

SUMMARY
Histone post-translational modifications (PTMs) are important genomic regulators often studied by chromatin immunoprecipitation (ChIP), whereby their locations and relative abundance are inferred by antibody capture of nucleosomes and associated DNA. However, the specificity of antibodies within these experiments have not been systematically studied. Here, we use histone peptide arrays and internally calibrated ChIP (ICeChIP) to characterize 52 commercial antibodies purported to distinguish the H3K4 methylforms (me1, me2, and me3, with each ascribed distinct biological functions). We find that many widely-used antibodies poorly distinguish the methylforms and that high- and low-specificity reagents can yield dramatically different biological interpretations, resulting in substantial divergence from the literature for numerous H3K4 methylform paradigms. Using ICeChIP, we also discern quantitative relationships between enhancer H3K4 methylation and promoter transcriptional output and can measure global PTM abundance changes. Our results illustrate how poor antibody specificity contributes to the “reproducibility crisis,” demonstrating the need for rigorous, platform-appropriate validation.
eTOC Paragraph:
Shah et al. evaluate 54 antibodies for specific H3K4 methylforms by internally calibrated chromatin immunoprecipitation (ICeChIP) and peptide arrays. Many of the most commonly-used antibodies poorly distinguish between methylforms, and high-specificity ICeChIP datasets show deviation from literature paradigms of H3K4 methylation biologybased on ChIP datasets generated with low-specificity antibodies.
INTRODUCTION
ChIP has contributed many seminal insights into histone PTM regulation and distribution (Barski et al., 2007; Guenther et al., 2007; Heintzman et al., 2007, 2009; Mikkelsen et al., 2007; Rada-Iglesias et al., 2011; Santos-Rosa et al., 2002; Schübeler et al., 2004; The ENCODE Project Consortium, 2012). However, ChIP interpretation relies on the assumption of near-perfect antibody specificity. The validity of this conjecture for the thousands of existing ChIP-seq datasets is uncertain, given that many commercial antibodies display considerable off-target binding in other experimental formats (Bock et al., 2011; Egelhofer et al., 2011; Fuchs et al., 2011; Grzybowski et al., 2015; Nishikori et al., 2012; Rothbart et al., 2015). Concerningly, apparent ChIP-seq replicates with different antibodies for a single PTM can radically differ, even within a single cell line and when using the highly standardized protocols of the ENCODE consortium (Figures 1 and S1), highlighting the role of antibodies in the scientific “reproducibility crisis” (Baker, 2015).
Figure 1. ENCODE ChIP-seq datasets display internal inconsistency and incongruity.
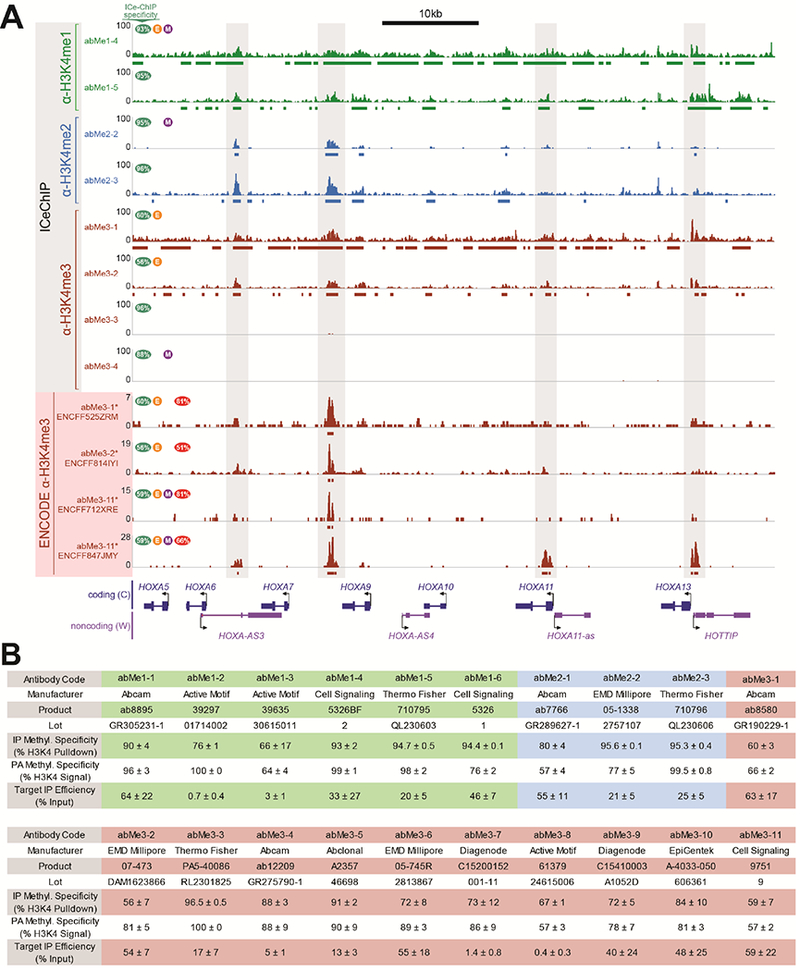
(A) ICeChIP-seq and ENCODE ChIP-seq tracks at distal HoxA cluster in K562 cells. Highly-specific antibodies reveal absence of H3K4me3; low-specificity antibodies detect appreciable signal from lower methyl forms. ENCODE tracks are reminiscent of ICeChIP tracks, but differ from one another and do not show true H3K4me3 signal. Green oval shows ICeChIP methylform specificity for each antibody, orange circle with E indicates antibody validated to ENCODE standards, purple circle with M indicates monoclonality, and red oval shows percentage of peaks in each ENCODE dataset found in all three of the other ENCODE datasets. Bars below tracks represent peaks. (B) Abbreviation codes, specificities in ICeChIP and peptide arrays, and target IP enrichments for antibodies referred to in the main text. Values represent average ± standard deviation.
Here, we have interrogated the specificity of antibodies targeting the three methylation states of lysine 4 on histone H3 (H3K4me1, H3K4me2, and H3K4me3), each ascribed distinct roles in chromatin regulation. H3K4me1 (~5–20% global abundance (LeRoy et al., 2013)) is thought to mark enhancers (Heintzman et al., 2007, 2009; Rada-Iglesias et al., 2011) and flanks promoters (Guenther et al., 2007). H3K4me2 (~1–4% global abundance (LeRoy et al., 2013)) is associated with tissue-specific transcription factor binding sites (Wang et al., 2014), enhancers (Rada-Iglesias et al., 2011), and promoter edges (Fang et al., 2010; Pekowska et al., 2010; Wang et al., 2014). H3K4me3 (~1% global abundance (LeRoy et al., 2013)) defines active transcriptional initiation at promoters (Guenther et al., 2007; Lauberth et al., 2013; Santos-Rosa et al., 2002; Schübeler et al., 2004; Vermeulen et al., 2007; Wysocka et al., 2006), and is also implicated in V(D)J recombination (Matthews et al., 2007), meiotic crossovers (Baudat et al., 2013), and pre-mRNA splicing (Bieberstein et al., 2012; Sims III et al., 2007). As many of these conclusions were drawn presuming that ChIP could discriminate between the three methylation states, we sought to systematically investigate the capacity of antibodies to do so.
To this end, we assessed the specificities of 52 commercial “ChIP grade” antibodies using histone peptide microarrays and ICeChIP (Figures S2 and S3). In the first approach, antibody is incubated with slide-immobilized peptides, and bound regions identified with a fluorescently-labeled secondary antibody (Figure 2A). Peptide microarray measurements allow simultaneous testing of a broad range of different off-target, on-target, and combinatorial PTMs (Bock et al., 2011; Egelhofer et al., 2011; Fuchs et al., 2011; Rothbart et al., 2015). The technique is considered the current gold standard of antibody characterization, but whether it recapitulates antibody performance in ChIP is unclear due to marked differences in experimental format (Uhlen et al., 2016). In contrast, ICeChIP uses DNA-barcoded semisynthetic nucleosome standards encompassing panels of histone PTMs directly spiked into a chromatin sample, allowing the measurement of antibody specificity in situ, and the determination of histone modification density (HMD), the absolute amount of PTM over a genomic interval (Figure 2B) (Grzybowski et al., 2015). However, each nucleosome standard must be independently synthesized, which is labor-intensive and technically challenging. Though peptide arrays and ICeChIP have been compared in a very limited way (Rothbart et al., 2015), the small scale of such studies precluded broader conclusions. Further, previous studies centered on antibody discrimination between different lysine residues (e.g. H3K4me3 vs. H3K9me3) rather than different methylation states of a single lysine (e.g. H3K4me2 vs. H3K4me3), the latter representing a potentially greater challenge. Integrating peptide array and ICeChIP analyses now enables us to critically evaluate antibodies and determine the extent of data transferability between each format.
Figure 2. Histone 3 lysine 4 (H3K4) antibodies display a range of methylform specificities.
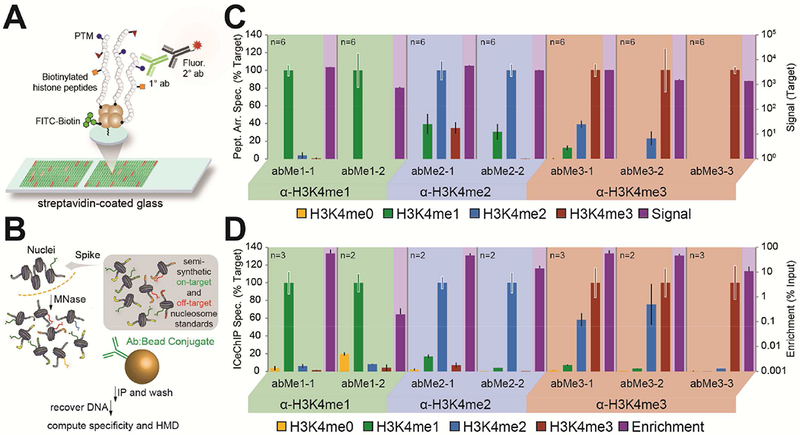
(A, B) Experimental workflows of (A) peptide arrays and (B) ICeChIP. (C, D) A representative selection of methylform binding (target relative to other forms on the left axis) by antibody from (C) peptide arrays and (D) ICeChIP is presented in bar graph form (extracted from the larger set of 52 antibodies: Figures S2 and S3). Purple bar represents raw fluorescence signal or ChIP enrichment, and maps to right axis (log10 scale). Black error bars represent SD of off-target specificity; coloured error bars represent average SD of on-target signal.
RESULTS
Antibody specificities range widely and often diverge across methods
A representative cohort from the 52 antibodies screened with both peptide array and ICeChIP (Figures S2 and S3) is shown in Figures 2C and 2D. High-specificity antibodies, with >90% aggregate methyl-specificity, were identified by both approaches (e.g. abMe1–1 and abMe3–3 in Figure 2C-D; Table S1), but notably, these reagents are often infrequently used (Table S1). When present, cross-reactivity most commonly occurred between states differing by a single methyl group (Figures S2 and S3), and was most severe for the anti-H3K4me3 antibodies. Remarkably, apparent specificity in peptide arrays and ICeChIP is only weakly correlated (R2 = 0.2337: Figure 3A) and is independent of both raw fluorescence in peptide arrays (Figure 3B) and IP enrichment in ICeChIP (Figure 3C), suggesting that antibody specificity trends are not driven by affinity alone. Notably, there was much greater platform disagreement for antibodies to H3K4me2 than for those to H3K4me1 or H3K4me3 (Figure S4A).
Figure 3. Antibodies can display different specificities in peptide arrays and ChIP.
