

Abstract
The development of redox-active metalloprotein catalysts is a challenging objective of de novo protein design. Within this Perspective we detail our efforts to create a redox-active Cu nitrite reductase (NiR) by incorporating Cu into the hydrophobic interior of well-defined three-stranded coiled coils (3SCCs). The scaffold contains three histidine residues that provide a layer of three nitrogen donors that mimic the type 2 catalytic site of NiR. We have found that this strategy successfully produces an active and stable CuNiR model that functions for over 1000 turnovers. Spectroscopic evidence indicates that the Cu(I) site has a lower coordination number in comparison to the enzyme, whereas the Cu(II) geometry may more faithfully reproduce the NiR type 2 center. Mutations at the helical interface successfully produce a hydrogen bond between an interfacial Glu residue and the Culigating His residue, which allows for the tuning of the redox potential over a 100 mV range. We successfully created constructs with as much as a 120-fold improvement from the original design by modifying the steric bulk above or below the Cu binding site. These systems are now the most active water-soluble and stable artificial NiR catalysts yet produced. Several avenues for improving the catalytic efficiency of later designs are detailed within this Perspective, including adjustment of their resting oxidation state, the use of asymmetric scaffolds to allow for single amino acid mutation within the second coordination sphere, and the design of hydrogen-bonding networks to tune residue orientation and electronics. Through these studies the TRI-H system has given insight into the difficulties that arise in creating a de novo redox active enzyme. Work to improve upon this model will provide strategies by which redox-active de novo enzymes may be tuned and detail how native enzymes accomplish catalytic efficiencies through proton gated redox catalysis.
Keywords: de novo, metalloenzyme, catalysis, nitrogen cycle, nitrite reductase, protein
The structure–function relationship is one of the most important questions that needs to be understood within the study of proteins. Put simply, researchers are working to determine what types and how much of particular structures are required for protein function and whether solely the amino acids of the active site are critical or if other components within the protein matrix infer long-range contributions to the active site (e.g., through a hydrogen-bonding network or pathways involved in substrate intake or product egress). These questions are particularly relevant to redox-active metalloenzymes, which also must control metal geometry across different active site oxidation states. The classical biochemistry approach to this question has been site-directed mutagenesis of native proteins to ascertain which amino acids are crucial to function, but recently protein redesign has taken a larger role in which one tries to build in function to an unrelated scaffold. With the aid of modern strategies using molecular evolution,1–3 significant progress has been made in catalyzing abiologic reactions using protein scaffolds. Within the realm of protein redesign, de novo protein design has proven to be an alternative and potent tool to strip away the evolutionary baggage that comes with native protein design.4–7 Researchers within this field attempt to insert function into a scaffold which does not exist in nature in order to test our understanding of the desired activity from a bottom-up rather than a top-down strategy. Recent estimates put the percentage of metalloproteins as high as 30–40% on the basis of an analysis of PDB submissions,8,9 and metalloproteins are known to play roles in critical biological systems such as nitrogen fixation10 and respiration;11 thus, it should be no surprise that their study makes up a sizable subsection of the de novo design field.12–15
Our laboratory has approached metalloprotein design using scaffolds based on an α helical three-stranded coiled coil or three helical bundle structures (TRI, CoilSer or α3D scaffolds) utilizing a heptad design (Table 1). Because the α-carbon atom of an amino acid is oriented 100° from the previous or subsequent residues, seven residue repeats allow for a nearly perfect helical coil that requires a small degree of supercoiling to be stable. Packing of the α helices creates a well-structured aggregate when hydrophobic residues (in our case Leu) are placed at the 1 (a) and 4 (d) heptad positions constructing a hydrophobic interior, while the remaining side chains utilize hydrophilic residues on the exterior to achieve proper salt bridging and solubility. These interior hydrophobic residues can then be mutated to metal binding residues to create a simplified metal binding site within a protein environment. By mutating internal residues to Cys (e.g., TRIL9C employs cysteine at the ninth position or the a site of the second heptad), we have exploited this unique environment to study the intricacies of heavy metal binding and to determine how one can control coordination numbers of these metals.16–25
Table 1.
Comparison of TRI Family Peptide Sequences Used as CA or CuNiR Mimics to the Sequence of the Original Scaffold Peptide
| peptidea | 1 | 2 | 9 | 16 | 23 | 30 |
|---|---|---|---|---|---|---|
| TRI | Ac-G | LKALEEK | LKALEEK | LKALEEK | LKALEEK | G-NH2 |
| TRIL9CL23H | Ac-G | WKALEEK | CKALEEK | LKALEEK | HKALEEK | G-NH2 |
| CSL9PenL23H | Ac-E | WEALEKK | PenAALESK | LQALEKK | HEALEHG | -NH2 |
| TRI-H | Ac-G | WKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 |
| TRI-EH | Ac-G | WKALEEK | LKALEEK | LKALEEE | HKALEEK | G-NH2 |
| TRI-H L19A | Ac-G | WKALEEK | LKALEEK | LKAAEEK | HKALEEK | G-NH2 |
The C and N termini of the peptides are amidated and acetylated, respectively. The first amino acid within each heptad is labeled with its overall position within the peptide.
Since these initial studies, our focus has shifted to that of mutating Leu layers to His for binding transition metals with the goal of creating de novo metalloenzymes. Incorporating Hg(II) into the cysteine layer and Zn(II) into the His layer of (TRIL9CL23H)3 gave HgSZnN(TRIL9CL23H)3 whose X-ray structure of the related CoilSer (CS) peptide (CSL9PenL23H) demonstrated that parallel 3SCCs that had Hg(II) bound to three S atoms and Zn(II) bound to three ε-N of histidine with an exogenous water could be prepared. In this case, the Hg(II) helped stabilize the assembly through strong HgII–S bonds, while the Zn(II) site closely resembled that observed for native carbonic anhydrases.26 This construct successfully created a de novo metallohydrolase which far outperformed any other synthetic model at the time, with rates for CO2 hydration within 2 orders of magnitude of those of native carbonic anhydrases.27 It was also shown that high activity could be obtained when the stabilizing Hg(II) was removed, demonstrating that a stabilizing structural site was not required. Further exploration of this construct varied the positioning of the Cys and His layers and determined that, while the Kd value for binding Zn(II) could be tuned by 1 order of magnitude and the pKa varied between 9.2 and 9.6, kcat/KM was largely unaffected, with small changes to KM being countered by changes in the opposite direction to kcat. This indicated that, while some aspects of the catalyst could be tuned via His positioning, it had very little effect on catalytic efficiency, and further constructs could use whatever positioning was necessary for secondary sphere design to be included. It also illustrated that much of the rate acceleration provided by the enzyme could be achieved just by mimicking the first coordination sphere of a native catalytic site independently from the utilized protein scaffold. Nevertheless, to achieve levels of catalysis approaching evolved enzyme activity, it was clear that engineering of the second and third coordination sphere environments was required. These studies were consistent with mutagenesis studies which showed that mutation of key acid base catalysts such as T199A decreased activity of the human carbonic anhydrase to levels nearly identical with those of our system.28,29
While these results were satisfying, the more challenging objective, developing redox catalysts, had not yet been achieved. Designing a metallohydrase such as carbonic anhydrase entails building a core suitable for a single metal oxidation state; however, many metalloenzymes require redox cycling during catalysis. De novo design of a metalloenzyme capable of redox coupling requires strategies which consider both oxidation states of the intended metal. One major advantage of synthetic proteins in comparison to smaller molecule mimics of active site structure is that the driving force for the formation of a stable scaffold can constrain environments around the first and second coordination spheres of a metal catalyst. Nonetheless, requiring a single molecule to accommodate two very different coordination environments provides an extra layer of complexity and may be related to why so few examples of de novo redox-capable nonheme-based metalloenzymes have been reported. A major exception to this statement is the Due Ferri family of proteins and work to create de novo proteins which bind FeS or dinuclear Mn clusters.30–32
An extreme example of the first coordination sphere changes associated with oxidation change is observed between Cu(I) and Cu(II). The cuprous ion has a strong preference for soft ligands and low coordination number, typically 2 or 3. In contrast, the cupric oxidation state is intermediate in its Lewis acidity and often is four- or five-coordinate. In these complexes there is typically a tetragonally distorted polyhedron with a plane of strong ligands and one or two donors at much longer distances. Despite the differences, Nature routinely uses copper trapped in coordination environments that are intermediate in structure between these two extremes in order to minimize inner-sphere reorganization energy associated with the oxidation reduction reactions. Thus, the challenge of developing mononuclear copper-based catalysts becomes an attractive target for the metalloprotein design community. This Perspective will focus on our work to model the type 2 copper center of Cu nitrite reductase (CuNiR) by creating the simplified scaffold TRIL23H3 ((TRI-H)3), which eschews the heavy metal binding site of TRIL9CL23H and substitutes Cu into the His3 layer, followed by investigations into how the catalytic efficiency of this construct can be tuned via long-range or secondary sphere effects.
CuNiR is an enzyme in the sequence of dissimilatory reduction of nitrate to dinitrogen, with the specific catalyzed reaction being the reduction of nitrite to nitric oxide. This conversion is achieved by coupling a type 1 copper electron transfer center to the type 2 copper catalytic center Cu(His3)(OH2). This latter center contains a distorted-tetrahedral environment, intermediate between the standard preferences of Cu(I) and Cu(II).33–36 The interplay between these two copper centers enables CuNiR to utilize gated catalysis in which nitrite binds to the oxidized type 2 copper center before electron transfer from the reduced type 1 copper center is possible. Secondary sphere amino acids around the Peptide type 2 copper catalytic center include Asp98 and His255, the former of which forms a hydrogen bond with both the reactant nitrite and product nitric oxide while His255 forms a hydrogen bond with Asp98, and both amino acids are part of a larger protein channel which connects the active site to solvent.37 Mutation of these amino acids decreases the efficiency of NiR activity ~100-fold, and their function has been linked to proton-coupled electron transfer.38,39 While our initial model could not take into account these long-range interactions, we were confident that the crystal structure of our previously reported carbonic anhydrase (HgIIS[ZnIIN(H2O)]-(CSL9CL23H)3+, where S and N subscripts denote the metal binding to Cys and His, respectively) mimic could be used as a starting point to understand how a copper ion would bind to a tris-histidine environment of the TRI peptides.
