

Summary
The dorsal raphe (DR) constitutes a major serotonergic input to the forebrain and modulates diverse functions and brain states including mood, anxiety, and sensory and motor functions. Most functional studies to date have treated DR serotonin neurons as a single population. Using viral-genetic methods, we found that subcortical- and cortical-projecting serotonin neurons have distinct cell body distributions within the DR and differentially co-express a vesicular glutamate transporter. Further, amygdala- and frontal cortex-projecting DR serotonin neurons have largely complementary whole-brain collateralization patterns, receive biased inputs from presynaptic partners, and exhibit opposite responses to aversive stimuli. Gain- and loss-of-function experiments suggest that amygdala-projecting DR serotonin neurons promote anxiety-like behavior, whereas frontal cortex-projecting neurons promote active coping in the face of challenge. These results provide compelling evidence that the DR serotonin system contains parallel sub-systems that differ in input and output connectivity, physiological response properties, and behavioral functions.
In brief:
Two highly complementary parallel serotonergic neuronal pathways in the brain projecting from the dorsal raphe to cortical and subcortical regions of the brain are each activated by reward but show opposite responses to aversive stimuli.
Introduction
The serotonin system powerfully modulates physiology and behavior in health and disease. It is the most widely used pharmacological target for treating depression and anxiety (Belmaker and Agam, 2008; Ravindran and Stein, 2010), and depression has become the leading cause of disability worldwide (World Health Organization, 2017). However, a physiological and circuitry-based theory of how the serotonin system is organized to carry out its diverse functions remains elusive (Dayan and Huys, 2015; Muller and Jacobs, 2010).
Serotonergic fibers originate from a few discrete nuclei in the brainstem but innervate the entire mammalian brain (Steinbusch, 1981). The dorsal raphe nucleus (DR) contains ~35% of ~26,000 serotonin-producing neurons in the mouse brain and is the predominant source of serotonergic innervation of the forebrain (Ishimura et al., 1988). Despite a large body of literature (Muller and Jacobs, 2010), a consensus on the primary functions of the DR serotonin system is lacking. For example, recent studies on the effects of acute activation of DR serotonin neurons have reported divergent findings, including reinforcement (Liu et al., 2014), promotion of waiting for delayed reward rather than reinforcement (Fonseca et al., 2015; Miyazaki et al., 2012), promotion of anxiety-like behaviors and suppression of locomotion (Teissier et al., 2015; Urban et al., 2016), and suppression of locomotion without effects on reinforcement or anxiety-like behaviors (Correia et al., 2017). While different behavioral assays and activation methods may contribute to these conflicting results, they may also stem from treating the DR serotonin system as a monolithic whole.
Accumulating evidence points to physiological and molecular heterogeneity within the DR serotonin system (Calizo et al., 2011; Cohen et al., 2015; Fernandez et al., 2016; Okaty et al., 2015). Based on retrograde tracing DR subregions may preferentially project to different targets (reviewed in Waselus et al., 2011). Whole-brain input mapping suggested heterogeneity of DR serotonin neurons has also been implicated with respect to presynaptic inputs (Weissbourd et al., 2014). Recent studies have begun to target DR subpopulations by utilizing optogenetic activation of serotoninergic terminals at specific targets (Marcinkiewcz et al., 2016) or genetic intersection combined with chemogenetic perturbations (Niederkofler et al., 2016). However, given the scale and complexity, comprehensive characterizations that integrate anatomy, physiology, and function are essential for understanding how DR serotonin system is organized to modulate diverse physiological and behavioral functions. Here, we used a combination of viral-genetic approaches to dissect the DR serotonin system with respect to projection targets, synaptic input, physiological responses, and behavioral functions.
Results
DR Serotonin Neurons that Project to Specific Targets Have Stereotyped Locations
To determine if the spatial distribution of DR serotonin neurons correlates with their projection targets, we performed retrograde tracing combined with serotonin marker staining at the DR. We sampled eight brain regions previously reported to be heavily innervated by DR projections (Allen Brain Atlas, 2017; Oh et al., 2014; Vertes, 1991): the paraventricular hypothalamic nucleus (PVH), central amygdala (CeA), lateral habenula (LHb), dorsal lateral geniculate nucleus (dLGN), olfactory bulb (OB), orbitofrontal cortex (OFC), piriform cortex (PIR), and entorhinal cortex (ENT) (n=4 per region). We injected HSV-Cre (Neve et al., 2005), which transduces neurons via their axon terminals, unilaterally into these regions in Ai14 tdTomato Cre reporter mice (Madisen et al., 2010) (Figure 1A; Figure S1A). Co-staining of DR sections with tryptophan hydroxylase 2 (Tph2) revealed that serotonin neurons projecting to specific output sites have stereotyped cell body locations in the DR. Specifically, serotonin neurons projecting to subcortical regions localized more in the dorsal DR, whereas those that project to the OB and three cortical areas preferentially localized in the ventral DR (Figure 1B).
Figure 1. Spatial Organization of DR Serotonin Neurons according to Axonal Projections and Vglut3 Co-expression.

(A) Schematic of retrograde labeling and 3D reconstruction of spatial locations of DR serotonin neurons. Cyan and red dots represent injection sites of HSV-Cre in Ai14 mice. Anti-Tph2 staining was performed on consecutive coronal sections containing DR (star). The positions of Tph2+/tdTomato+ cells were recorded in confocal images. Each section was registered to the Allen reference brain and reconstructed in 3D. DBSCAN was applied for spatial clustering to generate a 3D surface based on the location of Tph2+ neurons (STAR Methods).
(B) Representative coronal confocal sections of the DR. Magenta, retrogradely labeled cells from eight projecting sites; yellow, anti-Tph2 staining. Dashed line, aqueduct border. Scale, 100 μm. Left insets, high-magnification images of neurons marked by arrows in individual channels. Scale, 25 μm. Right insets (top, coronal view; bottom, sagittal view): yellow, cyan, and red structures represent 3D surface of the clusters of, respectively, all DR Tph2+ neurons; those that project to PVH/CeA/LHb/dLGN; or those that project to OB/PIR/OFC/ENT. Scale, 500 μm.
(C) Merged surface view of the DRTph2→SC (subcortical) cluster (cyan), →AC (anterior cortex) cluster (red), and →ENT cluster (brown) in coronal (C1) and sagittal (C2) view. Scale, 200 μm. C3, coronal projection showing the location of individual cells from the DRTph2→SC and →AC groups. C4, densities of DRTph2→SC and →AC neurons along D–V axis. The two clusters exhibit the same line density at a horizontal plane 3742 μm ventral to the brain surface (dashed line). 74% of DRTph2→SC neurons were dorsal to this plane, whereas 80% of DRTph2→AC neurons were ventral to this plane.
(D) Representative coronal confocal sections of DR showing anti-Tph2 staining in Vglut3-Cre/Ai14 mice (green), which express tdTomato in Vglut3+ cells (red).
(E) Coronal (E1) and sagittal (E2) view integrating projection-defined clusters and the cluster of Tph2+&Vlgut3+ neurons (yellow, 1730±219.8 neurons; n=3). Scale, 200 μm.
(F) Representative coronal confocal sections of the DR showing retrograde labeling neurons from eight brain regions (magenta, pseudo-colored from rabies-derived GFP), anti-Tph2 staining (green), and tdTomato from Vglut3-Cre+ neurons (red). Scale, 100 μm. Insets: magnified images showing the neurons indicated with arrows in individual channels. Scale, 25 μm.
(G) The proportion of GFP, Tph2, and Vglut3 triple positive neurons in GFP+/Tph2+ neurons for 8 projection brain regions.
In this and all subsequent figures, abbreviations for anatomical regions can be found in Methods. Axis labels: A, anterior; P, posterior, D, dorsal; V, ventral; M, medial; L, lateral. Error bars, SEM.
See Figure S1 for related data, and Table S4 for cell numbers labeled by retrograde tracers.
For quantitative analysis, we developed an algorithm to register all DR-containing histological sections to the Allen Institute reference brain (Figure 1A, right panels; Figure S1B–E; STAR Methods; Xiong et al., 2018). We then created clusters using the combined data from four brains with the same injection sites (Figure 1B, right insets). We found that serotonin neurons projecting to the four subcortical sites appeared largely overlapping, which we combined to produce a subcortical cluster (DRTph2→SC) (Figure 1C). Likewise, serotonin neurons projecting to the OB, PIR, and OFC also exhibited considerable overlap, which we combined to produce an anterior cortical cluster (DRTph2→AC). ENT-projecting serotonin neurons tended to distribute more caudally than other populations. DRTph2→SC and DRTph2→AC clusters preferentially occupied dorsal and ventral DR, respectively, albeit with partial overlaps that accounted for 17.4% of the DRTph2→SC volume and 39.6% of the DRTph2→AC volume (Figure 1C3–C4, Figure S1E–H). We also performed dual-color retrograde tracing to determine if individual DR serotonin neurons target to multiple areas. While the low efficiency of co-labeling even for two tracers sequentially injected at the same site prevented us from making strong conclusions, our data was nevertheless consistent with OFC-projecting DR serotonin neurons had more overlap with OB-projecting ones than with CeA-projecting ones (Figure S1I). These data demonstrate that the cell bodies of DR serotonin neurons are organized according to their projection patterns (see Movie S1 for a summary).
DR Serotonin Neurons that Co-express Vglut3 Preferentially Project to Cortical Regions
~60% of DR serotonin neurons co-express the vesicular glutamate transporter Vglut3 in the rat (Gras et al., 2002) and co-release glutamate in the mouse (Liu et al., 2014; Sengupta et al., 2017). To investigate the distribution of dual-transmitter-containing (DRVglut3&Tph2) neurons, we crossed Vglut3-Cre mice (Grimes et al., 2011) with the Ai14 reporter and stained DR sections with Tph2. We found that Vglut3 also subdivided the DR serotonin neurons roughly into dorsal and ventral compartments (Figure 1D). After registering three double-labeled brains to produce the DRVglut3&Tph2 cluster, we found that it had 66%±0.6% (mean±SEM) overlap with the DRTph2→AC but only 19%±1.1% overlap with the DRTph2→SC cluster (Figure 1E; Movie S1).
These data suggest that serotonin neurons that project to the OB and cortical regions more likely co-express Vglut3. To verify this, we injected GFP-expressing rabies virus as a retrograde tracer (Wickersham et al., 2007) into the above eight brain regions of Vglut3-Cre;Ai14 mice, and determined the percentage of GFP+/tdTomato+/Tph2+ triple-labeled cells within the GFP+/Tph2+ double-labeled population (Figure 1F). Indeed, many OB- and cortical-projecting serotonin neurons were Vglut3+, whereas subcortical-projecting serotonin neurons were mostly Vglut3− (Figure 1G). Thus, the DR serotonin system has a coordinated spatial and neurochemical organization with respect to its projection targets, although these subdivisions are not absolute.
OFC- and CeA-projecting DR Serotonin Neurons Have Largely Complementary Collateralization Patterns
A key question is to what extent serotonin neurons that project to one target region collateralize to other brain regions. Given the apparent distinction of cortical- and subcortical-projecting DR serotonin neurons (Figure 1), we used an intersectional strategy (Beier et al., 2015; Schwarz et al., 2015) to examine the collateralization patterns of OFC- and CeA- projecting ones as examples. Into Sert-Cre mice (Gong et al., 2007) that express Cre recombinase in cells expressing the serotonin transporter (Sert), we injected an axon-terminal-transducing AAV (Tervo et al., 2016) expressing Cre-dependent Flp recombinase (AAVretro-FLExloxP-Flp) at the OFC or CeA, and AAVs expressing Flp-dependent membrane tethered GFP (AAV-FLExFLP-mGFP) at the DR (Figure 2A). We then employed iDISCO+ (Renier et al., 2016) to clear the brain in a whole-mount preparation, registered the imaged volume from light-sheet imaging to the Allen reference brain, and examined resultant axonal arborization patterns (STAR Methods).
Figure 2. Distinct Collateralization Patterns of DRSert→OFC and DRSert→CeA Neurons.

(A) Schematic of viral-genetic tracing and whole-brain 3D imaging. w.d., working distance of the light-sheet microscope objective.