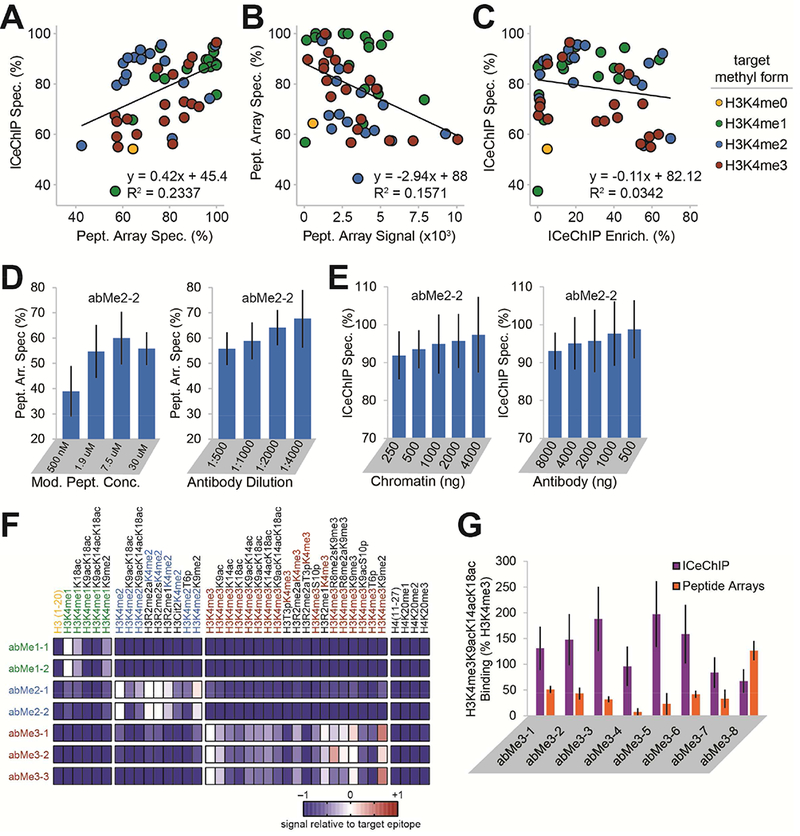
(A) Specificity computed for each antibody (of 52 tested) as target H3K4 methylform (indicated by dot colour) enrichment normalized to the sum of all H3K4 methylform enrichments. (B) Methylform specificity versus on-target signal in peptide arrays. (C) Methylform specificity versus on-target enrichment in ICeChIP. (D) Aggregate specificity in peptide arrays of abMe2-2, varying concentration of modified peptide (left) or antibody dilution (right). (E) Aggregate specificity of abMe2-2 in ICeChIP when varying amount of input chromatin (left) or amount of antibody (right). (F) Heatmap of peptide array antibody binding normalized to target for select combinatorial modifications (full peptide set detected in Figure S5). (G) Binding in ICeChIP and peptide arrays of selected anti-H3K4me3 antibodies to H3K4me3K9acK14acK18ac relative to singly-modified H3K4me3. All peptide arrays were conducted with six fluorescence measurements, and all ICeChIPs with one of each pulldown. Error bars represent SD.
We found that specificity in ICeChIP was not substantially affected by changes in relative methylform abundances for the antibodies screened (Figure S4B), suggesting that different chromatin abundances of the methylforms do not mask true antibody ChIP specificity. Yet, for approximately half of the antibodies screened in peptide arrays, changing the amount of epitope or antibody altered observed specificity (Figures 3D-E and S4C-D). We speculate that these differences in antibody specificities are the result of the different physical interactions underpinning the two methods: in peptide arrays, dilute antibody binds densely-packed epitope on a surface, whereas ICeChIP (and ChIP more generally) is the opposite. However, a complete understanding of these differences remains a challenge for future inquiry.
Peptide arrays permit simultaneous querying of combinations of H3K4 methylations with other PTMs (Bock et al., 2011; Egelhofer et al., 2011; Fuchs et al., 2011; Rothbart et al., 2015). In this context, many antibodies displayed reduced affinity for their target with flanking lysine acetylation (Figure 3F, all except abMe2–1 and abMe3–2; and Figure S5), which are thought to occasionally coexist (Taverna et al., 2006; Voigt et al., 2012). Yet in ICeChIP, we largely do not observe such reduced binding, with several antibodies displaying slightly elevated binding of H3K4me3K9acK14acK18ac nucleosomes relative to H3K4me3 alone (Figure 3G). Although these proximal modifications do impact apparent H3K4me3 capture in both platforms, the effects are subtle and poorly aligned between the two methods.
Antibodies with different off-target specificities yield materially different ICeChIP-seq profiles
We next examined 15 antibodies with a range of H3K4 methylform specificities on chromatin from K562 cells, a tier one ENCODE cell line (The ENCODE Project Consortium, 2012). Using our described method (Grzybowski et al., 2015), we isolated the on-target ChIP-seq signal for four antibodies to generate signal-corrected tracks (Figures 4A and S6A). As anticipated from its performance in both peptide arrays and ICeChIP-qPCR (Figure 2C-D), abMe3–2 captures substantial H3K4me2 (which is more abundant than H3K4me3) in ICeChIP-seq (Figure 4A). Consequentially, its distribution appeared more similar to that of high-specificity H3K4me2 than H3K4me3 antibodies (Figure 4B). Similar off-target capture issues were observed for all other low-specificity antibodies used for ICeChIP-seq (Figure S6).
Figure 4. Antibodies with different specificities yield markedly different ChIP-seq profiles in K562 cells.
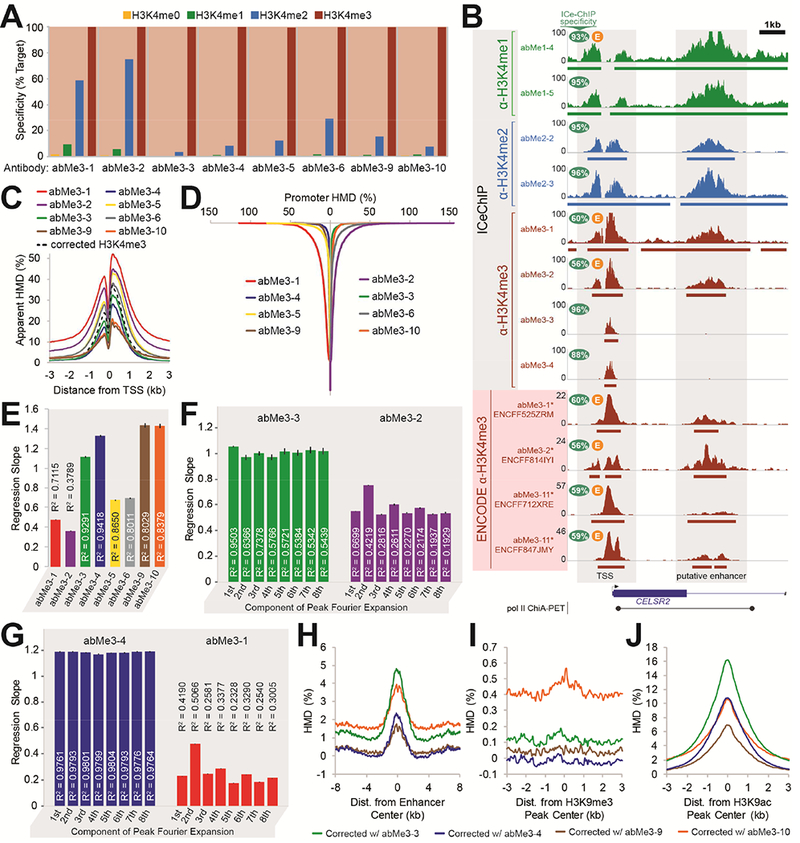
(A) Specificity profiles of anti-H3K4me3 antibodies measured by ICeChIP-seq (full range of specificities in Fig. S6A). (B) A representative chromosomal coordinate view showing several antibody ICeChIP-seq modification profiles and ENCODE project H3K4me3 modification profiles in K562 cells, with a putative promoter-enhancer connection (Li et al., 2012). Bars below tracks represent peaks. (C) Anti-H3K4me3 antibodies and signal-corrected H3K4me3 modification profiles contoured over all TSSs for all Refseq genes. (D) Average HMD measured by anti-H3K4me3 antibodies (sorted in descending order) at the +1 and +2 nucleosomes of genes with signal-corrected H3K4me3 HMD ≤ 0 (7,666 Refseq genes). Vertical axis represents position in sorted gene list. (E) Correlation between average HMD of signal-corrected H3K4me3 versus antibody-measured HMD at called peaks for each indicated antibody. Error bars represent 99.99% CI of regression slopes. (F) Correlation between pre-cosinusoidal factors of eight-component discrete Fourier cosine transform on antibody-measured peaks of signal-corrected H3K4me3 versus antibody-measured HMD for abMe3-3 (left) and abMe3-2 (right). Error bars represent 99.99% CI of regression slopes. (G) Correlation between pre-cosinusoidal factors of eight-component discrete Fourier cosine transform of measured HMDs by abMe3-3 versus abMe3-4 (left) or abMe3-1 (right), on peaks from abMe3-4 (left) or abMe3-1 (right). (H, I, J) Signal-corrected H3K4me3 modification profiles, generated from abMe1-5, abMe2-3, and the indicated H3K4me3 antibody, contoured over (H) stringently-defined enhancers, (I) H3K9me3 peaks, and (J) ENCODE H3K9ac peaks.
We then sought to determine if high-specificity and low-specificity antibodies had demonstrably different ChIP-seq profiles genome-wide. High-specificity and corrected H3K4me3 profiles are similar about transcription start sites (TSSs), whereas low-specificity antibodies show inflated apparent HMD, consistent with off-target signal leakage (Figure 4C). Strikingly, at TSSs with no measured H3K4me3 in the corrected profile, the high-specificity anti-H3K4me3 profiles display fewer genes with nonzero apparent HMD than do the low-specificity profiles (Figure 4D). Moreover, the HMD of peaks from high-specificity antibodies correlate more closely with the corrected profile than do low-quality antibodies (Figure 4E).
To compare the shapes of the ChIP-seq profiles, we applied a discrete cosine transform to the HMD distributions at called peaks genome-wide for both antibody and corrected profiles. This calculation allowed us to assess concordance of peak shape separately from HMD magnitude. The regression slope for the pre-trigonometric factors indicates concordance of HMD value, whereas the correlation coefficient indicates similarity of distribution shape (Video S1). For each term, the linear correlation with corrected profile is stronger and the slope closer to unity for high-versus low-specificity antibodies (Figure 4F), demonstrating that the shape and magnitude of high-specificity HMD profiles more closely resemble the signal-corrected profile. Similar comparisons between two additional high- or low-specificity antibodies for each methylform recapitulate these results (Figures 4G and S6B-C). Together, these data suggest that the profiles of high- and low-specificity antibodies are distinct, with different patterns genome-wide. Given that the most widely-used ChIP antibodies show poor methylform specificity (Table S1, Figures S2 and S3), conclusions drawn from datasets generated with these reagents should be tempered.
Beyond H3K4 methylform analysis, our ICeChIP spike-in pool also contained synthetic barcoded nucleosomes representing H3K9me1/2/3, H3K27me1/2/3, H3K36me3, H3K79me1/2/3, and H4K20me1/2/3 nucleosomes (Figure S6). With the exception of the low-specificity abMe1–3, the tested antibodies did not substantially capture PTMs on other lysines in histone H3, although we note several that showed substantial binding to H4K20me3 in either array testing (Figure S5) or ICeChIP (Figure S6). Off-target recognition of H4K20me3 is surprising given the low primary sequence similarity with H3K4, but such binding has previously been noted in qualitative peptide arrays (Bock et al., 2011). As H4K20me3 is relatively rare in rapidly dividing cells (Sanders et al., 2013), this cross-reactivity, though concerning, may be modest in impact.
Several antibodies displayed different sensitivity to flanking additional modifications in peptide arrays, allowing us to test whether those same patterns were apparent in ICeChIP-seq. On peptide arrays, abMe3–3 showed enhanced binding to H3K4me3 paired with H3K9me2 but reduced binding in combination with acetylation marks, whereas the opposite trend was seen for abMe3–9 and abMe3–10 (Figure S4). However, when signal-corrected tracks are generated with these antibodies, at stringently-defined enhancers, where H3 acetylation is expected, and H3K9me3 peaks, the differences between the profiles are small and often the opposite of what is predicted by peptide arrays (Figures 4H-I). Similarly, at ENCODE H3K9ac peaks, the profile corrected with abMe3–3 has ~10% higher apparent H3K4me3 HMD over abMe3-10 despite showing reduced capture of acetylated peptides in arrays (Figures 4J and S4). Collectively, these results suggest that biases in our ICeChIP analyses due to these combinatorial modifications are modest.