The divalent charge and Lewis acidity of Zn(II) resemble the cupric oxidation state, while the d10 electronic configuration mimicked Cu(I). Comparison of this Zn(II) binding site with the type 2 copper site of CuNiR from R. sphaeroides found that both metals were bound by three His residues via the ε nitrogens with water as a fourth coordinating ligand (Figure 1). This left us assured that the His3 site within TRI-H could make a suitable CuNiR mimic.40 We have previously shown that the driving force of hydrophobic packing within TRI constructs can be utilized to enforce otherwise disfavored metal geometries such as trigonal-planar HgIIS3 and so had reason to think that this strategy may be able to mimic the intermediate Cu binding geometry of native CuNiR.16,18
Figure 1.
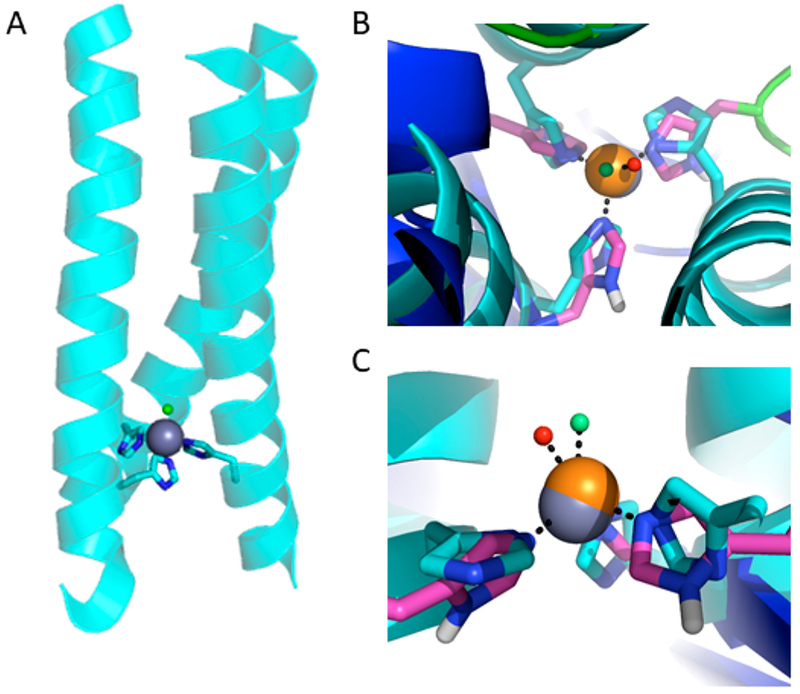
(A) Representation of the model for Cu(TRIH)3 based on the structure of HgIIS[ZnIIN(H2O)](CSL9CL23H)3+ (PDB 3PBJ). (B) View of the ZnII(H2O)His3 site along the pseudo-3-fold axis of HgIIS[ZnIIN(H2O)](CSL9CL23H)3+ (cyan), superimposed on the type 2 CuIIH2O(His)3 site in R. sphaeroides NiR (PDB 2DY2; backbone green, side chain pink). Coordinated water molecules are represented as spheres. (C) Side view of the same superposition in (B). Reprinted and adapted with permission from ref 40. Copyright 2012 Proceedings of the National Academy of Sciences of the United States of America.
X-ray crystallographic characterization of metalloprotein sites is presently the preferred method to deduce structural aspects of a catalytic center; however, this technique does not always bear the desired fruit. For example, crystals of the Cu(II) form of this protein have been prepared but rapidly degrade (through reduction) in the synchrotron beam. The Cu(I) proteins are air sensitive, making data collection more difficult. Thus, while now there are many structures22,41,42 of metals bound to the interior of three stranded coiled coils, we resorted to detailed spectroscopic characterization of the copper centers.
Cu(I) binding to this peptide was investigated with NMR and X-ray absorption spectroscopy (XAS).40 Two peaks were present in the aromatic region of the NMR spectrum and were assigned as the Nε and Nδ protons on His23. These peaks split upon Cu(I) binding, consistent with Cu(I) interacting with the His3 binding site as designed. The two peaks of the apo peptide were apparent in the presence of Cu(I) only at pHs below 4.45 on the basis of pH titration. XAS measurements were taken at pH 5.8 to investigate the geometry of the bound Cu(I). X-ray absorption near edge structure (XANES) analysis of the pre-edge 1s → 4p peak of Cu(I) is commonly used as an indicator of coordination number of the metal center, with greater intensities for lower coordination numbers and vice versa.43 Analysis of CuI(TRI-H)3 in this way suggested a three-coordinate Cu(I) complex, while extended X-ray absorption fine structure (EXAFS) analysis was modeled best as three Cu–N/O scatterers at a distance of 1.93 Å with long-distance scatterers indicating His ligation. We assigned this site as a distorted-trigonal-planar geometry on the basis of the larger than usual Debye–Waller factors of the His scatterers. The Cu(I) Kd value was determined by a competitive titration assay of disodium bathocuproinedisulfonate (Na2BCS) at pH 5.9 to be 3.1 pM.
Cu(II) binding was investigated via electronic absorption and electron paramagnetic resonance (EPR) spectroscopy. Absorption maxima of CuII(TRI-H)3 were consistent with a CuIIHis3 binding site with one or two waters, while EPR spectra indicated a five-coordinate structure.44,45 Hyperfine compression upon nitrite titration was similar to that observed in native CuNiR,46 and analysis of this titration gave a rough binding constant of 1.5–2.1 mM for nitrite binding to CuII(TRI-H)3.47 The Cu(II) Kd value was determined to be 40 nM at pH 5.9 by quenching of tryptophan fluorescence. Using the Nernst equation, this was combined with the Cu(I) Kd to calculate a reduction potential of +400 mV. Interestingly, this reduction potential is closer to that seen in native type 1 copper sites than to the type 2 copper site being modeled. This may be due to the trigonal geometry of Cu(I) stabilizing that oxidation state in comparison to native proteins. Thus, we had successfully created a peptide which bound both Cu(I) and Cu(II); however, the precise coordination environments for either oxidation level were not faithfully reproduced, as the Cu(I) form was too low coordinate and the Cu(II) protein was too high coordinate (Figure 2). Nevertheless, we felt that this system could serve as an important model for aqueous catalysis, as small-molecule models are often not water-soluble/-stable and studies of the native CuNiR are complicated by the presence of two distinct copper centers.
Figure 2.
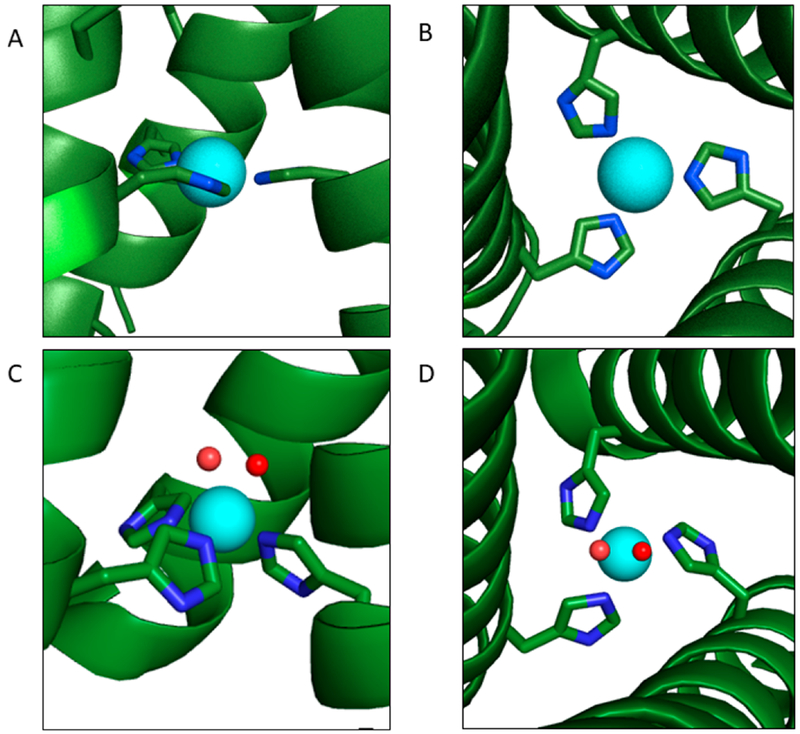
(A) Representation of the model for CuI(TRI-H)3 based on the structure of HgIIS[ZnIIN(H2O)](CSL9CL23H)3+ (PDB 3PBJ) and the XAS analysis of CuI(TRIH)3. (B) Bird’s eye view of (A). (C) Representation of the model for CuII(TRI-H)3 based on the structure of HgIIS[ZnIIN(H2O)](CSL9CL23H)3+ (PDB 3PBJ) and spectroscopic analysis of CuII(TRI-H)3. (D) Bird’s eye view of (C).
The most crucial test for any metalloenzyme mimic is its catalytic efficiency. Our assay for reactivity mixed CuII(TRI-H)3 with nitrite before initiating the reaction by addition of ascorbate as the sacrificial electron donor. Nitric oxide production was proved using a UV–vis spectroscopic assay that monitored the formation of FeEDTA(NO), while FTIR analysis of the headspace gas discounted the possibility that N2O was produced as a side product. The first-order rate constant was determined to be 4.6 × 10−4 s1 under catalytic conditions by monitoring the consumption of ascorbate (Figure 3). While this catalytic efficiency was certainly modest, this represented a stable and functional Cu(His)3 model in aqueous solution that can turn over more than 1000 times without obvious loss of activity and came with the added benefit that it only produced NO rather than the N2O side product that often accompanies nitrite reductase model catalysis. These observations made for a perfect starting point to enhance CuNiR activity within a simplified protein scaffold.
Figure 3.
