

Abstract
Background
Marfan syndrome (MFS) is an autosomal dominant connective tissue disorder caused by mutations in the FBN1 gene. Approximately 90% of classic MFS patients have a FBN1 mutation that can be identified by single-gene sequencing or gene-panel sequencing targeting FBN1. However, a small proportion of MFS patients carry a large genomic deletion in FBN1, which cannot be detected by routine sequencing. Here, we performed an MLPA (multiplex ligation-dependent probe amplification) test to detect large deletions and/or duplications in FBN1 and TGFBR2 in 115 unrelated Chinese patients with suspected MFS or early-onset aneurysm/dissection.
Results
Five novel large deletions encompassing a single exon or multiple exons in the FBN1 gene were characterized in five unrelated patients, of which four were proven by Sanger sequencing, and the breakpoints were identified. Three of them met the revised Ghent criteria when genetic results were not available, and the other two patients were highly suspected and diagnosed with MFS until the FBN1 deletions were identified.
Conclusions
Our finding expands the mutation spectrum of large FBN1 deletions and emphasizes the importance of screening for large FBN1 deletions in clinical genetic testing, especially for those with classic Marfan phenotype.
Electronic supplementary material
The online version of this article (10.1186/s40246-018-0178-y) contains supplementary material, which is available to authorized users.
Keywords: Marfan syndrome, MLPA, FBN1 gene, Deletion
Background
Marfan syndrome (MFS) is a connective tissue disorder with high clinical heterogeneity, mainly involving ocular, skeletal, and cardiovascular systems, with an estimated prevalence of 1:3000–1:5000 [1]. A large proportion of patients have visible signs, such as tall and slender stature, arachnodactyly, chest deformity, and scoliosis. Most patients have rapidly progressive myopia, and approximately 60% of affected individuals have ectopia lentis. However, cardiovascular abnormality might be the only defect in some MFS patients that is insidious and fatal.
MFS is caused by mutations in the FBN1 gene, which is located on chromosome 15q21.1 and encodes a 320-kDa extracellular matrix glycoprotein fibrillin-1 [2, 3], a major component of microfibrils. So far, more than 2500 mutations (HGMD Professional 2018.1 total) have been identified throughout FBN1, while missense mutations are the most common type [4, 5]. Sanger sequencing of FBN1 and panel sequencing including FBN1 as well as a number of other genes associated with inherited aortopathies are commonly used to identify mutations [6]; however, both of these methods have a limitation for detecting FBN1 large deletions (del) or duplications (dup), which have been reported in up to 7% of MFS patients [7].
Additionally, Loeys-Dietz syndrome (LDS), another inherited connective tissue disorder, which is caused mostly by TGFBR1 and TGFBR2 mutations, is often clinically indistinguishable from MFS [8]. However, up to now, no large genomic rearrangements in TGFBR1 or TGFBR2 have been reported in patients with aortic aneurysm/dissection and LDS features.
In this study, we performed a multiplex ligation-dependent probe amplification (MLPA) testing of FBN1 and TGFBR2 in 115 unrelated Marfan or early-onset aortopathy patients that were previously proven to be negative in a panel testing involving 15 genes associated with inherited aortopathy.
Results
A total of 115 patients with suspected MFS or early-onset aortic aneurysm/dissection, who had a negative result in a 15-gene panel testing, were included in this study and evaluated for gross deletions and duplications in FBN1 and TGFBR2 gene by MLPA assay. The baseline clinical characteristics are summarized in Table 1. Of all patients, 19 were classic MFS, which referred to those who met the Ghent criteria independent of genetic results, and 43 were suspected MFS, which referred to those with some positive signs (either aortic dilation or positive family history AND systemic score ≥ 3) but not meeting the criteria yet. Almost half of the patients had no other systemic abnormality except for aortic events. Five novel large deletions encompassing a single exon or multiple exons in the FBN1 gene were identified in five unrelated patients (Fig. 1, Table 2). Patients AD234, AD392, AD533-1, and AD680-1 harbored FBN1 deletions of exon 43, exon 56, exon 54, and exon 50, respectively, while patient AD437 had a large deletion encompassing exons 44–66 in FBN1. These data had been submitted to ClinVar (ClinVar accessions SCV000804313-000804317).