ICeChIP with high-specificity antibodies yields new quantitative insights into transcriptional control
Prior studies have relied on ChIP-seq without in situ antibody specificity information or calibration, so we next used our robust ICeChIP-seq datasets to critically re-evaluate previous findings and search for new biological insights. In particular, we chose to investigate distal enhancers and the promoters they regulate (Li et al., 2012). H3K4me3 is phenomenologically (Guenther et al., 2007; Lauberth et al., 2013; Santos-Rosa et al., 2002; Schübeler et al., 2004; Vermeulen et al., 2007) and biochemically (Lauberth et al., 2013; Vermeulen et al., 2007; Wysocka et al., 2006) associated with active promoters (Grzybowski et al., 2015), where it is flanked by the lower H3K4 methylforms; our present high-quality data recapitulates this general pattern (Figures 4C and S6G-L). H3K4me1 and H3K4me2 are canonically thought to be indicative of enhancers, but not of relative enhancer activity (Creyghton et al., 2010; Heintzman et al., 2007, 2009; Rada-Iglesias et al., 2011). There are scattered reports of H3K4me3 demarcating active enhancers (Pekowska et al., 2011), but the accumulated evidence suggests that H3K27ac, rather than H3K4me3, marks active enhancers (Creyghton et al., 2010; Heintzman et al., 2009; Rada-Iglesias et al., 2011). Our data confirm that H3K4me1 and H3K4me2 decorate stringently-defined enhancers; however, we detect little evidence for H3K4me3 at these sites (Figures 4H and S7). Importantly, though the high-specificity antibodies show little H3K4me3 at a putative enhancer, the low-specificity anti-H3K4me3 antibodies show substantial apparent H3K4me3 at such locations, as do the ENCODE H3K4me3 ChIP-seq tracks (Figures 4B and S7F). This artefactual capture, apparent in the low-specificity (but commonly used; see Table S1) anti-H3K4me3 antibodies (abMe3–1 and abMe3–2) and ENCODE data (some of which was performed with the same reagents), is attributable to signal leakage from lower methyl forms, which are abundant at enhancers.
Although there are some differences between datasets generated with different high-specificity antibodies (Figures 4C-F and 4H-J), they all indicate extremely low H3K4me3 levels at enhancers (Figure 4H, S7F). If some of the apparent signal inflation of abMe3-3 versus abMe3–4 (Figure 4H-J) was due to enhanced capture of H3K4me3 in the context of flanking acetylation (Figure 3G), these differences are quite modest. While this does not rule out the possibility that other proximal modifications could have more severe impacts on capture efficiency, leading to bias in the interpretation agnostic of such effects, for H3K4me and flanking lysine acetylation we observe a less severe dependence than anticipated.
We next investigated the relationship between enhancer H3K4 methylation and target gene expression, as defined by RNA Polymerase II ChIA-PET contacts (Li et al., 2012). Though we find that transcription from a given promoter modestly correlates with the average H3K4me1 HMD across contacting enhancers (Figure 5A; left), the sum of H3K4me1 HMD across all contacting enhancers correlates much more strongly (Figure 5A; center-left). Similar properties were observed for H3K4me2 (Figure 5B). We interpret these data to mean that the number and collective H3K4me1/me2 density of enhancers effectively predicts promoter activity, suggesting that enhancers may operate en masse rather than as isolated elements, and that the lower H3K4 methylforms may play some role in this process. Conversely, neither averages nor sums of enhancer H3K4me3 HMD correlated as well with gene expression (Figure 5C), nor did the ratio of enhancer H3K4me3 to H3K4me1 (Figure 5D), contrary to prior uncalibrated ChIP studies (Pekowska et al., 2011).
Figure 5. H3K4me1 and H3K4me2 HMD across enhancers contacting promoter regions is correlated to gene expression for all, metabolic, and developmental genes.
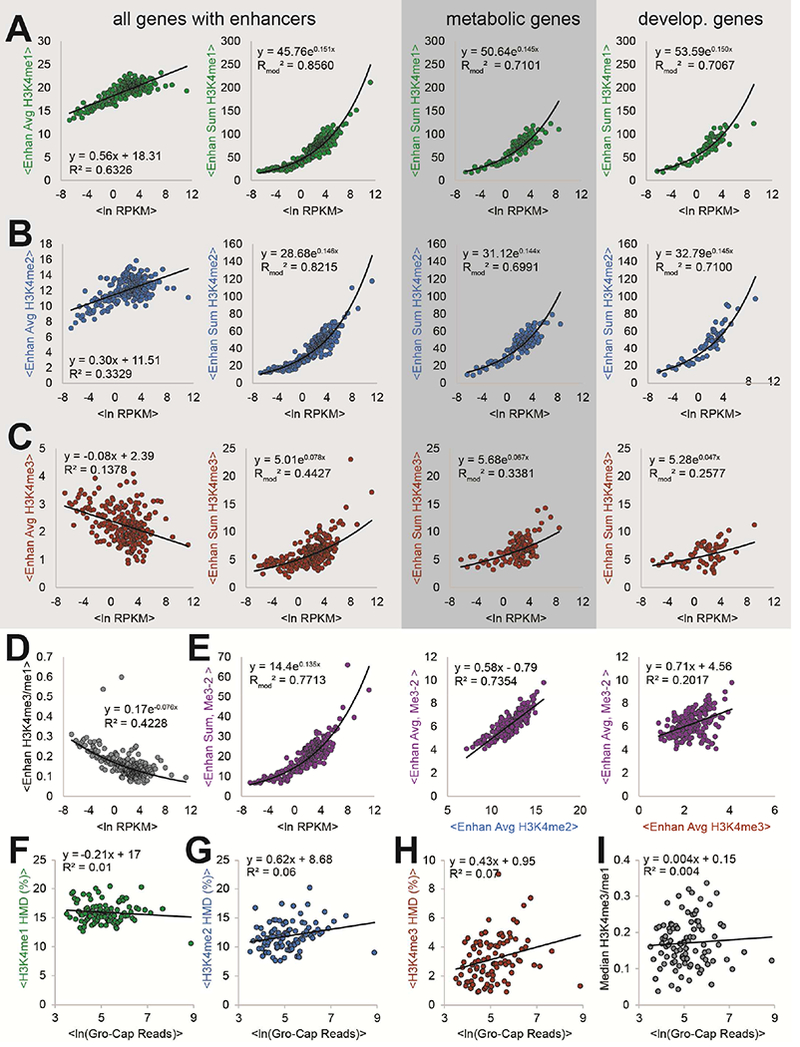
(A) H3K4me1, (B) H3K4me2, and (C) H3K4me3 HMD average (left) and sum (centre-left, centre-right, and right) across enhancers contacting associated promoter region (defined by pol II ChIA-PET contacts (Li et al., 2012)) versus transcriptional output (ln [RPKM]) for all (left and centre-left), metabolic (centre-right), and developmental (right) genes. HMD sum across enhancers versus ln (RPKM) for metabolic (centre-right) and developmental (right) genes (as defined by PANTHER GOSlims). (D) Ratio of enhancer H3K4me3 HMD to H3K4me1 HMD versus ln (RPKM). (E) abMe3-2 measured HMD sum (left) or average (centre and right) across enhancers versus ln RPKM (left), average corrected H3K4me2 enhancer HMD (centre), and average corrected H3K4me3 enhancer HMD. (F, G, H, I) Average transcript production, measured by average ln GRO-Cap reads, of unstable-unstable classified genes (Core et al., 2014) versus (F) average H3K4me1, (G) H3K4me2, (H) H3K4me3 HMD, and (I) median H3K4me3/H3K4me1 ratio. All scatterplots, unless otherwise noted, use corrected H3K4 methylation profiles and show binwise averages; bins contain fifty elements each and were created by sorting on ln (RPKM).
H3K4 methylation at enhancers is thought to primarily regulate cell-type specific and developmental genes (Heintzman et al., 2007, 2009; Rada-Iglesias et al., 2011). To investigate this, we compared gene expression and enhancer modification levels for metabolic, developmental, and multicellular system process-genes (Figures 5A-C and S7). Remarkably, our signal-corrected datasets showed no substantial differences between these gene ontology classes, indicating that enhancer-potentiated transcriptional activation may be more universal in mammalian gene expression than formerly appreciated (Heintzman et al., 2007, 2009; Rada-Iglesias et al., 2011).
To determine if low-specificity antibodies can materially affect these new observations, we analyzed the HMD sum across enhancers as measured by abMe3–2, which cross-reacts with H3K4me2 (Figures 2D, 4A, S2, and S6A). Here, the apparent H3K4me3 HMD sums at enhancers correlate strongly with gene expression (Figure 5E), unlike corrected and high-specificity H3K4me3 abMe3–3 sums (Figures 5E and S7). This apparent HMD at these loci is driven primarily by H3K4me2 rather than H3K4me3, so the low-specificity abMe3–2 incorrectly attributes this function to the latter PTM (Figure 5E). Importantly, other normalization methods with spike-in chromatin (Orlando et al., 2014), which normalize ChIP experiments but cannot control for specificity, would be similarly susceptible to this misleading artefact.
Revisiting literature enhancer mark paradigms with high-specificity antibodies
Multiple reports have implied a role for H3K4me3 at enhancers (Core et al., 2014; Pekowska et al., 2011), further suggesting that the H3K4me3:H3K4me1 ratio marks active enhancers (Pekowska et al., 2011). Our calibrated data, which enable meaningful ratiometric comparisons, show the opposite trend in K562 cells. Specifically, we find that the ratio of calibrated H3K4me3 to H3K4me1 is inversely related to enhancer activity (Figure 5D), consistent with our observation that enhancers lack substantial H3K4me3 (Figures 4G, 5C, and S7). The prior work relied upon an antibody (abMe3–1) for which two lots performed poorly in our study (Figures 2A, 4A-B, S2, and S3) (Pekowska et al. 2011); the substantial cross-reactivity we observe with H3K4me2, which is abundant at enhancers, may account for the disparity (Figure 5B). The use of crosslinking ChIP, which has been previously noted to reduce specificity (Fan and Struhl, 2009; Kasinathan et al., 2014; Teytelman et al., 2013), represents another potential source of the discrepancy. Regardless, several independent lines of evidence (Figures 5C-D and S7) lead us to conclude that the H3K4me3/H3K4me1 ratio is not positively correlated with enhancer activity in K562 cells, and, we suspect this to be more general.
Similarly, based on ENCODE ChIP-seq data, it has been suggested that H3K4me3 levels and the H3K4me3:H3K4me1 ratio at eRNA TSSs are positively correlated with eRNA transcription levels, as measured by GRO-Cap reads in K562 cells (Core et al., 2014). However, there are several potential issues with the ENCODE H3K4 methylation ChIP-seq datasets. Those for H3K4me3 in K562 cells (the only H3K4 methylation state with multiple independent datasets) display substantial divergence from one another (Figures 1, 4B, and S1A-B) and are all very different from our high-specificity ICeChIP-seq datasets (Figure S1C). This could be due to a wide variety of factors, including different antibody quality; sequencing depth; the use of crosslinked ChIP, which leads to greater off-target binding (Fan and Struhl, 2009; Kasinathan et al., 2014; Teytelman et al., 2013); sonication, which can generate a large size distribution of fragments and can damage epitopes (O’Neill and Turner, 2003); and the effect of single-end sequencing and read extension, which can result in oligonucleosome avidity distortion (Grzybowski et al., 2015). Conversely, our ICeChIP-seq datasets were generated with a native procedure, high sequencing depth, and by filtering out fragments with lengths greater than 200bp to avoid oligonucleosome avidity distortion. Whatever the cause, these differences lead to markedly different interpretations when coupled to readouts of eRNA in the same cell line (Core et al., 2014). We find that neither H3K4me1 (Figure 5F), H3K4me2 (Figure 5G), H3K4me3 (Figure 5H), nor the H3K4me3:H3K4me1 ratio (Figure 5I) is substantially correlated to the transcriptional level of eRNAs. This example highlights the need for ChIP-seq procedures that minimize off-target capture and underscores the pitfalls of treating ENCODE datasets as gold standards for these sorts of analyses.
