 −/− embryos at day E8.5. Scale bar, 1 mm.
- Hematoxylin and eosin (H&E) of heart from control (L/L) and knockout (L/L, cre) mice. Scale bar is 100 μm.
- Hematoxylin and eosin (H&E), and NADH and COX staining of skeletal muscle from control (L/L) and knockout (L/L, cre) mice. Scale bar is 100 μm.
- Changes in mitochondrial and nuclear DNA were determined by qPCR on DNA isolated from 4‐week‐old control (L/L, n = 3) and knockout (L/L, cre, n = 3) mouse hearts. Values are means ± SEM.
We crossed Elac2 +/loxP mice with transgenic mice expressing Cre recombinase under the control of the muscle creatinine kinase promoter (Ckmm‐cre) to produce heart‐ and skeletal muscle‐specific Elac2 knockout mice (Elac2 loxP/loxP,cre+). The Elac2 mice with Ckmm‐cre‐directed knockout have a short life span and die by 4 weeks with significant weight loss (Fig 1B) and profound cardiomyopathy, determined by increased heart size, histology, and echocardiography (Figs 1C–F and EV1B) as a result of ELAC2 loss (Fig 1D). In contrast, there were no dramatic defects in skeletal muscle (Fig EV1C). There were no changes in the mitochondrial and nuclear DNA levels (Fig EV1D). The early death of these mice compared to mice that die by 11 weeks due to impaired 5′ tRNA processing as a result of loss of the mitochondria‐specific RNase P catalytic subunit, MRPP3, indicated that this could be a consequence of combined effects on tRNA processing both in the nucleus and in mitochondria. Therefore, our analyses focused on both nuclear and mitochondrial RNA metabolism and their functional consequences.
ELAC2 is essential for 3′ tRNA processing but not for 5′ tRNA cleavage in mitochondria
ELAC2 has been shown to cleave the 3′ ends of mitochondrial tRNAs in vitro and by RNAi in cells 11, 12; however, the loss of ELAC2 has not been studied in vivo. Here, we show that knockout of ELAC2 in vivo causes dramatic loss of mature mitochondrial mRNAs such as mt‐Co1, mt‐Co2, mt‐Atp8/6, mt‐Nd4l/4, and mt‐Cyt b by 4 weeks of age (Fig 2A), as well as loss of mature tRNAs and rRNAs in mitochondria (Fig 2A) and an increase in unprocessed tRNAs. We validated the loss of mt‐mRNAs and rRNAs in the Elac2 knockout mice relative to controls by qRT–PCR (Fig 2B). Interestingly, we observed increased accumulation of the processing intermediates, including RNA19, but loss of longer polycistronic transcripts for other mRNAs in the Elac2 knockout mice relative to controls (Fig 2A), suggesting that the precursor transcripts are already processed by the functional RNase P complex at the 5′ end of most tRNAs, except for tRNAPhe, tRNAVal, and tRNALeu(UUR). Small RNA‐Seq identified accumulation of 3′ uncleaved mt‐tRNAs (Fig 2C), further identifying that ELAC2 is indispensable for 3′ tRNA processing.
Figure 2. Loss of ELAC2 causes impaired mitochondrial tRNA processing.
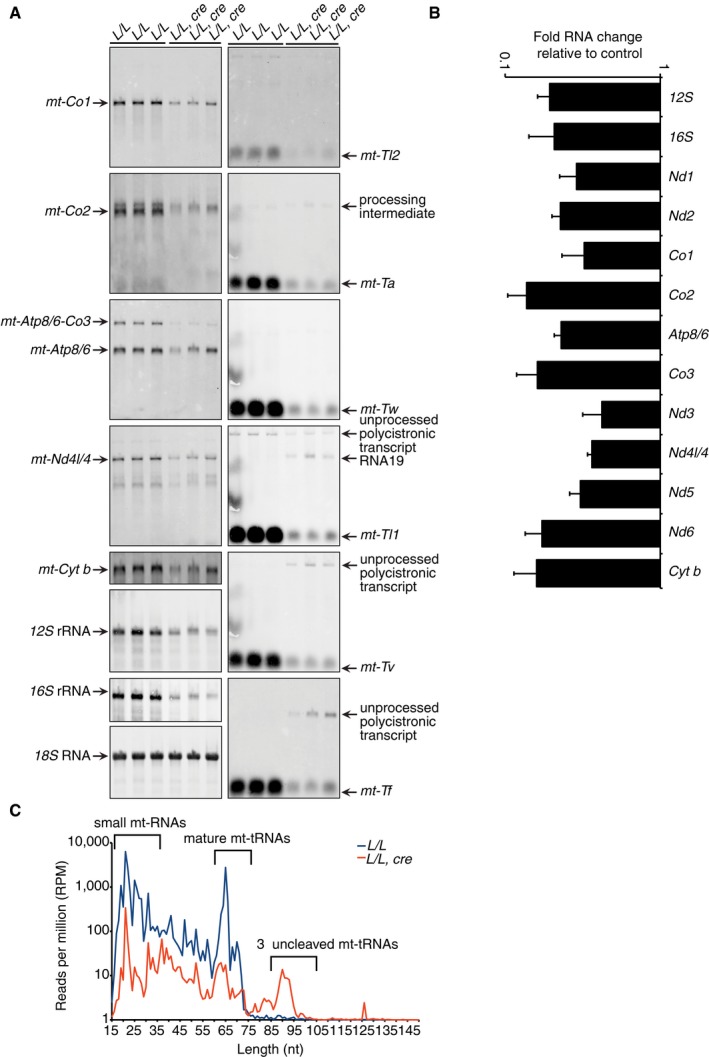
- The abundance of mature mitochondrial mRNAs, tRNAs, and rRNAs in hearts from 4‐week‐old control and knockout mice were analyzed by northern blotting. 18S rRNA was used as a loading control. The data are representative of results obtained from at least eight mice from each strain and three independent biological experiments.
- Mitochondrial RNAs were measured in total heart RNA from control (L/L) and knockout (L/L, cre) 4‐week‐old mice by qRT–PCR and normalized to 18S rRNA. Values are means ± SEM. All error bars are significant to P < 0.01 determined using the Student's t‐test.
- Size distribution of 5′ aligned reads from heart mitochondrial pre‐tRNAs from three control (L/L; blue) and three knockout (L/L, cre; red) mice. Significant accumulation of the 3′ uncleaved pre‐tRNAs was observed in the Elac2 knockout mice. The range of small mt‐RNAs, mature mt‐tRNAs (ranging from 58–75 nt), and precursor mt‐tRNAs are indicated.
Source data are available online for this figure.
Next, we carried out RNA‐Seq using total heart RNA from three control and three Elac2 knockout mice (Fig 3A) to identify the processing sites of ELAC2 across the entire mitochondrial transcriptome and the effects of impaired 3′ processing on the 5′ tRNA ends. We used this technique to exclude short RNAs, such as mature tRNAs, but instead to capture tRNAs that are part of unprocessed transcripts and by analyzing reads that span processed regions in the mitochondrial transcriptome we quantified regions that are increased in the Elac2 knockout mice where processing was impaired. In our RNA‐Seq datasets, we found an enrichment of reads across junctions that span all tRNA regions between the mRNA and rRNA genes, indicating an increase in precursor RNAs upon ELAC2 loss. Within the same datasets, we observed a significant reduction in mature mt‐RNAs (Fig 3A), validating our northern blot, qRT–PCR, and small RNA‐Seq findings (Fig 2A–C).
Figure 3. Transcriptome‐wide analyses of 3′ tRNA cleavage sites by RNA‐Seq.
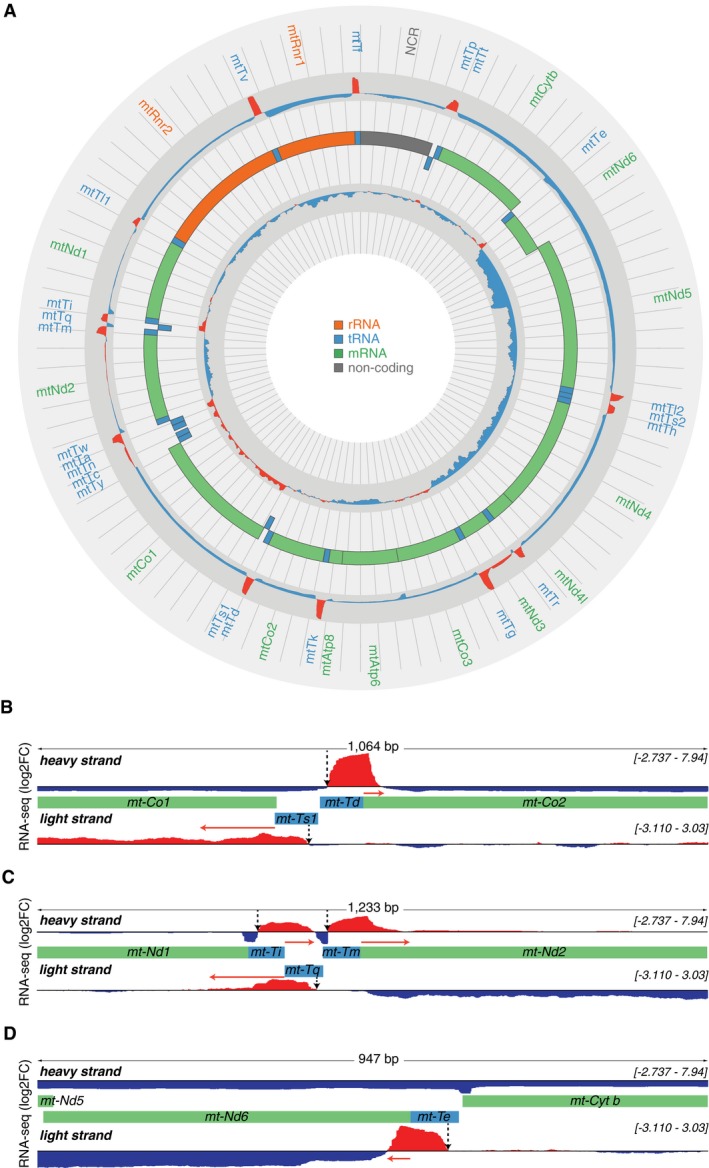
- A complete map of changes in mitochondrial RNA abundance determined by RNA‐Seq coverage (log2 fold change[KOmean/Ctrlmean]) from three control (L/L) and three knockout (L/L, cre) mice, on heavy (outer track) and light (inner track) strands. Increases are shown in red and decreases in blue. The mitochondrial genome is displayed in the central track; rRNAs are displayed in orange, mRNAs in green, tRNAs in blue and the non‐coding region (NCR) in gray. The log fold change scale for the heavy strand is −3 to 8.5 and for the light strand is −3.3 to 3.3.