(B) Overview of axonal projections from one representative brain each from the DRSert→OFC (blue) and DRSert→CeA (green) groups. Whole-mount imaging included the entire left hemisphere and the medial-most ~650 μm of the right hemisphere. Axes as indicated on right. See Movie S2 for a 3D rendering.
(C, D) Sagittal view of single 5-μm optical sections from eight individual brains registered to the Allen Institute common coordinate framework. Axons from DRSert→OFC (C1–C4; merged in C5) and DRSert→CeA (D1–D4; merged in D5) neurons are shown in green and blue, respectively.
(E) Sagittal brain atlas image (10 μm) from the Allen Institute that encompasses CeA and anterior insular cortex (AI). The red box indicates the displayed region of (C) and (D).</p/>(F) Coronal density maps of DRSert→CeA (left) and DRSert→OFC (middle) projections generated by voxel-wise dilation of axons. Right, a p-value map highlighting individual voxels with p<0.05 between groups. See Movie S3 for the fly-through of coronal maps of the brain rostral to DR.
(G) Heat map of relative labeling density (normalized to region volume and gross label content per brain) across 255 regions defined by the Allen Atlas. Additional columns show statistics for effect size, uncorrected p-values from two-tailed t-tests, and findings that pass false discovery rate at 10% (black bars). See Table S1 for the list of brain regions in the same sequence.
We analyzed the projection patterns of DR serotonin neurons labeled retrogradely from either the OFC or CeA (abbreviated as DRSert→OFC and DRSert→CeA neurons hereafter) in four brains each (Figure 2; Figure S2; Table S1). 3D rendering suggested that DRSert→OFC and DRSert→CeA axons exhibited complementary whole-brain projection patterns, preferentially innervating superficial (cortical) and deep (subcortical) regions (Figure 2B; Movie S2; Table S1). Examination in thin optical sections revealed the stereotypy of individual brains from the same group despite notable differences in labeling intensity (Figure 2C–E). For example, the OFC and OB were intensively innervated by DRSert→OFC axons but lacked DRSert→CeA axons (Figure S2A). By contrast, the CeA, PVH, ventral bed nucleus of stria terminalis (BNST), and substantia nigra were innervated by DRSert→CeA axons but lacked DRSert→OFC axons (Figure 2G; Figure S2B). DRSert→CeA axons also innervated the lateral amygdala, basolateral amygdala, and intercalated nucleus of the amygdala (Table S1). However, the nearby cortical amygdala was innervated mostly by DRSert→OFC axons (Figure S2A).
Whole-brain quantitative and statistical analyses of DRSert→OFC and DRSert→CeA axonal projections confirmed that the innervation patterns were largely complementary throughout the brain (Figure 2F–G; Movie S3; Table S1). Notably, many known targets of DR serotonin neurons (Allen Brain Atlas, 2017; Azmitia and Segal, 1978; Vertes, 1991) were not innervated by either of these subpopulations, including most of the striatum, thalamus, hypothalamus, somatosensory and motor cortex, and dorsal BNST (Table S1). Thus, the DR serotonin system contains at least two, and likely more, parallel sub-systems with distinct innervation patterns.
DRSert→OFC and DRSert→CeA Neurons receive biased input from specific nuclei
DR serotonin neurons as a whole receive monosynaptic inputs from diverse brain regions (Ogawa et al., 2014; Pollak Dorocic et al., 2014; Weissbourd et al., 2014). Given their parallel output organization, we next used cTRIO (cell-type-specific tracing the relationship between input and output) (Schwarz et al., 2015) to identify inputs to DRSert→OFC and DRSert→CeA neurons. We injected HSV-STOPflox-Flp unilaterally into the OFC or CeA, and AAVs carrying Flp-dependent constructs expressing TVA-mCherry (TC) fusion protein and rabies glycoprotein (G) into the DR of Sert-Cre mice. We then injected EnvA-pseudotyped, G-deleted, GFP-expressing rabies virus (RVdG) into the DR. Thus, only Sert-Cre+ neurons that project to the OFC or CeA could become starter cells for RVdG-mediated transsynaptic tracing (Figure 3A).
Figure 3. Biased Input Distributions for DRSert→OFC and DRSert→CeA Neurons.

(A) Schematic of cTRIO experiments.
(B) Confocal images of coronal sections containing the DR, showing OFC- and CeA-projecting serotoninergic starter cells (starter cell numbers for DRSert→OFC: 73±8.3, n=9 mice; for DRSert→CeA: 96±13.4, n=8 mice). Red, TVA-mCherry (TC) expression; green, GFP expression; cyan, anti-Tph2 staining. Scale, 100 μm. Insets: high magnification images of neurons indicated by arrows. Scale, 50 μm.
(C) Quantification of whole-brain inputs to DRSert→OFC and DRSert→CeA neurons (n=9, 8). Y-axis presents percentage of total inputs counted for each brain. Error bars, SEM. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 (multiple t-tests with Holm-Sidak correction).
(D) Representative GFP labeled ipsilateral input cells to DRSert→OFC and DRSert→CeA neurons. Scale, 250 μm.
See Figures S3 and Table S2 for related data.
Tph2 staining verified that starter cells were predominantly serotonin neurons (97%±2% DRSert→OFC and 94%±4% DRSert→CeA neurons were Tph2+; Figure 3B). While thousands of long-range input cells were identified in each experimental group, few GFP-labeled cells were found in the two controls: those without AAV expressing G and those using wild-type instead of Sert-Cre mice (Figure S3A–C), confirming specificity of input tracing.
To determine the long-range presynaptic partners for DRSert→OFC and DRSert→CeA neurons, we counted cell numbers in each of the 35 regions of interest (Table S2) (Weissbourd et al., 2014) from serial coronal sections. While presynaptic inputs to these two DR serotonin subpopulations originated from similar brain regions, there were striking quantitative differences. Specifically, DRSert→OFC neurons received proportionally more input from lateral hypothalamus (LHy), lateral habenula (LHb), and the majority of medulla nuclei. By contrast, DRSert→CeA neurons received significantly more input from CeA itself, BNST, PVH, and nucleus of the solitary tract (Figure 3C–D; Figure S3D). The marked enrichment of CeA input to DRSert→CeA neurons suggests strong DR-CeA reciprocal connectivity.
In summary, DRSert→OFC and DRSert→CeA neurons have largely complementary collateralization patterns (Figure 2) and receive quantitatively biased input from specific brain regions (Figure 3). Next, we examined if DRSert→OFC and DRSert→CeA neurons also exhibit different physiological response properties and behavioral functions.
DRSert→OFC and DRSert→CeA Neurons Are Both Activated by Reward But Show Opposite Responses to Punishment
Previous studies showed that DR serotonin neurons, when treated as a whole, were activated during reward consumption in freely moving mice (Li et al., 2016). However, single-unit recordings in head-fixed mice revealed heterogeneous responses of DR serotonin neurons to reward and punishment (Cohen et al., 2015). Since DRSert→OFC and DRSert→CeA neurons receive biased presynaptic inputs from different brain regions (Figure 3), it is possible that they respond differently to reward and punishment. To test this, we combined our viral-genetic strategy with fiber photometry (Gunaydin et al., 2014) by expressing genetically encoded Ca2+ indicator GCaMP6m (Chen et al., 2013) in DRSert→OFC or DRSert→CeA neurons. To compare to projection-specific DR serotonin neurons, we also expressed Cre-dependent GCaMP6m in the DR of Sert-Cre mice (“DRSert group”). We implanted an optical fiber into the DR at the GCaMP6m injection site through which we delivered 490-nm excitation light to monitor the activity of different serotonin neuron groups (Figure 4A1–C1), normalized by response to 405-nm control light to correct for movement artifact (Allen et al., 2017). We verified GCaMP6m expression and recording sites via post hoc histology (Figure 4A2–C2; Figure S4A1–C1). No task-evoked photometry signals were detected in DR serotonin neurons expressing GFP instead of GCaMP6m (Figure S4D).
Figure 4. DRSert→OFC and DRSert→CeA Neurons Are Both Activated by Reward but Show Opposite Responses to Punishment.

Fiber photometry recordings were performed on DRSert (A), DRSert→OFC (B) and DRSert→CeA (C) neurons.
(A1–C1) Schematic of viral injection and optical fiber (cyan) implantation.
(A2–C2) Confocal images of coronal sections showing fiber optic placement (dotted rectangle) and the expression of GCaMP6m (green) with Tph2 staining (red, A2’–C2’) in the DR. Vertical dashed lines represent the midline. Scale, 100 μm. Estimate of GCaMP6m+ serotonin neurons under optical fiber: DRSert group, 204±39.0, n=7 mice; DRSert→OFC group, 112±28.0, n=7 mice; DRSert→CeA group, 115±22.1, n=8 mice.
(A3–C3) Mean responses of individual mice to sucrose consumption after lever press. Time 0 is aligned to lick initiation (vertical dashed line). Red traces correspond to the mice shown in (A2–C2) and Figure S4. (A3’–C3’) Group data from all trials of individual mice showing quantification of the peak ΔF/F during sucrose water licking comparing to the ΔF/F at time 0 (paired t test; p<0.001 for all comparisons; n≥20 trials).
(A4–C4) Mean responses of individual mice from the three groups to electrical shock. Time 0 is aligned to onset of 1-sec electric shock delivery. (A4’–C4’) Group data from all the trials of individual mice showing quantification of the peak ΔF/F (positive extreme) and trough ΔF/F (negative extreme) recorded after electric shock delivery comparing to the ΔF/F at time (B4’, two-tailed paired t test, p<0.01 for all comparisons in inhibition. C4’, one-way ANOVA followed by multiple t-test, p<0.05 for all comparisons in activation. n=12 trials).
(D) Quantification of integrated ΔF/F signals between 0.13s to 0.65s before lever press (after subtracting integrated signals between 0.65s and 0.117s before lever press) comparing DRSert→OFC and DRSert→CeA neurons.
(E) Quantification of the peak ΔF/F recorded during sucrose water licking from DRSert→OFC and DRSert→CeA neurons.
(F) Quantification of the peak ΔF/F (negative or positive extreme) recorded after electric shock delivery from DRSert→OFC and DRSert→CeA neurons.
Error bars, SEM; n.s., not significant; *p<0.05; **p<0.01; ***p<0.001; ***p<0.001; ****p<0.0001. (For D–F, unpaired t-test; n=7, 8 mice for DRSert→OFC and DRSert→CeA groups, respectively).
See Figures S4 and Table S3 for related data.
To record reward responses, we trained mice to lever press for a sucrose water reward in a fixed-ratio paradigm. Each lever press led to one unit of sucrose water delivered from a nearby port, and water-restricted mice were allowed free access during the recording. Consistent with a previous report (Li et al., 2016), all recordings from the DRSert group showed an activation after the onset of licking, and the evoked activity persisted during the reward consumption period (Figure 4A3, A3’). The DRSert→OFC and DRSert→CeA groups showed similar activation pattern (Figure 4B3, B3’, C3, C3’, E; Figure S4A5–C5). Interestingly, when we aligned recording with the time of lever press, we found that in some mice, and more consistently in the DRSert→OFC group, serotonin neurons ramped up their activity prior to lever press (Figure S4A2–C3). Quantifications revealed significant activity increase prior to lever press in DRSert→OFC neurons compared to DRSert→CeA neurons (Figure 4D).
Next, we recorded responses to punishment by subjecting the same set of mice to mild electrical shocks. Remarkably, DRSert→OFC and DRSert→CeA neurons showed opposing responses to foot-shock. All seven mice from the DRSert→OFC group showed a long-lasting reduction of Ca2+ signals during and after the 1-sec foot-shock (Figure 4B4, B4’, E). By contrast, in all eight mice from the DRSert→CeA group, foot-shock induced a transient elevation of Ca2+ signals, followed by a small depression in a subset (Figure 4C4, C4’, E). The seven mice from the DRSert group showed more varied responses, including one that exhibited a biphasic response composed of a transient elevation followed by long-lasting depression (Figure 4A4, A4’).
These data indicate that DRSert→OFC and DRSert→CeA neurons respond similarly to reward, differently while anticipating an action that leads to reward, and oppositely to punishment. These findings suggest that previously described heterogeneous physiological responses of DR serotonin neurons to reward and punishment (Cohen et al., 2015) could result from recordings from projection-specific subpopulations.