Table 1.
The baseline clinical characteristics of the 115 unrelated patients
| Characteristics | Statistics (n = 115) |
|---|---|
| Age (years) | 29.4 ± 14.9 |
| Male gender | 87 (75.7%) |
| Primary diagnosis | |
| Marfan syndrome | 19 (16.5%) |
| Suspected Marfan syndrome | 43 (37.4%) |
| Thoracic aortic aneurysm and dissection | 53 (46.1%) |
Values are presented as mean ± SD or n (%)
Fig. 1.
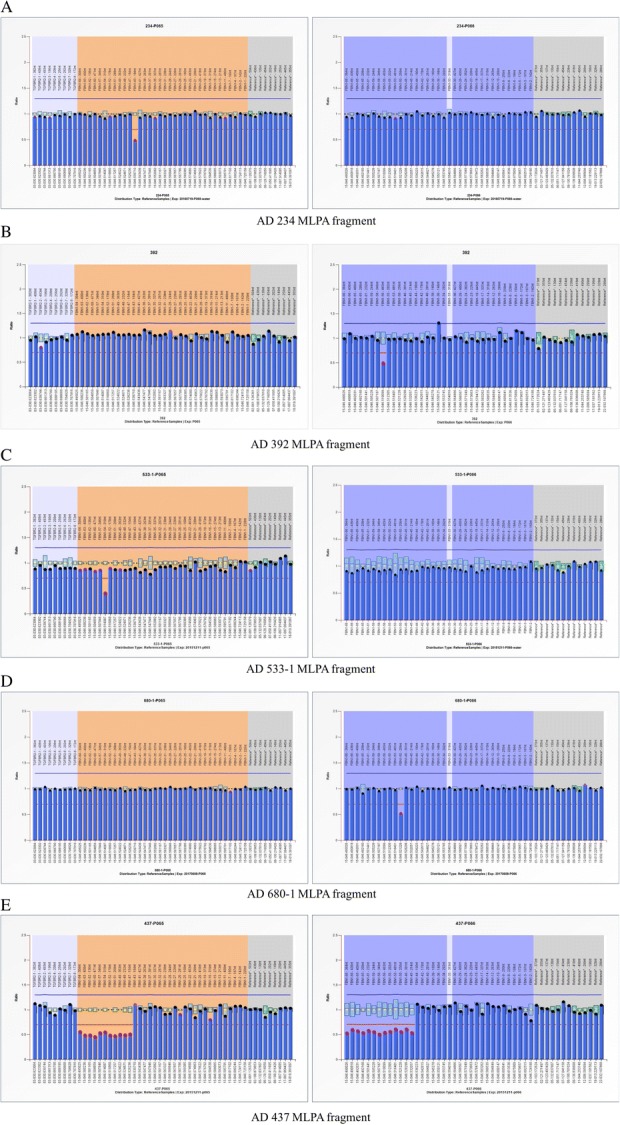
Results of semiquantitative MLPA. The results of MLPA for five patients. a Reduced relative peak areas of FBN1 exon 43 for patient AD234. b Reduced relative peak areas of FBN1 exon 56 for patient AD392. c Reduced relative peak areas of FBN1 exon 54 for patient AD533-1. d Reduced relative peak areas of FBN1 exon 50 for patient AD680-1. e Reduced relative peak areas of FBN1 exon 44–66 for patient AD437
Table 2.
Overview of cases with large deletions in FBN1 gene
| Patient No. | Age (y) | Deletion breakpoints | Deletion (FBN1 exon affected) | Phenotype |
|---|---|---|---|---|
| AD234 | 24 | g.48749026-48753819 | FBN1: exon 43 | Classic MFS |
| AD392 | 38 | g.48724560-48722281 | FBN1: exon 56 | Classic MFS |
| AD533-1 | 5 | g.48727672-48726338 | FBN1: exon 54 | Suspected MFS |
| AD680-1 | 14 | g.48734801-48730690 | FBN1: exon 50 | Suspected MFS |
| AD437 | 37 | NA | FBN1: exon 44–66 | Classic MFS |
All nucleotide positions are represented in relation to the human genome reference sequence (GRCh37/hg19), and position + 1 corresponds to the first nucleotide of the FBN1 reference sequence (GenBank NC_000015.9) at the genomic DNA (g) level
NA not available
To detect the breakpoints of deletions, we performed a long-range PCR followed by Sanger sequencing. Finally, the four single-exon deletions were all confirmed, and the breakpoints were found (Fig. 2). Regrettably, the deletion in AD437 could not be verified by the same method, since the mutated allele did not amplify well. Hence, we performed a quantitative PCR instead. Figure 3 shows that the quantity of genomic DNA from the proband amplified by primer pairs targeting exon 55 and exon 66 was half of that in the control samples, suggesting the true presence of a heterozygous deletion in this region.