- Genome browser view of the mean RNA‐Seq coverage (log2 fold change[KOmean/Ctrlmean]) showing the 3′ cleavage sites of mt‐tRNA Asp and mt‐tRNA SerAGY by ELAC2. Regions of interests are shown in green (for mRNAs) and blue (for tRNAs) boxes.
- Genome browser view of the mean RNA‐Seq coverage (log2 fold change [KOmean/Ctrlmean]) showing the 3′ cleavage sites of a cluster of tRNAs, mt‐tRNA Ile, mt‐tRNA Met, and mt‐tRNA Gln by ELAC2. Regions of interests are shown in green (for mRNAs) and blue (for tRNAs) boxes.
- Genome browser view of the mean RNA‐Seq coverage (log2 fold change[KOmean/Ctrlmean]) showing the 3′ cleavage site of mt‐tRNA Glu by ELAC2. Regions of interests are shown in green (for mRNAs) and blue (for tRNAs) boxes.
We identified impaired 3′ end processing across every mt‐tRNA irrespective of its position on the heavy or light strand, but not at the 5′ end, indicating that RNase P cleavage is unaffected by the loss of ELAC2 (Fig 3B–D). This is particularly evident in specific examples of a single tRNA that spans two mRNAs (Fig 3B), several tRNAs that span mRNAs (Fig 3C) or a tRNA encoded by the light strand (Fig 3D); in each case, we found increased accumulation of precursor transcripts at the 3′ end side of the tRNAs but not at the 5′ end. This is in stark contrast to the accumulation of precursor transcripts that span both the 5′ and 3′ ends of tRNAs in the absence of MRPP3 10, providing reciprocal validation of our previous finding that 5′ tRNA processing precedes 3′ tRNA cleavage.
Loss of ELAC2 causes impaired OXPHOS biogenesis and function
Next, we investigated the effects of ELAC2 loss on de novo mitochondrial translation, that is downstream of RNA processing, by measuring 35S‐methionine and cysteine incorporation to identify significant reduction in protein synthesis of all mitochondrially encoded proteins in the Elac2 knockout mice compared to controls (Fig 4A). Immunoblotting of mtDNA‐ and nuclear‐encoded polypeptide components of the electron translocating respiratory complexes revealed a significant decrease in their levels in the Elac2 knockout mice (Fig 4B) consistent with the reduction of the abundance and integrity of the intact mitochondrial respiratory complexes that we determined by immunoblotting following blue native polyacrylamide gel electrophoresis (BN–PAGE) (Fig 4C). Compromised OXPHOS biogenesis resulted in a significant reduction in mitochondrial oxygen consumption in the non‐phosphorylated, phosphorylated, and uncoupled respiration state of all three proton‐pumping complexes in the Elac2 knockout mice (Fig 4D). Therefore, loss of ELAC2 causes profound mitochondrial dysfunction through impaired OXPHOS biogenesis and oxygen consumption, further indicating that 3′ tRNA processing is non‐redundant in mitochondria and essential for energy production.
Figure 4. Loss of ELAC2 impairs mitochondrial biogenesis, mitoribosome assembly, and OXPHOS function.
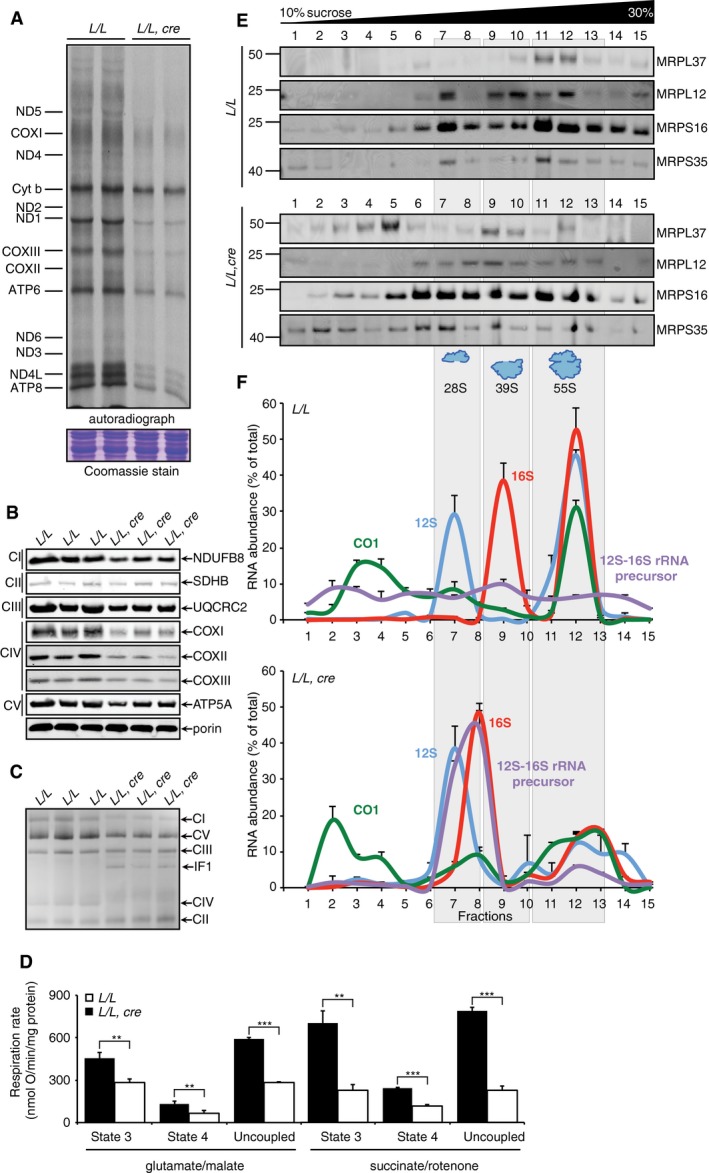
- In organello protein synthesis in heart mitochondria from 4‐week‐old control and knockout mice was measured by pulse incorporation of 35S‐labeled methionine and cysteine. Equal amounts of mitochondrial protein (50 μg) were separated by SDS–PAGE, stained with Coomassie to show equal loading, and visualized by autoradiography. Representative gels are shown of three independent biological experiments.
- Mitochondrial proteins (25 μg) from 4‐week‐old heart mitochondria from control and knockout mice were resolved on 4–20% SDS–PAGE gels and immunoblotted against antibodies to investigate the steady‐state levels of nuclear‐ and mitochondrial‐encoded proteins. SDHB was used as a loading control.
- Heart mitochondria (75 μg) from 4‐week‐old heart mitochondria from control and knockout mice were treated with 1% DDM and separated on a 4–30% BN–PAGE, and the respiratory complexes were immunoblotted using the OXPHOS BN–PAGE‐specific antibody.
- Loss of the ELAC2 causes profound reduction in mitochondrial respiration at complex I, II‐III, and IV in the knockout mice compared to controls. Non‐phosphorylating (state 4), phosphorylating (state 3), and uncoupled respiration in the presence of 0.5 μM CCCP was measured in heart mitochondria using an OROBOROS oxygen electrode using either pyruvate, glutamate, and malate or succinate as substrates in the presence of rotenone as indicated. L/L n = 4, L/L, cre n = 4; values are means ± SEM; **P < 0.01; ***P < 0.001, Student's t‐test.
- A continuous 10–30% sucrose gradient was used to determine the distribution of the small and large ribosomal subunit and the monosome in the control and knockout mice. Mitochondrial ribosomal protein markers of the small (MRPS35 and MRPS16) and large (MRPL37 and MRPL12) ribosomal subunits were detected by immunoblotting with specific antibodies. The data are representative of results from at least four independent biological experiments.
- The distribution of the mt‐Co1 mRNA, 12S and 16S rRNAs, and 12S‐16S rRNA junctions in sucrose gradients from L/L (top) and L/L, cre (bottom) 4‐week‐old mice was analyzed by qRT–PCR. The data are expressed as % of total RNA abundance and are representative of results from three independent biological experiments.
Mitochondrial gene expression is predominantly regulated at the post‐transcriptional level by RNA‐binding proteins 21 and impaired 5′ tRNA processing in Mrpp3 knockout mice caused a significant increase in nuclear‐encoded mitochondrial RNA‐binding proteins (mt‐RBPs) 10. Interestingly, immunoblotting for mt‐RBPs showed no change in response to impaired tRNA 3′ end processing between the Elac2 knockout and control mice (Fig EV2A), suggesting that the knockout mice are dying prematurely from other defects in addition to mitochondrial dysfunction. However, the levels of the mitochondrial proteases such as LONP1 and AFG3L2 were increased in the Elac2 knockout mice (Fig EV2B), indicating that there is retrograde signaling to the nucleus to upregulate these proteases in response to impaired OXPHOS biogenesis and function. These findings suggest that the initial stress response to imbalanced mitochondrial protein synthesis is increased protease activity, not upregulation of the mitochondrial gene expression machinery.
Figure EV2. Related to Fig 4: The effects of ELAC2 loss on RNA‐binding proteins.
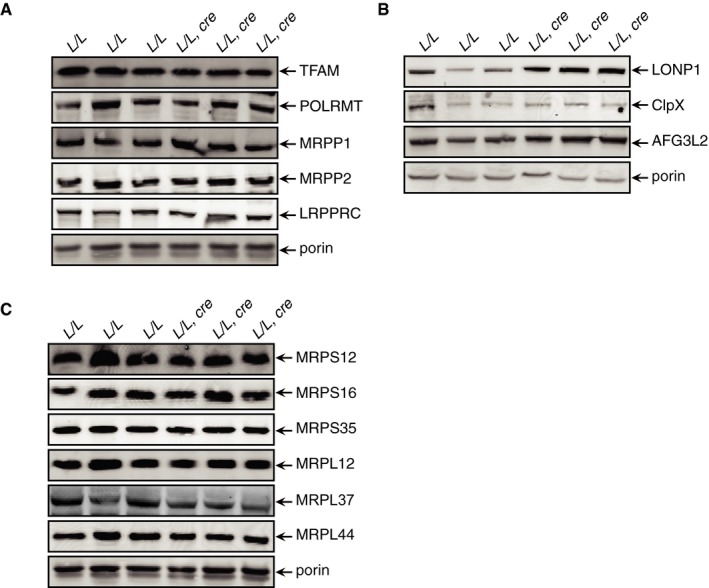
-
A–CThe levels of nuclear‐encoded mitochondrial RNA‐binding proteins (A), proteases (B), and mitochondrial ribosomal proteins (C) were measured by immunoblotting in heart mitochondria from 4‐week‐old control (L/L) and knockout (L/L, cre) mice. Porin was used as a loading control.