Both DRSert→OFC and DRSert→CeA Neurons Suppress Locomotion
To investigate the behavioral functions of DRSert→OFC and DRSert→CeA neurons, we employed two complementary approaches. In a gain-of-function approach, we expressed the hM3Dq chemogenetic activator (Armbruster et al., 2007) in DRSert→OFC or DRSert→CeA neurons by unilaterally injecting AAVretro-FLEx-Flp into the OFC or CeA of Sert-Cre mice, and Flp-dependent hM3Dq (Beier et al., 2017) into the DR in the experimental group. In the two control groups for each experimental group, we replaced either hM3Dq with GFP or Sert-Cre with wild-type mice (Figure 5A1, B1). Post hoc histology confirmed that hM3Dq expression was Cre-dependent and cells’ locations were consistent with previous results (Figure 5A2, B2). Whole-cell recordings of hM3Dq-expressing DR Sert-Cre+ neurons in brain slices validated that application of Clozapine N-oxide (CNO) enhanced the firing rate of hM3Dq-expressing but not control serotonin neurons (Figure S5A–C). In a loss-of-function approach, we conditionally knocked-out Tph2 from DRSert→OFC or DRSert→CeA neurons by bilaterally injecting AAVretro-Cre-2A-GFP into the projection site of Tph2flox/flox mice (Wu et al., 2012) 17 days prior to the onset of behavioral tests. As a control, we injected AAVretro-GFP instead (Figure 5C1, D1). Staining of Tph2flox/flox mice showed a lack of Tph2 protein in GFP+ DR neurons (Figure 5C2, D2) and significantly reduced terminal serotonin levels (Figure S5C–D), confirming the effectiveness of viral-mediated knockout. Although the vast majority of Tph2+ neurons that project to OFC and CeA were from DR, a small fraction were from median raphe (Figure S5E); thus median raphe serotonin neurons may have a small contribution to the loss-of-function phenotypes described below.
Figure 5. Chemogenetic Activation and Conditional Tph2 Knockout Reveal that Both DRSert→OFC and DRSert→CeA Neurons Suppress Locomotion.

(A, B) Chemogenetic activation of DRSert→OFC (A) and DRSert→CeA (B) neurons. (A1, B1) Schematic for experimental (Exp) groups. Conditions for the two controls (Ctrl) are listed below. (A2, B2) Confocal images of coronal sections showing the expression of hM3Dq-2A-mCherry (red), and co-labeling with Tph2 staining (green) in the DR. Dotted lines are aqueduct borders. Scale, 100 μm. Right, high magnification images of neurons indicated by arrows. Scale, 50 μm. See Table S4 for cell counts.
(C, D) DRSert→OFC (C) and DRSert→CeA (D) neurons were Tph2 depleted by bilaterally injecting AAVretro-Cre into OFC (C1) or CeA (D1) of the Tph2flox/flox mice. (C2, D2) Confocal images of coronal sections showing the expression of Cre-2A-GFP (green), almost all of which were negative from Tph2 staining (red) in the DR. (Cells counts: Exp DRSert→OFC group, 272±67.9, 98.6%±0.43% were Tph2−; Exp DRSert→CeA group, 331±76.6, 98.7%±0.33% were Tph2–. Ctrl DRSert→OFC group, 160±8.5, 16.7%±0.18% were Tph2–; Ctrl DRSert→CeA group, 177±42.2, 39.2%±0.16% were Tph2–; n=3 mice for all groups). Scale, 100 μm. Right, high magnification images showing neurons indicated by arrows. Scale, 25 μm.
(E) Chemogenetic activation of DRSertĺOFC neurons decreases distance traveled (one-way ANOVA followed by multiple t-tests; F(2, 31) = 4.42, t=2.672, 2.29. n= 9, 11, 14). *p<0.05.
(F) Conditionally knocking out Tph2 from DRSertĺOFC neurons does not have a significant effect on distance traveled (two-tail unpaired t-test, t=1.24, df=17; n=9, 10).
(G) Activation of DRSert→CeA neurons decreases distance traveled (one-way ANOVA followed by multiple t-test; F(2, 30) = 4.516; t=2.21, 2.83. n= 10, 11, 12). *p<0.05.
(H) Conditionally knocking out Tph2 from DRSert→CeA neurons increases distance traveled (two-tail unpaired t-test, t=5.07, df=14. n=8, 8). ***p<0.001. Error bars, SEM.
We subjected the same sets of mice to a series of behavioral paradigms known to engage the DR serotonin system in a fixed sequence (STAR Methods). We first quantified locomotion in the open field and found that chemogenetic activation of both DRSert→OFC and DRSert→CeA neurons significantly decreased locomotion compared with controls (Figure 5E, G). Conversely, Tph2 depletion from DRSert→CeA neurons significantly increased locomotion over controls, suggesting that serotonin is responsible for the locomotion suppression promoted by DRSert→CeA neurons (Figure 5H). However, Tph2 depletion from DRSert→OFC neurons did not affect locomotion (Figure 5F); one possibility is that the effect caused by activating this subpopulation involves glutamate, as most DRSert→OFC neurons were Vglut3+ (Figure 1G). Indeed, by combining channelrhodopsin-assisted circuit mapping, local field potential recording in OFC slices, and pharmacology, we found that DRSert→OFC fibers co-released serotonin and glutamate to modulate OFC network activity (Figure S6).
DRSert→CeA but not DRSert→OFC Neurons Promote Anxiety-like Behavior
Next, we tested the effects of manipulating DRSert→OFC and DRSert→CeA neurons on anxiety-like behavior. Excessive avoidance of the center in the open field or the open arms of the elevated plus maze (EPM) are widely used as indications of anxiety-like behavior. We found that activation of DRSert→OFC neurons did not affect center entry or center time in the open field, or open-arm entry or time in open arms of the EPM (Figure 6A, E). However, Tph2 depletion from DRSert→OFC neurons caused a significant decrease in center time and center entry compared with controls (Figure 6B), as well as a significant decrease in open-arm time in the EPM (Figure 6F). These data suggest that serotonin release in the DR→OFC projection has an anxiolytic effect.
Figure 6. DRSert→CeA but Not DRSert→OFC Neurons Promote an Anxiety-like State.

(A) Chemogenetic activation of DRSert→OFC neurons does not affect the number of center entries (A1) or time spent in the center (A2) of the open field (one-way ANOVA, A1, F(2, 31) = 1.23; A2, F(2, 31) = 0.757. n= 9, 11, 14).
(B) Conditionally knocking out Tph2 from DRSert→OFC neurons decreases the number of center entries (B1) and the time spent in the center (B2) (two-tail unpaired t-test; B1, t=2.22, df=17; B2, t=2.54, df=17. n=9, 10).
(C) Activation of DRSert→CeA neurons decreases the number of center entries (C1) and the time spent in the center (C2) (one-way ANOVA followed by multiple t-tests; C1, F(2, 30) = 6.54, t=3.02, 3.17; C2, F(2, 30) = 6.54, t=2.85, 3.30. n= 10, 11, 12).
(D) Conditionally knocking out Tph2 from DRSert→CeA neurons increases the number of center entries (D1), but not the time spent in the center (D2) (two-tail unpaired t-test; D1, t=4.31, df=14; D2, t=1.13, df=14. n=8, 8).
(E) Activation of DRSert→OFC neurons does not affect the number of entries to the open arm (E1) or the time spent in the open arm in the EPM (E2) (one-way ANOVA, E1, F(2, 31) = 0.290; E2, F(2, 31) = 0.341; n= 9, 11, 14).
(F) Conditionally knocking out Tph2 from DRSert→OFC neurons does not affect the number of open arm entries (F1), but increases the time spent in the open arm (F2) (two-tail unpaired t-test; F1, t=0.337, df=17; F2, t=2.45, df=17. n=9, 10).
(G) Activation of DRSert→CeA neurons decreases the number of open arm entries (G1), and decreases the time spent in the open arm (G2) (one-way ANOVA followed by multiple t-tests; G1, F(2, 30) = 7.59, t=3.00, 3.60.; G2, F(2, 30) = 3.43, t=1.51, 2.60. n= 10, 11, 12. n.s., p=0.142).
(H) Conditionally knocking out Tph2 from DRSert→CeA neurons does not affect the number of open arm entries (H1), but increased the time spent in the open arm (H2) (two-tail unpaired t-test; H1, t=0.045, df=14; H2, t=2.55, df=14. n=8, 8). *p<0.05; **p<0.01; ***p<0.001. Error bars, SEM.
By contrast, activation of DRSert→CeA neurons promoted anxiety-like behavior, causing significantly decreased center entry and center time in the open field (Figure 6C) and activation significantly decreased open arm entry and open arm time (Figure 6G) compared to all controls except one case. Conversely, Tph2 depletion from DRSert→CeA neurons appeared anxiolytic, causing a significant increase in center entry in the open field (Figure 6D) and open arm time in EPM (Figure 6H). Taken together, these data suggest that DRSert→CeA neurons are anxiogenic.
We also used auditory fear conditioning to test the effect of manipulating DRSert→CeA and DRSert→OFC neurons. Neither activation nor Tph2 depletion of DRSert➔OFC neurons affected fear learning and memory recall (Figure S7A, B, E, F). By contrast, activation of DRSert→CeA neurons significantly increased overall freezing time to the conditioned tone in fear learning and memory recall (Figure S7C, D). However, fear learning and memory were not affected by Tph2 depletion in DRSert→CeA neurons (Figure S7G, H), suggesting that other pathways may compensate for the loss of DRSert→CeA activity in fear learning and memory.
DRSert→OFC but not DRSert→CeA Neurons Enhance Active Coping
Finally, we asked whether DRSert→CeA and DRSert→OFC neurons could modulate coping behavior in a 2-day forced-swim test. Immobility and struggle (escape behavior) in the forced-swim test are often used as indications of passive and active coping in the face of challenge, respectively. During chemogenetic experiments, CNO was administrated only before the day-2 test. Whereas chemogenetic activation of DRSert→CeA neurons did not affect immobility (Figure 7C), Tph2 depletion significantly reduced immobility during the forced-swim test (Figure 7D); this suggests that DRSert→CeA neurons inhibit escape behavior, although we cannot rule out this being a secondary effect on movement (Figure 5H).
Figure 7. DRSert→OFC but Not DRSert→CeA Neurons Promote Escape Behavior in the Forced-Swim Test.

(A) Activation of DRSert→OFC neurons decreases the immobility time on Day 2 testing session (one-way ANOVA followed by multiple t-tests; F(2, 31) = 6.84; t=2.97, 3.24. n= 9, 11, 14). **p<0.01. See Figures S7I for related data.
(B) Conditionally knocking out Tph2 from DRSert→OFC neurons increases the immobility time (two-tail unpaired t-test, t=2.21, df=17. n=9, 10). *p<0.05.
(C) Activation of DRSert→CeA neurons does not affect the immobility time (one-way ANOVA; F(2, 29) = 2.50. n= 10, 11, 11).
(D) Conditionally knocking out Tph2 from DRSert→CeA neurons decreases the immobility time (two-tail unpaired t-test; t=3.58, df=14. n=8, 8). **p<0.01. Error bars, SEM.
(E) Summary of gain- and loss-of-function results.
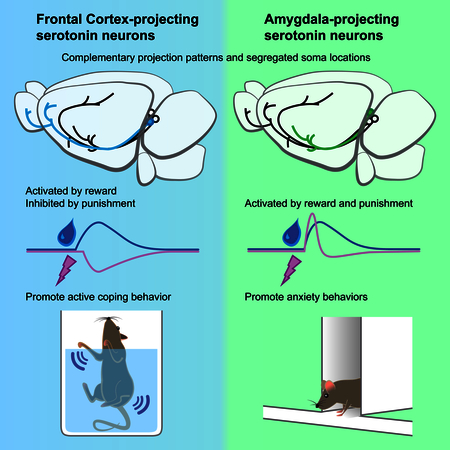
Remarkably, activation of DRSert→OFC neurons significantly enhanced escape behavior (decreased immobility) after CNO application on the day-2 test (Figure 7A, Figure S7I). Moreover, Tph2 deletion from DRSert→OFC neurons increased immobility (Figure 7B). Thus, both gain- and loss-of-function experiments indicate that DRSert→OFC neurons promote escape behavior in the forced-swim test. Taken together, these data suggest that activation of DRSert→OFC (but not DRSert→CeA) neurons promotes active coping in the face of challenge, and that serotonin release is necessary for this effect.