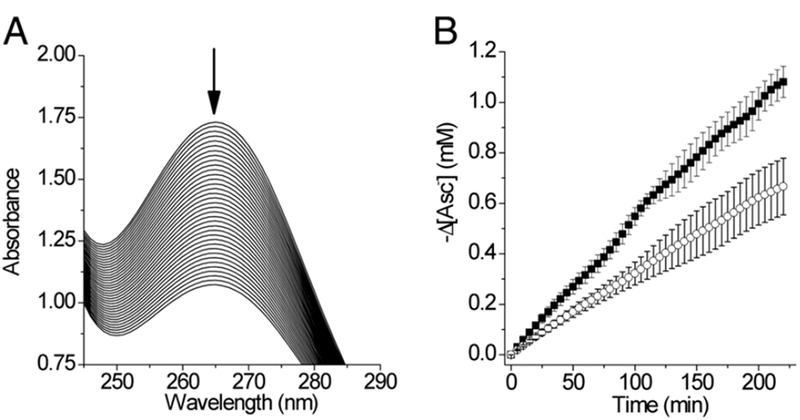
(A) UV spectra of a solution of sodium ascorbate and sodium nitrite (pH 5.9) collected every 5 min, containing CuII(TRI-H)32+ (0.180 mM) and apo-(TRI-H)3 (0.090 mM). (B) (■) decrease of [Asc−] vs time in samples, as in (A), and (◯) samples containing only apo-(TRI-H)3 (0.090 mM), pH 5.8. Reprinted with permission from ref 40. Copyright 2012 Proceedings of the National Academy of Sciences of the United States of America
Our next study sought to determine the effect on catalysis and redox potential of modulating electrostatics near the active site of Cu(TRI-H)3. This was spurred on by a comparison of the native proteins peptidylglycine α-hydroxylating monooxygenase (PHM) and the aforementioned CuNiR.48 Both proteins contain a Cu(His)3 structure, but one center is catalytic while the other serves solely as an electron transfer site to a separate catalytic species. Thus, structurally related Cu(His)3 centers can differ in function due to the surrounding protein environment. In addition, while systematic studies on how one can tune redox potentials had been done on heme proteins and cupredoxins, at the time no such study had been done on type 2 copper sites, though few would deny their importance to biology. Utilizing our simplified CuNiR model (the first of its kind to be fully characterized in both oxidation states), we sought to study the effects of modulating electrostatics free from the convoluting factors of native proteins. To this end, we synthesized and characterized a series of proteins, given in Table 2, with mutations to amino acids on the helical interface to modulate charges between 9 and 12 Å from the Cu(His)3 active site. We also hoped to investigate possible interactions between these remote amino acids and the His residues directly bound to Cu (Figure 4).49
Table 2.
TRI Family Peptide Sequences Used in this Study
| peptidea | 1 | 2 | 9 | 16 | 23 | 30 | Δcharge |
|---|---|---|---|---|---|---|---|
| TRI-H | Ac-G | WKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 | |
| TRI-HK22Q | Ac-G | WKALEEK | LKALEEK | LKALEEQ | HKALEEK | G-NH2 | –3 |
| TRI-EH | Ac-G | WKALEEK | LKALEEK | LKALEEE | HKALEEK | G-NH2 | –6 |
| TRI-EHE27K | Ac-G | WKALEEK | LKALEEK | LKALEEE | HKALKEK | G-NH2 | 0 |
| TRI-EHE27Q | Ac-G | WKALEEK | LKALEEK | LKALEEE | HKALQEK | G-NH2 | –3 |
| TRI-EHK24Q | Ac-G | WKALEEK | LKALEEK | LKALEEE | HQALEEK | G-NH2 | –9 |
| TRI-EHK24E | Ac-G | WKALEEK | LKALEEK | LKALEEE | HEALEEK | G-NH2 | –12 |
The C and N termini of the peptides are amidated and acetylated, respectively. The first amino acid within each heptad is labeled with its overall position within the peptide.
Figure 4.
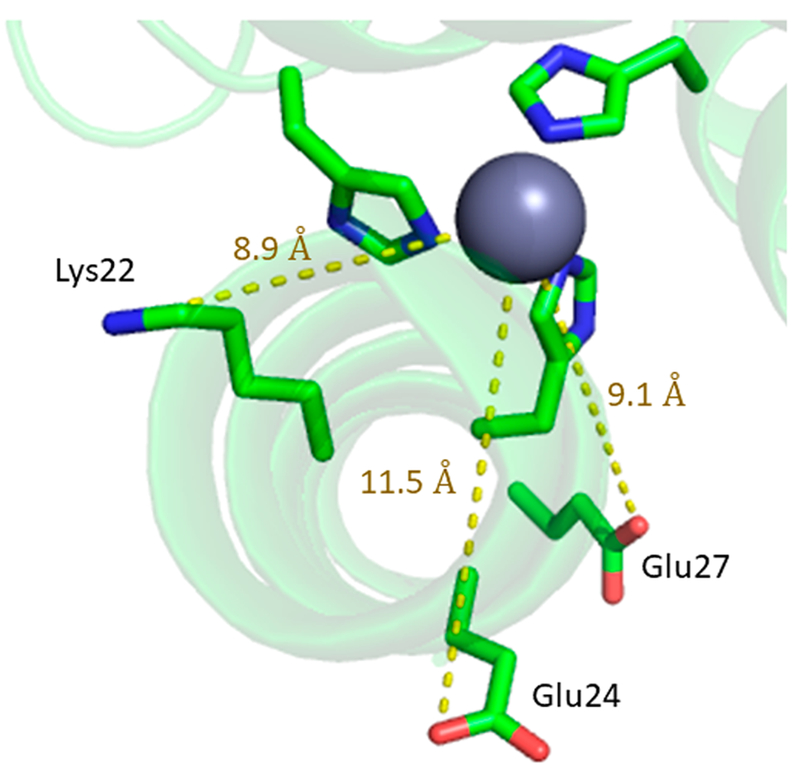
Pymol illustration of HgIIS[ZnIIN(H2O)](CSL9CL23H)3+ (PDB 3PBJ) to show the distance between Lys22, Glu24, or Glu27 and the His3-bound metal.
The first mutant analyzed (designated TRI-EH) converts the positively charged lysine in position 22 to a negatively charged glutamate, making the 3SCC more negative by 6 charge units. The pH titration of CuII(TRI-H)3 had two apparent pH-dependent regions, with a lower pH transition associated with Cu(II) binding to the peptide (being complete by pH 5.7), while a higher pH event (with a pKa of 8.5) was assigned to the deprotonation of a copper-bound water to hydroxide. When CuII(TRI-EH)3 was examined, the same pH-dependent Cu binding was seen; however, the water to hydroxide conversion was shifted to higher pH (pKa = 9.86) (Scheme 1). Furthermore, an additional pH-dependent conversion with a pKa of 6.33 was discovered. This intermediate pH event was assigned to deprotonation of Glu22, allowing the formation of a hydrogen bond with the proton of His23 (Scheme 1) This second pH process is not observed when the lysine in the 24th position rather than the lysine in the 22nd position is mutated to Glu (TRI-HK24E), which is consistent with this interpretation. Comparison of TRI-H and TRI-EH via XAS analysis of Cu(I) and EPR analysis of Cu(II)-bound constructs showed no large differences in the geometry of the two copper binding sites at pH 5.8, though a decreased XANES pre-edge peak and decreased Debye–Waller factors in the EXAFS of TRI-EH indicated a slight change from a more T shaped geometry toward trigonal planar. Further studies on the effect of changing charge near the active site were done with TRI-EH as the parent peptide of the series, rather than TRI-H, as this peptide allows for a larger range of charge differences (Table 2).
Scheme 1. :
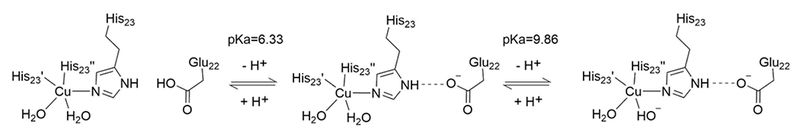
Illustration of Deprotonation Events Hypothesized in TRI-EH titration
On examination of the series in Table 2 at pH 7.4, the Cu(I) Kd value was found to decrease in a linear manner by 2 orders of magnitude as the charge of the peptide was adjusted from 0 (TRI-EHE27K) to –12 (TRI-EHK24E), while the Cu(II) affinity remained largely unchanged (Figure 5). For Cu(I) the affinities ranged from pico- to femtomolar regardless of pH (5.8 or 7.4), whereas the cupric dissociation constants were between nano- and micromolar values at pH 7.4 and decreased by 2 orders of magnitude as the pH decreased (Figure 5). Redox potentials calculated on the basis of these affinities were found to vary by as much as 100 mV at a specified pH on going across the range of peptides. As the Cu(II) binding constant was relatively invariant, the change in potential primarily reflects the destabilization of the Cu(I) peptides as the negative charge increases. In contrast, because the binding affinity for Cu(I) to each peptide was essentially invariant with pH, the ~+100 mV shift in potential observed for each peptide on going from pH 7.5 to 5.8 is a consequence of the destabilization of the Cu(II) protein under more acidic conditions. That reduction potential was linked to pH was thought to be a consequence of the proposed hydrogen bond between Glu22 and His23 within the TRI-EH construct family.
Figure 5.