Loss of 3′ tRNA processing impairs mitoribosome assembly
Reduced mitochondrial protein synthesis in the Elac2 knockout mice prompted us to investigate the effects of ELAC2 loss on the steady‐state levels of mitoribosomal proteins (Fig EV2C). Surprisingly, we found that the levels of both small and large ribosomal proteins were not different between the Elac2 knockout and control mice (Fig EV2C), consistent with the levels of the mt‐RBPs (Fig EV2A). These findings are in contrast to the decreased levels of mitoribosomal proteins identified in Mrpp3 knockout mice where 5′ tRNA processing was impaired 10. Next, we analyzed how impaired 3′ tRNA processing affected the assembly and stability of the mitochondrial ribosome. We separated mitochondrial lysates on 10–30% sucrose gradients and analyzed the distribution of small and large ribosomal subunits by immunoblotting using specific antibodies (Fig 4E). Loss of 3′ tRNA processing impaired assembly of the 55S monosome, and the small and large ribosomal subunit proteins migrated closer to the top of the gradient in the Elac2 knockout mice (Fig 4E). The migration of the small ribosomal subunit proteins was particularly disrupted, suggesting that these proteins cannot associate with the unprocessed 12S rRNA. In contrast, the large ribosomal subunit proteins still formed a large molecular weight complex indicating that a subcomplex of the large ribosomal subunit could form. This is consistent with a previous observation when 5′ tRNA processing was impaired upon MRPP3 loss in mice 10, corroborating our findings that the unprocessed 16S rRNA transcript could stimulate early assembling large ribosomal proteins to form a subcomplex; however, the formation of the mature large subunit could not be finalized.
Next, we investigated the distribution of mitochondrial transcripts within the ribosomal fractions by qRT–PCR of each fraction (Fig 4F) to determine the distribution of the unprocessed transcripts within the sucrose gradient in control mice. The distribution of the 12S and 16S rRNA aligned with protein markers of the small and large ribosomal subunits, respectively (Fig 4F). Interestingly, the 16S rRNA was redistributed in the Elac2 knockout mice to less dense fractions of the sucrose gradient that co‐migrated with the unprocessed polycistronic transcripts that contain both the 12S and 16S rRNAs (Fig 4F) and indicated reduced assembly of 55S ribosomes. In the control mice, the mt‐Co1 mRNA was present in the pool of free mt‐mRNAs in the earlier fractions of the gradient and with the ribosomal fractions; however, upon ELAC2 loss, mt‐Co1 was redistributed to the top of the gradient because correctly assembled mitoribosome subunits that recruit mRNAs are lacking in these mice (Fig 4F). In addition, the unprocessed transcript containing the 12S and 16S rRNA co‐migrated with the large subcomplex detected in the sucrose gradient immunoblots from the Elac2 knockout mice (Fig 4E), but this unprocessed transcript was lacking in the control mice (Fig 4F). This finding is consistent with the large ribosomal subunit subcomplex that is assembled on the 12‐16S rRNA precursor transcript when MRPP3 is lost in mice, validating our conclusions that the assembly of the large ribosomal subunit is co‐transcriptional 10.
Combined defects in nuclear and mitochondrial gene expression in the absence of ELAC2 cause early‐onset death
Mutations in ELAC2 have been identified to cause severe mitochondrial disease involving cardiomyopathy by disruption of mitochondrial tRNA cleavage 22. However, the role of ELAC2 on nuclear RNA processing and their contribution to cardiomyopathy and disease have never been investigated, as it has been assumed that ELAC2 loss predominantly affects mitochondrial function. Therefore, we used RNA‐Seq to analyze the differential nuclear gene expression between Elac2 knockout mice compared to controls (Dataset EV1). We used these datasets to dissect the differences in gene expression of the dually targeted ELAC2 protein that regulates both nuclear and mitochondrial tRNA processing and compared them to RNA‐Seq datasets from mice lacking the mitochondria‐targeted MRPP3 protein that regulates tRNA processing only in mitochondria (Appendix Figs S1–S3). We identified the unique changes that are present in the Elac2 or Mrpp3 knockout mice along with the genes that are commonly altered between both Elac2 and Mrpp3 knockout mice relative to their respective control mice, which are a result of global OXPHOS dysfunction. Gene ontology analyses of the gene expression changes that were unique to the Elac2 knockout mice were identified for cellular compartments, biological processes, and molecular functions. The unique major changes in the Elac2 knockout mice in the cellular compartments GO are in the nucleus, cytosolic ribosomes, nuclear, and spliceosomal snRNP complexes, consistent with its localization in the nucleus. The changes that were in common with the Mrpp3 knockout mice were in mitochondria‐related genes affecting OXPHOS function and mitochondrial morphology consistent with ELAC2's dual localization to mitochondria and the nucleus (Appendix Fig S1).
The unique changes for biological processes in the Elac2 knockout mice predominantly involved nuclear non‐coding RNA metabolism, RNA processing, and the biogenesis of ribonuclear complexes, consistent with its nuclear localization where it is thought to also have a role in tRNA processing. In contrast, the changes that were common to both Elac2 and Mrpp3 knockout mice in biological processes were all related to metabolic changes in catabolism, carbon one metabolism, OXPHOS, apoptosis, and RNA regulation, in line with their role in mitochondrial RNA processing and the dire consequence of its loss on mitochondrial OXPHOS and biogenesis (Appendix Fig S2). The changes for molecular function that were common to both the Elac2 and Mrpp3 knockout mice related to RNA‐binding, nuclease activity, and protein binding that relate to their core functions in RNA‐binding and processing and their association with other proteins as components of granules or spliceosomal RNP complexes (Appendix Fig S3). Taken together, we identified that ELAC2 loss results in significant and unique defects in nuclear gene expression that are related to nuclear RNA processing, splicing, and cytoplasmic translation, in addition to changes in mitochondrial function that are a consequence of OXPHOS defects and are commonly found in mouse models where only mitochondrial RNA‐binding proteins have been knocked out 10. Our results indicate that the combined defects in impaired RNA processing of both nuclear and mitochondrial regulatory RNAs cause the early‐onset cardiomyopathy and death in our mice and possibly in patients with ELAC2 mutations.
ELAC2 loss impairs nuclear tRNA processing and causes imbalanced levels of regulatory non‐coding RNAs
The role of ELAC2 in nuclear RNA processing has not been investigated in vivo previously. To identify the in vivo role of the dually localized ELAC2 in nuclear‐encoded RNA processing, we investigated how its loss affected non‐coding and regulatory RNA metabolism by small RNA‐Seq (sRNA‐Seq). We also carried out microRNA sequencing (miRNA‐Seq) to identify changes in miRNAs, small RNAs, and tRNA fragments (tRFs) (Fig 5). We identified that the largest changes were in nuclear and mitochondrial DNA‐encoded tRNAs, although there were significant changes in other small RNA types including microRNAs (miRNAs), small nuclear RNAs (snRNAs), and particularly in small nucleolar RNAs (snoRNAs) (Fig 5A). Analyses of the tRNA levels identified that most nuclear‐encoded mature tRNA levels were significantly decreased and their precursors were increased, identifying ELAC2 as the major enzyme involved in processing of the 3′ end of nuclear tRNAs in vivo (Fig 5B). There was clear impairment of 3′ end processing in nuclear tRNAs in the Elac2 knockout mice (Fig 5C). Individual analyses of nuclear tRNAs identified the increase in the majority of tRNA precursors (Fig 5D), such as that shown for tRNA Asp(GTC) and tRNA Gly(GCC) (Fig 5E), and a dramatic decrease in their mature levels (Fig EV3A). We validated the effects on mature nuclear tRNAs by northern blotting where we observed significant decrease in the tRNA Asp(GTC), tRNA Gly(GCC), but not for one member of the tRNA Lys(TTT) family (Fig 5F), whereas other members and families such as the tRNA Lys(CTT) were significantly decreased (Fig EV3A and Dataset EV2). Investigation of precursor tRNAs identified significant accumulation of tRNAs retaining their 3′ trailer regions in the Elac2 knockout mice (Fig 5G), indicating that ELAC2 is essential for their processing in the nucleus. Our findings identify ELAC2 as a major enzyme required for 3′ end processing of most tRNAs in the nucleus. In addition, we show that ELAC2 is required for mascRNA processing from the Malat1 transcript, as mascRNA is absent in the Elac2 knockout mice (Fig EV3B), confirming that ELAC2 is essential for regulatory ncRNA processing both in the nucleus and in mitochondria.
Figure 5. ELAC2 is required for nuclear tRNA processing.
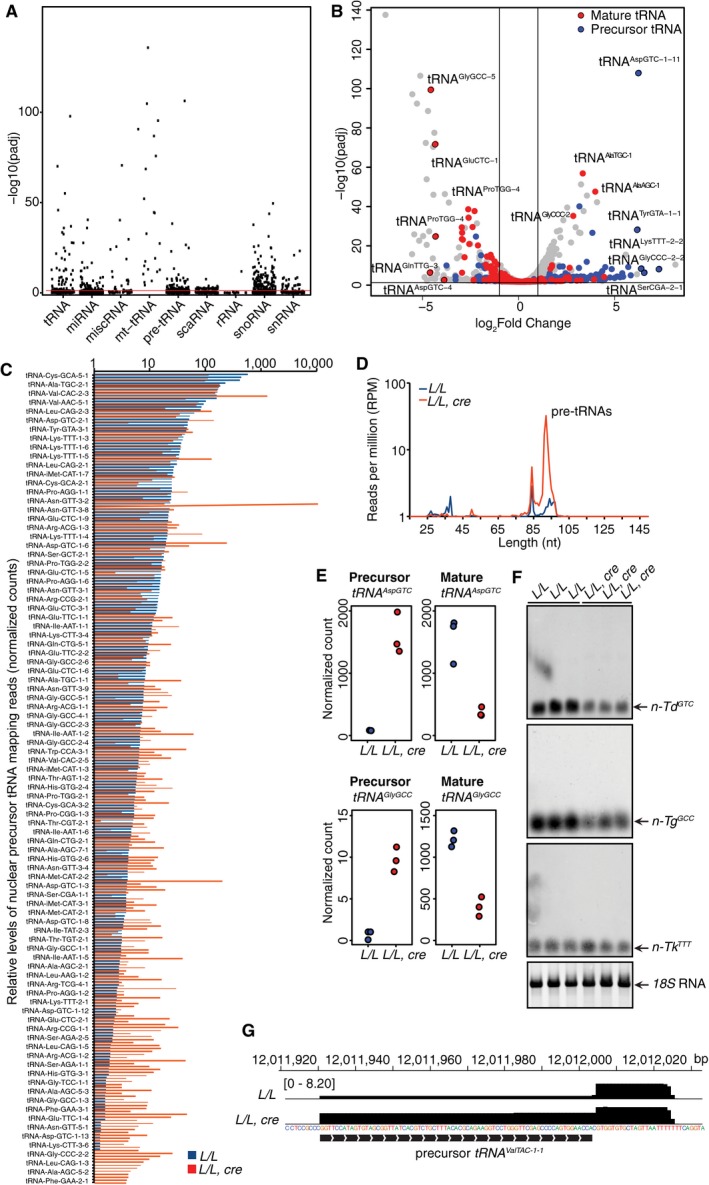
- Scatter plot of the significance of small RNA differential expression changes (−log10 adjusted P‐value).