Examining catalytically dead MLL3/4 mutants with high-specificity antibodies and ICeChIP
To further investigate enhancer biology, we conducted ICeChIP-seq in R1 mouse embryonic stem cells (mESCs) with wild-type (WT) and catalytically dead MLL3/4 mutants (dCD MLL3/4) reported to have markedly reduced H3K4me1 global abundance (Dorighi et al., 2017). Sequencing confirms the high specificity of abMe1–6, both relative to H3K4 methylforms and cross-lysine reactivity (Figure 6A). Globally, we observe that WT H3K4me1 abundance is consistent with other global abundance measurements of this PTM in mESCs (Voigt et al., 2012) and we observe roughly three-fold loss of H3K4me1 in dCD mESCs relative to WT, measured either as proportion of nucleosomes or integrated HMD (Figure 6B), confirming that abMe1–6 is specific enough to detect such global abundance differences.
Figure 6. Highly-specific anti-H3K4me1 ICeChIP-seq can reveal differences between MLL3/4 WT and /catalytically dead cell lines.
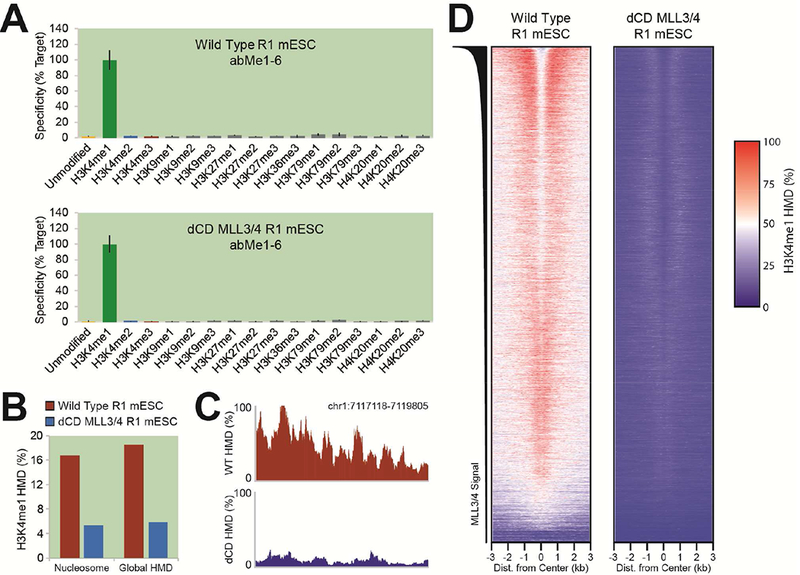
(A) Specificity of H3K4me1 ICeChIP-seq in WT and dCD MLL3/4 R1 mESCs. (B) Global H3K4me1 abundances, as proportion of nucleosomes (left) and globally integrated HMD (right). (C) A representative genome browser view of H3K4me1 HMD in WT and dCD MLL3/4 R1 mESCs near an enhancer (Dorighi et al., 2017). (D) Heatmap of H3K4me1 HMD about enhancers in WT and dCD MLL3/4 R1 mESCs, sorted by MLL3/4 ChIP-seq signal (Dorighi et al., 2017).
These datasets further serve to highlight the importance of calibration for ChIP-seq. ICeChIP-seq genome browser views (Figure 6C) and heatmaps (Figure 6D) of H3K4me1 about enhancer centers for WT and dCD lines show a much more pronounced difference between the two lines than previously reported (Dorighi et al., 2017), likely due to inappropriate assumptions inherent in normalization of uncalibrated data. These discrepancies emphasize the importance both of the absolute quantification offered by ICeChIP and its ability to provide robust quantification amidst to global changes of histone modification abundances, as with these lines.
Reexamining other H3K4 methylform paradigms with high-specificity antibodies
Beyond enhancers, the H3K4 methylforms have been broadly correlated with transcription factor binding. It has been suggested that the H3K4me3 and H3K4me1 profiles are similar in both shape and magnitude between both genic and intergenic TBP sites (Koch et al., 2011). Although we cannot confirm the lot of abMe3–1 used in these prior experiments is identical to ours, we recapitulate their results, where the apparent H3K4me3 distribution with abMe3–1 is comparable in shape and magnitude at both genic and intergenic TBP sites (Figure 7A, abMe3–1, grey profile). However, two lots of abMe3–1 show substantial cross-reaction with H3K4me2 (Figure 4A-B, S2, and S3), and its apparent binding profile appears entirely attributable to this methylform. Our H3K4me2 HMD profiles all look quite similar at genic and intergenic TBP sites, whereas the calibrated H3K4me3 distributions are distinct, with little H3K4me3 at the intergenic sites (Figure 7A, red lines). This demonstrates that specificity information within the ChIP experiment is essential for reliable interpretation, as without it, seemingly incorrect conclusions are drawn about the H3K4 methylation state at TBP sites (Koch et al., 2011).
Figure 7. Use of low- vs. high-specificity reagents in the literature may yield demonstrably different biological interpretations for a broad range of proposed paradigms.
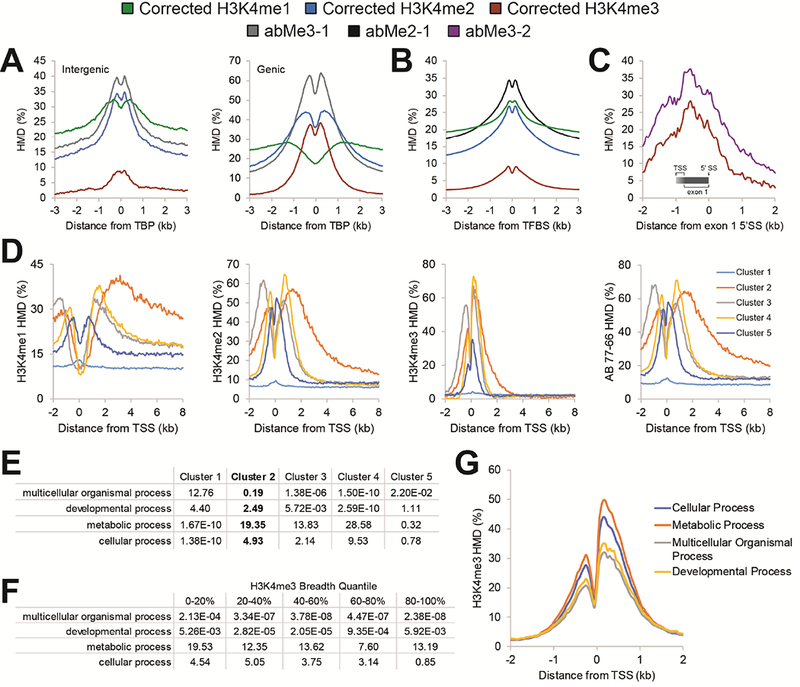
(A) Apparent HMD profiles of H3K4me1, H3K4me2, H3K4me3 and abMe3-1 about intergenic (left) and genic (right) TATA-binding protein (TBP) sites that have been previously described (Koch et al., 2011). (B) Apparent HMD profiles of H3K4me1, H3K4me2, H3K4me3, and abMe2-1 about transcription factor binding sites (TFBS) that have been previously described (Wang et al., 2014). (C) Apparent HMD profiles of H3K4me3 and abMe3-2 about the first exon splice site (SS) for transcripts with a first exon between 750-1000 nucleotides in length. Gradient indicates the region in which the TSS of this set of genes could be. (D) Apparent HMD profiles about TSS for H3K4me1, H3K4me2, H3K4me3, and abMe2-1. Clusters were generated using k-means clustering of HMD distribution about TSS (Pekowska et al., 2010). (E) -Ln (p) of gene ontology enrichment for the clusters profiled in panel (D). (F) -Ln (p) of gene ontology enrichment for genes by quantile of H3K4me3 peak breadth at said gene. (G) Corrected H3K4me3 profiles about TSSs by gene ontology classes in K562 cells.
We employed a similar analysis as another transcription factor study, in which it was proposed that H3K4me2 is the predominant marker of transcription factor binding sites (TFBS) (Wang et al., 2014). The prior H3K4me2 dataset (Barski et al., 2007; Wang et al., 2014) is derived from abMe2–1. The apparent H3K4me2 HMD around TFBS with this antibody is greater than that of the signal-corrected H3K4me1 HMD (Figure 7B), which would appear to support previous conclusions. However, we observe that abMe2–1 cross-reacts with H3K4me1 (Figures S2, S3, and S6), and when we instead examine the corrected distributions, we find that H3K4me1 HMD is actually higher than H3K4me2 HMD about TFBS (Figure 7B). This suggests that TFBS and the surrounding areas actually have slightly more H3K4me1 than H3K4me2, and that the latter does not uniquely demarcate these sites.
In addition to enhancers and TFBS, H3K4 methylation is thought to serve biological roles within gene bodies. As an example, there are reportedly two H3K4me3 peaks of comparable magnitude flanking the first exon of genes: the first (canonical) at the TSS (Guenther et al., 2007; Santos-Rosa et al., 2002), and the second atop the 5’-spice site that defines the end of the first exon (Bieberstein et al., 2012). These observations were based on re-analysis of ENCODE data from K562 cells. Our studies with the same reagent (abMe3–2) indicate its considerable cross-reactivity with H3K4me2 (Figures 2, S2, S3, and S6). When we conduct similar analyses on genes with the first exon between 750–1000nt (the first length at which the two putative peaks clearly resolve in the original study (Bieberstein et al., 2012)), we fail to see such a peak at the first exon-intron boundary (Figure 7C), indicative of no H3K4me3 enrichment specific to this splice site. In addition to the above concerns regarding the ENCODE datasets, the previous report used raw H3K4me3 sequencing reads from ENCODE (Bieberstein et al., 2012), which would not accommodate any differences in nucleosome density at the TSS and the first splice site, whereas ICeChIP (and many conventional ChIP) datasets are normalized to input density and are therefore largely independent of such differences (Grzybowski et al., 2015). In this example as well, our high-quality ICeChIP datasets yield different biological interpretations than those proposed in the literature.
Another such example can be found in analysis of the distribution of H3K4me2 over gene bodies. It has been reported that H3K4me2 is highly elevated over the gene body of tissue-specific, immune system process genes in CD4+ T-cells (Pekowska et al., 2010). When we apply the same procedure to identify such genes in K562 cells, we find there is indeed a cluster of genes with somewhat elevated H3K4me2 across the entire gene body (Figure 7D, Cluster 2), though it appears less dramatic and spread-out than may be expected from prior studies. However, we also see that H3K4me1 is more highly spread-out and elevated over this gene class (Figure 7D), reminiscent of their description of the H3K4me2 distribution (Pekowska et al., 2010). We also note that the antibody used by the prior study, abMe2–1, produces results more similar, but not identical results in our analyses (Figure 7D). abMe2–1 displayed some cross-reactivity to H3K4me1 (Figures 2, S2, S3, and S6; abMe2-1), and is likely further compromised by the greater relative abundance (2–10 fold across a variety of cell types) of H3K4me1 over H3K4me2 (LeRoy et al., 2013). We also find that this cluster of genes that display the described gene body enrichment profile is, in K562 cells, highly enriched for metabolic processes and not as enriched for cell-type specific processes as previously described (Figure 7E). Thus, while the differences between our findings and prior reports may be attributable to antibody quality, ChIP procedure, or cell type, the former is likely the most consequential.
Finally, we examined the role of H3K4 methylation domain breadth at gene promoters. It has been proposed that broad H3K4me3 domains mark cell identity genes across a range of cell types, including K562 cells, driving transcriptional constancy (Benayoun et al., 2014; Chen et al., 2015; Dahl et al., 2016). To critically assess this phenomenon with our datasets, we analysed the enriched gene ontology classes in K562 cells across different quantiles of H3K4me3 peak breadth. To our surprise, we instead found that metabolic genes were the most enriched class (Figure 7F) and that metabolic processes have, on average, a broader peak structure at TSSs (Figure 7G), suggesting that the proposed role of broad H3K4me3 domains does not apply to K562 cells. As the conclusions in previous publications were largely based on the ENCODE H3K4me3 ChIP-seq tracks, which we have found to be substantially different from our datasets in K562 cells (and indeed, from each other), it is possible that prior interpretations were similarly compromised by antibody quality.