Discussion
DR serotonin neurons project broadly and modulate diverse functions. It has been unclear how they are anatomically and functionally organized. Here we provide evidence for the existence of parallel sub-systems that differ in input and output connectivity, physiological response properties, and behavioral functions.
Anatomical Organization of the DR Serotonin System
Previous anterograde and retrograde tracing suggested that the DR is organized along both the anterior-posterior (Abrams et al., 2004; Commons, 2015) and dorsal-ventral axes (Lowry et al., 2005; Muzerelle et al., 2016), and that individual DR neurons can send collaterals to two or more separate brain regions (Gagnon and Parent, 2014). However, different studies have not fully agreed upon the details of the topography due to technical limitations. For anterograde tracing, the resolution is limited by the spread of injected dyes or the genetic access to subtypes of DR serotonin neurons. For retrograde labeling, each study usually focused on a small subset of targets. Collateralization patterns are poorly understood because most data were collected by injecting two retrograde tracers into two pre-specified brain regions and observing double- labeled DR serotonin neurons, which provides limited insight into the overall extent of the collateralization.
Our retrograde tracing combined with image registration provides a more comprehensive view of the spatial organization of DR serotonergic projections in the mouse (Figure 1). With the exception of ENT-projecting DR serotonin neurons, which tended to localize to caudal DR, we did not find a prominent anterior-posterior topographic. We did find that ventral and dorsal serotonin neurons preferentially innervated cortical and subcortical targets, respectively, confirming and extending previous reports (Lowry et al., 2005; Muzerelle et al., 2016; Prouty et al., 2017). We further uncovered a strong correlation between cortical projections and Vglut3 co-expression in DR serotonin neurons, and demonstrated that OFC-projecting serotonin neurons co-release glutamate to regulate cortical activity (Figure S6). An interesting speculation is that glutamate co-release in cortex allows the DR serotonin system to regulate cortical circuits on a more rapid time scale via ionotropic glutamate receptors, whereas release of serotonin alone in subcortical circuits primarily serves a slower modulatory function via metabotropic receptors. Indeed, ionotrophic serotonin receptors are preferentially expressed in cortical and hippocampal interneurons (Barnes and Sharp, 1999).
Viral-genetic labeling in combination with whole-mount imaging revealed for the first time whole-brain collateralization patterns of projection-defined DR serotonin neurons, showing that DRSert→OFC and DRSert→CeA axons innervate largely complementary targets (Figure 2; Figure S2; Movies S2, S3). These analyses further indicate that the collateralization of individual DR serotonin neurons can be extremely broad (e.g., DRSert→OFC neurons also innervate OB anteriorally and ENT posteriorally) yet highly specific (e.g., DRSert→OFC neurons innervate cortical amygdala but avoid the nearby basolateral amygdala). These collateralization data comprise a map indicating which brain targets are likely coordinately or differentially modulated by serotonin.
cTRIO analyses further revealed that DRSert→OFC and DRSert→CeA neurons receive biased input from specific brain regions (Figure 3). Thus, the input-output architecture of the DR serotonin system differs from that of the locus coeruleus norepinephrine system with more homogeneous input and output (Schwarz et al., 2015), and resembles more closely those of the midbrain dopamine systems (Beier et al., 2015; Lerner et al., 2015), with biased input and segregated output. We want to emphasize that each sub-system we characterized may still be heterogeneous in their composition. For example, DRSert→OB neurons may be better described by a two-cluster model (STAR Method). Future systematic analyses utilizing the methods employed here, supplemented by high-resolution tracing of the axonal arborizations of individual serotonin neurons, will provide a more complete understanding of how the ~9,000 DR serotonin neurons differentially innervate target fields to modulate diverse physiological functions.
Distinct Functions of the DRSert→OFC and DRSert→CeA Sub-systems
We used chemogenetic activation as a gain-of-function and Tph2 depletion as a loss-of-function approach to functionally dissect projection-specific DR serotonin neurons. Because gain-of-function experiments alone may not reflect physiological functions of the system under manipulations, we consider our conclusions stronger if loss- and gain-of-function experiments generate opposite effects. There are advantages to using Tph2 depletion instead of chemogenetic silencing as a loss-of-function approach. First, chemogenetic silencing requires a higher CNO concentration than does chemogenetic activation; as the active component of CNO may be clozapine (Gomez et al., 2017), which engages several serotonin receptors at high concentrations (Meltzer, 1994), this strategy may ectopically affect serotonin-related behavior. Second, since a large fraction of DR serotonin neurons likely co-releases glutamate, Tph2 depletion specifically addresses the function of serotonin in these neurons. A caveat is that this manipulation is irreversible, and compensatory changes may occur in the circuit during the time between AAVretro-Cre injection and behavioral testing; thus, this loss-of-function strategy may not reveal the full function of serotonin release.
The DR serotonin system has been consistently shown to negatively regulate locomotion (Correia et al., 2017; Teissier et al., 2015; Whitney et al., 2016). Our data (Figure 5) suggest that the inhibitory effect of DR serotonin neurons on locomotion is at least partly mediated by serotonin release from DRSert→CeA neurons. Glutamate released from DRSert→OFC neurons may also contribute to this effect.
The role of serotonin in anxiety-like behavior in the cortex has been extensively studied with pharmacological and genetic manipulations of serotonin receptors (Albert et al., 2014). However, given the opposing roles of different serotonin receptors and their complex expression patterns in excitatory and inhibitory cortical neurons, it is difficult to predict the effect of cortical serotonin release on anxiety. We found that while chemogenetic activation of DRSert→OFC neurons did not significantly affect anxiety-like behavior, conditional Tph2 depletion in these neurons enhanced anxiety-like behavior (Figure 6). Since DRSert→OFC neurons mostly co-express Vglut3 (Figure 1) and can co-release glutamate (Figure S6), one interpretation is that activation of DRSert→OFC neurons results in release of both glutamate and serotonin that have opposing effects on anxiety-like behavior. When serotonin is selectively removed from this pathway, anxiety-like behaviors were promoted, suggesting that cortical serotonin release suppresses anxiety. In contrast, both gain- and loss-of-function experiments indicated that DRSert→CeA neurons promote anxiety-like behavior (Figure 6 and Figure S7). DRSert→CeA neurons collateralize to other amygdala nuclei, BNST, and PVH (Figure 2), all of which have been implicated as anxiety-related regions (Calhoon and Tye, 2015). Moreover, cTRIO analysis revealed that CeA and BNST provided particularly strong input to DRSert→CeA neurons compared to DRSert→OFC neurons (Figure 3). Thus, our data demonstrate that DRSert→CeA neurons promote anxiety-like behavior, likely involving reciprocal connections between the DR and CeA/BNST.
A recent large-scale meta-analysis provided strong evidence for the efficacy of selective serotonin reuptake inhibitors in treating depression (Cipriani et al., 2018). Immobility in the forced-swim test represents a passive coping strategy in the face of challenge and is often used to indicate a depression-like state in rodent models (Petit-Demouliere et al., 2005). Chemogenetic activation of DR serotonin neurons has recently been reported to reduce immobility in the forced-swim test (Teissier et al., 2015). Both our gain- and loss-of-function manipulations in the forced-swim test suggest that DRSert→OFC neurons, but not DRSert→CeA neurons, mediate this effect (Figure 7). Optogenetic activation of prefrontal cortical neuron terminals in the DR has previously been shown to promote active coping (Warden et al., 2012). Furthermore, frontal cortex inputs to the DR preferentially synapse onto serotonin neurons that are localize in the ventral DR (Weissbourd et al., 2014), where DRSert→OFC neurons also localize (Figure 1). Thus, DRSert→OFC neurons and frontal cortex→DRSert neurons may constitute a reciprocal loop to promote active coping in the face of challenge.
Technical difficulties make obtaining fiber photometry data during the above behaviors modulated by DRSert→OFC and DRSert→CeA neurons difficult. Nevertheless, the differential response properties of DRSert→OFC and DRSert→CeA neurons in the context of reward and punishment are consistent with their behavioral functions, and functions described by previous studies. For example, DRSert→OFC neurons appear to encode positive valence; they are also activated in anticipation of an action that leads to reward. We speculate that both properties may contribute to a behavioral role of promoting active coping in the face of challenge (Figure 7) and waiting for delayed reward (Miyazaki et al., 2012). Their inhibition by punishment could play a role in depression-like states induced by aversive stimuli. On the other hand, activation of DRSert→CeA neurons by punishment is consistent with a behavioral function of promoting anxiety, a brain state often equated to anticipation of future punishment.
In conclusion, our behavioral analyses demonstrate that anatomically-segregated DR subsystems have distinct, and sometimes even opposing, functions (Figure 7E). Thus, DR serotonin neurons should no longer be viewed as a monolithic population. We have provided means to further dissect the complexity of the DR serotonin system; such an endeavor will advance our understanding of neuromodulation in health and aid our development of effective therapies for brain disorders such as anxiety and depression.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Liqun Luo (lluo@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All procedures followed animal care and biosafety guidelines approved by Stanford University’s Administrative Panel on Laboratory Animal Care and Administrative Panel of Biosafety in accordance with NIH guidelines. For anatomical experiments (Figures 1–3), male and female mice aged 8–20 weeks on a CD1 and C57BL/6J mixed background were used. The Ai14 tdTomato Cre reporter mice (JAX Strain 7914), Vglut3-Cre (also known as Slc18a8-Cre; JAX Strain 18147), and Sert-Cre (MMRRC, Stock #017260-UCD) were used where indicated. For all other experiments (Figures 4–7), male mice aged 8–12 weeks on the C57BL/6J background were used when the experiments started; these mice had no prior procedure except for viral injection or/and fiber implantation. Animals were randomly assigned to experimental groups. Tph2flox/flox was obtained from Qi Wu (Wu et al., 2012). All mice used in fiber photometry recording were group housed with littermates, and they were trained and tested in the lever-pressing experiments before the foot shock experiment. All male mice used in gain- and loss-of-function behavioral experiments were test naïve for individual behavioral paradigms, and were individually housed with one female partner. Mice were housed in plastic cages with disposable bedding on a 12 hours light/dark cycle with food and water available ad libitum, except when placed on water restriction. Experiments were done during the light phase.
METHOD DETAILS
Stereotaxic Surgeries
Mice were anesthetized either with ketamine/dexmedetomidine mixture (Figure 1 and Figure 3) or 1.5%–2.0% isoflurane (Figure 2 and Figure 4–7) and placed in a stereotaxic apparatus (Kopf Instruments). For virus injection, the following coordinates (in mm) were used: +4.0 AP, 0.75 ML, –1.5 DV for OB; +2.6 AP, 1.7 ML, –1.7 DV for OFC; + 1.2 AP, 2.8 ML, –4.5 DV for PIR; –3.3 AP, 4.5 ML, –4.5 DV for ENT; 0.2 AP, 0.6 ML, –4.9 DV for PVH; –1.05 AP, 2.86 ML, –4.55 DV for CeA; –1.4 AP, 0.4 ML, –2.6 DV for LHb; –2.3 AP, 2.6 ML, - 2.7 DV for dLGN; –4.3AP, 1.10 ML, –2.85 DV for DR, with 20° ML angle. (AP is relative to bregma; DV is relative to the brain surface when AP is –1.0). After surgery, mice recovered on a heated pad until ambulatory and then returned to their homecage.
Viruses
Viruses with the following volumes and titers were injected and the injection time lines were listed in Table S4:
-
HSV-hEF1a-cre, 2 × 109 infectious units/ml;
OB, 500 nl; OFC, 750 nl; PIR, 500nl; ENT, 500 nl; PVH, 300 nl; CeA, 300 nl; LHb, 300 nl; dLGN, 500 nl.
-
HSV-hEF1a-LS1L-FLPo, 5 × 109 infectious units/ml;
OFC, 750 nl; CeA, 300 nl.
-
eGFP-expressing G-deleted Rabies Virus, 1 × 109 gc/ml;
OB, 500 nl; OFC, 750 nl; PIR, 500 nl; ENT, 500 nl; PVH, 300 nl; CeA, 300 nl; LHb, 300 nl; dLGN, 500 nl.