Fig. 2.
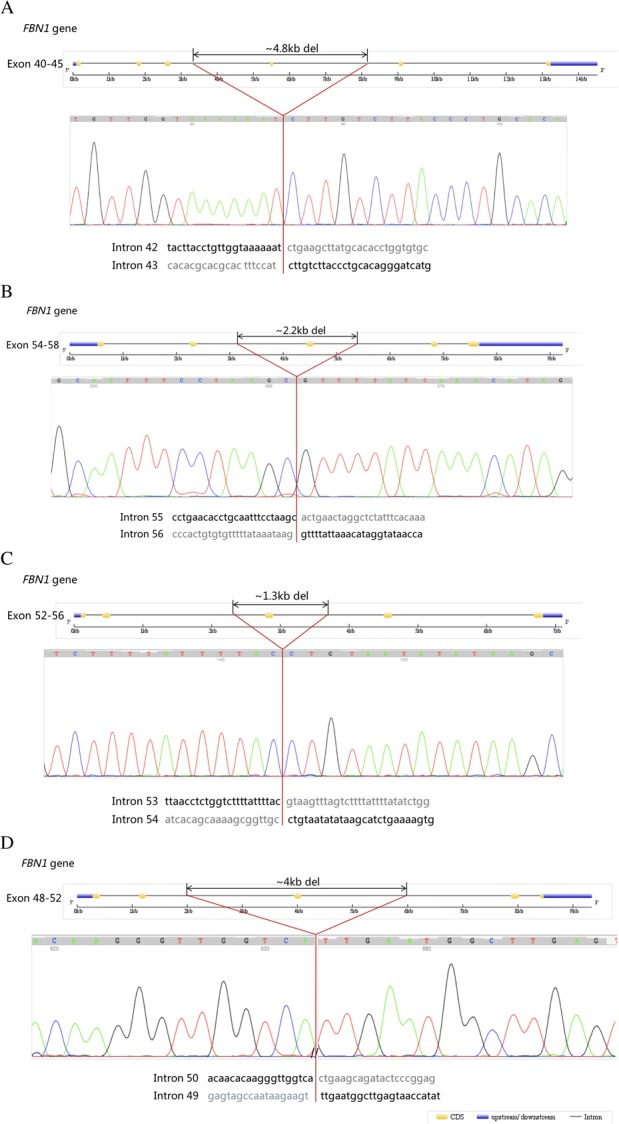
Sequences of PCR products spanning the breakpoint junctions of the four single exon deletions. a ~ 4.8 kb deletion encompassing exon 43 in patient AD234. b ~ 2.2 kb deletion encompassing exon 56 in patient AD392. c ~ 1.3 kb deletion encompassing exon 54 in patient AD533-1. d ~ 4.0 kb deletion encompassing exon 50 in patient AD680-1
Fig. 3.