DISCUSSION
Methodological strengths and limitations of peptide arrays and ICeChIP
The largest concern with poor-quality antibodies is that off-target binding will lead to erroneous biological interpretation. In conventional ChIP, with no effective metrics to assess antibody specificity in situ, the researcher is effectively blind to this pitfall, potentially compromising their results. Peptide arrays present the only practical way to broadly examine the impact of flanking combinatorial PTMs and have predictive value for other epitope-dense experimental formats, such as immunoblotting (Rothbart et al., 2012). However, our results suggest that peptide arrays, though commonly used for ChIP antibody validation (Bock et al., 2011; Egelhofer et al., 2011; Fuchs et al., 2011; Rothbart et al., 2015), often fail to accurately reflect antibody performance within ChIP experiments, either for methylform specificity or the impact of combinatorial PTMs. We have begun to examine the physical underpinnings of these differences, but given the distinct experimental formats, they are unlikely reducible to a single concrete principle and in any case, are largely immaterial to the practical matter: that peptide arrays are inappropriate for predicting antibody performance in ChIP.
ICeChIP is not without its limitations. The specificity information afforded by ICeChIP is restricted to the breadth of the semisynthetic nucleosomal standards available. However, these standards, particularly those bearing combinatorial modification patterns, are laborious to construct. If there is a discrepancy between datasets at loci that potentially bear combinatorial modifications, without these additional standards, it is difficult to assess which view is correct. For example, we see modest differences between datasets generated with different highly-specific H3K4me3 antibodies (Figure 4H-J) even when the measurement error is reduced by the massive signal averaging implicit in metanalysis. These apparent differences are attributable to several possible sources: differential sensitivity to flanking modifications (either increasing affinity, thereby artifactually inflating the HMD, or the converse); differential off-target nucleosome capture of marks not represented in the panel of nucleosomal standards deployed; and for individual loci, input and IP sampling error can also drive more pronounced peak shape and height differences.
Further, even if a broad range of nucleosome standards bearing combinatorial modifications were constructed, the analysis of histone modifications at co-modified loci would not be straightforward. It is possible, for example, that at a given locus, there are two sub-populations of cells with different PTM states that the two PTMs do not actually coexist on the same nucleosomes. To evaluate this possibility, a sequential ICeChIP protocol would need to be developed, sequentially selecting for nucleosomes with each PTM. However, to date, the sequential ChIP protocols in the literature (Bernstein et al., 2006; Kinkley et al., 2016; Mikkelsen et al., 2007; Seenundun et al., 2010; Sen et al., 2016; Weiner et al., 2016) have tended towards denaturative, crosslinked protocols with the questionable specificity and IP enrichment inherent in crosslinked ChIP (Fan and Struhl, 2009; Kasinathan et al., 2014; Teytelman et al., 2013). To this end, sequential native ICeChIP remains an active area of study for us, but nonetheless, represents a present limitation of the method.
Beyond combinatorial modifications, ICeChIP is limited in its ability to accurately assess nucleosome-depleted regions. At such regions, input coverage is sparse, leading to low sampling and high uncertainty in HMD values. Though in principle this could be addressed by higher sequencing depth, the relevance of the histone modification density at locations with such low nucleosome occupancy would be questionable. Additionally, ICeChIP assumes that native nucleosomes are stable enough to survive the ChIP protocol, but it has been previously observed certain histone variants and modifications may reduce nucleosome stability (Jin and Felsenfeld, 2007; Neumann et al., 2009). If these nucleosomes are unstable during the ChIP experiment, then that may result in artifactually reduced representation in the IP, whereas the DNA will still exist in the input, resulting in deflated apparent HMD.
In this study, we reduced the impact of variability of input preparation, cellular heterogeneity and authentic biological differences between samples by performing the bulk of comparative immunoprecipitations side-by-side from the same pool of input. In other contexts, these factors could become significant contributors to apparent signal.
We often use signal correction in order to more effectively isolate on-target signal from the antibody-measured signal, which is a convolution of on- and off-target binding. Yet such signal correction is not strictly necessary. Indeed, because signal correction uses multiple antibodies to compute a given modification track, the track will often have greater uncertainty than ICeChIP-seq with a single antibody. We use signal correction for making more nuanced and accurate comparisons in the aggregate, where many loci are being treated and analyzed as one dataset. In these analyses, the error is reduced by averaging. However, when examining individual loci, where the error in a signal-corrected track is more substantial, it may be better to use a single high-quality antibody for the most accurate view of mark distributions.
It is important to note that these limitations also exist with uncalibrated ChIP. However, in that approach, the researcher is completely blind to the questions of specificity and accurate quantification, whereas ICeChIP at least offers some information to that end. Despite its limitations, ICeChIP represents a powerful tool to enable more quantitative studies of histone PTMs.
Discrepancies with the literature due to antibody and ChIP quality
Here, we have used our ICeChIP datasets to critically re-examine ENCODE project datasets and other H3K4-methylform paradigms related to transcriptional control. As disagreement between our data and prior literature could reflect cell-type specific differences, we have focused on findings proposed as general features of mammalian chromatin. The examples we have presented here comment on the role of antibodies and ChIP-seq procedures generally in the widely-publicized biological “reproducibility crisis” (Baker, 2015). For a variety of potential reasons, particularly antibody specificity, several of the interpretations currently in the literature are not recapitulated by the high-quality ICeChIP datasets we have produced herein, casting some doubt on the many thousands of existing datasets that currently exist for histone PTMs across a wide range of organisms and cell types, and their use to draw a great many biological conclusions. In several instances, we were able to reproduce the phenomena reported with our K562 ICeChIP datasets using the same antibody catalogue numbers. However, in each of these cases, the precise interpretation was flawed owing to off-target antibody capture, which the authors could not possibly have known at the time due to inadequate validation criteria.
This set of discrepancies makes a powerful argument for in situ metrics of antibody specificity within ChIP experiments as distinct from spike-in normalization for the purposes of comparison (Al-Sady et al., 2013; Grzybowski et al., 2015; Orlando et al., 2014). It is unfortunately commonplace for authors to omit the specific antibody lot numbers used, but if distinctions between our data and the literature arise from lot-to-lot variation (Nishikori et al., 2012) this is equally troubling with regard to the scientific reproducibility crisis (Baker, 2015).
Although it is impractical to perform similar analyses of the thousands of papers in the literature that have used the antibodies described here in ChIP experiments, we fear that what we have discovered for a small selection of H3K4 methylation paradigms may represent a larger problem for the field. Furthermore, while we focus here on the specificity problems for antibodies raised to H3K4-methylforms, our ongoing (and comparably extensive) studies of other “PTM-specific” antibodies show similar promiscuity issues (data not shown), and a dose of skepticism for precise conclusions drawn from uncalibrated ChIP with many of these reagents is similarly warranted.
Our results strongly indicate that the field needs to establish and adopt more rigorous quality control standards for ChIP reagents to ensure more robust and reproducible data in the future (Table S2; Video S2). Crucially, this includes more careful validation of ChIP antibodies, ideally by direct testing to panels of related internal nucleosome standards that encompass the broadest achievable range of possible cross-reactivities in a ChIP setting (Grzybowski et al., 2015). Apart from calibration, we propose that the norms of ChIP-seq data publication should include clear indication of antibody catalogue and lot numbers, sequencing of input, and quantitative analysis rather than use of called peaks, which reduces quantitative data to a mere binary. Different protocols can also affect the specificity of the ChIP-seq experiment, and though ICeChIP effectively accommodates for this variation (Grzybowski et al., 2015), we also suggest the use of native ChIP rather than the oftentimes far more noisy, low-efficiency, idiosyncratic and artefact-prone cross-linked ChIP with sonication for accessible histone tails (Fan and Struhl, 2009; Kasinathan et al., 2014; Teytelman et al. 2013). As a whole, our study demonstrates both the danger of using unvalidated antibodies in ChIP and the power of calibrated ChIP to robustly measure histone PTMs and drive new biological discovery.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alex Ruthenburg (aruthenburg@uchicago.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human female K562 cell lines were grown at 37°C wit h 5% CO2 and 95% humidity in Dulbecco’s Modified Eagle Media (DMEM, Gibco) supplemented with 10% (v/v) HyClone FBS Characterized U.S. and 1x Penicillin/Streptomycin (Gibco). Cells were seeded into vented flasks to a density of 200,000 cells per mL of culture and were passaged at 1–2 million cells per mL of culture. K562 cell lines were authenticated by ATCC and found to be a 100% match with the reference cell line. mESCs were cultured as previously described (Dorighi et al., 2017).
METHOD DETAILS
Octamer and Nucleosome Preparation
Octamers were prepared as described (Grzybowski et al., 2015; Ruthenburg et al., 2011). Recombinant core histones were expressed in BL21 (DE3) with pRARE2 and mixed to equimolarity with the relevant semisynthetic histones in freshly prepared filter sterilized Unfolding Buffer (50 mM Tris-HCl pH 8.0, 6.3 M Guanidine-HCl, 10 mM 2-mercaptoethanol, 4 mM EDTA) to a final concentration of ≥ 1 mg histone per mL. The histone reconstitution was then added to 3500 MWCO SnakeSkin dialysis tubing (Pierce) and dialyzed overnight at 4°C against 500– 1000 volumes of filter sterilized Refolding Buffer (20 mM Tris-HCl pH 7.5, 2 M NaCl, 5 mM DTT, 1 mM EDTA).
After dialysis, the histone mixture was centrifuged at 18,000 g for 1 hour at 4°C, and subjected to gel filtration chromatography (Superdex 200 10/300 GL, GE Healthcare, resolved with Refolding Buffer). Each fraction that displayed a peak on the UV chromatogram was analysed by SDS-PAGE (22 mA current in 1x Laemmli Buffer for 70 minutes), stained with SYPRO Ruby (BioRad) per manufacturer instructions, and imaged with a 610BP emission filter at 600V PMT setting. Octamer fractions with equimolar quantities of each core histone were pooled and concentrated (Amicon Ultra-4 Centrifugal Filters, 10,000 MWCO, Millipore) to 5–15 μM octamer (measured spectroscopically).
Nucleosomes were reconstituted onto 147bp DNAs composed of the core Widom 601 sequence (Lowary and Widom, 1998) modified with a 22bp barcode on each end, with each barcode composed of two distinct 11 bp sequences not found in the human or mouse genomes. The DNA and octamer were mixed to a final concentration of 1 μM each in 2 M NaCl, and then dialyzed in dialysis buttons (Hampton Research) and a 10,000 MWCO SnakeSkin dialysis membrane (Pierce) against 200 mL of Refolding buffer for 10 minutes. Dialysis then continued as 2L of Buffer 10 (20 mM Tris-HCl pH 7.5, 1 mM EDTA, 1mM DTT) was added (flow rate 2–2.5 mL per minute).
Dialyzed samples were diluted with an equal volume of Nucleosome Dilution Buffer (20 mM Sodium Cacodylate pH 7.5, 10% v/v glycerol, 1 mM EDTA, 10 mM 2-mercaptoethanol, Filter Sterilized), and 1 μl was analysed by native PAGE (100 V in 1x TBE for 30 minutes), stained with SYBR Gold in 1xTBE for one hour, and visualized with a UV transilluminator gel imager. Fractions containing nucleosomes and minimal free DNA were pooled and diluted to a working concentration of ~ 1 nM nucleosome with filter sterilized Nucleosome Storage Buffer (10 mM Sodium Cacodylate pH 7.5, 100 mM NaCl, 50% v/v glycerol, 1 mM EDTA, 1x Protease Inhibitor Cocktail [1 mM PMSF, 1mM ABESF, 0.8 μM aprotinin, 20 μM leupeptin, 15 μM pepstatin A, 40 μM bestatin, 15 μM E-64 from a 200x DMSO stock]) and stored at −20°C.
Peptide Microarrays
Peptide microarrays were fabricated using an Aushon 2470 microarrayer and used as described (Cornett et al., 2017; Rothbart et al., 2015). Briefly, antibodies were diluted according to the manufacturers recommended western blot concentration (unless otherwise indicated) in Array Hybridization Buffer (PBS [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.6], 5% BSA, 0.1% Tween-20) and 500 μL (5 μL for 48-well format) was hybridized onto a peptide microarray for 1 hour at 4°. Slides were washed in PBS and probed with a fluorescently labelled secondary antibody (Life Technologies A-21244 or A-21235). Microarrays were scanned using an Innopsys InnoScan 110AL microarray scanner and analysed using ArrayNinja (Dickson et al., 2016). Specificity was calculated as described below for ICeChIP data from the raw fluorescent signal.