-
EnvA-pseudotyped, eGFP-expressing G-deleted Rabies (RVdG), 1 × 109 gc/ml;
DR, 500 nl.
-
AAVretro-CAG-FLExloxP-Flp, 6.9 × 1012 gc/ml;
OFC, 750 nl; CeA, 300 nl.
-
AAV8-hSyn1-FLExFRT-mGFP, 2.9 × 1013 gc/ml;
DR, 500 nl.
-
AAV5-CAG-FLExFRT-TC, 2.6 × 1012 gc/ml, AAV8-CAG-FLExFRT-G, 1.3 × 1012 gc/ml;
DR, 500 nl;
-
AAV1-hEF1a- FLExloxP-GCaMP6m, 1 × 1013 gc/ml;
DR, 500 nl;
-
AAV8-CAG- FLExFRT-GCaMP6m, 1.8 × 1013 gc/ml;
DR, 500 nl;
-
AAV8-hsyn1-FLExFRT-hM3Dq-mCherry, 6.2 × 1012 gc/ml;
DR, 500 nl;
-
AAVretro-CMV-Cre-2A-eGFP, 8.7 × 1012 gc/ml;
OFC, 750 nl; CeA, 300nl.
-
AAVDJ-EF1a-DIO-hM3D(Gq)-mCherry, 4.83 × 1013 gc/ml;
DR, 750 nl;
-
AAVDJ-EF1-DIO-hChR2(H134R)-EYFP, 5.27 × 1013 gc/ml
DR, 500 nl.
Note that we used three different viruses for retrograde labeling: HSV, AAVretro, and RVdG. Table S4 listed the number of infected DR serotonin neurons via output sites from these three viruses in various experiments utilizing these viruses, as well as times from viral injections to data collection. In general AAVretro yields higher efficiency, which is the reason why we switched to AAVretro after it was introduced in 2016.
Histology and Imaging
Animals were perfused transcardially with phosphate buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Brains were dissected, post-fixed in 4% PFA for 12–24 hours in 4 ºC, then placed in 30% sucrose for 24–48 hours. They were then embedded in Optimum Cutting Temperature (OCT, Tissue Tek) and stored at –80ºC until sectioning. For the antibody staining in Figure 1, 50-μm sections containing DR were collected onto Superfrost Plus slides to maintain the anterior to posterior sequence. All working solutions listed below included 0.2% NaN3 to prevent microbial growth. Slides were then washed 3×10 min in PBS and pretreated overnight with 0.5 mM SDS at 37ºC. Slides were then blocked for 4 hours at room temperature in 10% normal donkey serum (NDS) in PBS with 0.3% Triton-X100 (PBST), followed by incubation in primary antibody (Novus, rabbit anti-Tph2) diluted 1:1000 in 5% NDS in PBST for 24 hours at RT. After 3×10 min washes in PBS, secondary antibody was applied for 6 hours at room temperature (donkey anti-rabbit, Alexa-647 or Alexa-488, Jackson ImmunoResearch), followed by 3×10 min washes in PBST. Slides were then stained for NeuroTrace Blue (NTB, Invitrogen). For NTB staining, slides were washed 1×5 min in PBS, 2×10 min in PBST, incubated for 2–3 hours at room temperature in (1:500) NTB in PBST, washed 1×20 min with PBST, and 1×5 min with PBS. Sections were additionally stained with DAPI (1:10,000 of 5 mg/mL, Sigma-Aldrich) in PBS for 10–15 min and washed once more with PBS. Slides were mounted and coversliped with Fluorogel (Electron Microscopy Sciences). After that, the slides were then imaged using a Zeiss 780 confocal microscope, and images were processed using NIH ImageJ software. After that, whole slides were then imaged with a 5x objective using a Leica Ariol slide scanner with the SL200 slide loader.
For long-range tracing analysis in cTRIO experiments (Figure 3), consecutive 50-μm coronal sections covering the whole brain (with the exception of sections including the DR) were collected and NTB stained as described above. For DR-containing slices in Figure 3–7, staining was applied to floating sections. Primary antibodies (Novus, rabbit anti-Tph2, 1:1000; Rockland, rabbit anti-RFP, 1:1000; Abcam, goat anti-Tph2, 1:500; Aves Labs Inc., anti-GFP, 1:2000) were applied for 48 hours and secondary antibodies for 12 hours at 4ºC.
For serotonin staining, animals were perfused transcardially with modified aCSF containing (in mM): 225 sucrose, 119 NaCl, 2.5 KCL, 1 NaH2PO4, 4.9 MgCl2, 0.1 CaCl2, 26.2 NaHCO3, 1.25 glucose, 3 kynurenic acid, and 1 Na-ascorbate (all chemicals were from Sigma, St Louis, MO, USA), followed by 4% paraformaldehyde (PFA). Brains were dissected, post-fixed in 4% PFA for 12 hours at 4 ºC, and placed in 30% sucrose for 24–48 hours at 4 ºC. They were then embedded in Optimum Cutting Temperature (OCT, Tissue Tek) and stored at in the –80ºC freezer until sectioning. 30-μm sections containing OFC and CeA were collected every three slices as floating sections. And then they were blocked for 4 hours at room temperature in 10% normal donkey serum (NDS) in PBS with 0.3% Triton-X100 (PBST). Primary antibody (ImmunoStar, rabbit anti-5HT, 20080) was diluted 1:5000 in 5% NDS in PBST, and incubated for 48 hours at 4 ºC and secondary antibodies 12 hours at 4ºC.
2D Registration
For 2D registration (Figure 1 and S1), whole-slide images of scanned slides were imported into custom Matlab software to segment images into individual brain sections based on the NTB stain. To accelerate processing, the full resolution images (xy-resolution =1.29 μm/pixel) were initially down-sampled by a factor of 32 in both x- and y- dimensions. Segmentation included the application of a mask fit to the edge of each section to remove all image features outside the section. Background subtraction and contrast enhancement of the NTB channel were then applied. The processed NTB images for each section were then serially analyzed using a combination of automated and manual methods. To estimate the sectioning angle differences between individual samples and the Allen reference brain (Allen Institute for Brain Science, 2015, http://brain-map.org/api/index.html), we assumed a parallel sectioning angle for each brain. To generate a standard atlas accounting for different sectioning angles, the atlas was rotated, re-sectioned into coronal slices, and slices were re-indexed in order. The histological sequence of each sample was compared to the newly generated re-sectioned atlas slices. Every third slice of the experimental brain was automatically selected for quantitative evaluation of the cutting angle difference (severely damaged slices were skipped). Images from each group were first introduced to the same coordinates using a similarity transformation estimated by the Umeyama method based on contour point correspondence generated by Shape Context. Slices were further rescaled in the horizontal and vertical directions to accommodate the global deformation. Features identified by Histogram of Oriented Gradients (HOG) were then extracted from both images, and the L2 norm of HOG difference was used as the similarity metric. The difference between two images is measured as a scalar, which is the summation of the HOG difference over all blocks. Matching slice index differences of half brains were used to determine the cutting angle. Matching slice indices were then interpolated linearly to identify the best matching atlas section for each sample slice in the experimental brain. All the experimental slices were registered non-rigidly to their computed corresponding slice in the optimally rotated atlas to build a pixel-wise mapping from the 2D slice sequence to the reference volume. We augmented the Markov random field (MRF) approach to model brain tissue coherency. We made further improvements based on the data-specific properties of our experimental dataset, including segmenting the aqueduct with a convolutional neural network and locally warping it with a thin plate spline (TPS). For a more detailed description of this procedure see Xiong et al. (2018).
3D Reconstruction and Clustering of DR Neurons
To construct the volume presenting DR serotonin neurons, slices containing the Tph2-positive neurons from four animals were registered to Allen’s reference atlas (4698 ± 376.6 per brain, caudal DR was excluded). The line connecting the highest and lowest points of the aqueduct was defined as the midline, with a “zero” value along the medial-lateral axis. To reflect the bilateral symmetry of the DR serotonin system, mirror images were created for each cell across the midline plane (Figure S1A). All the cells’ 2D positions were determined automatically by custom Matlab program employing k-means, followed by 3D registration. DBSCAN was performed to cluster the combined data and establish a 3D surface of the DR serotonin system that covered ~97% of Tph2-positive neurons using custom software. Delaunay Triangulation was then performed on the clusters outputted from DBSCAN to define the boundaries. Catmull Clark subdivision was then applied to the boundary to finalize the shape of 3D clusters.
For each brain with specific retrograde injections and Tph2/Vglut3 dual-labeled neurons, the 2D positions of the neurons were determined manually and registered to the same reference atlas, allowing cross-comparison of the data from the DR of different brains. DBSCAN was performed to cluster individual groups and establish each 3D surface. To find a good pair of parameters, we explored the entire parameter space and each pair’s respective resulting cluster boundaries by generating 10,000 pairs of parameters for each set of data. We then extracted three features (number of clusters, average density, and coverage) to evaluate the groups resulting from each parameter selection. We plotted the relationship between average density and coverage of all the pairs from the ten datasets that generate a >60% coverage of the total cells. We found that, for nine out of ten datasets, one cluster had the same expressive power as multiple clusters and the only sub-system that might be better described as two clusters is DRSert→OB. Thus we decide to choose one cluster representation for each dataset, since the main purpose of this visualization attempt is to show the spatial distribution proportion of each projection defined serotonin neurons. We also noted that six out of ten of these datasets have a “knee” around 85% coverage that describes the best point in terms of a tradeoff between coverage and density. We therefore used the pair of parameters that have the largest average density when the coverage is around 85%. MinPints, minimum number of points required to form a dense region. Eps, epsilon.
| MinPints | Eps | Cluster # | Density | Coverage | |
|---|---|---|---|---|---|
| DRSert→PVH | 9 | 165 | 1 | 0.0018 | 85.96% |
| DRSert→CeA | 11 | 177 | 1 | 0.0012 | 82.97% |
| DRSert→LHb | 8 | 183 | 1 | 0.0010 | 79.91% |
| DRSert→dLGN | 12 | 201 | 1 | 0.0010 | 87.64% |
| DRSert→PIR | 14 | 157 | 1 | 0.0030 | 82.08% |
| DRSert→OFC | 10 | 130 | 1 | 0.0035 | 83.46% |
| DRSert→ENT | 11 | 193 | 1 | 0.0015 | 81.24% |
| DRSert→OB | 8 | 176 | 1 | 0.0015 | 86.03% |
| DRSert→OB | 14 | 195 | 2 | 0.0020 | 80.15% |
| DRSert→subcortical area | 16 | 149 | 1 | 0.0023 | 81.93% |
| DRSert→anterior cortical area | 13 | 150 | 1 | 0.0023 | 82.17% |
Further processing followed the procedures described above. To test whether the two-component model we proposed based on the raw data can hold regardless of the parameter choice, we examined the subcortical and cortical groupings that resulted from the randomly chosen 10,000 pairs of parameters. We sampled a random cortical grouping with a random subcortical grouping 10,000 times and calculated the respective overlap percentage. To ensure each randomly sampled grouping were plausible, we only used groupings that had more than 60 percent coverage. We found that the mean overlap percentage was 22.25% ± 4.01% (mean ± standard deviation). Additionally there exists no grouping of subcortical and cortical that has more than 30% overlap. Thus, we conclude that the positions of serotonin neurons with subcortical and cortical projections are largely separate regardless of the various boundaries that are possible.
Cell Density and Line Density
Cell density at location D, denoted as D(x, y, z), is a function that calculates cell numbers located in a 3D coordinate system defined by x (axis M→L), y (axis D→V), and z (axis A→P). Define N (x0, y0, z0) to be the number of cells located in space defined by x ≤ x0, y ≤ y0, z ≤ z0.