- Volcano plot from the small RNA‐Seq dataset (−log10 adjusted P‐value versus log2 fold change) highlighting the mature and precursor tRNA mapping reads.
- Average relative abundance (normalized counts) of precursor tRNA mapping reads in L/L and L/L, cre mice (n = 3).
- Average length distribution of reads with 5ʹ ends mapped to the RNase P cleavage site of nuclear tRNAs in L/L and L/L, cre mice (reads per million, RPM; n = 3).
- Normalized read counts for precursor and mature mapped reads for tRNAAspGTC and tRNAGlyGCC in all samples.
- The abundance of nuclear tRNAs in hearts from 4‐week‐old L/L and L/L, cre mice was analyzed by northern blotting similarly as shown in Fig 2A, and 18S rRNA was used as a loading control. Nuclear precursor tRNAs were too low to detect by northern blotting and were identified by RNA‐Seq.
- Normalized coverage profile (reads per million, RPM) of pre‐tRNAValTAC−1‐1 demonstrating increased unprocessed transcripts in the knockout mice.
Source data are available online for this figure.
Figure EV3. Related to Figs 5 and 6: Loss of Elac2 causes a primarily transcriptional response.
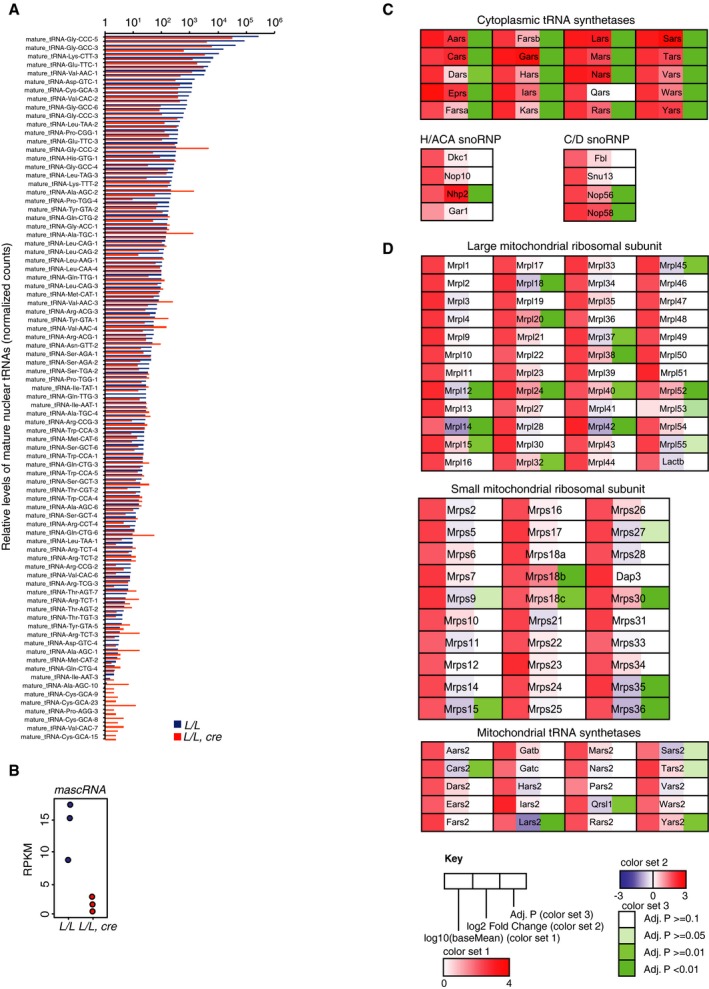
-
A–DReads mapping from the small RNA‐Seq datasets were summarized to identify that mature tRNA sequences were reduced in most cases in the Elac2 knockout mice (A), loss of the mascRNA derived from Malat1 (B), a strong coordinated increase in cytoplasmic tRNA synthetases and several snoRNP components (C). Differential expression analyses on the RNA‐Seq datasets identified an increased transcriptional regulation of mitochondrial ribosomal mRNAs and tRNA synthetases (D). Taken together, these data show that loss of ELAC2 and consequent impaired processing of non‐coding RNAs causes a compensatory increase in ribosomal proteins and tRNA synthetases in response to reduced mature tRNA levels.
Impaired tRNA processing stimulates C/D box snoRNA production to compensate for decreased translation
We investigated the consequences of reduced nuclear tRNA levels on cytoplasmic protein synthesis by de novo labeling using 35S cysteine and methionine to find ~50% reduction in translation upon ELAC2 loss in vivo (Fig 6A), indicating that this contributes to the premature death of the mice by 4 weeks of profoundly dilated cardiomyopathy. Sucrose gradient separation of cytoplasmic ribosomes from cell lysates followed by northern blotting to identify the effects on translation and polysome formation further indicated that ELAC2 loss led to decreased protein synthesis because of reduced polysome formation (Fig 6B), consistent with accumulation of precursor tRNAs that cannot be used for protein synthesis.
Figure 6. ELAC2 regulates the levels of regulatory ncRNAs.
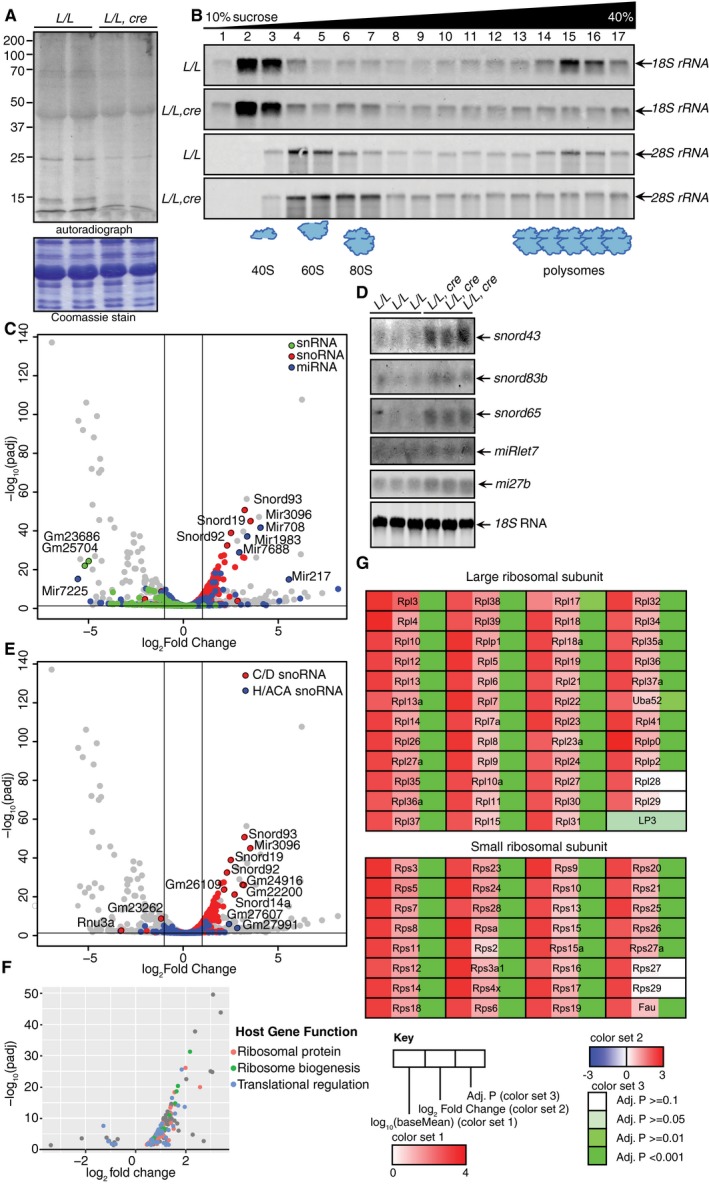
- De novo cytoplasmic protein synthesis was measured in 4‐week‐old L/L and L/L, cre mice using 35S‐labeled cysteine and methionine, and 15 μg of protein lysates was resolved by SDS–PAGE. Coomassie‐stained gels were used to show equal loading of protein lysates.
- Cytoplasmic ribosomes were resolved on 10–40% sucrose gradients and the RNAs associated with each fraction were used to detect the distribution of the 28S and 18S rRNAs by northern blotting.
- Volcano plot from the small RNA‐Seq dataset (−log10 adjusted P‐value versus log2 fold change) of small RNA mapping reads, highlighting snRNAs (green), snoRNAs (red), and miRNAs (blue).
- The abundance of snoRNAs in hearts from 4‐week‐old L/L and L/L, cre mice as analyzed by northern blotting and 18S rRNA was used as a loading control.
- Volcano plot from the small RNA‐Seq dataset (−log10 adjusted P‐value versus log2 fold change) of small RNA mapping reads, highlighting C/D box (red) and H/ACA box (blue) snoRNAs.
- Volcano plot (−log10 adjusted P‐value versus log2 fold change) of snoRNA host genes, highlighted according to function (ribosomal protein in red, ribosome biogenesis in green, and translation regulation in blue).
- Large and small ribosomal subunit mRNAs and their relative abundance, differential expression, and significance.
Source data are available online for this figure.