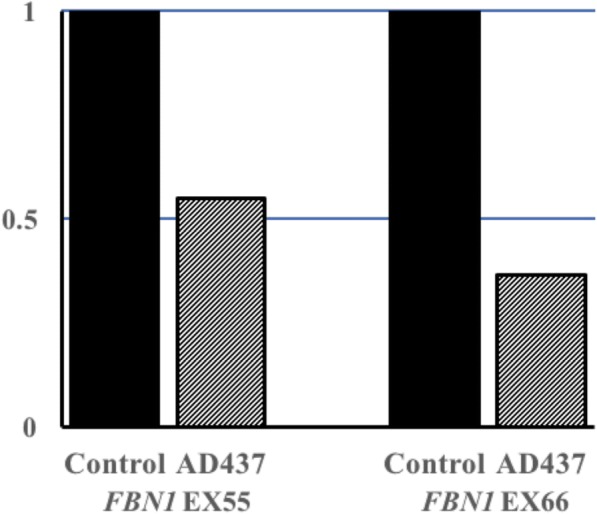
Verification of gross deletions in AD437 by quantitative PCR. The bar graph shows the relative ratio of DNA from AD437, indicating the presence of a heterozygous deletion in the region
All five FBN1 large deletion carriers had multiple system deformities. Information on the clinical manifestation of the disease and family history is summarized in Table 3. Three patients (AD234, AD392, and AD437) were classic MFS, while the other two (AD533-1 and AD680-1) both had a systemic score of 6 but did not meet the criteria yet when genetic results were not available, probably due to their young ages. Combined with genetic results, these two patients were eventually diagnosed with MFS. Notably, patient AD437 had a gross deletion involving the last 23 exons, but there was no significant difference in the severity of clinical phenotypes when compared with the other four single-exon deletion carriers.
Table 3.
The information of patients’ clinical manifestation and family history
| Patients | |||||
|---|---|---|---|---|---|
| AD234 | AD392 | AD533-1 | AD680-1 | AD437 | |
| Age (y) | 24 | 38 | 5 | 14 | 37 |
| Gender | Male | Female | Male | Male | Female |
| Height (cm) | 178 | 167 | 120 | 180 | 177 |
| Weight (kg) | 70 | 58 | 23 | 52 | 71 |
| Cardiovascular system | |||||
| Aortic diameter (cm) | 5.6 | 3.5 | 3.3 | 3.2 | 4.7 |
| Z-score | 8.8 | 2.0 | 6.8 | 2.1 | 6.2 |
| Aortic dissection | Y | N | N | N | Y |
| Skeletal system | |||||
| Pectus carinatum deformity | Y | NA | Y | N | Y |
| Wrist and thumb signs | Y | Y | Y | Y | Y |
| Scoliosis or thoracolumbar kyphosis | N | N | N | Y | N |
| Joint hypermobility | NA | Y | N | N | N |
| Reduced upper segment/lower segment ratio AND increased arm/height | N | N | NA | Y | N |
| Hindfoot deformity | NA | Y | N | N | NA |
| Ocular | |||||
| Ectopia lentis | N | Y | N | N | NA |
| Myopia/strabismus | Y | Y | Y | Y | N |
| Other features | |||||
| Skin striae | N | Y | N | N | Y |
| Family history | Y | NA | N | N | Y |
Y presence of criterion, N absence of criterion, NA not available
Discussion
Marfan syndrome has a highly variable manifestation, from a mild phenotype to early-onset and rapidly progressive MFS. Cardiovascular abnormality could be the only defect in some affected individuals. According to the 2010 revised Ghent criteria, in the absence of family history, the combination of aortic root dilation (Z ≥ 2)/dissection and identification of a causal FBN1 mutation was sufficient to establish a diagnosis of MFS [9]. Accordingly, we performed an MLPA assay to screen for FBN1 and TGFBR2 large genomic rearrangements not only in the diagnosed/suspected MFS patients but also in those early-onset aneurysm/dissection patients with minor skeletal and ocular involvement, who had a negative result in a 15-gene panel testing associated with heritable aortopathy.
Finally, five patients with large FBN1 deletions were identified in our cohort. All five patients had multiple systemic deformities, and three of them met the 2010 Ghent criteria when genetic results were not available, while the other two met the diagnostic criteria until FBN1 gross deletions were detected, probably due to their young ages. Meanwhile, no gross deletions/duplications were identified in patients with only aortic aneurysm/dissection but without other systemic involvement. This result supported the hypothesis that FBN1 gross deletions usually lead to classic MFS [10–12].