Cell linear density along y at location y0 is defined as
N(y0) refers to the number of cells at location defined by y ≤ y0, i.e. ventral to the brain surface at y0μm.
iDISCO-based Whole-Brain Axon Tracing
Brains were perfused, dissected, and processed according to the iDISCO+ pipeline as previously described (Renier et al., 2016). Whole brains were processed in 5-ml volumes, labeled with a 1:2000 dilution of anti-GFP antibody (Aves, GFP-1020) for 10 days and secondary Alexafluor 647 (Jackson Immunoresearch) for 7 days. Images were collected with a LaVision Lightsheet UltramicroscopeII at 0.8X magnification using 640 nm and 488 nm imaging lasers and a z-step size of 3 μm. The working distance of the microscope allowed visualization of the right hemisphere of each brain in the sagittal plane with an approximate imaging depth of 6 mm. The image stack of GFP+ axons in the 640-nm channel was first processed with a series of high-pass filters to reduce background noise and striping artifacts generated by shadows from the lightsheet. A 2D pixel classifier was trained in Ilastik using 2–5 images from each of 8 brains. Autofluorescent fiber tracts were separated from labeled axons with a second pixel classifier. Contiguous 3D objects were classified in Matlab according to volume, solidity, orientation, intensity, and proximity to remove artifacts with similar properties. The image stack of autofluorescence in the 488 nm channel was aligned to a reference brain generated by serial two photon tomography that was co-registered to the Allen Institute’s Common Coordinate Framework (CCF) (Zeng, 2018). Subsequently, the processed stack of axons was transformed to the same coordinates. Registration and transformation were performed using the Elastix toolbox (Klein et al., 2010; Shamonin et al., 2013). Voxels classified as axons were equally thresholded in all brains and counted by regions as described in the 2017 CCF. Within the Allen’s hierarchy of brain areas, regions distinguished solely by layers or anatomical location were collapsed into their “parent” region (e.g., Layers 1–6 of both dorsal and ventral anterior cingulate area are labeled as “anterior cingulate area”). These decisions were made prior to analysis and were agreed upon by four separate anatomical experts. Reported values of axonal labeling density for individual brain regions were normalized both to the volume of the region itself and the total labeling density for that sample to eliminate variability due to injection volume. Regions are sorted by divided based on comparing mean intensity between groups, and values are then sorted by their second principal component. Fiji and Imaris software were used to generate images.
cTRIO Experiments
Mice were anaesthetized with 65 mg/kg ketamine and 13 mg/kg xylazine (Lloid Laboratories) via intra-peritoneal injection and injected with 500 nl of a 1:1 mixture of AAV8 CAG-FLExFRT-G and AAV5 CAG-FLExFRT-TC into the DR, and also injected with either 750 nl HSV-STOPflox-Flp into ipsilateral OFC or 300 nl into ipsilateral CeA using coordinates described above. After recovery, mice were housed in a BSL2 facility. Two weeks later, 500 nl RVdG was injected into the DR using the procedure described above. After recovery, mice were housed in a BSL2 facility for 5 days before euthanasia.
Cell counting was performed manually using Fiji. For quantifications of subregions, boundaries were based on the Allen Institute’s reference atlas (Lein et al., 2007) with consultation of Franklin and Paxinos (2013). The infralimbic cortex and medulla are as defined in the Allen atlas; for medulla, sections anterior to the appearance of the DR were omitted due to possible local background (Figure S3). For counts of thalamic subregions, we were conservative while counting sections that border midbrain nuclei, so our counts may underestimate posterior thalamic subregions. We did not adjust for the possibility of double counting cells from consecutive sections, which would result in overestimates/tion, the extent of which would depend on the size of the cells in the regions quantified.
Fiber Photometry
For virus injection and fiber optic cannula implantation, mice were anesthetized with 1.5%–2.0% isoflurane. For DRSert group, AAV1-hEF1a- FLExloxP-GCaMP6m was injected into the DR (500nl). For DRSert→OFC and DRSert→CeA group, AAVretro-CAG-FLExloxP-Flp was injected into the OFC (750nl) or the CeA (300 nl) bilaterally, followed by AAV8-CAG-FLExFRT-GCaMP6 injection into the DR (500 nl). For fiber photometry experiments, a fiber optic cannula was implanted over the DR through the same hole used during the virus injection. To reduce autofluorescent artifacts and maximize light collection, cannulae (special order from Doric Lenses) were fabricated using 0.48 NA 400 μm BFH48–400 fibers, non-fluorescent epoxy and metal 2.5 mm ferrules. Cannulae were fixed to the skull using dental cement (Parkell, C&B metabond). After surgery, mice recovered on a heated pad until ambulatory and then returned to their homecage. Each groups comprised two cohorts.
Fiber photometry was performed using modulated 405 nm and 490 nm LEDs (Thorlab, M405F1 and M490F3). The light path was coupled to a 0.53-NA, 400-μm optical fiber patch cord, which was then coupled to the fiber implant in each mouse. Behavioral data from the operant system was synchronized to the fluorescence data using a TTL pulse at the start of each session. Signals were digitized using a digital signal acquisition board, demultiplexed using a software lock-in amplifier, and then low-pass filtered to 30 Hz before saving to disk at 381 samples/s. At the start of each recording session, fluorescence values in the 490-nm and 405-nm channels were approximately matched to ensure accurate fitting and subtraction during analysis. Three weeks after viral injection and fiber implantation, and before recording during behavior, mice were screened for high GCaMP expression. For the lever-pressing experiments, GCaMP-expressing mice were trained to drink in the operant box after lever pressing. After reaching proficiency, mice were tested with the following protocol: after 48 hours of water-restriction, mice were allowed to lever press to obtain 5% sucrose reinforcements for at least 20 trials. The fluorescence signals, reinforcements, and licks were recorded throughout the session. For the foot-shock experiments, GCaMP-expressing mice were first habituated to the shock box for 15 minutes. 24 hours later, mice were exposed to 12 delivered 0.5-mA, 1 s electric shocks in random interval in a 40min session. The start of each shock was marked by a TTL pulse.
Fiber photometry data were analyzed in MATLAB. Each channel was loaded and resampled to 3.81 Hz. The 405-nm channel was scaled to the 490-nm channel using a least-squares fit, and then ΔF(t)/F0 = (F490(t) – scaledF405(t)) / scaledF405(t) was computed and smoothed with a 1.9 s moving average filter. Finally, the median ΔF(t)/F0 from the 5-min baseline period (prior to licking or stimulus delivery in each experiment) was subtracted from the entire trace.
Patch-clamp Whole-Cell Recording
To determine how hM3Dq expressing and non-expressing DR serotonin neurons respond to CNO application, we injected AAVDJ-EF1a-DIO-hM3D(Gq)-mCherry into the DR of 8-week male Sert-Cre mice. Three weeks later, acute coronal sections encompassing the DR were sliced for electrophysiological experiments according to previously described methods (Ren et al., 2011). Briefly, adult mice were deeply anesthetized with intraperitonial (i.p.) injection of avertin (300 mg/kg) and transcardially perfused with ~5 ml of ice-cold oxygenated solution containing (in mM) 225 sucrose, 119 NaCl, 2.5 KCl, 1 NaH2PO4, 4.9 MgCl2, 0.1 CaCl2, 26.2 NaHCO3, 1.25 glucose, 3 kynurenic acid, and 1 Na-ascorbate (all chemicals were from Sigma, St Louis, MO, USA). Mice were then rapidly decapitated and whole brains were dissected into ice-cold oxygenated slicing solution containing (in mM) 110 choline chloride, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.3 NaH2PO4, 1.3 Na-ascorbate, 0.6 Na-pyruvate, 20 glucose, and 25 NaHCO3 (saturated with 95% O2 and 5% CO2). Coronal brain sections (250 μm thick) containing DR were cut with a vibratome (VT1000s, Leica, Nussloch, Germany). Slices were incubated for at least 1 hr at 34°C in oxygenated artificial cerebrospinal fluid (aCSF) containing (in mM) 125 NaCl, 2.5 KCl, 2 CaCl2, 1.3 MgCl2, 1.3 NaH2PO4, 1.3 Na-ascorbate, 0.6 Napyruvate, 20 glucose, and 25 NaHCO3. They were then transferred to a recording chamber on an upright Olympus fluorescent microscope equipped with differential interference contrast optics (DIC, COHU 4915–2000). During recording, slices were submerged and superfused (2 ml/min) with artificial cerebral spinal fluid (aCSF) at room temperature (22–24°C). Whole-cell recordings from DR serotonin neurons were obtained under visual control via DIC microscopy. Recording pipettes (4–7 MΩ) were backfilled with internal solution containing (in mM) 130 K-gluconate, 10 HEPES, 0.6 EGTA, 5 KCl, 3 Na2ATP, 0.3 Na3GTP, 4 MgCl2, and 10 Na2phosphocreatine (pH 7.2–7.4). Current-clamp recordings were carried out with a computer-controlled amplifier (MultiClamp 700B, Molecular Devices); neurons were held at –58 mV. Traces were low-pass filtered at 2.6 kHz and digitized at 10 kHz (DigiData 1440, Molecular Devices). Data were acquired by Clampex 10.4 and analyzed using Clampfit 10.4 software (Molecular Devices). To quantify action potential firing frequency, at least five minutes of baseline activity were collected from each cell. 10 μM CNO was added into the aCSF perfusion circulation for 3 min, after which it was replaced by normal aCSF. Drug effects were measured by recording traces 5 min before and 10 min after drug perfusion. Action potential firing frequency was counted by the “event detection” function of Clampfit 10.4. Control recordings were performed as described above, but from tdTomato+ neurons from Sert-Cre;Ai14 mice.
Local Field Potential Recordings and Current Source Density Analysis of Optogenetically-Evoked Responses
To examine how activation of serotonergic fibers modulates network activity in the orbital frontal cortex (OFC), we injected AAVDJ-EF1-DIO-hChR2(H134R)-eYFP into the DR of 8 weeks male Sert-Cre mice. Five months later, acute slices containing the OFC were collected in accordance with the procedures described in (Makinson et al., 2017). Mice were anesthesized with pentobarbital (50 mg/kg) and perfused with ice-cold sucrose buffer containing (in mM): 234 sucrose, 2.5KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, and 11 glucose, equilibrated with 95% O2 and 5% CO2, pH 7.4. Mice were decapitated, the brain dissected, and coronal slices (400 μm) were collected using a Leica VT1200S vibratome. Slices were transferred to oxygenated aCSF (containing in mM: 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2,2 CaCl2, 26 NaHCO3), bubbled continuously with 95% O2 and 5% CO2, and incubated at 32°C for 1 hour. Then, slices were incubated at room temperature for 1–5 hours before being placed in an interface recording chamber, in which they were continuously perfused with oxygenated aCSF (30–32°C) at a flow rate of 2–3ml/min.
A linear silicon multichannel probe (16 channels, 100 μm inter-electrode spacing, NeuroNexus Technologies) was placed in the OFC perpendicular to the laminar plane, such that the electrode array collected local field potentials (LFPs) from each cortical lamina. An optical fiber was situated immediately medial to the recording array directly above layer II/III to deliver 10 ms pulses of blue light (25 mW). Additionally, a bipolar tungsten electrode was placed in layer V to deliver electrical pulses (0.1 ms, 100 uA) to evoke synaptic responses and confirm the efficacy of drugs altering synaptic neurotransmission. Signals from all sixteen channels were digitized at 25 kHz using a 0–3000 Hz band-pass filter and amplified and stored using a RZ5D processor multichannel workstation (Tucker-Davis Technologies). A current source density (CSD) analysis was performed by calculating the second spatial derivative of the LFP (Nicholson and Freeman, 1975). This was used to estimate the magnitude, direction, and location of synaptic current recorded in the LFPs. When net positive current enters a cell, this creates an extracellular negativity reflected in a current “sink,” and appears as a negative deflection in the CSD. Conversely, current “sources” indicate net negative current flowing into a cell and will create positive CSD responses.
To measure the baseline response, light pulses were delivered once a minute to activate ChR2 in DR fibers in the OFC, and LFPs were collected and averaged over at least 5 trials. To determine the contribution of different neurotransmitters to light-evoked LFPs, drugs were bath applied for at least 30 minutes, and light responses were collected for another 5 trials. We utilized a cocktail of 5HT antagonists including GR 113808 (10 μM), Metergoline (10 μM), and Ondansetron (1 μM), and a separate cocktail of ionotropic glutamate receptor antagonists DNQX (25 μM) and CPP (1 μM). To avoid capturing a light artifact in our measurements, we extracted the average amplitude of the CSD over a 100 ms window measured 160 ms following the end of the light pulse. Control experiments in inactivated slices revealed that the light artifact coincided in time with the light pulse. LFP and CSD plots were generated using custom MATLAB scripts. Statistics were performed using SPSS 22 Statistics. For normal data, paired t tests were used to compare CSD amplitudes between baseline and drug conditions. For data without a normal distribution, a Wilcoxon Signed Rank test was used.