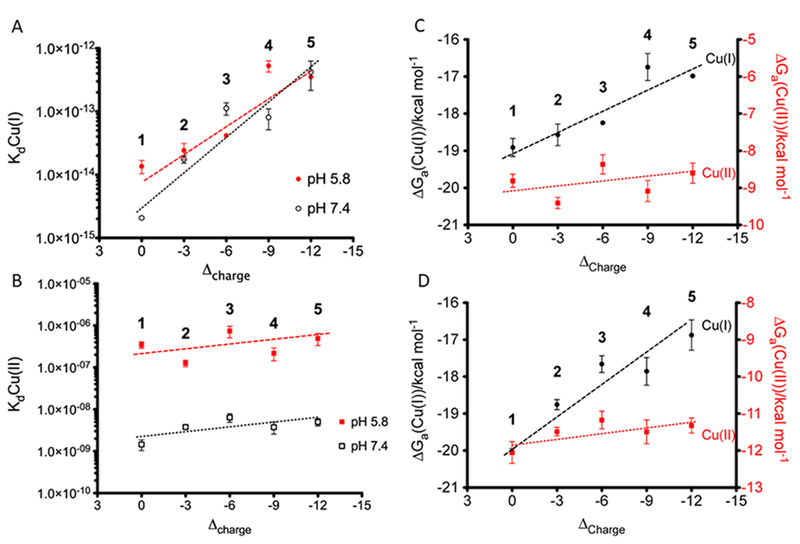
(A) Cu(I) dissociation constants and (B) Cu(II) dissociation constants with respect to ▵charge at pH 5.8 and 7.4. It should be noted that the y axes have log scales. Free energies of binding for Cu(I) and Cu(II) at (C) pH 5.8 and (D) pH 7.4. Peptides: (1) TRI-EHE27K; (2) TRI-EHE27Q; (3) TRI-EH; (4) TRI-EHK24Q; (5) TRI-EHK24E. Reprinted and adapted with permission from ref 49. Copyright 2013 American Chemical Society.
We originally hypothesized that the reduction potential would become less positive in this series of peptides as the negative charge increased around the active site, since Cu(II) binding would be tighter; however, the Cu(II) affinity was hardly modified. Nonetheless, at a fixed pH the reduction potential did become less positive upon an increase in charge because Cu(I) binding was destabilized. The fact that changes to positions 24 and 27 below the His layer affect the Cu(I) affinity in a way that does not follow expectations from electrostatics (increasing positive charge increases Cu(I) affinity) suggests that these changes reflect hydrogen bonding and salt bridging effects between surface residues, leading to changes in Cu(I) geometry which affect affinity. We postulated that an induced rack effect like that thought to exist within cupredoxins may be in play for some of these constructs, with greater effects being seen on Cu(I) geometry, as its structure was thought to be more rigid than that of Cu(II) in our constructs. While the trend in reduction potential across this series is the same at pH 5.8 and 7.4, there is a reduction potential shift to more positive values at lower pH. This is because the Cu(I) state binding is not pH dependent while the Cu(II) affinity decreases at low pH. The 100 mV change over 1.6 pH units is consistent with a change of one proton per electron in this range. Both the UV–vis and EPR spectral parameters associated with the Cu(II) chromophore shifted over this range in a manner that was inconsistent with protonation of a hydroxide bound to copper but rather with the protonation of a H-bonded side chain carboxylate with the pKa value reported above.
Finally, assaying NiR activity across this series, we found a 4-fold increase as the negative charge was increased from 0 to –12. A correlation between increasing reduction potential and decreasing NiR rate was thus discovered (Figure 6). While it is tempting to think that this correlation would be due to electron transfer rates, this is unlikely due to our assay conditions, in which ascorbate is in massive excess and electron transfer is, thus, extremely rapid. Instead we believe that this correlation may be related to changing reorganization energy requirements as the construct cycles between three-coordinate Cu(I) and four- to five-coordinate Cu(II). In summary, this study demonstrated that metal binding affinities and reduction potentials could be markedly modified by second and third coordination sphere modifications and that a hydrogen-bonding residue could be designed to interact with the Cu(His)3 binding site; however, these changes had little effect on catalysis. Furthermore, while satisfying explanations for the observed results could be extracted through using multiple spectroscopic techniques, the collected data were disappointing from a design perspective, as one hoped to alter structure and physical properties more predictively.
Figure 6.
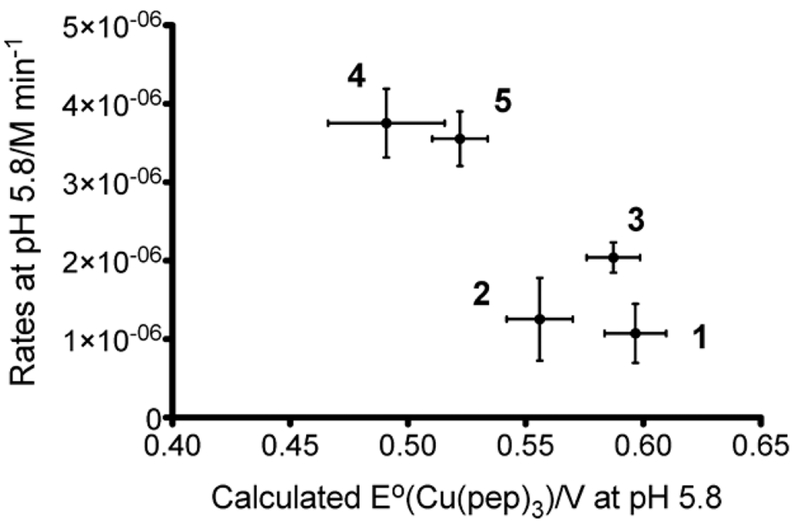
Rates at pH 5.8 in relation to the calculated reduction potentials at pH 5.8. Peptides: (1) TRI-EHE27K; (2) TRI-EHE27Q; (3)TRI-EH; (4) TRI-EHK24Q; (5) TRI-EHK24E. Reprinted with permission from ref 49. Copyright 2013 American Chemical Society
Clearly, the electrostatic environment around the catalytic center in a protein is important for defining activity. To evaluate our designed center more carefully, we used 2D IR techniques to study a carbonyl bound to CuI(TRI-H)3.50 This strategy serves as a means of probing the energetic fields around the metal binding site to investigate what strategies might be used to increase NiR efficiency to a greater degree. Modulation of these fields is thought to be an important factor in catalysis, and investigating the effect of the protein on the excited state of CO can give hints into how a transition state (itself an excited state) could be affected by the protein scaffold. Also, a plethora of studies of CO bound to heme have shown CO bound to a transition metal to be a sensitive probe of structure and dynamics.51
In collaboration with the Kubarych group, we examined the ultrafast dynamic evolution of 2D IR signals of the carbon monoxide adduct of CuI(TRI-H)3.50 These studies revealed a shift in vibrational anharmonicity with a half-life of around 2 ps. Two transitions were observed, the first being the ground to first excited state (the 0–1 transition), which red shifts, and the transition of the first to second excited state (the 1–2 transition), which blue shifts. The excited-state shift (1–2) was larger than that corresponding to the ground-state transition (0–1). Similar behavior was also observed in CuI(CO)(TRI-H K22Q K24Q)3, in which the Lys residues adjacent to the His23 binding site are converted from positive amino acids to neutral amino acids. These observations show that this feature is a general feature of TRI peptides and is not isolated to a single peptide sequence. This feature was not, however, observed within a small-molecule iron–carbonyl complex studied in earlier work, suggesting that it is related to the protein environment conferred by this 3SCC.52
Several computational modeling techniques were explored to understand this behavior. A comparison of the stretching frequency of Cu(I)(CO)(TRI-H)3 to native type 2 copper systems as well as three- or four-coordinate histidyl model complexes proved that our construct was most consistent with a four-coordinate CuI(CO)His3 binding site (Table 3). Thus, the effect of the local environment on this nonequilibrium relaxation was examined using a model consisting of a copper ion with three 5-ethylimidazole ligands as well as a carbonyl as a minimal model of the active site of CuI(CO)(TRI-H)3. The crystal structure of the Zn site within our previously described carbonic anhydrase model was used for starting coordinates of this site before geometry optimization in Gaussian 09 using the B3LYP functional and 6-311G(d,p) basis set. Unfortunately, this model was unable to explain the feature seen in experiments. The full de novo enzyme was next modeled using QM/MM via the ONIOM method to investigate the effects of steric and electrostatic interactions arising from the peptide scaffold. A 6° decrease in the Cu–C–O bond angle was observed in this model upon excitation, in comparison to the small-molecule QM model, in which a less than 1° decrease was seen. Calculated frequencies of such a bending motion were consistent with the effect seen within 5 ps. These studies demonstrate the importance of the overall protein environment around the metal active site and illustrate why this de novo approach provides a useful scaffold for homogeneous catalytic reactions of this sort.
Table 3.
Comparison of CO Stretching Frequencies for (CO)Cu(I)(TRI-H)3 and Several Literature Values
Further investigation of this bond angle deformation found the electric field of the peptide caused a distortion with in the histidines bound to the Cu rather than directly causing the vibrational coupling to the Cu–C–O bond angle. DFT on the small-molecule model mentioned above using fixed His–Cu dihedral angles determined that while the Cu–C–O bond angle was unaffected by His–Cu dihedral angle in the vibrational ground state, it was very sensitive in a vibrational excited state (Figure 7). Further, the fact that electrostatics and His distortion are linked was determined to be due to a 3.6 D dipole moment over the entire histidine ring. Thus, it seems possible that His dihedral angles could influence the potential energy surface of the NiR transition state and, therefore, reaction kinetics. These findings suggested that long-range changes could have large effects on catalysis such as mutations above or below the His plane. Furthermore, these observations help us to understand how remote mutations, such as K22E that forms H bonds to the bound histidines, can influence metal ion stability and reduction potentials of the system.
Figure 7.
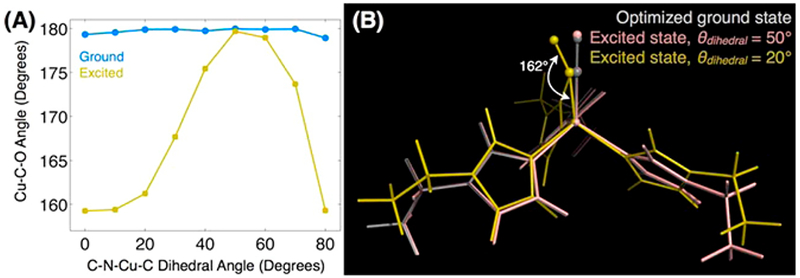
Propensity for excited-state structural changes appears to be controlled by the geometry of the histidine’s coordination with copper. Results of DFT calculations of the small Cu(EtIm)3CO cluster in the absence of the protein matrix show that (A) the optimized Cu–C–O bond angle is insensitive to the Cδ–Nε– Cu– C dihedral angle in the CO vibrational ground state. In the excited state, however, the bond angle and tilt become acutely sensitive to the histidine coordination geometry. (B) Structures selected from the full dihedral angle scan with dihedral angles of 20° and 50° illustrate the link between the histidine coordination and the copper-ligand bonding. This geometrical distortion only occurs in the CO excited state, suggesting the inadequacy of ground-state equilibrium structures to reveal reactivity. Reprinted with permission from ref 50. Copyright 2013 American Chemical Society.