Although gross genomic rearrangement within the FBN1 gene only contributed to a small proportion of MFS genetic causes (1.8–2.9%) (UMD, http://www.umd.be/FBN1/; HGMD, http://www.hgmd.cf.ac.uk/ac/gene.php?gene=FBN1), it was important to identify the pathogenic mutation to afford the patient an opportunity for prenatal testing and preimplantation genetic diagnosis (PGD). FBN1 mutations could be identified by sequencing in most Marfan patients (up to 93% in classic Marfan patients) [13]. However, Sanger sequencing and next-generation sequencing are commonly used in clinical genetic testing and are limited in their ability to detect large deletions and duplications. MLPA is a commonly used method to screen large del/dup, commercially, easily, and rapidly. In our cohort, 5 out of 62 patients (8.1%) with diagnosed or suspected MFS but with negative results in panel sequencing had large FBN1 deletions, which proved it to be efficacious and cost-effective to screen for FBN1 large genomic rearrangement in those MFS patients with multiple systemic involvements and a negative FBN1 sequencing result.
Since MLPA and SNP (single-nucleotide polymorphism) arrays are more applicable in clinical genetic testing, increasing gross FBN1 genomic deletions/duplications has been reported (summarized in Table 4), but until now, there has been no definite and conclusive genotype-phenotype correlation. Current studies reveal that the whole gene deletion of FBN1 did not lead to a more severe phenotype [12], and in-frame deletion involving exon 24–53 seemed to result in a high risk of early-onset and rapidly progressive form of MFS [11, 14–16].
Table 4.
Overview of MFS cases with gross deletions in FBN1 gene
| Variation | Patient | Reference PMID (year) | ||
|---|---|---|---|---|
| Deletion (FBN1 exon affected) | Affected domains | Age (y) | Phenotype in papers | |
| Single-exon deletion | ||||
| FBN1:g.46,701,985_46,728,871 (Ex1) | – | 25 | Classic MFS | 17492313 (2002) |
| FBN1:Ex1 | – | NA | Classic MFS | 24501682 (2013) |
| FBN1:Ex1 | – | NA | Classic MFS | 24793577 (2014) |
| FBN1:Ex2 | – | 52 | Classic MFS | 11700157 (2001) |
| FBN1:Ex3 | 1st EGF-like | NA | MFS | 21907952 (2011) |
| FBN1:Ex6 | 3rd EGF-like | 49 | Potential MFS | 28842177 (2017) |
| FBN1:c.3603_3668 del (Ex29) | 18th cbEGF-like | After birth | Neonatal MFS | 10441700 (1999) |
| FBN1:Ex30 | 19–20th cbEGF-like | < 1 | Suspected Beals-Hecht syndrome | 25944730 (2015) |
| FBN1:Ex32 | 21–22th cbEGF-like | 1 | Neonatal MFS | 18412115 (2008) |
| FBN1:Ex36 | 25–26th cbEGF-like | NA | Classic MFS | 19839986 (2009) |
| FBN1:g.48,749,026_48,753,819 del (Ex43) | 7th TB, 29th cbEGF-like | 24 | Classic MFS | In this study |
| FBN1:g.48,734,801-48,730,690 del (Ex50) | 35th cbEGF-like | 14 | MFS | In this study |
| FBN1:Ex52 | 8th TB, 36th cbEGF-like | 40 | Classic MFS | 11700157 (2001) |
| FBN1:g.48,727,672-48,726,338 del (Ex54) | 37–38th cbEGF-like | 5 | MFS | In this study |
| FBN1:g.48,724,560_48,722,281 del (Ex56) | 39–40th cbEGF-like | 38 | Classic MFS | In this study |
| Multi-exon deletion | ||||
| FBN1:Ex1–5 | 1–3rd EGF-like | 27 | Classic MFS | 21936929 (2011) |
| FBN1:g.46,580,456_46,883,035 (Ex1-16) | 1–3rd EGF-like, 1st TB, 4–10th cbEGF-like | 40 | Classic MFS | 17492313 (2002) |
| FBN1:Ex1–36 | 1–3rd EGF-like, 4–26th cbEGF-like, 1–5th TB | 15 | Classic MFS | 28842177 (2017) |
| FBN1:g.48,890,962_48,922,918 (Ex2-4) | 1–2nd EGF-like | 32 | Classic MFS | 29850152 (2018) |
| FBN1:Ex6–65 | 3rd EGF-like, 4–47th cbEGF-like, 1–9th TB | NA | Classic MFS | 24793577 (2014) |