ICeChIP
ICeChIP was performed as described (Grzybowski et al., 2015), based on the Brand et al. (Brand et al., 2008) native ChIP protocol with internal standards added. Briefly, cell pellets were washed twice with 5 mL of PBS, then washed twice with 5 ml of filter sterilized Buffer N (15 mM Tris-HCl pH 7.5, 15 mM NaCl, 60 mM KCl, 8.5% w/v Sucrose, 5 mM MgCl2, 1 mM CaCl2, 1 mM DTT, 200 μM PMSF, 50 μg/mL BSA, 1x Roche Protease Inhibitor Cocktail), with each wash consisting of complete resuspension of the pellet, centrifugation at 500 g for 5 minutes at 4°C, and removal of supernatant. The washed pellet was then resuspended in at least 2 packed cell volumes (PCV) of Buffer N and mixed with 1 volume of Lysis Buffer (Buffer N supplemented with 0.6% NP-40 Substitute) and incubated on ice for 10 minutes to lyse cells.
The crude nuclei were spun down at 500 g for 5 minutes at 4°C before being resuspended in at least 6 packed nuclear volumes (PNV) of Buffer N and applied to the top of 7.5 mL of filter sterilized Sucrose Cushion N (15 mM Tris-HCl pH 7.5, 15 mM NaCl, 60 mM KCl, 30% w/v Sucrose, 5 mM MgCl2, 1 mM CaCl2, 1 mM DTT, 200 μM PMSF, 50 μg/mL BSA, 1x Roche Protease Inhibitor Cocktail) in a 15 ml centrifuge tube, then spun down at 500 g for 12 minutes at 4°C in a swinging-bucket rotor. The supernatant was discarded, and the pellet resuspended in ~ 2 PNV of Buffer N.
The nucleic acid content of the nuclei per unit volume was quantified by diluting 2 μL of nuclei suspension into 48 μL of 2 M NaCl, water-bath sonicating to solubilize DNA, and spectroscopically measuring nucleic acid concentration by Nanodrop (where one A280nm = 50 ng/μL chromatin). After accounting for the 25-fold dilution of the measurement sample, the concentration of the nuclei was adjusted to 1 μg/μL of chromatin. Nuclei were dispensed to 100 μL aliquots, flash frozen, and stored at −80°C prior to use.
For use, nuclei aliquots were thawed and spiked with ~ 1 μl of each barcoded nucleosome standard per 50 μg of chromatin. This suspension was then mixed by pipette, transferred to a new tube, and warmed to 37°C for 2 minutes. 1 unit of micrococcal nuclease (MNase, Worthington) per 4.375 μg of chromatin was added, and samples incubated at 37°C while shaking at 900 rpm for 12 minutes. Digestions were stopped by adding 1/9 volume of filter sterilized 10x MNase Stop Buffer (100 mM EDTA, 100 mM EGTA) while slowly vortexing, and nuclei lysed by adding 5 M NaCl to a final concentration of 600 mM while slowly vortexing. 66 mg of HAP resin (BioRad, CHT™ Ceramic Hydroxyapatite, Type I, 20 um) per 100 μg of chromatin digested was rehydrated with 200 μl of filter sterilized HAP Buffer 1 (5 mM Sodium Phosphate pH 7.2, 600 mM NaCl, 1 mM EDTA, 200 μM PMSF) per 100 μg of chromatin digested. Lysed nuclei were centrifuged at 18,000 g for 1 minute to pellet insoluble nuclear debris, and the soluble fraction added to the rehydrated HAP resin and incubated for 10 minutes at 4°C with rotation.
After incubation, the HAP resin slurry was added to a centrifugal filter unit (Millipore Ultrafree MC-HV Centrifugal Filter 0.45 μm) and spun at 1000 g for 30 seconds at 4°C. The HAP resin left on the filter unit was then washed 4 times with 200 μL HAP Buffer 1, and 4 times with 200 μl filter sterilized HAP Buffer 2 (5 mM Sodium Phosphate pH 7.2, 100 mM NaCl, 1 mM EDTA, 200 μM PMSF) by spinning at 1000 g for 30 seconds at 4°C. HAP resin was eluted into a clean tube with th ree 100 μl solutions of filter sterilized HAP Elution Buffer (500 mM Sodium Phosphate pH 7.2, 100 mM NaCl, 1 mM EDTA, 200 μM PMSF). The nucleic acid content of the elution was then quantified by Nanodrop, and the chromatin concentration adjusted to 20 ng/μl with filter sterilized ChIP Buffer 1 (25 mM Tris pH 7.5, 5 mM MgCl2, 100 mM KCl, 10% v/v glycerol, 0.1% v/v NP-40 Substitute) with 100 μg/ml of BSA.
With the exception of the Koide Lab 304M3B antibody, each ICeChIP was conducted with 12.5 μl of Protein A Dynabeads (Invitrogen) and 3 μg of antibody unless a different antibody amount was noted, in which case the amount of magnetic beads used was scaled linearly with the amount of antibody. The Protein A Dynabeads for each ICeChIP were washed with 50 μl of ChIP Buffer 1 by use of a magnetic rack, then resuspended in 50 μL of ChIP Buffer 1. In a separate set of tubes, 3 μg of each antibody was diluted to 100 μl with ChIP Buffer 1. The antibody and Protein A Dynabead suspensions were combined and incubated on a rotator at 4°C for at l east 1 hour, then washed with 200 μl of ChIP Buffer 1 by use of a magnetic rack and resuspended in 50 μl of ChIP Buffer 1.
After antibody washing, 150 μl of the diluted chromatin elution was added to each antibody-bead conjugate and incubated for 15 minutes on a rotator at 4°C. Beads were then washed twice with filter sterilized ChIP Buffer 2 (25 mM Tris pH 7.5, 5 mM MgCl2, 300 mM KCl, 10% v/v glycerol, 0.1% v/v NP-40 Substitute) and once with filter sterilized ChIP Buffer 3 (10 mM Tris pH 7.5, 250 mM LiCl, 1 mM EDTA, 0.5% Sodium Deoxycholate, 0.5% v/v NP-40 Substitute), with a wash consisting of removal of the existing supernatant by use of a magnetic rack, resuspension into 150 μl of buffer, transfer to a new siliconized tube, and incubation on the rotator for 10 minutes at 4°C. After these washes, the supernatant was removed, the beads resuspended in ChIP Buffer 1, transferred to a new siliconized tube, rinsed once with 200 μl of TE before being resuspended in 50 μl of ChIP Elution Buffer and incubated at 55°C for 5 minutes.
After incubation, the supernatant was transferred to a new set of siliconized tubes, and the beads discarded. To each supernatant was then added 2 μl of 5 M NaCl, 1 μl of 500 mM EDTA, and 1 μl of 10 mg/mL Proteinase K. 15 μl of Input DNA was also diluted to 50 μl with 35 μl of ChIP Elution Buffer (50 mM Tris pH 7.5, 1 mM EDTA, 1% w/v SDS, Filter Sterilized) and was supplemented with 2 μL of 5 M NaCl, 1 μL of 500 mM EDTA, and 1 μL of 10 mg/mL Proteinase K. The IP elutions and diluted input were then incubated at 55°C for 2 hours for a Proteinase K di gestion. After digestion, the DNA was purified by adding 1.5 volumes of Serapure HD (1:50 dilution of Sera-Mag SpeedBeads [Fisher], 20% PEG-8000, 2.5 M NaCl, 10 mM Tris pH 7.5, 1 mM EDTA, 0.05% Tween-20, Filter Sterilized prior to addition of SpeedBeads), incubating at room temperature for 15 minutes, then collecting the beads on a magnetic rack, washing twice with 150 μl of 70% ethanol, and eluting into 50 μl ddH2O, which was then recovered and stored at −20°C.
DNA Quantification and Analysis by qPCR
To assess antibody specificity, the relative amounts of DNA pulled-down from the targeted versus the off-target modified nucleosome standards was characterized by qPCR. qPCR was conducted using TaqMan Gene Expression Master Mix (Applied Biosystems) using the primers and hydrolysis probe listed (Table S3). These primers and probe for the barcoded sequences were previously qPCR validated for effectiveness and quality (Grzybowski et al., 2015). Primers were used at 900 nM; hydrolysis probe at 250 nM, in the TaqMan Gene Expression Master Mix (Applied Biosystems). The qPCR program was run at 95°C for 1 0 minutes, followed by 40 cycles, each consisting of 15 seconds at 95°C followed by 1 minute at 60°C and concluding with a plate read.
Cq values were analysed using the ΔΔCq method. Briefly, the Cq values for each target for each sample were averaged together to obtain the mean Cq value. Enrichment for each barcode was then computed as Enrichment = 2Cqinput – Cqip * 10, accounting for the 10-fold dilution of Input relative to IP and multiplying by 100% for Enrichment as a percentage of target. Off-target binding to alternate PTMs were computed by normalizing each enrichment to that of the on-target PTM: referred to as “Specificity (% Target)”. Overall specificity was computed by dividing the enrichment of the target PTM by the sum of the enrichments for all H3K4 methylforms (i.e. H3K4me0 + H3K4me1 + H3K4me2 + H3K4me3); referred to as “Aggregate Specificity.”
Illumina Library Preparation
Illumina libraries were prepared as described (Grzybowski et al., 2015), with minor modifications. Briefly, Serapure purified DNA was quantified using Quant-iT™ PicoGreen (Thermo Fisher) as per manufacturer instructions. Libraries were then generated from up to 10 ng of each DNA sample (input or IP) with the NEBNext Ultra II DNA Library Prep kit (New England Biolabs) per manufacturer instructions. The DNA content of each library was then quantified and pooled for Illumina sequencing. Cluster generation and paired-end sequencing was conducted using standard Illumina protocols by the University of Chicago Genomics Facility on the Illumina NextSeq. Multiplexing was conducted to yield approximately 150 million reads for H3K4me1 antibodies or low-specificity H3K4me2 antibodies, approximately 100 million reads for high-specificity H3K4me2 antibodies or low-specificity H3K4me3 antibodies, and approximately 50 million reads for high-specificity H3K4me3 antibodies. One replicate of each antibody was sequenced.
QUANTIFICATION AND STATISTICAL ANALYSIS
To align reads, a reference genome was first created, consisting of the human genome (GRCh38/hg38) or the mouse genome (mm9) appended respectively by the sequences of each of the nucleosome standard barcodes. Reads were then mapped to the appropriate reference genome using Bowtie2 using the sensitive pre-set and end-to-end alignment options (Langmead and Salzberg, 2012). Using SAMTools (Li et al., 2009), any reads which were not paired, not mapped in a proper pair, or mapped with a map quality < 20 were discarded to prevent low-quality reads from impacting downstream analyses. Reads were then flattened to create a single mapping from each matched pair of reads by retaining only one fragment per pair, and any mappings with lengths >200bp were also discarded to ensure only mononucleosomes were being analyzed (Grzybowski et al., 2015). Bedgraphs of genome coverage were then generated using BEDTools (Quinlan and Hall, 2010), and IP / input genome coverage bedgraphs were merged using BEDTools (Quinlan and Hall, 2010). The sum of reads across ladder members for each nucleosomal standard was computed for each sample and HMD bedgraphs were then generated from the merged bedgraphs using awk to apply the following formula: HMD (%) = 100% * (IPlocus/Inputlocus) / (IPbarcode/Inputbarcode). Error and 95% confidence intervals were computed with Poisson statistics and error propagation from the merged bedgraphs using awk to apply the following formula: 95CI Error (%) = 1.96 * HMD (%) * sqrt (1/IPlocus + 1/Inputlocus). Bigwig files were generated for visualization using the bedGraphToBigWig tool (Kent et al., 2010).
Correction was conducted using the antibodies AB 8895 (abMe1–1), AB 7766 (abMe2– 1), AB 12209 (abMe3–4), and AB 8580 (abMe3–1), unless otherwise noted. Correction was done using our previously-described method (Grzybowski et al., 2015) against H3K4me1, H3K4me2, H3K4me3, and H4K20me3 off-target binding. Briefly, measured HMD by each antibody can be described by a vector M, and the measured specificities by each antibody described by a square matrix S. Then, we can state, if other off-target binding is negligible, that the correct HMDs for H3K4me1, H3K4me2, H3K4me3, and H4K20me3 can be expressed by the vector C such that M=CS. As such, the vector C can be computed as CSS−1 = C = MS−1. The elements of S−1 were then used to compute the HMD and Error of the corrected profiles using awk to linearly combine the AB 8895 (abMe1–1), AB 7766 (abMe2–1), AB 12209 (abMe3–4), and AB 8580 (abMe3–1) profiles.