Next, we investigated the changes in the small non‐coding RNAs and identified that small nucleolar RNAs (snoRNA) and miRNAs were increased in the absence of ELAC2, whereas small nuclear RNAs (snRNAs) were decreased (Fig 6C). We validated our findings from the sRNA‐Seq by northern blotting, showing that the levels of snoRNAs, involved in rRNA processing and modification, as well as specific miRNAs were increased (Fig 6D). Next, we examined the effects of ELAC2 on the specific classes of snoRNAs and we identified a clear enrichment of the C/D snoRNAs but not H/ACA snoRNAs (Fig 6E), suggesting that loss of ELAC2 affects their levels and is important for pre‐rRNA processing and methylation of C/D snoRNA targets. Examination of the host genes of differentially expressed C/D snoRNAs in our RNA‐Seq datasets revealed that cytoplasmic ribosomal mRNAs along with ribosome biogenesis and translation‐related mRNAs are significantly upregulated in the Elac2 knockout mice (Figs 6F and EV3C), as a compensatory measure to decreased cytoplasmic protein synthesis. We identified that the expression of almost all large and small cytoplasmic ribosome coding mRNAs is significantly increased (Fig 6G), while mRNAs encoding mitochondrial ribosomal proteins have a less uniform response (Fig EV3D), identifying a transcriptional upregulation of cytoplasmic protein synthesis in response to decreased translation as a consequence of impaired nuclear and mitochondrial tRNA processing.
tRFs and termination‐associated RNAs—novel transcripts produced from pre‐tRNAs
Next, we examined tRNA fragment (tRF) expression, as recent studies have shown that these sRNAs are abundant and can have important functions in translation and mRNA stability 5. They are classified as primarily derived from the 5′ ends of mature tRNAs (tRF‐5s), the 3′ ends of mature tRNAs (tRF‐3s), or the 3′‐trailers of pre‐tRNAs (tRF‐1s). ELAC2 is known to generate a tRF‐1, known as tRF‐1001, from tRNA‐Ser‐TGA 6; however, the global importance of ELAC2 in tRF production has not been examined before. In our data (Dataset EV3), the pool of tRF‐5s was dominated by 28‐ and 31‐nt 5′ fragments of tRNA‐Gly‐GCC, with 3′ cleavage sites located within the anticodon stem and anticodon loop, respectively, which were greatly reduced in the Elac2 knockout mice (Fig EV4A). tRF‐5s have been presumed to be produced from mature tRNAs; however, they could in principle be derived from RNase P cleaved pre‐tRNAs without 3′‐end cleavage. Loss of ELAC2 and subsequent decrease in the majority of tRF‐5s indicates that they are produced from mature tRNAs (Fig 7A). Although the abundance of most tRF‐5s was reduced in the knockout mice, there were particular fragments that instead showed robust increases, including 23‐ and 32‐nt fragments of tRNA‐Val‐AAC/TAC and 18‐nt fragments of tRNA‐Ser‐GCT, tRNA‐His‐GTG, and tRNA‐Lys‐TTT (Dataset EV2). This indicates that either this subset of tRF‐5s can be derived from pre‐tRNAs or that they are preferentially stabilized in the stress conditions generated upon loss of ELAC2, possibly via association with argonaute proteins as has been previously observed 23.
Figure EV4. Related to Fig 7: Loss of Elac2 causes varied effects on tRNA fragment pools and increases in pre‐tRNA‐derived small RNAs.
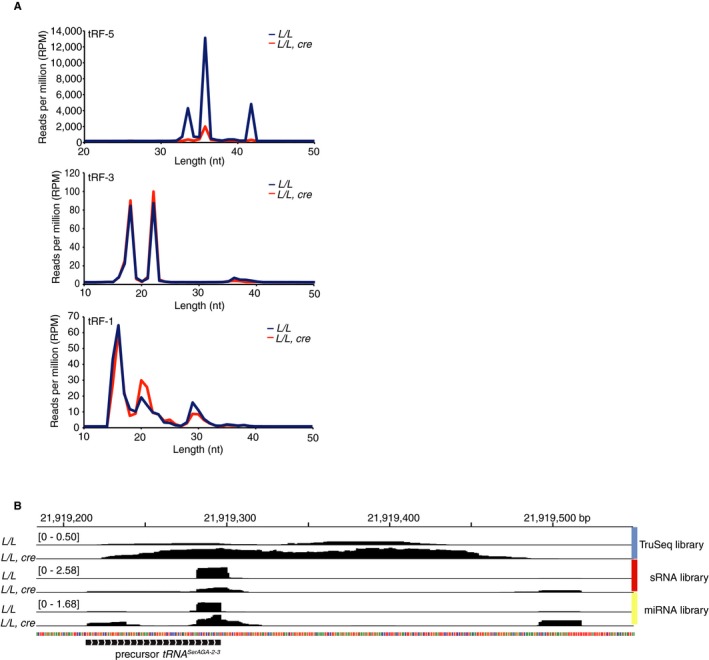
-
A, BWe identified that tRNA fragments derived from the 5′ and 3′ ends of mature tRNAs (tRF‐5 and tRF‐3, respectively) and 3′ pre‐tRNA trailers (tRF‐1) showed differential changes (A) and increases in pre‐tRNA‐derived small RNA associated with Pol III termination sites, including those derived from secondary termination sites from long pre‐tRNA transcripts (B).
Figure 7. Increased levels of specific pre‐tRNAs drive the production of miRNAs and taRNAs.
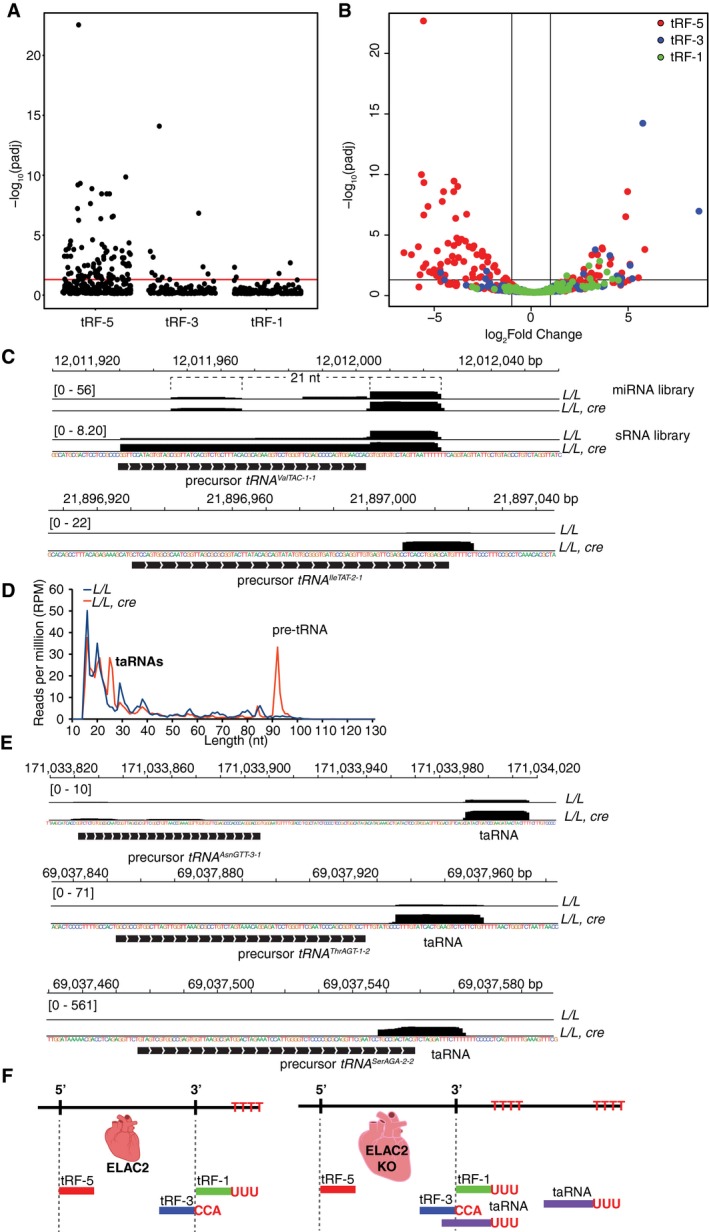
- Scatter plot of the significance of tRF‐5, tRF‐3, and tRF‐1 differential expression changes (−log10 adjusted P‐value).
- Volcano plot (−log10 adjusted P‐value versus log2 fold change) of differential expression changes for tRF‐5s (red), tRF‐3s (blue), and tRF‐1s (green).
- Normalized coverage profile (reads per million, RPM) of pre‐tRNAValTAC−1‐1 in miRNA (top panel) and sRNA libraries (bottom panel) illustrating the presence of a putative tRNA‐derived miRNA and miRNA* product.
- Average length distribution of reads mapped to nuclear tRNA loci, including 100 nt up‐ and downstream, in L/L and L/L, cre mice (reads per million, RPM; n = 3).
- Normalized coverage profiles (reads per million, RPM) of pre‐tRNAThrAGT−1‐2, pre‐tRNASerAGA−2‐2, and pre‐tRNAAsnGTT−3‐1 from the small RNA‐Seq dataset, illustrating the presence of taRNAs with 5′ ends up‐ and downstream of ELAC2 cleavage sites.
- Schematic diagram of the pathways producing tRFs and new taRNAs in L/L and L/L, cre mice.
In both the knockout and wild‐type mice, 18‐ and 22‐nt fragments were the most abundant 3′ tRNA fragments detected and, surprisingly, the abundance of tRF‐3 fragments was not altered as much as the tRF‐5s in the Elac2 knockout mice (Figs 7A and EV4A). The robust presence of tRF‐3s, which possess 3′‐CCAs acquired after 3′ end processing, indicates alternative pathways exist that are capable of 3′ cleavage in an ELAC2‐independent manner, which are already known to exist in yeast 24, although these cannot compensate for the loss of ELAC2 as the processing of the majority of nuclear tRNAs is compromised. Further insights into this process were found by analyzing the tRF‐1 fragment distribution in the Elac2 knockout mice. The most abundant tRF‐1s were predominantly 16, 20–21, and 29 nt in size and were mostly unchanged between the knockout and control mice (Figs 7A and B, and EV4A). However, there appear to be at least two cases where a pre‐tRNA‐encoded miRNA has increased expression in the knockout mice. One is located at the tRNA‐Ile‐TAT‐2‐1 locus (Fig 7C), which has been previously identified as a Dicer‐dependent miRNA cleaved from an alternate conformation of the pre‐tRNA 25. The other is located within the tRNAVal‐TAC‐1‐1 locus, where there are two fragments: a 21‐nt fragment immediately downstream of the tRNA 3′ end and an internal 21‐nt fragment of lower abundance. The latter fragment may be a miRNA* product given its size (Fig 7C). Both of these products have elevated abundance in the Elac2 knockout mice, likely because in the absence of ELAC2, these pre‐tRNAs are processed by the miRNA‐processing pathway.