Drug Administration:
Clozapine N-oxide (CNO; Cayman Chemical, Item No. 12059) was dissolved in 0.4% DMSO and 0.9% NaCl. We administered CNO by intraperitoneal injection with a lower dose (1 mg/kg) and gave it to all the groups of animals to diminish the possibility that the behavioral effects were caused by a CNO side effect rather than serotonin neuron activation. To achieve 1 mg/kg body weight when CNO was injected 40 min before the onset of behavioral tests (Teissier et al., 2015). According to the most recent report regarding the CNO and clozapine’s influence on locomotion/anxiety related behavior test, there is no side effect when the dose of CNO is 1 mg/kg, but significant side-effect when the dose of CNO is 10 mg/kg. Only ~2% of CNO converts to clozapine after injection (Gomez et al., 2017), which means that the clozapine concentration in our experiments is likely to be ~0.02 mg/kg, far below 1 mg/kg clozapine that has been reported to elevate locomotor activity in the open field test.
Behavioral Assays
The behavioral assays described below are in the order of assay performance. Each assay was separated from the previous one by at least 5 days. All the mice that survived the surgery were included in the data analysis. One mouse died before all the assays were completed. Each groups comprised two to three cohorts.
Open field.
The open field apparatus consisted of a 50 cm x 50 cm clear Plexiglas arena. The intensity of the ambient light in the OFT is 199.3±0.5 LUX. Mice were acclimated to the experimental test room for at least 30 min prior to testing. To start a session, a mouse was placed in the center of the arena and allowed to freely explore for 10 minutes with video recording (Prut and Belzung, 2003). The total distance traveled (m), time spent in center (25 cm × 25 cm) (s) and center entries were automatically quantified by software (Biobserve).
Elevated plus maze.
The elevated plus maze apparatus consisted of two open and two closed arms extending out from a central platform. Each arm of the maze was 30 cm long and 5 cm wide. The maze surface was 85 cm above the floor. Each mouse was placed in the same position on the open arm of the maze at the beginning of the assay (facing the center) and allowed to explore the apparatus for 5 minutes (Walf and Frye, 2007). The number of open and closed arm entries as well as the total time spent in open and closed arms were automatically quantified by software (Biobserve).
Auditory fear conditioning.
The protocol was modified from previous studies (Do-Monte et al., 2015). Mice were habituated to the conditioning chamber and tones for 15 min per day for 3 days. On the fourth day, animals in gain-of-function experiments received a CNO injection 40 min before fear conditioning. The fear conditioning chamber consisted of a square cage (18 × 18 × 30 cm) with a floor wired to a shock generator and a scrambler, surrounded by an acoustic chamber (Coulbourn Instruments). We used two tones in a differential auditory fear conditioning protocol (CS+: 4 kHz, 30 sec, ~75 dB and CS–: 16 kHz, 30 sec, ~75 dB). The protocol consisted of 4 baseline tones (2 CS+, 2 CS–, interleaved), followed by interleaved presentations of 8x CS+ that co-terminated with a 1 s, 0.25 or 0.50 mA foot shock for gain- and loss-of-function experiments, respectively, and 4x CS– that were not paired with a foot shock. During a 1-day memory retrieval session, animals returned to the conditioning chamber and were presented with interleaved 8x CS+ and 4x CS–.
Forced-swim test.
Mice were placed for 6 min in a plastic cylinder (height: 25 cm; diameter: 18.5 cm) filled with water (15±1°C) to a depth of 14 cm. The water depth was adjusted so that the animals were forced to swim or float without their hind limbs touching the bottom. The sessions were videotaped and independently analyzed by two researchers blind to genotype. Duration of immobility (the time during which the subject made only the minimal movements necessary to keep their heads above water) was scored by averaging the results from the two researchers. A two-day forced-swim test was applied. For gain-of-function experiments, mice only received one CNO injection 40 min before the 2nd day test.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of anatomical, fiber photometry, electrophysiological recording and behavior tests data is described in corresponding section of text and methods details. All statistical tests and data analyses were performed using MATLAB and GraphPad Prism. Data were expressed as means ± SEMs in figures and text. Group differences were detected using either one-way analysis of variance (ANOVA), or with two-way ANOVA, both followed by Holm-Sidak test. According to histogram of dependent variable, the distributions of data meet normality assumption. Bartlett’s test shows no significant difference among variance of groups. Multiple comparisons were corrected when appropriate by adjusting pvalues using the Holm-Sidak method. Significance was defined as p<0.05. Sample sizes were chosen based on those used in previous papers. Full details of each statistical test used (tests, statistics, significance levels, sample sizes, animal numbers, SEMs, and degrees of freedom) are described in each figure legend, except for Figure 4 and S4, which are listed in Table S3.
Supplementary Material
Figure S1. Spatial Organization of DR Serotonin Neurons with Respect to Axonal Projections, Related to Figure 1. (A) Examples of labeled neurons at eight sites of injecting HSV-Cre into Ai14 mice (yellow arrows). Scale, 500 μm. (B) Schematic of data collection for 3D reconstruction of DR serotonin neurons. After Tph2 staining, the positions for all the positive neurons were recorded. The line connecting the highest and lowest points of the aqueduct (AQ; dashed ovals) was defined as the midline, and a mirror image was created for each cell across the midline (reflecting the bilateral symmetry of DR serotonin neurons), and then the two mirror images were merged (left four panels). The “zero” along the dorsal-ventral axis refers to the lowest point of AQ for individual slices during data collection. In registered brains, “zero” refers to the brain surface (right panel). (C) Coronal (left) and sagittal (right) view of individual cells’ location (cyan) from four subcortical projecting DR serotonin neuron groups. Yellow represent the surface of the cluster that include all Tph2+ cells. (D) Coronal (left) and sagittal (right) view of individual cells’ location (red) from OB and three cortical projecting DR serotonin neuron groups. (E) Quantification of the vertical distance between cells from either DRTph2→SC group (cyan) or DRTph2→AC group (red) and a plane 3742 μm ventral from the brain surface. (Unpaired twotailed test, t=4.43, df=2286). ****p<0.01. Error bars, SEM. (F) Coronal (left) and sagittal (right) view of individual cells’ location from a combination of PVH-, CeA-, LHb-, and dLGN-projecting DR serotonin groups (cyan) and a combination of OB-, PIR-, OFC- and ENT- projecting DR serotonin groups (red). Dashed line, 3752 μm below the brain surface. (G) Quantification of cell density along the dorsal-ventral axis. Dashed line shows where the two clusters share the same line density at 3752 μm ventral to the brain surface. The probability for cyan and red cells being dorsal to the 3752-μm deep plane is 75.08% and 15.67%, respectively. (H) Quantification of the vertical distance between cells from either of the two clusters in F and 3752-μm deep plane. (Unpaired two tailed test, t=30.9, df=2958). ****p<0.01. Error bars, SEM. (I) Quantification of dual-site retrograde tracing. HSV-Cre was injected into OFC of the three groups of Ai14 mice (n=3), and AAVretro-CAG-GFP was injected into OFC, OB, or CeA for individual group. Anti-Tph2 staining was performed on consecutive coronal sections containing DR. The proportion of Tph2, GFP, and tdTomato triple positive neurons in GFP+/Tph2+ or tdTomata+/Tph2+ neurons for the three co-injection experiments are listed on the y-axis.
Figure S2. Select Target Regions Highlight the Complementary Nature of Axonal Projections from DRSert→CeA and DRSert→OFC Neurons, Related to Figure 2. (A) DRSert→OFC axons heavily innervate OFC, OB, and the cortical amygdala (CoA) (within the yellow outlines). DRSert→CeA axons largely avoid these regions. (B) DRSert→CeA axons target the CeA as expected, but are also found in PVH and BNST while DRSert→OFC axons are largely absent from these regions. Images of axons are aligned to each brain’s own autofluorescence and both channels are matched to the Allen Common Coordinate Framework. All sections are sagittal with 20 μm in z; scale, 400 μm.
Figure S3. Control Experiments and Additional Images for cTRIO, Related to Figure 3. (A) Schematic of control groups for cTRIO experiments applied on DR. Wild type (WT) instead of Sert-Cre mice were used in No Cre Ctrl, and AAV8-FLExFRT-G was omitted in No G Ctrl. (B) Schematic of brain regions quantified for input neurons. Regions approximately 1 mm anterior and posterior to the center of the DR were excluded from analysis due to local background labeling from rabies virus (see Weissbourd et al., 2014), including median raphe (MR), ventral tegmental area (VTA), substantia nigra pars compacta (SNc), interpeduncular nucleus (IPN), periaqueductal gray (PAG), locus coeruleus (LC) and several medulla nuclei. (C) Quantification of long-distance background infection in control groups. Y-axis presents total input cell numbers, and X-axis lists group names. n=9, 8, 4, 4, 4, 4. Error bars, SEM. (D) Representatives of GFP labeled ipsilateral input cells in PVH (at four coronal section planes from anterior to posterior; left panels) and in medulla nuclei and DCN (right panels) to OFC- and CeA-projecting DR serotonin neurons. Scale, 250 μm.
Figure S4. Summary of Optical Fiber Placement and Representative Traces for Fiber Photometry Recording, Related to Figure 4. Fiber photometry recording was performed on DRSert (A), DRSert→OFC (B), and DRSert→CeA (C) neurons expressing GCaMP6m, and on DRSert neurons expressing eGFP (D). (A1, B1, C1) Schematic DR diagram showing the end of optic fiber placement (oval) for each mouse. AQ, aqueduct. Note that we adjusted the fiber implant positions to maximize DRSert→OFC and DRSert→CeA neurons based on data from Figure 1. (A2, B2, C2) Mean responses of individual mice to lever press. Time 0 is aligned to lever press (vertical dashed line). Time window of −0.117s to −0.65s is in shaded grey (a), and time window of −0.65s to −0.13s is in shaded pink (b). (A3, B3, C3) Group data from all the trials of individual mice showing the quantification of the mean ΔF/F during “window a” compared to the mean ΔF/F during “window b”. A3, Signal of three mice from DRSert group was significantly increased (p<0.0001). B3, Signal of four mice from DRSert→OFC group was significantly increased (p<0.01), and one significantly decreased (p<0.05). C4, Signal of one DRSert→CeA group was significantly decreased (p<0.05). (two-tail paired t test). See Table S3 for detailed data. (A4, B4, C4) Example responses to sucrose consumption observed in one mouse from the DRSert (A4), DRSert→OFC (B4), or DRSert→CeA (C4) group. Time 0 is aligned to the initiation of sucrose water licking. (A5, B5, C5) Example of responses to foot-shock observed in one mouse each from the DRSert (A5), DRSert→OFC (B5), or DRSert→CeA (C5) group. Time 0 is aligned to the onset of foot-shock (yellow shadow covered the foot-shock duration). Solid and dotted lines represent mean ± SEM. (D1) Schematic of viral injection and optical fiber implantation of the eGFP control group. (D2) Confocal images of coronal sections showing fiber optic placement (dotted rectangle) and the expression of eGFP (green) in the DR. Vertical dashed lines represent the midline. Scale, 100 μm. (D3) Mean responses of individual mice (n=6, each with a different color) to sucrose consumption after lever press. Time 0 is aligned to lick initiation (vertical dashed line). (D4) Mean responses of individual mice from to electrical shock. Time 0 is aligned to onset of 1-sec electric shock delivery.