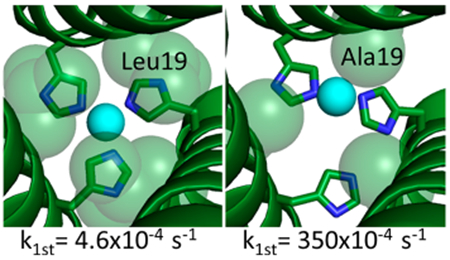
Having explored the effect of mutations at the helical interface and determined via 2D IR of CuI(CO)(TRI-H)3 that long-range interior mutations may be able to change the energetic landscape of the active site, we turned our attention to mutations in the interior of the 3SCC. Through work with Cd(II) binding to Cys3 sites within the TRI system, we had determined that steric changes around the metal binding site within 3SCCs can be used to control the coordination number of the bound metal.20,23,58–61 Thus, we sought to test if similar control could be used on a Cu(His)3 site and then evaluate the effect of such control on NiR activity. Mutations were made to the 19 and 26 leucine layers that are positioned directly above and below the CuHis3 layer, respectively (Table 4). Ile and D-Leu were substituted for L-Leu above the Cu(His)3 site (19th position) to test the effect of increased steric bulk in this position, and Ala or Asp was incorporated either above or below the catalytic center (19th or 26th positions) to test the effect of decreased steric bulk or the addition of an amino acid capable of hydrogen bonding which may assist in catalysis (Figure 8).62
Table 4.
TRI-H Family Peptide Sequences Used in the Study of the Effects of Internal Mutationsa
| peptideb | 1 | 2 | 9 | 16 | 23 | 30 |
|---|---|---|---|---|---|---|
| TRI-H | Ac-G | WKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 |
| L19I | Ac-G | WKALEEK | LKALEEK | LKAIEEK | HKALEEK | G-NH2 |
| L19DL | Ac-G | WKALEEK | LKALEEK | LKADLEEK | HKALEEK | G-NH2 |
| L19A | Ac-G | WKALEEK | LKALEEK | LKAAEEK | HKALEEK | G-NH2 |
| L26A | Ac-G | WKALEEK | LKALEEK | LKALEEK | HKAAEEK | G-NH2 |
| L19D | Ac-G | WKALEEK | LKALEEK | LKADEEK | HKALEEK | G-NH2 |
| L26D | Ac-G | WKALEEK | LKALEEK | LKALEEK | HKADEEK | G-NH2 |
The first row is the parent TRI-H, sequence while subsequent rows give modified forms of TRI-H.
The C and N termini of the peptides are amidated and acetylated, respectively. The first amino acid within each heptad is labeled with its overall position within the peptide.
Figure 8.
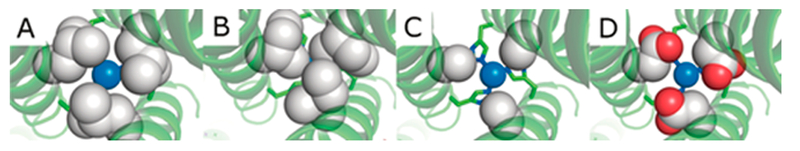
PyMol models of TRIW-H (A) L19I, (B) L19, (C) L19A, and (D) L19D showing the change in steric packing above the copper center made on the basis of the crystal structure of ZnIINHgIIS(CSL9PenL23H)3+ (PDB code 3PBJ) or ZnII(H2O)(GRAND-CSL12AL16C)3− (PDB code 5KB2). Peptide strands are represented by helices, and His23 side chains are represented by sticks. Residues on the 19th position and Cu(I) are shown in spheres to illustrate the space-filling above the copper center; Cu is shown in blue and O in red. Reprinted with permission from ref 62. Copyright 2018 Angewandte Chemie International Edition.
Comparing the first-order rate constants for all the constructs listed, we determined they fell into two distinct groups: L19I and L19DL showed no effect on NiR efficiency, while L19A, L26A, L19D, and L26D all increased the NiR efficiency between 60 and 75 times greater than that of the original TRI-H. The fastest derivative (L19A) had a catalytic efficiency of 3.5 × 102 s1. Michaelis–Menten kinetic analysis of L19A proved it had the highest reported catalytic efficiency within homogeneous aqueous conditions at 1.0 M−1 s−1.
Due to the rate at which Cu(II) is reduced under our assay conditions, the prevailing thought has been that Cu(I) is the resting oxidation state during first-order rate kinetic analysis. Thus, we measured CuI(peptide)3 XAS to determine if structural changes were the cause of the kinetic differences seen. Surprisingly, XANES and EXAFS analysis found that the enhanced NiR constructs could be divided into two groups. L26A and L26D had Cu–N distances (1.91 Å) and 1s → 4p peak intensity consistent with three-coordinate Cu(I) like not only the original TRI-H but also the L19I and L19DL variants. L19A and L19D, variants in which the steric encumbrance above the Cu(His)3 site was expected to decrease, instead had Cu–N distances (1.86–1.88 Å) and 1s → 4p peak intensity more consistent with two-coordinate Cu(I). Surprisingly, it seemed that decreasing steric bulk above the Cu had decreased rather than increased the coordination number of the Cu(His)3 site, counter to what one would predict on the basis of the previously mentioned CdIICys3 studies. Rather than allowing a water to enter the helical interior and bind, it appears that steric bulk above the Cu(His)3 forces the Cu(I) to remain three-coordinate and decreasing this steric bulk allows the Cu(I) to go into a more preferred two-coordinate geometry.
While an interesting revelation, it was apparent that this shift to a more two-coordinate geometry was unlikely to explain the increased NiR efficiency of L19A and L19D ,as it was not observed in L26A and L26D, which had almost identical catalytic efficiency increases. Closer examination of the 1s → 4p transition found that, while L26A and L26D had lower intensity than L19A and L19D, all four had similarly red shifted energies in comparison to the constructs that did not have improved NiR rates from 8982.8 to 8983.6 eV (Figure 9). Previous studies have shown that the Cu(I) coordination geometry can influence not only the intensity but also the energy of this transition, which may explain this energy shift. Another intriguing possibility is that this energy shift belies a change in the net electron density of the bound Cu(I). This would fit well with the results from our collaboration with the Kubarych laboratory, in which it was thought possible that long-range interior mutations could affect the potential energy surface of the NiR transition state. While an interesting possibility, these constructs will require further testing to determine exactly what factors are in play to cause such a large jump in NiR activity.
Figure 9.
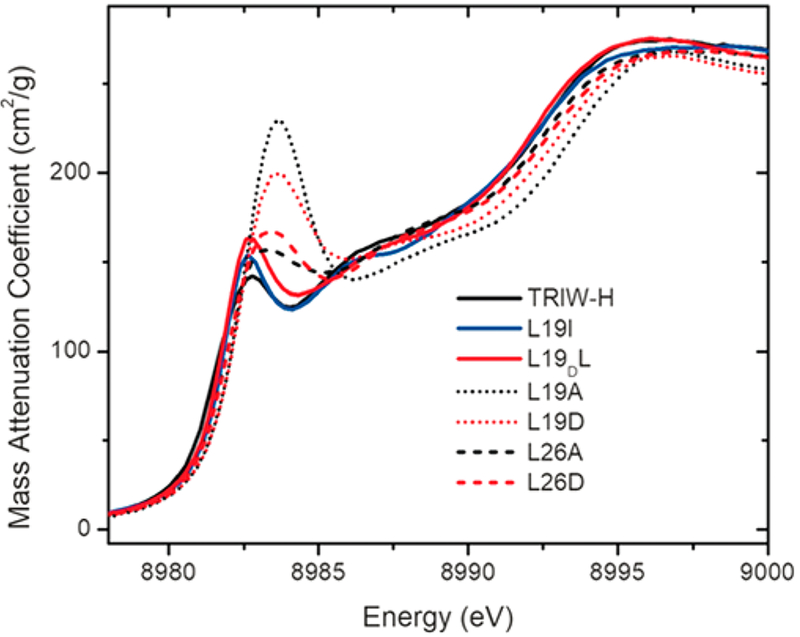
XANES of Cu(I)-bound constructs reported within ref 62, measured at pH 5.8. Reprinted with permission from ref 62. Copyright 2018 Angewandte Chemie International Edition.
Thus, we have shown that a protein designed to accomplish carbonic anhydrase activity can perform nitrite reduction simply by replacing Zn(II) with Cu(II). Spectroscopic investigations of the Cu binding environment in this family of constructs reveals a Cu(I) coordination of His3 when the second coordination sphere is sterically demanding or His2 with a protonated imidazole in the absence of sterically bulky nearby resides above the His3 layer (Figure 10). In contrast, assignment of the Cu(II) coordination geometry is not differentiated between four-coordinate (His3(H2O)) and five-coordinate (His3(H2O)2) geometries (Figure 10). We have recently obtained Cu(II) XAS data of a variety of TRI-H constructs which we hope will allow clarification of the Cu(II) coordination assignment, but at the moment this structure remains ambiguous. CuNiR maintains a CuHis3(H2O) environment regardless of oxidation state, which suggests that one of the limiting factors in our constructs may be an increased reorganization energy in comparison to the native system. There is, however, evidence which would suggest that this is not a dominant factor in the difference between our systems and CuNiR, as direct reduction of the Cu(II) centers by ascorbate in the absence of nitrite is much faster than the catalytic rate. The resting state of our system is the Cu(I) protein, as the assay our laboratory uses requires a large excess of ascorbate. Thus, the rate-limiting step must be associated with the Cu(I) nitrite complex since the conversion of CuII(TRI-H)3 to CuI(TRI-H)3 is fast.