| FBN1:Ex13–49 | 7–34th cbEGF-like, 3–7th TB | 5 | MFS | 18412115 (2008) |
| FBN1:Ex24–26 | 14–16th cbEGF-like | After birth | Neonatal MFS | 20455198 (2010) |
| FBN1:Ex33–38 | 21–26th cbEGF-like, 6th TB | 1 | Neonatal MFS | 24199744 (2014) |
| FBN1:Ex34–43 | 23–29th cbEGF-like, 6–7th TB | 22 | Classic MFS | 19863550 (2010) |
| FBN1:Ex37–65 | 26–47th cbEGF-like, 3–9th TB | NA | Classic MFS | 24793577 (2014) |
| FBN1:Ex42–43 | 7th TB, 29th cbEGF-like | > 46 | Classic MFS | 11710961 (2001) |
| FBN1:Ex44–46 | 29–31th cbEGF-like | > 6 | Childhood onset MFS | 11710961 (2001) |
| FBN1:Ex44–66 | 29–47th cbEGF-like, 8–9th TB | 37 | Classic MFS | In this study |
| FBN1:Ex48–53 | 33–37th cbEGF-like, 8th TB | 15 | Neonatal MFS | 28842177 (2017) |
| FBN1:Ex49–50 | 34–35th cbEGF-like | 3 | Neonatal MFS | 28842177 (2017) |
| FBN1:Ex50–63 | 35–46th cbEGF-like, 8–9th TB | 65 | MFS | 19659760 (2009) |
| FBN1:Ex58–63 | 41–46th cbEGF-like | 17 | Juvenile onset classic MFS | 17189636 (2007) |
| FBN1:c.7456_7821 del* (Ex61–64) | 43–46th cbEGF-like | 48 | Classic MFS | 1631074 (1994) |
| Whole gene deletion | ||||
| FBN1:Ex1–66 | Full gene | 16 | Incomplete MFS | 20478419 (2010) |
| FBN1:Ex1–66 | Full gene | 42 | Classic MFS | 21936929 (2011) |
| FBN1:Ex1–66 | Full gene | 15 | Classic MFS | 21936929 (2011) |
| FBN1:Ex1–66 | Full gene | 12 | Classic MFS | 21936929 (2011) |
| FBN1:Ex1–66 | Full gene | 41 | MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 39 | MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 16 | MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 13 | MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 27 | MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 21 | MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 34 | MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 5 | Potential MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 13 | Potential MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 8 | Potential MFS | 21063442 (2011) |
| FBN1:Ex1–66 | Full gene | 13 | Classic MFS | 22260333 (2012) |
| FBN1:g.48,931,968_51,102,375 (Ex1–66) | Full gene | 14 | MFS | 27615407 (2016) |
NA not available
*The deletion was represented as nt. 4762_5127 in partial cloned sequence of FBN1 (PMID:1852207), and it was converted into its standardized nomenclature in accordance with HGVS (Human Genome Variation Society), in which the position + 1 corresponds to the A of the ATG start codon of the mRNA reference sequence (GenBank NM_000138) at the cDNA (c) level. Except for this, all of the other nucleotide positions and patient phenotypes were shown as it was reported in the reference article
Conclusions
In summary, our data expand the number of large FBN1 deletions and emphasize that screening for gross deletions in FBN1 genes is necessary for clinically suspected MFS patients, especially in those who have a negative result in conventional sequencing methods.
Methods
Participants
Patients with MFS or early-onset aortopathy were referred for a genetic test from the Center of Vascular Surgery in Fuwai Hospital and Department of Cardiovascular Surgery in Xiangya Hospital Central South University. Of these, 115 patients in whom no causal mutation was identified in a 15-gene panel associated with heritable aortopathy, including ACTA2, CLO3A1, FBN1, FBN2, MYH11, MYLK, NOTCH1, PRKG1, SKI, SLC2A10, SMAD3, SMAD4, TGFB2, TGFBR1, and TGFBR2, were enrolled in this study to screen for FBN1 and TGFBR2 large del/dup.