There are several loci that produce an increased abundance of small RNAs that resemble tRF‐1s, in that they contain 3′ poly(U) motifs that align with the Pol III termination site, but have 5′ ends that do not align with the ELAC2 cleavage site. Some of these loci have secondary downstream poly(T) stretches that appear capable of terminating Pol III and producing long pre‐tRNAs that we have called termination‐associated RNAs (taRNAs). We identified increased levels of taRNAs while analyzing precursor tRNAs (Fig 7D). For example, the tRNA‐Asn‐GTT‐3‐1 locus produces a ~200‐nt pre‐tRNA, as well as a 26‐ to 27‐nt fragment at the second downstream poly(T) region located ~110‐nt downstream of the tRNA 3′ end (Fig 7E). Additionally, the Ser‐AGA‐2‐3 locus produces a pre‐tRNA that spans over 300 nt (Fig EV4B), as observed with RNA‐Seq, in addition to tRF‐5s, tRF‐3s, and a 26‐ to 27‐nt termination‐associated RNA (taRNA) fragment at terminating at a downstream poly(T) site. Other pre‐tRNAs produce taRNAs that include sections of the 3′ tRNA acceptor stem as well as the 3′ pre‐tRNA trailer. For example, tRNA‐Ser‐AGA‐2‐2 expresses a 25‐ to 29‐nt fragment that contains a 3′ poly(U) but its 5′ terminal nucleotide located 11 nt upstream of the tRNA 3′ end (Fig 7E). Both of these types of fragments are increased in abundance in the Elac2 knockout mice presumably due to the increase in unprocessed pre‐tRNA available resulting from loss of Elac2; however, their 5′ ends must be generated by another mechanism. All of these taRNAs have consistent sizes (25–27 nt) and appear to be products of Pol III termination due to the alignment of their 3′ ends within poly(T) genomic stretches. This suggests that they may be protein footprints of the Lupus autoantigen (La) protein that binds at the 3′ poly(U) stretch at the ends of Pol III‐generated transcripts (Fig 7F). The lack of Elac2 may prevent the release of La from the pre‐tRNA transcript, resulting in the fragment seen at Ser‐AGA‐2‐2; however, these RNAs are also observed far downstream of tRNA3′ ends indicating a potential secondary function of Elac2 in La release. La protein has previously been shown to stabilize pre‐tRNA structure to prevent refolding and processing via the miRNA pathway 26; however, our results indicate that in some circumstances it might also act as a molecular ruler to direct cleavage in adjacent 5′ regions.
Discussion
We have investigated the role of ELAC2 in vivo for the first time to identify its molecular targets and consequences of its loss. Our findings show that ELAC2 acts as the sole mitochondrial RNase Z and that it also has a major role in the processing of nuclear tRNAs in vivo. The specific deletion of ELAC2 in the hearts of mice caused a profound cardiomyopathy and premature death by 4 weeks of age. The dual localization of ELAC2 in the nucleus and mitochondria and its activity in both evidently contributes to this premature death, since deletion of the mitochondria‐specific endonuclease subunit of the mitochondrial RNase P complex, MRPP3, that cleaves the 5′ of mt‐tRNAs causes cardiomyopathy and death by 11 weeks of age 10. It is possible that a distinct mitochondrial RNase P enzyme has evolved to cope with the complexity of the polycistronic mitochondrial transcripts and extensive 5′ leaders, as 5′ tRNA processing precedes 3′ tRNA processing 10. ELAC2 performs a similar function by cleaving the 3′ tRNA ends of already 5′‐processed transcripts that are likely structurally similar to those in the nucleus. We show that ELAC2 is required for the cleavage of all mt‐tRNAs at their 3′ ends, although this process is preceded by the RNase P activity at the 5′ ends, since we do not observe large polycistronic transcripts accumulating within mitochondria nor impairment of 5′ tRNA processing. Impaired 3′ tRNA processing led to loss of mitochondrial protein synthesis in the absence of mature tRNAs, and consequently, the biogenesis and function of the mitochondrial ribosomes and OXPHOS system were severely diminished. We observed similar defects when we deleted Mrpp3 in mice 10, but in contrast to this model of impaired 5′ tRNA processing, loss of ELAC2 did not alter the levels of mt‐RBPs nor mitoribosomal proteins, which were dramatically increased in the Mrpp3 knockout mice. This is likely because the mice die very young and there is insufficient time for the activation of retrograde responses to mitochondrial dysfunction, as identified in mice beyond 10 weeks 27. Alternatively, the combined consequences of impaired tRNA processing in both mitochondria and the nucleus may activate different pathways to those in the Mrpp3 knockout mice that do not require transcriptional upregulation of mitochondrial RBPs.
Here, we show that ELAC2 is responsible for the processing of most nuclear‐encoded tRNAs and validate that ELAC2 cleaves the mascRNA from its Malat1 transcript as previously shown in vitro 28. Loss of ELAC2 leads to a profound increase in nuclear pre‐tRNAs and a reduction in mature tRNAs that is sufficient to decrease cytoplasmic protein synthesis significantly and reduce polysome formation. Interestingly, in response to impaired 3′ tRNA processing in the nucleus and reduced translation we identified a specific upregulation of C/D box snoRNAs as an alternate mode to attempt to compensate for impaired cytoplasmic protein synthesis. Their expression correlates with that of their host genes, all of which are increased to upregulate protein synthesis and ribosomal assembly. This also likely contributes to the transcriptional upregulation of both cytoplasmic and mitochondrial translation factors and ribosomal proteins, all of which are incapable of overcoming the dramatic loss of mature tRNAs. Furthermore, our findings indicate that ELAC2 plays a downstream role in snoRNA‐mediated RNA modification in the nucleolus.
Here, we show that the loss of ELAC2 has a significant effect on the processing of pre‐tRNAs into miRNAs and tRNA fragments, both of which are important for regulating gene expression. 5′ tRF from tRNA‐Val has been previously reported to be induced under multiple stress conditions in HepG2 cells including nutrient deficiency, heat shock, hypothermia, and hypoxia 29, and the 32‐nt Val fragment detected in our dataset contains the CU box motif (SCUBYC) required for Ybx1 binding but lacks the 5′ TOG motif needed for G‐quadruplex formation and stress granule production 30. A tRNA‐His fragment has previously been observed bound to the ribosome in yeast 31, and a 5′ Lys fragment was increased upon respiratory syncytial virus infection in A549 and small alveolar epithelial cells 32. No functions have been reported for tRF‐5‐Ser‐GCT; however, it has been reported that short 5′ tRFs require a 3′‐GG dinucleotide to inhibit translation in human cells 33, a feature present at the 3′ ends of the Ser‐GCT and His‐GTG fragments. tRFs have also been reported to associate with Argonaute proteins and appear to possess the ability to silence gene expression at the RNA level 23. The coordinated upregulation of ribosomal and translation‐related mRNAs in the absence of ELAC2 may be the result of the loss of tRF‐mediated gene silencing or translation inhibition. This is an attractive idea as it would constitute a signaling mechanism to coordinate the expression of tRNAs with their cognate partners, the ribosome, and tRNA synthetases.
Our identification of a new class of tRNA fragments, the taRNAs, is intriguing as their 5′ ends appear to be primarily determined by the distance from the Pol III termination site since they are of consistent size whether they are derived from the distal ends of long pre‐tRNAs terminating at secondary downstream termination sites or at primary termination sites of short pre‐tRNAs. Their alignment to Pol III transcript ends implicates Pol III termination and the La protein in their biogenesis, in concert with as yet unknown nuclease to generate the 5′ ends. Alterations to La function, and particularly release of La from precursor transcripts, may also be responsible for the changes observed for snoRNA and miRNAs, since La is known to associate with DGCR8 and participates in miRNA biogenesis 34, and it is also reported to associate with RNase P to target the exosome to snoRNAs for degradation 35. It is possible that ELAC2 is also involved in snoRNA degradation, which could explain their increased abundance upon ELAC2 loss. Alternatively, impaired tRNA processing may be sequestering a proportion of La by preventing its removal from precursors, with downstream effects for non‐tRNA processes involving La.
In conclusion, we identify that RNA processing in vivo is essential for gene expression via the regulation of non‐coding regulatory RNAs and the biogenesis of the nuclear and mitochondrial proteomes. The coordination between RNA processing and gene expression highlights the importance of miRNAs, snoRNAs, and new taRNAs for gene expression and survival.
Materials and Methods
Animals and housing
Elac2 transgenic mice on a C57BL/6N background were generated by the European Mouse Mutant Archive, EMMA (Biomodels, Austria). The neomycin cassette was removed by mating Elac2 +/loxP‐neo mice with transgenic mice ubiquitously expressing Flp recombinase. The resulting Elac2 +/loxP mice were mated with mice ubiquitously expressing Cre recombinase to generate heterozygous knockout Elac2 +/− mice that were bred with each other to identify that the homozygous loss of Elac2 was embryonic lethal. Heart‐ and skeletal muscle‐specific knockout mice were generated by crossing Elac2 loxP/loxP mice with transgenic mice expressing Cre under the control of the muscle creatinine kinase promoter (Ckmm‐cre). Double heterozygous mice (Elac2 loxP/+, +/Ckmm) were mated with Elac2 loxP/loxP mice to generate heart‐specific knockout (Elac2 loxP/loxP, +/Ckmm) and control mice (Elac2 loxP/loxP). Mice were housed in standard cages (45 cm × 29 cm × 12 cm) under a 12‐h light/dark schedule (lights on 7 am to 7 pm) in controlled environmental conditions of 22 ± 2 °C and 50 + 10% relative humidity and fed a normal chow diet (Rat & Mouse Chow, Specialty Foods, Glen Forrest, Western Australia), and water was provided ad libitum. The study was approved by the Animal Ethics Committee of the UWA and performed in accordance with Principles of Laboratory Care (NHMRC Australian code for the care and use of animals for scientific purposes, 8th Edition 2013).
Mitochondrial isolation
Mitochondria were isolated from homogenized hearts and isolated by differential centrifugation as described previously 10, 36.
Sucrose gradient fractionation
Sucrose gradient fractionation was carried out as described previously 10, 36.
Immunoblotting
Specific proteins were detected using rabbit polyclonal antibodies against: POLRMT (produced in‐house, diluted 1:1,000), TFAM (ab131607), MRPP1 (HPA036671 Sigma), MRPP2 (SAB1405920, Sigma), ELAC2 (10061‐1‐AP), LRPPRC (sc66844, Santa Cruz), MRPL44 (16394‐1‐AP), MRPL37 (15190‐1‐AP) (1:1,000), MRPL12 (14795‐1‐AP) (1:500), MRPS35 (16457‐1‐AP) (1:1,000), MRPS16 (16735‐1‐AP) (Proteintech), Clpx (Sigma—Prestige Antibodies HPA040262, diluted 1:500), Lonp (15440‐1‐AP), YME1L1 (ab170123), OMA‐1 (ab104316) Abcam, diluted 1:500), and mouse monoclonal antibodies against: β‐actin (ab8226), porin (ab15895), total OXPHOS cocktail antibody (ab110412), NDUFA9 (ab14714), SDHA (ab14715), UQCRC2 (ab14745), COXI (ab14705), COXII (ab198286), COXIII (ab110259), COXIV (ab14744), ATP5a (ab14748), ATP inhibitory factor 1 (ab110277) (Abcam, diluted 1:1000), in Odyssey blocking buffer (Li‐Cor). IR Dye 800CW goat anti‐rabbit IgG or IRDye 680LT goat anti‐mouse IgG (Li‐Cor) secondary antibodies were used, and the immunoblots were visualized using an Odyssey Infrared Imaging System (Li‐Cor).