Figure 10.
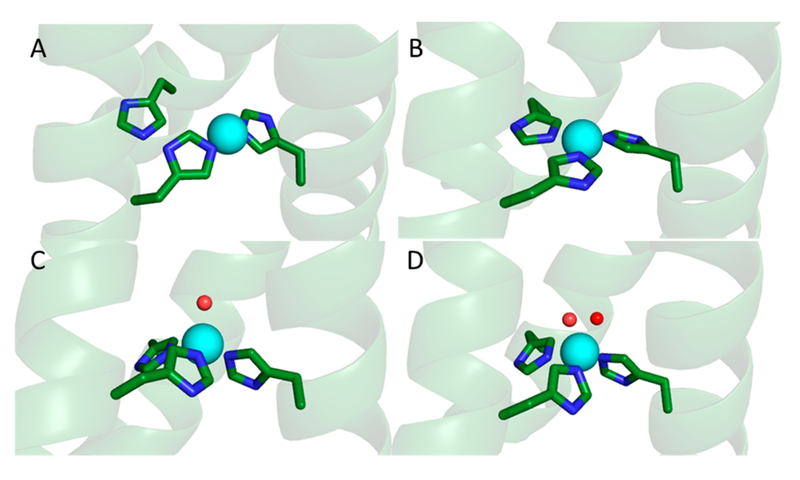
(top) Representations of the (A) two-coordinate or (B) three-coordinate model for CuI(TRI-H)3 based on the structure of HgIIS[ZnIIN(H2O)](CSL9CL23H)3+ (PDB 3PBJ). (bottom) Representations of the (C) four-coordinate or (D) five-coordinate model for CuII(TRI-H)3 based on the structure of HgIIS[ZnIIN(H2O)]-(CSL9CL23H)3+ (PDB 3PBJ).
Whether the rate-determining process is nitrite binding, proton transfer, product release (Scheme 2), or some combination of these factors cannot presently be differentiated. We have observed similar catalytic efficiencies for constructs which are dominated by two-coordinate Cu(I) geometry and those with three-coordinate Cu(I) geometry in comparing second coordination sphere mutations above or below the CuHis3 layer, suggesting that rearrangement of the first coordination sphere is not limiting the reaction under the present conditions. Finally, we have determined that, regardless of Cu(I) coordination, the addition of a ligand such as CO can force the coordination number to 4, suggesting that NO2− could do the same, which may explain why the initial number of ligands bound to Cu(I) before nitrite is present may not explain the reaction profiles.
Scheme 2. :
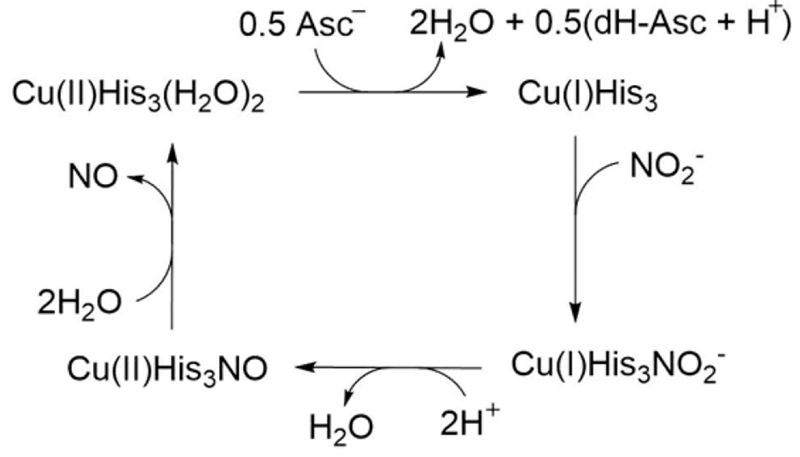
Illustration of Nitrite Reductase Assay Used for the TRI-H System
Comparing the pseudo-first-order rate constants of all the TRI-H constructs reviewed here (Figure 11), we see that, although mutations to the helical interface had measurable effects on reduction potential, their effect on NiR activity is minimal. Interior mutations to amino acids above or below the His3 binding site, however, can increase the NiR activity as much as 75-fold. That the L19I construct has only a small effect on NiR activity while L19A or L19D increases the NiR activity by 50–75-fold is consistent with decreasing steric hindrance being the key to improved NiR activity. Using this increase in catalytic efficiency, we have started to utilize more detailed kinetic studies to determine how one would best improve the catalytic efficiency further. Michaelis–Menten kinetic analysis of TRI-H L19A revealed that the calculated Michaelis constant (KM) of this system was 240 mM, while previous studies calculated a KM value for CuNiR of 35 μM.63This difference of 4 orders of magnitude in substrate affinity appears to be a major limitation of this artificial system. The origin of this difficulty for substrate recognition may be in part due to the comparative resting states for the native CuNiR (a Cu(II) enzyme) and our designed proteins (Cu(I) enzymes). Analysis of nitrite binding to CuII(TRI-H)3 via EPR gives a rough estimate of 1.5–2.1 mM for the dissociation constant of nitrite for our system in the cupric state. In theory, having the nitrite binding form be Cu(II) rather than Cu(I) for the TRI peptides could lead to a 200-fold increase in catalytic efficiency. That this shift would not improve the KM value to that of the native system is likely due to protein–substrate interactions which are present in CuNiR but have not been incorporated into our model complex. Furthermore, nitrite tends to bind to Cu(I) as an N-bound form, whereas the nitrite oxygen atoms are preferred ligands for the cupric ion.64–66 Reorientation of the substrate on the copper could lead to further rate enhancement, though a more recent computational study postulates that nitrite flips orientation after Cu(II) is reduced in native CuNiR, which would mean that changing the resting oxidation state will only improve the catalytic efficiency due to differences in substrate binding affinity.66 We are currently exploring assays that allow us to use a CuII(TRI-H)3 enzyme rather than the current assay’s limitation to the cuprous resting state.
Figure 11.
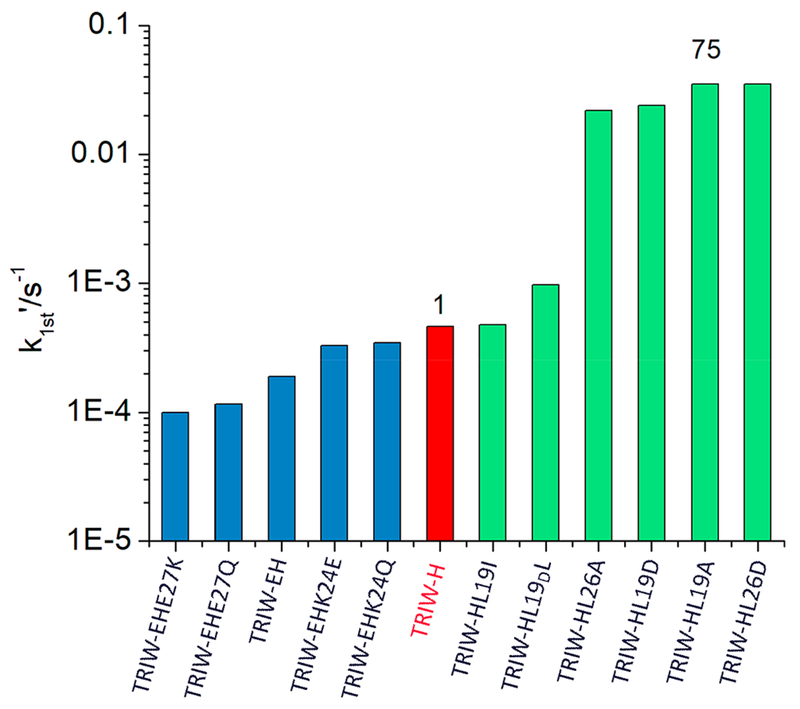
Pseudo-first-order rate constants at pH 5.8 for the original NiR model TRIW-H (red), the series of exterior mutations reported in ref 49 (blue), and the series of interior mutation reported in ref 62 (green). The relative efficiencies of TRIW-H and TRIW-HL19A are included above those constructs for context. This figure was adapted with permission from ref 62. Copyright 2018 Angewandte Chemie.
In addition to optimizing the resting state oxidation level, the incorporation of a nearby acid/base catalyst is an obvious approach to improve catalytic efficiency. This strategy emulates native CuNiR, which has an Asp and His in the second coordination sphere that proves crucial for proton-gated electron transfer and catalysis.65 We described above our first attempt to incorporate such an interaction by introducing Asp at the 19th or 26th positions, either above or below our CuHis3 layer, but the fact that Ala in those positions gave similar improvements in catalytic efficiency indicates that the Asp was unlikely to be functioning as intended. There are two issues with the present designs that probably will need to be addressed to optimize the proper function of a carboxylate acid/base catalyst. These factors are the chelate ring structure introduced when a carboxylate residue is incorporated in close proximity to the copper and the symmetric nature of the 3SCC. The heptad repeat separates potential hydrophobic layers by either two (abcd) or three (defga) intervening residues. Simultaneous mutation of adjacent a and d positions, as in L19DL23H, places two metal binding ligands in close proximity, effectively forming an internal chelating group. While such a structure strongly stabilizes the metal protein complex, it leads to the wrong first coordination sphere environment with a chelating, rather than pendant, carboxylate in addition to the desired imidazole. Thus, carboxylate side chains may need to be incorporated in the sequence at positions other than a or d to be effective. Native proteins exploit dissymmetry to polarize an active site; however, homotrimeric 3SCCs yield metal environments that are symmetric because each peptide is identical. Therefore, one can only design a second coordination sphere that has zero or three aspartates in a layer adjacent to the metal binding site. These three Asp residues within a single layer cannot yield asymmetric polarization at the active site. Even worse, they may directly interact to form hydrogen bonds that could perturb the side chain pKas and which could leave no Asp residue free to interact with the bound nitrite as intended. We have attempted to work with the symmetric nature rather than against it by introducing Arg residues above or below the His layer, with the idea that the positive charges of these residues would draw in the negatively charge nitrite. This strategy appears to have been somewhat successful with first-order rate constants of 0.057 and 0.046 s−1 for L19R and L26R, respectively, in comparison to 0.035 s−1 for our previously most active construct L19A, but it seems clear that directing the orientation of a secondary coordination sphere amino acid will be a high priority within future designs.