Multiplex ligation-dependent probe amplification (MLPA)
MLPA assays were performed to detect FBN1 and TGFBR2 large deletions or duplications using the commercially available SALSA MLPA kits P065 and P066 (MRC-Holland, Amsterdam, The Netherlands), which contained probes for all exons of FBN1 and TGFBR2. According to the manufacturer’s instructions, a total of 100–200 ng of genomic DNA of each patient was used for hybridization, and amplification products from each MLPA assay were separated by capillary electrophoresis on an ABI 3500XL Dx Genetic Analyzer (Life Technologies, USA). The results were analyzed using Coffalyser software. Deletions and duplications with deviations more than 30% were suspected as significant alterations.
Sanger sequencing
To verify the results of MLPA and identify the breakpoints of the deletions, we performed a long-range PCR and subsequent Sanger sequencing. Primers flanking the predicted deletions were designed and LA Taq Hot Start Version kit (Takara, Japan) was used in the PCR system with the following cycling process: 5-min initial denaturation at 96 °C, 30 cycles of 10 s at 98 °C, and 15 min at 68 °C, finished by a 10-min final extension step at 72 °C. Then, the products were detected through agarose gel electrophoresis and sequenced by the inner primers on the ABI 3730XL Genetic Analyzer.
Quantitative PCR
Quantitative PCR (qPCR) with the SYBR green reporter dye was performed to quantify relative target gene regions copy number in genomic DNA, and housekeeping gene GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as the reference gene. The primer pairs were designed by Primer3 Input (http://bioinfo.ut.ee/primer3-0.4.0/) (see Additional file 1). All qPCRs were performed using 2 × SYBR FAST qPCR Kit Master Mix (KAPA Biosystems, America) with the QuantStudio 6 Flex Real-Time PCR System.
Additional file
Table S1. The quantitative PCR primer pairs for AD437. (PDF 132 kb)
Acknowledgements
We thanked all the subjects who participated in this study.
Funding
This work was supported by the grant of CAMS Initiative for Innovative Medicine (2016-I2M-1-016) and the grant from the Youth Foundation of Fuwai Hospital, National Center for Cardiovascular Disease, China (NO. 2016-F05).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- del
Deletion
- dup
Duplication
- FBN1
Fibrillin-1
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HGMD
Human Gene Mutation Database
- HGVS
Human Genome Variation Society
- LDS
Loeys-Dietz syndrome
- MFS
Marfan syndrome
- MLPA
Multiplex ligation-dependent probe amplification
- NA
Not available
- qPCR
Quantitative PCR
- SNP
Single-nucleotide polymorphism
- UMD
Universal Mutation Database
Authors’ contributions
HY coordinated the project and wrote the manuscript. YM and KZ performed MLPA and quantitative PCR. ML, XS, and FL recruited patients and collected clinical information. YZ and GZ were in charge of sample handling and quality control. LW and CS were in charge of the clinical evaluation and sample management. ZZ designed the project and revised the manuscript. All authors had read and approved the final manuscript.
Ethics approval and consent to participate
Everyone accepting the genetic test was adequately informed and signed a consent form. The study was approved by the ethics committee of Fuwai Hospital (Approval No. 2017-877) and adhered to the Declaration of Helsinki.
Consent for publication
Everyone accepting the genetic test signed a consent form and agreed to allow their anonymized samples and genetic results to be used for further research studies and publications.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hang Yang, Email: chrisyang_1999@163.com.
Yanyun Ma, Email: myy717@126.com.
Mingyao Luo, Email: luomingyao@fuwai.com.
Kun Zhao, Email: zhaokun_fw@163.com.
Yinhui Zhang, Email: zyh0863@163.com.
Guoyan Zhu, Email: zhugy525888@foxmail.com.
Xiaogang Sun, Email: xiaogangsun2006@vip.sina.com.
Fanyan Luo, Email: drlfy@163.com.
Lin Wang, Email: wanglin79922@csu.edu.cn.
Chang Shu, Email: changshu01@fuwaihospital.org.
Zhou Zhou, Email: zhouzhou@fuwaihospital.org.