Peak calling was conducted for all H3K4 methylation antibodies using Macs2 using the bdgpeakcall command (Zhang et al., 2008), with the input being the HMD bedgraphs computed for each sample. To compute average HMD across a series of intervals, a “double mapping” procedure was used. First, the HMD bedgraph was mapped onto 1 bp windows made for each interval using BEDTools (Quinlan and Hall, 2010). Then, the mapped windows were mapped onto the original intervals using BEDTools (Quinlan and Hall, 2010). This procedure ensured that the degree of overlap of the interval with each value of the HMD bedgraph was accounted for in the mapping procedure. Using this double-mapping procedure, the average HMD and average 95% CI Error of each called peak was computed. At this point, those peaks with greater average HMD than average 95% CI Error were selected as “high-confidence” peaks. All subsequent peak analyses were conducted with these “high-confidence” peaks. For the H3K9me3 antibody, peak calling was conducted using Macs2 using the bdgbroadcall command (Zhang et al., 2008), with the input being the HMD bedgraph. These peaks were treated as the H3K9me3 broad peaks. Peak HMD correlations (Figures 4D and S6), were conducted by computing average HMD as measured by antibody and corrected profile across antibody-measured peaks and subsequently correlating these computed average HMDs using R, forcing through origin. Stringently defined enhancers were defined as those that are not overlapping with a Refseq promoter and have a transcription factor binding site (Wang et al., 2014), GRO-Cap TSS (Core et al., 2014), ATAC-seq peak (Buenrostro et al., 2015), and ENCODE H3K27ac peak, FAIRE-seq peak, DNase HS site, and P300 peak (The ENCODE Project Consortium, 2012)) which make contact with at least one promoter by pol II ChIA-PET (Li et al., 2012).
For Fourier analyses (Figures 4F, S1, and S6), the 1200bp region centered upon each peak centre was sectioned into eight 150bp windows using BEDTools (Quinlan and Hall, 2010). For each window, the average HMD as measured by the antibodies or corrected profile to be used, depending on the analysis employed, was computed as above. The eight windows were then assembled into eight-element vectors for each peak interval, and the pretrigonometric factors of a Fourier Discrete Cosine Transform computed on these vectors using Mathematica 10.2 with the command FourierDCT.The pretrigonometric factors were then correlated using R for each of the eight components, forcing through origin.
Profiles of HMD distributions about features including transcription start sites, first exons, and TBP sites were generated using HOMER annotatePeaks (Heinz et al., 2010). Gene ontology was conducted using HOMER findGO (Heinz et al., 2010). Gene ontology terms were largely classed into the overarching PANTHER GOSlim terms (Mi et al., 2013).
Integrated genome-wide HMD was computed by computing average of HMD across all base-pairs in genome. Nucleosome global modification abundance was computed as ratio of total genomic IP to input reads divided by ratio of barcode IP to input reads, much like computation of locus-specific HMD. The integrated genome-wide HMD represents the proportion of the genome that has the modification of interest; the nucleosome global modification abundance represents the proportion of nucleosomes bearing the modification of interest. These two would be equivalent if nucleosomes were uniformly distributed about the genome, but are otherwise not necessarily equivalent.
Statistical details of experiments can be found in the relevant figure legends. Linear correlations with R were forced through origin for more appropriate slope comparison.
DATA AND SOFTWARE AVAILABILITY
The ICeChIP-seq datasets generated in this study have been deposited in the Gene Expression Omnibus under accession number GSE103543.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H13K4me1 antibody Abcam ab8895, Lot Number GR305231-1. | Abcam | Cat#ab8895, RRID:AB_306847 |
| Anti-H3K4me1 antibody Abclonal A2355, Lot Number 46694. | Abclonal | Cat#A2355 |
| Anti-H3K4me1 antibody Abclonal A2355, Lot Number 46695. | Abclonal | Cat#A2355 |
| Anti-H3K4me1 antibody Active Motif 39297, Lot Number 01714002. | Active Motif | Cat#39297, RRID:AB_2615075 |
| Anti-H3K4me1 antibody Active Motif 39297, Lot Number 21008001. | Active Motif | Cat#39297, RRID:AB_2615075 |
| Anti-H3K4me1 antibody Active Motif 39635, Lot Number 30615011. | Active Motif | Cat#39635 |
| Anti-H3K4me1 antibody Cell Signaling 5326, Lot Number 1. | Cell Signaling | Cat#5326, RRID:AB_10695148 |
| Anti-H3K4me1 antibody Cell Signaling 5326BF, Lot Number 2. | Cell Signaling | Cat#5326BF, RRID:AB_2616017 |
| Anti-H3K4me1 antibody Diagenode C15310037, Lot Number A399-001. | Diagenode | Cat#C15310037 |
| Anti-H3K4me1 antibody Diagenode C15410037, Lot Number A1657D. | Diagenode | Cat#C15410037 |
| Anti-H3K4me1 antibody Diagenode C15410194, Lot Number A1862D. | Diagenode | Cat#C15410194, RRID:AB_2637078 |
| Anti-H3K4me1 antibody Diagenode C15410194, Lot Number A1863-001D. | Diagenode | Cat#C15410194, RRID:AB_2637078 |
| Anti-H3K4me1 antibody EMD Millipore 07-436, Lot Number DAM1687548. | EMD Millipore | Cat#07-436, RRID:AB_310614 |
| Anti-H3K4me1 antibody EpiGentek A-4031-050, Lot Number 606359. | EpiGentek | Cat#A-4031–050 |
| Anti-H3K4me1 antibody RevMAb 31-1046-00, Lot Number P-01-00415. | RevMAb | Cat#31-1046-00, RRID:AB_2716371 |
| Anti-H3K4me1 antibody Thermo Fisher 710795, Lot Number QL230603. | Thermo Fisher | Cat#710795, RRID:AB_2532764 |
| Anti-H3K4me1 antibody Thermo Fisher 720072, Lot Number RB226262. | Thermo Fisher | Cat#720072, RRID:AB_2532793 |
| Anti-H3K4me2 antibody Abcam ab32356, Lot Number GR253788-9. | Abcam | Cat#ab32356, RRID:AB_732924 |
| Anti-H3K4me2 antibody Abcam ab7766, Lot Number GR289627-1. | Abcam | Cat#ab7766, RRID:AB_2560996 |
| Anti-H3K4me2 antibody Abclonal A2356, Lot Number 46696. | Abclonal | Cat#A2356 |
| Anti-H3K4me2 antibody Abclonal A2356, Lot Number 46697. | Abclonal | Cat#A2356 |
| Anti-H3K4me2 antibody Active Motif 39141, Lot Number 01008001. | Active Motif | Cat#39141, RRID:AB_2614985 |
| Anti-H3K4me2 antibody Active Motif 39679, Lot Number 15515008. | Active Motif | Cat#39679 |
| Anti-H3K4me2 antibody Cell Signaling 9725, Lot Number 9. | Cell Signaling | Cat#9725, RRID:AB_823530 |
| Anti-H3K4me2 antibody Diagenode C15200151, Lot Number 001−11. | Diagenode | Cat#C15200151 |
| Anti-H3K4me2 antibody Diagenode C15310035, Lot Number A391-001. | Diagenode | Cat#C15310035 |
| Anti-H3K4me2 antibody Diagenode C15410035, Lot Number A9360014P. | Diagenode | Cat#C15410035 |
| Anti-H3K4me2 antibody EMD Millipore 051338, Lot Number 2757107. | EMD Millipore | Cat#05-1338, RRID:AB_1977248 |
| Anti-H3K4me2 antibody EMD Millipore 07-030, Lot Number DAM1479603. | EMD Millipore | Cat#07-030, RRID:AB_11213050 |
| Anti-H3K4me2 antibody Epicypher 13-0013, Lot Number 14247001. | Epicypher | Cat#13-0013, RRID:AB_ |
| Anti-H3K4me2 antibody EpiGentek A-4032-050, Lot Number 606360. | EpiGentek | Cat#A-4032-050 |
| Anti-H3K4me2 antibody Thermo Fisher 49-1004, Lot Number A391001161216. | Thermo Fisher | Cat#49-1004, RRID:AB_2533855 |
| Anti-H3K4me2 antibody Thermo Fisher 710796, Lot Number QL230606. | Thermo Fisher | Cat#710796, RRID:AB_2532765 |
| Anti-H3K4me2 antibody Thermo Fisher 720073, Lot Number QL226263. | Thermo Fisher | Cat#720073, RRID:AB_2532794 |
| Anti-H3K4me3 antibody Abcam ab12209, Lot Number GR275790-1. | Abcam | Cat#ab12209, RRID:AB_442957 |
| Anti-H3K4me3 antibody Abcam ab8580, Lot Number GR190229-1. | Abcam | Cat#ab8580, RRID:AB_306649 |
| Anti-H3K4me3 antibody Abcam ab8580, Lot Number GR273043-4. | Abcam | Cat#ab8580, RRID:AB_306649 |
| Anti-H3K4me3 antibody Abclonal A2357, Lot Number 46698. | Abclonal | Cat#A2357 |
| Anti-H3K4me3 antibody Abclonal A2357, Lot Number 46699. | Abclonal | Cat#A2357 |
| Anti-H3K4me3 antibody Active Motif 39159, Lot Number 12613005. | Active Motif | Cat#39159, RRID:AB_2615077 |
| Anti-H3K4me3 antibody Active Motif 61379, Lot Number 24615006. | Active Motif | Cat#61379 |
| Anti-H3K4me3 antibody Cell Signaling 9727, Lot Number 2. | Cell Signaling | Cat#9727, RRID:AB_561095 |
| Anti-H3K4me3 antibody Cell Signaling 9751, Lot Number 9. | Cell Signaling | Cat#9751, RRID:AB_2616028 |
| Anti-H3K4me3 antibody Diagenode C15200152, Lot Number 001-11. | Diagenode | Cat#C15200152 |
| Anti-H3K4me3 antibody Diagenode C15410003, Lot Number A1052D. | Diagenode | Cat#C15410003, RRID:AB_2616052 |
| Anti-H3K4me3 antibody Diagenode C15410003, Lot Number A5051-001P. | Diagenode | Cat#C15410003, RRID:AB_2616052 |
| Anti-H3K4me3 antibody EMD Millipore 05-745R, Lot Number 2813867. | EMD Millipore | Cat#05-745R, RRID:AB_1587134 |
| Anti-H3K4me3 antibody EMD Millipore 07-473, Lot Number DAM1623866. | EMD Millipore | Cat#07-473, RRID:AB_1977252 |
| Anti-H3K4me3 antibody Epicypher 13-0004, Lot Number 13171001. | Epicypher | Cat#13-0004 |
| Anti-H3K4me3 antibody EpiGentek A-4033-050, Lot Number 606361. | EpiGentek | Cat#A-4033-050 |
| Anti-H3K4me3 antibody RevMAb 31-103900, Lot Number P-09-00676. | RevMAb | Cat#31-1039-00, RRID:AB_2716373 |
| Anti-H3K4me3 antibody Thermo Fisher PA5-40086, Lot Number RL2301825. | Thermo Fisher | Cat#PA5-40086, RRID:AB_2608316 |
| Anti-H3K4me3 antibody Koide Lab 304M3B, Lot Number 040416AG. | Koide Lab | Cat#304M3B |
| Anti-H3K9me3 antibody Koide Lab 309M3B, Lot Number 072913TH. | Koide Lab | CAT#309M3B |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Semi-synthetic H3K4me1 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K4me1 Ladders |
| Semi-synthetic H3K4me2 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K4me2 Ladders |
| Semi-synthetic H3K4me3 Nucleosomes | Ruthenburg Lab | H3K4me3 Ladders |
| Semi-synthetic H3K9me1 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K9me1 Ladders |
| Semi-synthetic H3K9me2 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K9me2 Ladders |
| Semi-synthetic H3K9me3 Nucleosomes | Ruthenburg Lab | H3K9me3 Ladders |
| Semi-synthetic H3K27me1 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K27me1 Ladders |
| Semi-synthetic H3K27me2 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K27me2 Ladders |
| Semi-synthetic H3K27me3 Nucleosomes | Ruthenburg Lab | H3K27me3 Ladders |
| Semi-synthetic H3K36me3 Nucleosomes | Ruthenburg Lab | H3K36me3 Ladders |
| Semi-synthetic H3K79me1 Nucleosomes | Ruthenburg Lab | H3K79me1 Ladders |
| Semi-synthetic H3K79me2 Nucleosomes | Ruthenburg Lab | H3K79me2 Ladders |
| Semi-synthetic H3K79me3 Nucleosomes | Ruthenburg Lab | H3K79me3 Ladders |
| Semi-synthetic H3K4me1 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K4me1 Ladders |
| Semi-synthetic H3K4me1 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K4me1 Ladders |
| Semi-synthetic H3K4me1 Nucleosomes | Synthesized by Epicypher, Inc., Reconstituted by Ruthenburg Lab | H3K4me1 Ladders |
| Critical Commercial Assays | ||
| EpiTitan™ Histone Peptide Array | Epicypher, Inc. | EpiTitan™ |
| EpiTitrate™ Histone Peptide Array | Epicypher, Inc. | EpiTitrate™ |
| NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® | New England Biolabs | Cat#E7645 |
| Deposited Data | ||
| ICeChIP-seq Datasets in K562 Human Cells | This Paper | GEO: GSE103543 |
| ICeChIP-seq Datasets in WT R1 mouse embryonic stem cells | This Paper | GEO: GSE103543 |
| ICeChIP-seq Datasets in dCD MLL3/4 R1 mouse embryonic stem cells | This Paper | GEO: GSE103543 |
| Experimental Models: Cell Lines | ||
| Human: K562 Cells | ATCC | CCL-243 |
| Mouse: WT R1 mESC | Wysocka Lab | None |
| Mouse: dCD MLL3/4 R1 mESC | Wysocka Lab | None |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| See Primer Table | This Paper | Primer Table |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtiebio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| BEDTools | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| Mathematica 10 | Wolfram | https://www.wolfram.com/mathematica/ |