Outside of the active site, we have successfully designed a putative hydrogen-bonding residue which can interact with the metal binding His residues in TRI-EH, though this had little effect on the catalytic efficiency. We are currently exploring the use of histidine residues with methylation on either Nδ or Nε to investigate whether the effects observed in TRI-EH are caused by a hydrogen bond between the mutated Glu22 and His23, but assuming this proves to be the case, it presents some exciting opportunities. Hydrogen-bonding networks are a common theme in native proteins and are utilized by nature to direct amino acid orientation and fine-tune electronic and/or redox properties. Including hydrogen bonding into our own designs would allow us as chemists to fine-tune parameters in a similar fashion. This is particularly meaningful when it comes to directing amino acid orientation which allows for much more precise designs of second coordination sphere amino acids within these constructs. Admittedly, the symmetric nature of the 3SCC-based constructs presented in this review make such detailed hydrogen bond networking difficult or impossible, and so we have begun exploring His3 sites and NiR activity within the three-helical bundle peptide α3D. Just as for the TRI peptides, we have observed excellent CO2 hydration activity with Znα3DH3 which has a Zn(II) bound to three histidines and a water.67 Thus, we may be able to achieve comparable activities for NiR activity when Cu(II) is substituted into this system and then more easily introduce asymmetry around the active center than is possible in our current 3SCC constructs.
The Tri-H peptides illustrate how metalloprotein design rapidly led to the most active artificial CO2 hydration catalysts. By substitution of the metal from Zn(II) to Cu(I), highly stable constructs that have the highest catalytic efficiency and turnovers for artificial catalysts in aqueous media for nitrite reductase reactions have been achieved, illustrating a possible mechanism for organisms using native proteins to adapt to a new environment by switching metals in a pre-existing metal binding site in order to gain function. This work exemplifies one of the greatest benefits of the bottom-up approach of de novo design: once a working model is obtained, there are always a variety of branching design pathways one can take to optimize the construct further. Once the desired activity was identified, single site mutations in the second coordination sphere led to activities that span a more than 500-fold range. Exploring new possibilities such as modification of the first coordination sphere or local control of acid/base residues will then give us insight into the native system’s requirements for high catalytic efficiency. Thus, we think it is clear that the TRI-H CuNiR model and related designs will continue to be an important strategy for exploring type 2 copper centers within a simplified protein environment.
ACKNOWLEDGMENTS
V.L.P. thanks the National Institutes of Health for financial support of this research (ES012236). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Joo H; Lin Z; Arnold FH Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature 1999, 399, 670–673. [DOI] [PubMed] [Google Scholar]
- (2).Jeschek M; Reuter R; Heinisch T; Trindler C; Klehr J; Panke S; Ward TR Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537, 661–665. [DOI] [PubMed] [Google Scholar]
- (3).Dydio P; Key HM; Nazarenko A; Rha JY-E; Seyedkazemi V; Clark DS; Hartwig JF An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. [DOI] [PubMed] [Google Scholar]
- (4).Huang P-S; Boyken SE; Baker D The coming of age of de novo protein design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]
- (5).Joh NH; Wang T; Bhate MP; Acharya R; Wu Y; Grabe M; Hong M; Grigoryan G; DeGrado WF De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 2014, 346, 1520–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mills JH; Khare SD; Bolduc JM; Forouhar F; Mulligan VK; Lew S; Seetharaman J; Tong L; Stoddard BL; Baker D Computational Design of an Unnatural Amino Acid Dependent Metalloprotein with Atomic Level Accuracy. J. Am. Chem. Soc 2013, 135, 13393–13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Clark KM; Yu Y; Marshall NM; Sieracki NA; Nilges MJ; Blackburn NJ; van der Donk WA; Lu Y Transforming a Blue Copper into a Red Copper Protein: Engineering Cysteine and Homocysteine into the Axial Position of Azurin Using Site-Directed Mutagenesis and Expressed Protein Ligation. J. Am. Chem. Soc 2010, 132, 10093–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Andreini C; Bertini I; Rosato A Metalloproteomes: A Bioinformatic Approach. Acc. Chem. Res 2009, 42, 1471–1479. [DOI] [PubMed] [Google Scholar]
- (9).Andreini C; Cavallaro G; Lorenzini S; Rosato A MetalPDB: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2012, 41, D312–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hoffman BM; Lukoyanov D; Yang Z-Y; Dean DR; Seefeldt LC Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yoshikawa S; Shimada A Reaction Mechanism of Cytochrome c Oxidase. Chem. Rev 2015, 115, 1936–1989. [DOI] [PubMed] [Google Scholar]
- (12).Yu F; Cangelosi VM; Zastrow ML; Tegoni M; Plegaria JS; Tebo AG; Mocny CS; Ruckthong L; Qayyum H; Pecoraro VL Protein Design: Toward Functional Metalloenzymes. Chem. Rev 2014, 114, 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mocny CS; Pecoraro VL De Novo Protein Design as a Methodology for Synthetic Bioinorganic Chemistry. Acc. Chem. Res 2015, 48, 2388–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).DeGrado WF; Summa CM; Pavone V; Nastri F; Lombardi A De Novo Design and Structural Characterization of Proteins and Metalloproteins. Annu. Rev. Biochem 1999, 68, 779–819. [DOI] [PubMed] [Google Scholar]
- (15).Zastrow ML; Pecoraro VL Designing functional metalloproteins: From structural to catalytic metal sites. Coord. Chem. Rev 2013, 257, 2565–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dieckmann GR; McRorie DK; Tierney DL; Utschig LM; Singer CP; O’Halloran TV; Penner-Hahn JE; DeGrado WF; Pecoraro VL De Novo Design of Mercury-Binding Two- and Three-Helical Bundles. J. Am. Chem. Soc 1997, 119, 6195–6196. [Google Scholar]
- (17).Dieckmann GR; McRorie DK; Lear JD; Sharp KA; DeGrado WF; Pecoraro VL The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. J. Mol. Biol 1998, 280, 897–912. [DOI] [PubMed] [Google Scholar]
- (18).Farrer BT; Harris NP; Balchus KE; Pecoraro VL Thermodynamic Model for the Stabilization of Trigonal Thiolato Mercury(II) in Designed Three-Stranded Coiled Coils. Biochemistry 2001, 40, 14696–14705. [DOI] [PubMed] [Google Scholar]
- (19).Ghosh D; Lee K-H; Demeler B; Pecoraro VL Linear Free-Energy Analysis of Mercury(II) and Cadmium(II) Binding to Three-Stranded Coiled Coils. Biochemistry 2005, 44, 10732–10740. [DOI] [PubMed] [Google Scholar]
- (20).Iranzo O; Cabello C; Pecoraro VL Heterochromia in Designed Metallopeptides: Geometry-Selective Binding of Cd(II) in a De Novo Peptide. Angew. Chem. Int. Ed 2007, 46, 6688–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tebo AG; Hemmingsen L; Pecoraro VL Variable primary coordination environments of Cd(II) binding to three helix bundles provide a pathway for rapid metal exchange. Metallomics 2015, 7, 1555–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ruckthong L; Peacock AFA; Pascoe CE; Hemmingsen L; Stuckey JA; Pecoraro VL d-Cysteine Ligands Control Metal Geometries within De Novo Designed Three-Stranded Coiled Coils. Chem. - Eur. J 2017, 23, 8232–8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ruckthong L; Deb A; Hemmingsen L; Penner-Hahn JE; Pecoraro VL Incorporation of second coordination sphere d-amino acids alters Cd(II) geometries in designed thiolate-rich proteins. JBIC, J. Biol. Inorg. Chem 2018, 23, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ghosh D; Pecoraro VL Understanding Metalloprotein Folding Using a de Novo Design Strategy. Inorg. Chem 2004, 43, 7902–7915. [DOI] [PubMed] [Google Scholar]
- (25).Touw DS; Nordman CE; Stuckey JA; Pecoraro VL Identifying important structural characteristics of arsenic resistance proteins by using designed three-stranded coiled coils. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 11969–11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hakansson K; Carlsson M; Svensson LA; Liljas A Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J. Mol. Biol 1992, 227, 1192–1204. [DOI] [PubMed] [Google Scholar]
- (27).Zastrow ML; Peacock AFA; Stuckey JA; Pecoraro VL Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat. Chem 2012, 4, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krebs JF; Ippolito JA; Christianson DW; Fierke CA Structural and functional importance of a conserved hydrogen bond network in human carbonic anhydrase II. J. Biol. Chem 1993, 268, 27458–27466. [PubMed] [Google Scholar]
- (29).Liang Z; Xue Y; Behravan G; Jonsson B-H; Lindskog S Importance of the conserved active-site residues Try7, Glu106 and Thr199 for the catalytic function of human carbonic anhydrase II. Eur. J. Biochem 1993, 211, 821–827. [DOI] [PubMed] [Google Scholar]