References
- 1.Judge DP, Dietz HC. Marfan’s syndrome. Lancet (London) 2005;366(9501):1965–1976. doi: 10.1016/S0140-6736(05)67789-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117(21):2802–2813. doi: 10.1161/CIRCULATIONAHA.107.693523. [DOI] [PubMed] [Google Scholar]
- 3.Maddox BK, Sakai LY, Keene DR, Glanville RW. Connective tissue microfibrils. Isolation and characterization of three large pepsin-resistant domains of fibrillin. J Biol Chem. 1989;264(35):21381–21385. [PubMed] [Google Scholar]
- 4.Groth KA, Von Kodolitsch Y, Kutsche K, Gaustadnes M, Thorsen K, Andersen NH, Gravholt CH. Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet Med. 2017;19(7):772–777. doi: 10.1038/gim.2016.181. [DOI] [PubMed] [Google Scholar]
- 5.Dordoni C, Ciaccio C, Santoro G, Venturini M, Cavallari U, Ritelli M, Colombi M. Marfan syndrome: report of a complex phenotype due to a 15q21.1 contiguos gene deletion encompassing FBN1, and literature review. Am J Med Genet A. 2017;173(1):200–206. doi: 10.1002/ajmg.a.37975. [DOI] [PubMed] [Google Scholar]
- 6.Yang H, Luo M, Fu Y, Cao Y, Yin K, Li W, Meng C, Ma Y, Zhang J, Fan Y, et al. Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci Rep. 2016;6:33002. doi: 10.1038/srep33002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lerner-Ellis JP, Aldubayan SH, Hernandez AL, Kelly MA, Stuenkel AJ, Walsh J, Joshi VA. The spectrum of FBN1, TGFbetaR1, TGFbetaR2 and ACTA2 variants in 594 individuals with suspected Marfan syndrome, Loeys-Dietz syndrome or thoracic aortic aneurysms and dissections (TAAD) Mol Genet Metab. 2014;112(2):171–176. doi: 10.1016/j.ymgme.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 8.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 9.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–485. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 10.Furtado LV, Wooderchak-Donahue W, Rope AF, Yetman AT, Lewis T, Plant P, Bayrak-Toydemir P. Characterization of large genomic deletions in the FBN1 gene using multiplex ligation-dependent probe amplification. BMC Med Genet. 2011;12:119. doi: 10.1186/1471-2350-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Wu W, Lu C, Liu Y, Wang R, Si N, Liu F, Zhou J, Zhang S, Zhang X. Gross deletions in FBN1 results in variable phenotypes of Marfan syndrome. Clin Chimica Acta. 2017;474:54–59. doi: 10.1016/j.cca.2017.08.023. [DOI] [PubMed] [Google Scholar]
- 12.Hilhorst-Hofstee Y, Hamel BC, Verheij JB, Rijlaarsdam ME, Mancini GM, Cobben JM, Giroth C, Ruivenkamp CA, Hansson KB, Timmermans J, et al. The clinical spectrum of complete FBN1 allele deletions. Eur J Human Genet. 2011;19(3):247–252. doi: 10.1038/ejhg.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radonic T, de Witte P, Groenink M, de Bruin-Bon RA, Timmermans J, Scholte AJ, van den Berg MP, Baars MJ, van Tintelen JP, Kempers M, et al. Critical appraisal of the revised Ghent criteria for diagnosis of Marfan syndrome. Clin Genet. 2011;80(4):346–353. doi: 10.1111/j.1399-0004.2011.01646.x. [DOI] [PubMed] [Google Scholar]
- 14.Singh KK, Elligsen D, Liersch R, Schubert S, Pabst B, Arslan-Kirchner M, Schmidtke J. Multi-exon out of frame deletion of the FBN1 gene leading to a severe juvenile onset cardiovascular phenotype in Marfan syndrome. J Mol Cell Cardiol. 2007;42(2):352–356. doi: 10.1016/j.yjmcc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Liu W, Schrijver I, Brenn T, Furthmayr H, Francke U. Multi-exon deletions of the FBN1 gene in Marfan syndrome. BMC Med Genet. 2001;2:11. doi: 10.1186/1471-2350-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apitz C, Mackensen-Haen S, Girisch M, Kerst G, Wiegand G, Stuhrmann M, Niethammer K, Behrwind G, Hofbeck M. Neonatal Marfan syndrome: unusually large deletion of exons 24-26 of FBN1 associated with poor prognosis. Klinische Padiatrie. 2010;222(4):261–263. doi: 10.1055/s-0030-1247510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The quantitative PCR primer pairs for AD437. (PDF 132 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.