| R version 3.2.3 - “Wooden Christmas Tree” | The R Foundation for Statistical Computing | https://www.r-project.org/ |
| Other | ||
Supplementary Material
Highlights:
54 commercial H3K4 methylform antibodies are tested with ICeChIP and peptide arrays
Peptide arrays are poorly predictive of antibody performance in ChIP contexts
High-specificity antibodies reveal quantitative relationships in enhancer biology
Many literature H3K4 methylation paradigms are flawed, often due to poor antibodies
ACKNOWLEDGMENTS
We wish to thank P. Faber and H. Whitehurst in the University of Chicago Functional Genomics Facility for Illumina sequencing. We also thank the following for donating reagents used in this study: Abclonal (six antibodies), Active Motif (six antibodies), Cell Signaling Technology (three antibodies), and Diagenode (three antibodies). We also thank Dr. Chuck Epstein of the Broad Institute for his generous gift of five antibodies used by the ENCODE Consortium. We further thank Dr. Joanna Wysocka of Stanford University for her generous gift of mESC cell lines. We also wish to thank Claire Kokontis and Lindsay Stolzenburg for their helpful comments on this manuscript. This study was supported by the National Institutes of Health under award numbers R44-HG008907 to Z-W.S. and M-C.K., R00-CA181343 to S.B.R., R35-GM124736 to S.B.R., and R01-GM115945 to A.J.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare competing financial interests. EpiCypher is a commercial developer / supplier of platforms similar to those used in this study: peptide arrays (i.e. EpiTitrate™ and EpiTitan™) and ICe-ChIP barcoded nucleosomes (i.e. SNAP-ChIP™ and CAP-ChIP™ : both under license from the University of Chicago [Patent #US20160341743]). R.N.S., A.T.G., A.J.R., and S.B.R. have served in a compensated consulting role to Epicypher; A.T.G and A.J.R, hold partial intellectual property rights to ICeChIP as inventors.
REFERENCES
- Al-Sady B, Madhani HD, and Narlikar GJ (2013). Division of Labor between the Chromodomains of HP1 and Suv39 Methylase Enables Coordination of Heterochromatin Spread. Mol. Cell 51, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M (2015). Reproducibility crisis: Blame it on the antibodies. Nat. News 521, 274. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Baudat F, Imai Y, and de Massy B (2013). Meiotic recombination in mammals: localization and regulation. Nat. Rev. Genet 14, 794–806. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, et al. (2014). H3K4me3 Breadth Is Linked to Cell Identity and Transcriptional Consistency. Cell 158, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 125, 315–326. [DOI] [PubMed] [Google Scholar]
- Bieberstein NI, Carrillo Oesterreich F, Straube K, and Neugebauer KM (2012). First Exon Length Controls Active Chromatin Signatures and Transcription. Cell Rep. 2, 62–68. [DOI] [PubMed] [Google Scholar]
- Bock I, Dhayalan A, Kudithipudi S, Brandt O, Rathert P, and Jeltsch A (2011). Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics 6, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M, Rampalli S, Chaturvedi C-P, and Dilworth FJ (2008). Analysis of epigenetic modifications of chromatin at specific gene loci by native chromatin immunoprecipitation of nucleosomes isolated using hydroxyapatite chromatography. Nat. Protoc 3, 398–409. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, et al. (2015). Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat. Genet 47, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, and Lis JT (2014). Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet 46, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornett EM, Dickson BM, and Rothbart SB (2017). Analysis of Histone Antibody Specificity with Peptide Microarrays. JoVE J. Vis. Exp e55912–e55912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY, et al. (2016). Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BM, Cornett EM, Ramjan Z, and Rothbart SB (2016). ArrayNinja: An Open Source Platform for Unified Planning and Analysis of Microarray Experiments. Methods Enzymol 574, 53–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD, Garcia BA, Adelman K, and Wysocka J (2017). Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 66, 568–576.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, et al. (2011). An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol 18, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, and Struhl K (2009). Where Does Mediator Bind In Vivo? PLOS ONE 4, e5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, Lan F, Mei P, Yuan G-C, Lian C, et al. (2010). Human LSD2/KDM1b/AOF1 Regulates Gene Transcription by Modulating Intragenic H3K4me2 Methylation. Mol. Cell 39, 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs SM, Krajewski K, Baker RW, Miller VL, and Strahl BD (2011). Influence of Combinatorial Histone Modifications on Antibody and Effector Protein Recognition. Curr. Biol 21, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzybowski AT, Chen Z, and Ruthenburg AJ (2015). Calibrating ChIP-Seq with Nucleosomal Internal Standards to Measure Histone Modification Density Genome Wide. Mol. Cell 58, 886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, and Young RA (2007). A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell 130, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, and Felsenfeld G (2007). Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 21, 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinathan S, Orsi GA, Zentner GE, Ahmad K, and Henikoff S (2014). High-resolution mapping of transcription factor binding sites on native chromatin. Nat. Methods 11, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkley S, Helmuth J, Polansky JK, Dunkel I, Gasparoni G, Fröhler S, Chen W, Walter J, Hamann A, and Chung H-R (2016). reChIP-seq reveals widespread bivalency of H3K4me3 and H3K27me3 in CD4+ memory T cells. Nat. Commun 7, 12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch F, Fenouil R, Gut M, Cauchy P, Albert TK, Zacarias-Cabeza J, Spicuglia S, de la Chapelle AL, Heidemann M, Hintermair C, et al. (2011). Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat. Struct. Mol. Biol 18, 956–963. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, and Roeder RG (2013). H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation. Cell 152, 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, DiMaggio PA, Chan EY, Zee BM, Blanco MA, Bryant B, Flaniken IZ, Liu S, Kang Y, Trojer P, et al. (2013). A quantitative atlas of histone modification signatures from human cancer cells. Epigenetics Chromatin 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. (2012). Extensive Promoter-Centered Chromatin Interactions Provide a Topological Basis for Transcription Regulation. Cell 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276, 19–42. [DOI] [PubMed] [Google Scholar]
- Matthews AGW, Kuo AJ, Ramón-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, et al. (2007). RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, and Thomas PD (2013). Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc 8, 1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, Noort J van, Rhodes D, and Chin JW (2009). A Method for Genetically Installing Site-Specific Acetylation in Recombinant Histones Defines the Effects of H3 K56 Acetylation. Mol. Cell 36, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikori S, Hattori T, Fuchs SM, Yasui N, Wojcik J, Koide A, Strahl BD, and Koide S (2012). Broad Ranges of Affinity and Specificity of Anti-Histone Antibodies Revealed by a Quantitative Peptide Immunoprecipitation Assay. J. Mol. Biol 424, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LP, and Turner BM (2003). Immunoprecipitation of native chromatin: NChIP. Methods 31, 76–82. [DOI] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq Normalization Reveals Global Modulation of the Epigenome. Cell Rep. 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Pekowska A, Benoukraf T, Ferrier P, and Spicuglia S (2010). A unique H3K4me2 profile marks tissue-specific gene regulation. Genome Res. 20, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, Imbert J, Andrau J-C, Ferrier P, and Spicuglia S (2011). H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 30, 4198–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Lin S, Britton L-M, Krajewski K, Keogh M-C, Garcia BA, and Strahl BD (2012). Poly-acetylated chromatin signatures are preferred epitopes for site-specific histone H4 acetyl antibodies. Sci. Rep 2, srep00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, Dickson BM, Raab JR, Grzybowski AT, Krajewski K, Guo AH, Shanle EK, Josefowicz SZ, Fuchs SM, Allis CD, et al. (2015). An Interactive Database for the Assessment of Histone Antibody Specificity. Mol. Cell 59, 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ, et al. (2011). Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 145, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders YY, Liu H, Zhang X, Hecker L, Bernard K, Desai L, Liu G, and Thannickal VJ (2013). Histone Modifications in Senescence-Associated Resistance to Apoptosis by Oxidative Stress. Redox Biol. 1, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NCT, Schreiber SL, Mellor J, and Kouzarides T (2002). Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411. [DOI] [PubMed] [Google Scholar]
- Schübeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, Leeuwen F van, Gottschling DE, O’Neill LP, Turner BM, Delrow J, et al. (2004). The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 18, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seenundun S, Rampalli S, Liu Q-C, Aziz A, Palii C, Hong S, Blais A, Brand M, Ge K, and Dilworth FJ (2010). UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 29, 1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Block KF, Pasini A, Baylin SB, and Easwaran H (2016). Genome-wide positioning of bivalent mononucleosomes. BMC Med. Genomics 9, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ III, Millhouse S, Chen C-F, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, and Reinberg D (2007). Recognition of Trimethylated Histone H3 Lysine 4 Facilitates the Recruitment of Transcription Postinitiation Factors and Pre-mRNA Splicing. Mol. Cell 28, 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li H, Baker L, Boyle J, Blair LP, Chait BT, et al. (2006). Yng1 PHD Finger Binding to H3 Trimethylated at K4 Promotes NuA3 HAT Activity at K14 of H3 and Transcription at a Subset of Targeted ORFs. Mol. Cell 24, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teytelman L, Thurtle DM, Rine J, and Oudenaarden A. van (2013). Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci 110, 18602–18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Bandrowski A, Carr S, Edwards A, Ellenberg J, Lundberg E, Rimm DL, Rodriguez H, Hiltke T, Snyder M, et al. (2016). A proposal for validation of antibodies. Nat. Methods 13, 823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WWMP, van Schaik FMA, Varier RA, Baltissen MPA, Stunnenberg HG, Mann M, and Timmers HTM (2007). Selective Anchoring of TFIID to Nucleosomes by Trimethylation of Histone H3 Lysine 4. Cell 131, 58–69. [DOI] [PubMed] [Google Scholar]
- Voigt P, LeRoy G, Drury WJ III, Zee BM, Son J, Beck DB, Young NL, Garcia BA, and Reinberg D (2012). Asymmetrically Modified Nucleosomes. Cell 151, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li X, and Hu H (2014). H3K4me2 reliably defines transcription factor binding regions in different cells. Genomics 103, 222–228. [DOI] [PubMed] [Google Scholar]
- Weiner A, Lara-Astiaso D, Krupalnik V, Gafni O, David E, Winter DR, Hanna JH, and Amit I (2016). Co-ChIP enables genome-wide mapping of histone mark co-occurrence at single-molecule resolution. Nat. Biotechnol 34, 953–961. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. (2006). A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442, 86–90. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.