- (30).Chino M; Maglio O; Nastri F; Pavone V; DeGrado WF; Lombardi A Artificial Diiron Enzymes with a De Novo Designed Four-Helix Bundle Structure. Eur. J. Inorg. Chem 2015, 2015, 3371–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Roy A; Sarrou I; Vaughn MD; Astashkin AV; Ghirlanda G De Novo Design of an Artificial Bis[4Fe-4S] Binding Protein. Biochemistry 2013, 52, 7586–7594. [DOI] [PubMed] [Google Scholar]
- (32).Olson TL; Espiritu E; Edwardraja S; Canarie E; Flores M; Williams JC; Ghirlanda G; Allen JP Biochemical and spectroscopic characterization of dinuclear Mn-sites in artificial four-helix bundle proteins. Biochim. Biophys. Acta, Bioenerg 2017, 1858, 945–954. [DOI] [PubMed] [Google Scholar]
- (33).Iwasaki H; Noji S; Shidara S Achromobacter cycloclastes Nitrite Reductase The Function of Copper, Amino Acid Composition, and ESR Spectra1. J. Biochem 1975, 78, 355–361. [DOI] [PubMed] [Google Scholar]
- (34).Libby E; Averill BA Evidence that the Type 2 copper centers are the site of nitrite reduction by Achromobacter cycloclastes nitrite reductase. Biochem. Biophys. Res. Commun 1992, 187, 1529–1535. [DOI] [PubMed] [Google Scholar]
- (35).Kukimoto M; Nishiyama M; Murphy MEP; Turley S; Adman ET; Horinouchi S; Beppu T X-ray Structure and Site-Directed Mutagenesis of a Nitrite Reductase from Alcaligenes Faecalis S-6: Roles of Two Copper Atoms in Nitrite Reduction. Biochemistry 1994, 33, 5246–5252. [DOI] [PubMed] [Google Scholar]
- (36).Murphy MEP; Turley S; Adman ET Structure of Nitrite Bound to Copper-containing Nitrite Reductase from Alcaligenes faecalis: MECHANISTIC IMPLICATIONS. J. Biol. Chem 1997, 272, 28455–28460. [DOI] [PubMed] [Google Scholar]
- (37).Antonyuk SV; Strange RW; Sawers G; Eady RR; Hasnain SS Atomic resolution structures of resting-state, substrate-and product-complexed Cu-nitrite reductase provide insight into catalytic mechanism. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 12041–12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kataoka K; Furusawa H; Takagi K; Yamaguchi K; Suzuki S Functional Analysis of Conserved Aspartate and Histidine Residues Located Around the Type 2 Copper Site of Copper-Containing Nitrite Reductase. J. Biochem 2000, 127, 345–350. [DOI] [PubMed] [Google Scholar]
- (39).Boulanger MJ; Kukimoto M; Nishiyama M; Horinouchi S; Murphy MEP Catalytic Roles for Two Water Bridged Residues (Asp-98 and His-255) in the Active Site of Copper-containing Nitrite Reductase. J. Biol. Chem 2000, 275, 23957–23964. [DOI] [PubMed] [Google Scholar]
- (40).Tegoni M; Yu F; Bersellini M; Penner-Hahn JE; Pecoraro VL Designing a functional type 2 copper center that has nitrite reductase activity within α-helical coiled coils. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 21234–21239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ruckthong L Crystallographic Comparison of Tris-thiolate Sites in Designed Proteins to Control Metal Geometries; Ph.D. Thesis, University of Michigan, Ann Arbor, MI, 2016. [Google Scholar]
- (42).Ruckthong L; Zastrow ML; Stuckey JA; Pecoraro VL A Crystallographic Examination of Predisposition versus Preorganization in de Novo Designed Metalloproteins. J. Am. Chem. Soc 2016, 138, 11979–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kau LS; Spira-Solomon DJ; Penner-Hahn JE; Hodgson KO; Solomon EI X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen. J. Am. Chem. Soc 1987, 109, 6433–6442. [Google Scholar]
- (44).Prenesti E; Daniele PG; Berto S; Toso S Spectrum–structure correlation for visible absorption spectra of copper(II) complexes showing axial co-ordination in aqueous solution. Polyhedron 2006, 25, 2815–2823. [Google Scholar]
- (45).Fittipaldi M; Wijma HJ; Verbeet MP; Canters GW; Huber M Electronic structure of the two copper sites in nitrite reductase by 9 and 95 GHz EPR on cavity mutants. Appl. Magn. Reson 2006, 30, 417. [Google Scholar]
- (46).Olesen K; Veselov A; Zhao Y; Wang Y; Danner B; Scholes CP; Shapleigh JP Spectroscopic, Kinetic, and Electrochemical Characterization of Heterologously Expressed Wild-Type and Mutant Forms of Copper-Containing Nitrite Reductase from Rhodobacter sphaeroides 2.4.3. Biochemistry 1998, 37, 6086–6094. [DOI] [PubMed] [Google Scholar]
- (47).Yu F De novo Designed Metallopeptides with a Type 2 Copper Center: A Structural and Functional Model for Copper Nitrite Reductase; Ph.D. Thesis, University of Michigan, Ann Arbor, MI, 2014. [Google Scholar]
- (48).Chufán EE; Prigge ST; Siebert X; Eipper BA; Mains RE; Amzel LM Differential Reactivity between Two Copper Sites in Peptidylglycine α-Hydroxylating Monooxygenase. J. Am. Chem. Soc 132, 15565–15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yu F; Penner-Hahn JE; Pecoraro VL De Novo-Designed Metallopeptides with Type 2 Copper Centers: Modulation of Reduction Potentials and Nitrite Reductase Activities. J. Am. Chem. Soc 2013, 135, 18096–18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ross MR; White AM; Yu F; King JT; Pecoraro VL; Kubarych KJ Histidine Orientation Modulates the Structure and Dynamics of a de Novo Metalloenzyme Active Site. J. Am. Chem. Soc 2015, 137, 10164–10176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lim M; Hamm P; Hochstrasser RM Protein fluctuations are sensed by stimulated infrared echoes of the vibrations of carbon monoxide and azide probes. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 15315–15320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jackson CS; Schmitt S; Dou QP; Kodanko JJ Synthesis, Characterization, and Reactivity of the Stable Iron Carbonyl Complex [Fe(CO)(N4Py)](ClO4)2: Photoactivated Carbon Monoxide Release, Growth Inhibitory Activity, and Peptide Ligation. Inorg. Chem 50, 5336–5338. [DOI] [PubMed] [Google Scholar]
- (53).Zhang H; Boulanger MJ; Mauk AG; Murphy MEP Carbon Monoxide Binding to Copper-Containing Nitrite Reductase from Alcaligenes faecalis. J. Phys. Chem. B 2000, 104, 10738–10742. [Google Scholar]
- (54).Boswell JS; Reedy BJ; Kulathila R; Merkler D; Blackburn NJ Structural Investigations on the Coordination Environment of the Active-Site Copper Centers of Recombinant Bifunctional Peptidylglycine α-Amidating Enzyme. Biochemistry 1996, 35, 12241–12250. [DOI] [PubMed] [Google Scholar]
- (55).Jaron S; Blackburn NJ Does Superoxide Channel between the Copper Centers in Peptidylglycine Monooxygenase? A New Mechanism Based on Carbon Monoxide Reactivity. Biochemistry 1999, 38, 15086–15096. [DOI] [PubMed] [Google Scholar]
- (56).Blackburn NJ; Pettingill TM; Seagraves KS; Shigeta RT Characterization of a carbon monoxide complex of reduced dopamine beta-hydroxylase. Evidence for inequivalence of the Cu(I) centers. J. Biol. Chem 1990, 265, 15383–15386. [PubMed] [Google Scholar]
- (57).Himes RA; Park GY; Barry AN; Blackburn NJ; Karlin KD Synthesis and X-ray Absorption Spectroscopy Structural Studies of Cu(I) Complexes of HistidylHistidine Peptides: The Predominance of Linear 2-Coordinate Geometry. J. Am. Chem. Soc 2007, 129, 5352–5353. [DOI] [PubMed] [Google Scholar]
- (58).Lee K-H; Matzapetakis M; Mitra S; Marsh ENG; Pecoraro VL Control of Metal Coordination Number in de Novo Designed Peptides through Subtle Sequence Modifications. J. Am. Chem. Soc 2004, 126, 9178–9179. [DOI] [PubMed] [Google Scholar]
- (59).Lee K-H; Cabello C; Hemmingsen L; Marsh ENG; Pecoraro VL Using Nonnatural Amino Acids to Control Metal-Coordination Number in Three-Stranded Coiled Coils. Angew. Chem., Int. Ed 2006, 45, 2864–2868. [DOI] [PubMed] [Google Scholar]
- (60).Peacock AFA; Hemmingsen L; Pecoraro VL Using diastereopeptides to control metal ion coordination in proteins. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 16566–16571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Peacock AFA; Stuckey JA; Pecoraro VL Switching the Chirality of the Metal Environment Alters the Coordination Mode in Designed Peptides. Angew. Chem., Int. Ed 2009, 48, 7371–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Koebke KJ; Yu F; Salerno E; Stappen CV; Tebo AG; Penner-Hahn JE; Pecoraro VL Modifying the Steric Properties in the Second Coordination Sphere of Designed Peptides Leads to Enhancement of Nitrite Reductase Activity. Angew. Chem., Int. Ed 2018, 57, 3954–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Abraham ZH; Smith BE; Howes BD; Lowe DJ; Eady R R pH-dependence for binding a single nitrite ion to each type-2 copper centre in the copper-containing nitrite reductase of Alcaligenes xylosoxidans. Biochem. J 1997, 324, 511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Tocheva EI; Eltis LD; Murphy MEP Conserved Active Site Residues Limit Inhibition of a Copper-Containing Nitrite Reductase by Small Molecules. Biochemistry 2008, 47, 4452–4460. [DOI] [PubMed] [Google Scholar]
- (65).Ghosh S; Dey A; Sun Y; Scholes CP; Solomon EI Spectroscopic and Computational Studies of Nitrite Reductase: Proton Induced Electron Transfer and Backbonding Contributions to Reactivity. J. Am. Chem. Soc 2009, 131, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Li Y; Hodak M; Bernholc J Enzymatic Mechanism of Copper-Containing Nitrite Reductase. Biochemistry 2015, 54, 1233–1242. [DOI] [PubMed] [Google Scholar]
- (67).Cangelosi VM; Deb A; Penner-Hahn JE; Pecoraro VL A De Novo Designed Metalloenzyme for the Hydration of CO2. Angew. Chem. Int. Ed 2014, 53, 7900–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]