DNA quantification
Real‐time PCR was conducted on 90 ng of DNA isolated from mouse hearts using primers for Rnr2 and β‐2 microglobulin to determine mitochondrial and nuclear DNA levels, respectively (GeneWorks). Amplification was conducted using a Rotor‐Gene Q (Qiagen) using SensiMix SYBR mix (Bioline).
RNA isolation, northern blotting, and qRT–PCR
RNA was isolated from total hearts or heart mitochondria using the miRNeasy Mini kit (Qiagen) incorporating an on‐column RNase‐free DNase digestion to remove all DNA. RNA (5 μg) was resolved on 1.2% agarose formaldehyde gels, then transferred to 0.45‐μm Hybond‐N+ nitrocellulose membrane (GE Lifesciences), and hybridized with biotinylated oligonucleotide probes specific to mouse mitochondrial mRNAs, rRNAs, and tRNAs 37. Hybridizations were carried out overnight at 50°C in 5× SSC, 20 mM Na2HPO4, 7% SDS, and 100 μg/ml heparin, followed by washing. The signal was detected using streptavidin‐linked infrared‐labeled antibody (diluted 1: 2,000 in 3× SSC, 5% SDS, 25 mM Na2HPO4, pH 7.5) using an Odyssey Infrared Imaging System (Li‐Cor), respectively. Complementary DNA (cDNA) was prepared using the QuantiTect Reverse Transcription Kit (Qiagen) and used as a template in the subsequent PCR that was performed using a Rotor‐Gene Q using SensiMix SYBR mix (Bioline) and normalized to 18S rRNA.
RNA‐Seq and alignments
Strand‐specific RNA sequencing was performed on total RNA from three control and three Elac2 knockout mice using the Illumina NextSeq 500 platform, according to the Illumina TruSeq RNA‐Seq, small RNA‐Seq, and miRNA protocols. We used RNA‐Seq to investigate nuclear and mitochondrial mRNA and rRNA changes using the standard TruSeq library preparation and also to investigate precursor RNA accumulation as we have previously 10. RNA sequencing libraries for small RNA‐Seq were constructed using the Illumina Small RNA Sample Prep Kit, with a size selection step for 15‐ to 100‐nt RNAs and were used to analyze tRNAs and other non‐coding RNAs. miRNA libraries were constructed according to the Illumina miRNA protocol and were used to analyze miRNAs, small RNAs, and tRNA fragments. Although there are approaches used for tRNA sequencing via demethylation of RNA prior to sequencing, this method is no more quantitative than small RNA‐Seq and we have found that it is more effective to sequence small RNA‐Seq libraries to high depth without prior manipulation of the RNA samples. This also enables matched RNA samples to be used for small RNA‐Seq, miRNA‐Seq, and standard RNA‐Seq in parallel, which enables reliable comparisons across different libraries.
small RNA
Raw reads were trimmed of adapter sequences (5′‐AGATCGGAAGAGCACACG‐TCTGAACTCCAGTCAC‐3′) with cutadapt v1.14 38, discarding reads with a trimmed length of less than 15 nt. Only reads with successfully trimmed adapters were retained for subsequent analysis. Trimmed reads for technical replicates were pooled for each sample and alignment to the NumtS‐masked mouse genome (mm10) plus a set of mature tRNA sequences, using Bowtie 2 v2.2.9 39 with the parameters: ‐D 20 ‐R 3 ‐N 0 ‐L 15 ‐i S,1,0.50 ‐k 100 (equivalent to the –very sensitive preset, with the seed substring shortened to 15 nt and up to 100 alignments reported). The mature tRNA sequences were derived from the genomic tRNA database (GtRNAdb) 40, by removing intronic sequences, adding a 3′‐CCA to each tRNA and a 5′ G nucleotide to each histidine tRNA, and collapsing the set to unique sequences. In addition, 11 tRNA loci possessed genome‐encoded CCA trinucleotides immediately downstream of what would be the discriminator base of the mature sequence, as well as degenerate, or extremely proximal or distal downstream Pol III transcription termination sites. Cleavage of precursor tRNA 3′ ends by mammalian RNase Z is inhibited by the presence of a 3′‐CCA 41, particularly if the 3′ trailer is very short or very long, therefore these loci appear to be less likely of producing mature tRNAs. In addition, most of these loci have low scores on gtRNAdb and several possess sequences that, if expressed and successfully processed into mature tRNAs, would be identical to other tRNAs in the set of unique sequences, making the set non‐unique, and as such, these tRNAs were excluded from the unique mature set (but retained as part of the precursor annotations).
Prior to differential expression, a single alignment for each read was retained according to the alignment scores of the highest scoring primary and secondary alignments, and their mapping locations (secondary alignments with scores lower than the primary were not considered). All primary alignments with a higher alignment score than any secondary alignments were retained. Primary alignments with one or more secondary alignments with equal scores (ambiguous multimappers) were retained if none mapped to precursor or mature tRNA sequences, or the mitochondrial chromosome. Those that did map to a tRNA sequence were retained according to their mapping locations: if all alignments mapped either to precursor or mature tRNA, then the primary precursor or mature alignment was retained, respectively; if the alignments were mapped to both precursor and mature tRNA, but nowhere else, then a mature tRNA alignment was randomly selected (to improve the specificity of precursor alignments); if the alignments mapped to a precursor or mature tRNA and chrM, a mitochondrial alignment was randomly retained (under the rationale that a mitochondrial tRNA is the most likely source due to the high amount of mitochondrial RNA in cardiomyocytes); if the equally scoring alignments mapped to multiple locations on chrM, or if they mapped to other nuclear genomic locations in addition to tRNAs, they were excluded from further analysis. Gene‐level read counts were produced using featureCounts v1.5.1 42 (–largestOverlap ‐s 1) with the GENCODE vM14 annotation limited to small ncRNA annotations (miRNA, snoRNA, snRNA, sRNA, scaRNA, scRNA, rRNA, and misc_RNA) combined with the GtRNAdb‐derived precursor and mature tRNA annotations, and an in‐house mitochondrial annotation. Differential expression was performed with DESeq2 v1.16.1 43 and visualized with ggplot2 44 in the R statistical environment.
TruSeq RNA‐Seq
Raw reads from technical replicates for each sample were pooled and aligned to the NumtS‐masked mouse genome (mm10) with HISAT2 v2.0.5 45 using default parameters. Alignments were filtered to retain only properly paired, primary alignments with SAMtools v1.5 46. Read pairs that aligned to the mitochondria were realigned to the mitochondrial genome sequence with the –no‐spliced‐alignment parameter enabled, to avoid introducing spurious splice junctions into mitochondrial alignments. The mitochondrial and nuclear alignments were combined, and gene‐level counts were produced with featureCounts v1.5.1 42 (–largestOverlap ‐s 2) using the GENCODE vM14 annotation and an in‐house mitochondrial annotation. Differential expression analysis was performed with DESeq2 v1.16.1 43 and visualized with ggplot2 v2.2.1 44 and PathVisio v3.2.4 47. Mitochondrial genome coverage across both strands (raw and normalized) of three Elac2 knockout (L/L, cre) and three control (L/L) mice for the TruSeq library is shown in Dataset EV4.
tRF analysis
Raw paired‐end reads from the miRNA sequencing library were merged with BBMap v37.02 (sourceforge.net/projects/bbmap), removing the adapter sequences, and technical replicates were pooled. Merged and pooled reads with a length of at least 15 nt were aligned the mouse genome (mm10) with Bowtie 2 v2.2.9 39 with the parameters: ‐D 20 ‐R 3 ‐N 0 ‐L 15 ‐i S,1,0.50 ‐k 100 (equivalent to the –very sensitive preset, with the seed substring shortened to 15 nt and up to 100 alignments reported). In order to account for the presence of non‐template 3′‐CCA found on tRF‐3s, all unaligned and non‐exactly aligning reads were extracted and cutadapt v1.1.4 used to detect and trim up to a single ‐CCA triplet anchored at the 3′ end. Trimmed reads were subsequently aligned to the mouse genome and filtered for exact alignments as above. Multimapping reads (those with equally top scoring primary and secondary alignments) that mapped to genomic tRNA loci were retained only if all equally scoring secondary alignments were also mapped to tRNA loci from which the primary alignment was retained. Primary alignments were also retained for multimappers outside of tRNA loci and non‐multimappers. tRNA fragments were identified according to the mapped position of their 5′ and 3′ terminal nucleotides: tRF‐5s by the 5′ terminal nucleotide of a read mapping to the 5′ end of a tRNA; tRF‐3s by the 3′ end from the CCA‐trimmed data mapping to the 3′ end of a tRNA; and tRF‐1s if the 5′ end mapped to a tRNA 3′+1 position. Reads from these sets of alignments were extracted and collapsed by sequence to identify the most abundant fragments, irrespective of mapping location (ambiguous in the case of multimappers). Potential source loci were identified by scanning tRNA sequences using the putative tRF sequences with scan_for_matches and examining coverage of these with the full set of exact alignments.
Translation assays
In organello translation, assays were carried out in isolated heart mitochondria as described before 10, 36.
Blue native PAGE
BN–PAGE was carried out using isolated mitochondria from hearts as described previously 48. BN–PAGE gels were analyzed by transferring to PVDF and immunoblotting against the respiratory complexes.
Respiratory chain function and complex activity
The mitochondrial oxygen consumption flux was measured with an Oxygraph‐2k (Oroboros Instruments), as previously described 10, 36, 48.
Histology
Mouse hearts were fixed with 10% neutral‐buffered formalin for 24 h and stored in phosphate‐buffered saline or 70% ethanol. Tissues were embedded in paraffin, sectioned using a microtome transferred to positively charged slides. Slides were heated for 2 h at 60°C and treated with xylene, xylene and ethanol (1:1), and decreasing concentrations of ethanol (100, 95, 80, 60%) before they were washed in distilled H2O (dH2O). The H&E staining was performed as described before 49. Skeletal muscle was frozen in OCT, and the tissue was sectioned and stained for complex I and IV activity using NADH and cytochrome c oxidase as substrates. Coverslips were attached using DPX mounting media (Scharlau), and images were acquired using a Nikon Ti Eclipse inverted microscope using a Nikon 20× objective.
Echocardiography
Echocardiography was performed on mice under light methoxyflurane anesthesia with the use of an i13L probe on a Vivid 7 Dimension (GE Healthcare) as described previously 27.
Data availability
The transcriptomic data from this publication have been deposited to the Gene Expression Omnibus database https://www.ncbi.nlm.nih.gov/geo/ and assigned the identifier GSE111228.
Author contributions
OR and AF conceived the project. OR and AF designed the experiments. SJS, GR, TRR, KP, JAE, IK, LH, A‐MJS, HMV, LCH, OR, and AF conducted and analyzed the experiments. SS and IK also carried out the bioinformatic analyses. LH, OR, and AF provided funding and supervision. SS, OR, and AF wrote the manuscript and the other authors approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Source Data for Figure 2A
Source Data for Figure 5F
Source Data for Figure 6D
Acknowledgements
This project was supported by fellowships and project grants from the National Health and Medical Research Council (APP1058442, APP1045677, APP1041582, APP1023460, APP1005030, APP1043978 to AF and OR), Australian Research Council (to AF and OR), the Cancer Council of Western Australia (to OR). SS was supported by a UWA Postgraduate scholarship, and KLP was supported by NHMRC Dora Lush and AMDF scholarships. We thank AGRF for RNA sequencing. We thank the Wellcome Trust Sanger Institute Mouse Genetics Project (Sanger MGP) and its funders for providing the mutant mouse line (Allele: Elac2<tm1a(EUCOMM)Wtsi>), and the European Mouse Mutant Archive (http://www.infrafrontier.eu) partner University of Veterinary Medicine, Vienna, Austria, for rederivation and breeding of the mouse mutant strain carrying the floxed allele (Elac2<tm1c(EUCOMM)Wtsi>).
EMBO Reports (2018) 19: e46198
References
- 1. Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS (2010) Non‐coding RNAs: regulators of disease. J Pathol 220: 126–139 [DOI] [PubMed] [Google Scholar]
- 2. Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A (2007) The expanding RNA polymerase III transcriptome. Trends Genet 23: 614–622 [DOI] [PubMed] [Google Scholar]
- 3. Altman S, Kirsebom L, Talbot S (1995) tRNA: structure, biosynthesis, and function.
- 4. Chen Y, Chen Y, Beck A, Beck A, Davenport C, Davenport C, Chen Y, Shattuck D, Tavtigian S, Shattuck D et al (2005) Characterization of TRZ1, a yeast homolog of the human candidate prostate cancer susceptibility gene ELAC2 encoding tRNase Z. BMC Mol Biol 6: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maraia RJ, Lamichhane TN (2010) 3′ processing of eukaryotic precursor tRNAs. WIREs RNA 2: 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee YS, Shibata Y, Malhotra A, Dutta A (2009) A novel class of small RNAs: tRNA‐derived RNA fragments (tRFs). Genes Dev 23: 2639–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gustafsson CM, Falkenberg M, Larsson N‐G (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85: 133–160 [DOI] [PubMed] [Google Scholar]
- 8. Montoya J, Ojala D, Attardi G (1981) Distinctive features of the 5′‐terminal sequences of the human mitochondrial mRNAs. Nature 290: 465–470 [DOI] [PubMed] [Google Scholar]
- 9. Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135: 462–474 [DOI] [PubMed] [Google Scholar]
- 10. Rackham O, Busch JD, Matic S, Siira SJ, Kuznetsova I, Atanassov I, Ermer JA, Shearwood A‐MJ, Richman TR, Stewart JB et al (2016) Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep 16: 1874–1890 [DOI] [PubMed] [Google Scholar]
- 11. Lopez Sanchez MI, Sanchez MIGL, Mercer TR, Davies SM, Davies SMK, Shearwood A‐MJ, Nygård KKA, Nygård KK, Richman TR, Mattick JS et al (2011) RNA processing in human mitochondria. Cell Cycle 10: 2904–2916 [DOI] [PubMed] [Google Scholar]
- 12. Brzezniak LK, Bijata M, Szczesny RJ, Stepien PP (2011) Involvement of human ELAC2 gene product in 3′ end processing of mitochondrial tRNAs. RNA Biol 8: 616–626 [DOI] [PubMed] [Google Scholar]
- 13. Reinhard L, Sridhara S, Hallberg BM (2017) The MRPP1/MRPP2 complex is a tRNA‐maturation platform in human mitochondria. Nucleic Acids Res 45: 12469–12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rackham O, Mercer TR, Filipovska A (2012) The human mitochondrial transcriptome and the RNA‐binding proteins that regulate its expression. WIREs RNA 3: 675–695 [DOI] [PubMed] [Google Scholar]
- 15. Rossmanith W (2011) Localization of human RNase Z isoforms: dual nuclear/mitochondrial targeting of the ELAC2 gene product by alternative translation initiation. PLoS ONE 6: e19152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Park CB, Asin‐Cayuela J, Cámara Y, Shi Y, Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument‐Bromage H, Tempst P et al (2007) MTERF3 is a negative regulator of mammalian mtDNA transcription. Cell 130: 273–285 [DOI] [PubMed] [Google Scholar]
- 17. Metodiev MD, Spåhr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J, Habermann B, Larsson N‐G, Ruzzenente B (2014) NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet 10: e1004110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Cámara Y, Milenkovic D, Zickermann V, Wibom R, Hultenby K et al (2012) LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J 31: 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Metodiev M, Lesko N, Park C, Camara Y, Shi Y, Wibom R, Hultenby K, Gustafsson C, Larsson N (2009) Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab 9: 386–397 [DOI] [PubMed] [Google Scholar]
- 20. Cámara YY, Asin‐Cayuela JJ, Park CBC, Metodiev MDM, Shi YY, Ruzzenente BB, Kukat CC, Habermann BB, Wibom RR, Hultenby KK et al (2011) MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab 13: 527–539 [DOI] [PubMed] [Google Scholar]
- 21. Liu G, Mercer TR, Shearwood A‐MJ, Siira SJ, Hibbs ME, Mattick JS, Rackham O, Filipovska A (2013) Mapping of mitochondrial RNA‐protein interactions by digital RNase footprinting. Cell Rep 5: 839–848 [DOI] [PubMed] [Google Scholar]
- 22. Haack TB, Kopajtich R, Freisinger P, Wieland T, Rorbach J, Nicholls TJ, Baruffini E, Walther A, Danhauser K, Zimmermann FA et al (2013) ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet 93: 211–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haussecker D, Huang Y, Lau A, Parameswaran P, Fire AZ, Kay MA (2010) Human tRNA‐derived small RNAs in the global regulation of RNA silencing. RNA 16: 673–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skowronek E, Grzechnik P, Spaeth B, Marchfelder A, Kufel J (2014) tRNA 3′ processing in yeast involves tRNase Z, Rex1, and Rrp6. RNA 20: 115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R (2008) Mouse ES cells express endogenous shRNAs, siRNAs, and other microprocessor‐independent, Dicer‐dependent small RNAs. Genes Dev 22: 2773–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hasler D, Lehmann G, Murakawa Y, Klironomos F, Jakob L, Grässer FA, Rajewsky N, Landthaler M, Meister G (2016) The lupus autoantigen la prevents mis‐channeling of tRNA fragments into the human microRNA pathway. Mol Cell 63: 110–124 [DOI] [PubMed] [Google Scholar]
- 27. Perks KL, Ferreira N, Richman TR, Ermer JA, Kuznetsova I, Shearwood A‐MJ, Lee RG, Viola HM, Johnstone VPA, Matthews V et al (2017) Adult‐onset obesity is triggered by impaired mitochondrial gene expression. Sci Adv 3: e1700677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilusz JE, Freier SM, Spector DL (2008) 3′ end processing of a long nuclear‐retained noncoding RNA yields a tRNA‐like cytoplasmic RNA. Cell 135: 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fu H, Feng J, Liu Q, Sun F, Tie Y, Zhu J, Xing R, Sun Z, Zheng X (2009) Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett 583: 437–442 [DOI] [PubMed] [Google Scholar]
- 30. Lyons SM, Gudanis D, Coyne SM, Gdaniec Z, Ivanov P (2017) Identification of functional tetramolecular RNA G‐quadruplexes derived from transfer RNAs. Nat Commun 8: 1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zywicki M, Bakowska‐Zywicka K, Polacek N (2012) Revealing stable processing products from ribosome‐associated small RNAs by deep‐sequencing data analysis. Nucleic Acids Res 40: 4013–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Q, Lee I, Ren J, Ajay SS, Lee YS, Bao X (2013) Identification and functional characterization of tRNA‐derived RNA fragments (tRFs) in respiratory syncytial virus infection. Mol Ther 21: 368–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sobala A, Hutvagner G (2013) Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biol 10: 26–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng Q, Yang H‐J, Yuan YA (2017) Autoantigen la regulates microRNA processing from stem‐loop precursors by association with DGCR8. Biochemistry 56: 6098–6110 [DOI] [PubMed] [Google Scholar]
- 35. Macias S, Cordiner RA, Gautier P, Plass M, Cáceres JF (2015) DGCR8 acts as an adaptor for the exosome complex to degrade double‐stranded structured RNAs. Mol Cell 60: 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Richman TR, Spåhr H, Ermer JA, Davies SMK, Viola HM, Bates KA, Papadimitriou J, Hool LC, Rodger J, Larsson N‐G et al (2016) Loss of the RNA‐binding protein TACO1 causes late‐onset mitochondrial dysfunction in mice. Nat Commun 7: 11884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rackham O, Davies SMK, Shearwood A‐MJ, Hamilton KL, Whelan J, Filipovska A (2009) Pentatricopeptide repeat domain protein 1 lowers the levels of mitochondrial leucine tRNAs in cells. Nucleic Acids Res 37: 5859–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet J 17: 10–12 [Google Scholar]
- 39. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Meth 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chan PP, Lowe TM (2016) GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44: D184–D189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nashimoto M (1997) Distribution of both lengths and 5′ terminal nucleotides of mammalian pre‐tRNA 3″ trailers reflects properties of 3″ processing endoribonuclease. Nucleic Acids Res 25: 1148–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- 43. Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ginestet C (2011) ggplot2: elegant graphics for data analysis. J Roy Stat Soc 174: 245–245 [Google Scholar]
- 45. Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Meth 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kutmon M, van Iersel MP, Bohler A, Kelder T, Nunes N, Pico AR, Evelo CTA (2015) PathVisio 3 ‐ an extendable pathway analysis toolbox. PLoS Comput Biol 11: e1004085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mourier A, Ruzzenente B, Brandt T, Kühlbrandt W, Larsson N‐G (2014) Loss of LRPPRC causes ATP synthase deficiency. Hum Mol Genet 23: 2580–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Richman TR, Ermer JA, Davies SMK, Perks KL, Viola HM, Shearwood A‐MJ, Hool LC, Rackham O, Filipovska A (2015) Mutation in MRPS34 compromises protein synthesis and causes mitochondrial dysfunction. PLoS Genet 11: e1005089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Source Data for Figure 2A
Source Data for Figure 5F
Source Data for Figure 6D
Data Availability Statement
The transcriptomic data from this publication have been deposited to the Gene Expression Omnibus database https://www.ncbi.nlm.nih.gov/geo/ and assigned the identifier GSE111228.