Figure S5. Functional Validation of hM3Dq and AAVretro-Cre in Serotonin Neurons. Related to Figure 5–7. (A) Sample traces showing that the addition of CNO to two acute brain slices from Sert-Cre mice transduced with AAV-FLExloxP-hM3Dq increased the firing rate of DR serotonin neurons (top trace, example from 7/9 slices) or transformed tonic firing to phasic firing (lower trace, example from 2/9 slices). (B) Left, grouped data from the experiments performed in A (two-tail paired t-test, **p<0.005, t=3.95, df=8. n=9, from 9 slices of 4 mice); right, grouped data from experiments performed in control Sert-Cre;Ai14 mice (two-tail paired t-test, n.s. not significant, t=0.050, df=6. n=7, from 6 slices of 2 mice). (C) Representative images of OFC, CeA, and hippocampus showing serotonin staining at terminals after AAVretro-Cre injection into either OFC or CeA of Tph2flox/flox mice. Scale, 50 μm. (D) Quantification of the fluorescence intensity in OFC and CeA slices, normalized to the staining intensity in hippocampal sections from each brain. n=3, 3, **p<0.01; ***p<0.001. (E) Quantification of OFC- (upper panel) and CeA- (lower panel) projecting serotonin neurons located in either DR or MR. AAVretro-Cre-2A-GFP was unilaterally injected into either OFC or CeA of Ai14 mice. tdTomato+/Tph2+ dual-labeled neurons in DR and MR were counted. n=3. Error bars, SEM.
Figure S6. DRSert→OFC fibers release serotonin and glutamate to modulate OFC network activity, related to Figures 1, 5–7. To examine how activation of DR fibers modulates neural activity in the OFC, we utilized mice expressing ChR2 in DRSert neurons for in vitro electrophysiology. Light-evoked local field potentials (LFPs) were recorded from OFC slices in an interface-recording chamber. Current source density (CSD) analysis was performed on LFP data to determine the location, magnitude, and direction of synaptic currents evoked by ChR2 activation in the OFC circuit in the absence or presence of cocktail of serotonin or ionotropic glutamate receptor blockers. (A) Upper panel: schematic of the OFC circuit including pyramidal neurons (Pyr), interneurons (Int), as well as the potential locations of serotonergic innervation by DR fibers (brown). Bottom panel: Experimental design of LFP recordings. A silicon multichannel probe was placed in the OFC (grey area outlined in the right schematic), while an optical fiber was situated medial to the recording array above layer II/III. Additionally, a bipolar tungsten electrode was placed in layer V to deliver electrical pulses (0.1 ms, 100 μA) to evoke synaptic responses and confirm the efficacy of drugs altering neurotransmission. (B) Representative LFP recording in response to a 10-ms/25-mW blue light pulse. (C) Representative CSD derived from the LFP recording. In response to the blue light pulse, a consistent response was observed involving prominent current sinks (net positive current flowing into the cell) in layer (L) II (orange star) and LV (green star), and a smaller source in LIII (net negative current flowing inside neurons; purple star). A 100-ms time window ~160 ms following the end of the light pulse (between dashed vertical lines) was used to quantify CSD and the effect of neurotransmitter blockers in D−G below. This window was chosen to avoid potential contamination from the light artifact, while ensuring that we could measure significant current sinks and sources (STAR Methods). To confirm that the responses we quantified were direct rather than via polysynaptic circuit activation, we applied 1 μM tetrodotoxin to block voltagegated Na+ channels with, and 100 μM 4-aminopyridine to partially block K+ channels (n=6 slices), and found that the responses persisted. (D) Representative responses of L2 sink, L3 source, and L5 sink in the absence (baseline [BL], solid trace) or presence (drug, dashed trace) of cocktail of serotonin receptor (HTR) antagonists GR 113808 (10 μM), Metergoline (10 μM), and Ondansetron (1 **M). (E) Representative responses of L2 sink, L3 source, and L5 sink in presence and absence of ionotropic glutamate receptor (iGluR) antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX, 25 μM) and 3-((+-)-2-carboxypiperazin-4-yl)-propyl-1- phosphonic acid (CPP, 1 μM). (F) Quantification of the CSD at baseline (BL, ChR2 stimulation alone) and in the presence of HTR antagonists (from 4 mice). While the L2 sink and L3 source were not affected, the L5 sink was significantly reduced (less negative, p = 0.008), suggesting serotonin release from DR fibers in the OFC contributes to activation of L5 OFC neurons. (G) Same as F, but comparing CSDs and in baseline and the presence of iGluR antagonists (from 5 mice). The L2 Sink shows a weak trend of a reduction (p = 0.073), but both the L3 Source and L5 Sink are significantly reduced in response to iGluR antagonists (p = 0.003 and p = 0.015 respectively). Given that ChR2-evoked responses were reduced both by serotonin receptor and iGluR antagonists, our data suggest that DR neurons co-release serotonin and glutamate in the OFC.
Figure S7. Functional Analysis of CeA- and OFC-projecting DR Serotonin Neurons in Fear Learning and Memory, Related to Figure 6, 7. (A) Chemogenetic activation of DRSert→OFC neurons does not affect freezing time to conditioned tone (CS+) during fear conditioning (one-way ANOVA, F[2, 31] = 1.28. n= 9, 11, 14). (B) Chemogenetic activation of DRSert→OFC neurons does not affect freezing time to CS+ during 1-day recall (one-way ANOVA, F[2, 31] = 1.195. n= 9, 11, 14). (C) Chemogenetic activation of DRSert→CeA neurons increases freezing time to CS+ during fear conditioning. ***p<0.001, ****p<0.0001 (one-way ANOVA followed by multiple t-tests; F[2, 29] = 18.0; t= 5.55, 4.70. n= 10, 11, 12). (D) Chemogenetic activation of DRSert→CeA neurons increases freezing time to CS+ during 1-day recall. **p<0.01, ***p<0.001 (one-way ANOVA followed by multiple t-tests, F[2, 29] = 9.61; t=4.58, 3.429. n= 10, 11, 12). (E) Conditionally knocking out Tph2 from DRSert→OFC neurons does not affect freezing time to CS+ during fear conditioning (two-tail unpaired t-test; t=1.47, df=17. n=9, 10). (F) Conditionally knocking out Tph2 from DRSert→OFC neurons does not affect freezing time to CS+ during 1-day recall (two-tail unpaired t-test; t=0.0624, df=17. n=9, 10). (G) Conditionally knocking out Tph2 from DRSert→CeA neurons does not affect freezing time to CS+ during fear conditioning (two-tail unpaired t-test; t=0.990, df=14. n=9, 10). (H) Conditionally knocking out Tph2 from DRSert→CeA neurons does not affect freezing time to CS+ during 1-day recall (two-tail unpaired t-test; t=0.685, df=14. n=9, 10). (I) Activation of DRSert→OFC neurons does not affect the immobility time in forced-swim test on day-1 training session (one-way ANOVA, F[2, 31] = 1.44, n= 9, 11, 14).
Movie S1: 3D Representation of DR Serotonin Neurons’ Spatial Organization with Respect to Axonal Projections and Vglut3 Co-Expression, Related to Figure 1.
Movie S2: 3D Rendering of DRSert→OFC and DRSert→CeA Axon Collateralization Patterns, Related to Figure 2B–D. Projecting rostrally passing through the lateral hypothalamus, the DRSert→OFC axons split into a medial branch that travels dorsally to medial frontal cortex; the rest fans out laterally to the cortex, with some projecting posteriorly to the ENT and others innervating the PIR and insular cortex anteriorly, eventually combining with the medial branch to jointly innervate other prefrontal areas and the OB. By contrast, the DRSert→CeA axons intensely innervated most amygdala nuclei and several hypothalamic nuclei, notably the ventrolateral preoptic nucleus, PVH, and the ventral BNST. Consistent with our retrograde tracing experiments, DRSert→OFC axons did not innervate most forebrain subcortical regions, whereas DRSert→CeA axons rarely invaded any cortical areas.
Movie S3: Fly-through Coronal Density Maps of DRSert→OFC (left) and DRSert→CeA (middle) Projections, and A P-value Map (right), Related to Figure 2F.
Table S1: Allen Brain Atlas IDs and Their Corresponding Names as Identified by the 2017 Common Coordinate Framework, Related to Figure 2. Regions were selected prior to analysis such that areas defined by individual layers (e.g., cortical layers I−VI), cell identity, and anatomical cardinal directions are collapsed into their parent region. Individual normalized regional densities for each brain are aligned to the heat maps from Figure 2G.
Table S2: Quantification of cTRIO: Number of Cells in Region (Proportion of Total), Related to Figure 3. For each sample, the raw cell count for each brain region is displayed in the left column, the fraction of total input for that brain region is displayed in the right column, and the total number of inputs is displayed at the bottom. Medulla nuclei were first calculated individually and then all collapsed into other medulla nuclei expect for the nucleus of the solitary tract (NST).
Table S3: Details of Statistical Analyses of Fiber Photometry Recording Data. Related to Figure 4 and Figure S4.
Table S4: Summary of Number of Cells Labeled by Retrograde Tracing. Related to Figure 1, Figure 5.
Dorsal raphe (DR) serotonin neurons are organized into parallel sub-systems
OFC- and CeA-projecting DR serotonin neurons have complementary collateralizations
OFC- and CeA-projecting DR serotonin neurons respond oppositely to aversive stimuli
OFC- and CeA-projecting DR serotonin neurons have distinct behavioral functions
Acknowledgement:
We thank E. Adams and M. Tessier-Lavigne for advice on iDISCO+, T. Davidson and K. Deisseroth for advice on fiber photometry, E. Steinberg and R. Malenka for advice on behavior, Q. Wu for Tph2flox/flox mice, A. Karpova for the AAVretro vector, Stanford, Salk, and UNC Viral Core for viruses, Allen Institute for the reference atlas and T. Gilbert for the advice on its use, M. Chen for code files for data analysis, K. Beier and G. Nachtrab for sharing AAVs, S. Bell for advice on 2D registration, and R. Malenka, A. Mizrahi, A. Shuster, L. DeNardo, J. Lui, M. Wagner, and W. Allen for critiques on the manuscript. This work was supported by BRAIN initiative grants (R01 NS104698, NeuroNex). L.L. is an HHMI investigator.
Abbreviations for anatomical regions
- AI
anterior insular cortex
- BLA
basolateral amygdala
- BNST
bed nucleus of the stria terminalis
- CeA
central amygdala
- CoA
cortical amygdala
- DB
nucleus of the diagonal band
- DCN
deep cerebellum nuclei
- dLGN
dorsal lateral geniculate nucleus
- ENT
entorhinal cortex
- LHb
lateral habenula
- LHy
lateral hypothalamus
- NST
nucleus of solitary tract
- OB
olfactory bulb
- OFC
orbitofrontal cortex
- PIR
piriform cortex
- PSTh
parasubthalamic nucleus
- PVH
paraventricular hypothalamus
- PVHd
paraventricular hypothalamus, descending division
- SI
substantia innominate
- SNc
substantia nigra compacta
- SNr
substantia nigra pars reticulate
- Sth
subthalamic nucleus
- VLPO
ventrolateral preoptic nucleus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Abrams JK, Johnson PL, Hollis JH, and Lowry CA (2004). Anatomic and functional topography of the dorsal raphe nucleus. Ann N Y Acad Sci 1018, 46–57. [DOI] [PubMed] [Google Scholar]
- Albert PR, Vahid-Ansari F, and Luckhart C (2014). Serotonin-prefrontal cortical circuitry in anxiety and depression phenotypes: pivotal role of pre- and post-synaptic 5-HT1A receptor expression. Front Behav Neurosci 8, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen Brain Atlas (2017). Projection :: Allen Brain Atlas: Mouse Connectivity.
- Allen Institute for Brain Science (2015). Allen Brain Atlas API.
- Allen WE, DeNardo LA, Chen MZ, Liu CD, Loh KM, Fenno LE, Ramakrishnan C, Deisseroth K, and Luo L (2017). Thirst-associated preoptic neurons encode an aversive motivational drive. Science 357, 1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, and Roth BL (2007). Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104, 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmitia EC, and Segal M (1978). An autoradiographic analysis of the differential ascending projections of the dorsal and median raphe nuclei in the rat. J Comp Neurol 179, 641–667. [DOI] [PubMed] [Google Scholar]
- Barnes NM, and Sharp T (1999). A review of central 5-HT receptors and their function. Neuropharmacology 38, 1083–1152. [DOI] [PubMed] [Google Scholar]
- Beier KT, Kim CK, Hoerbelt P, Hung LW, Heifets BD, DeLoach KE, Mosca TJ, Neuner S, Deisseroth K, Luo L, et al. (2017). Rabies screen reveals GPe control of cocaine-triggered plasticity. Nature 549, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, Gao XJ, Kremer EJ, Malenka RC, and Luo L (2015). Circuit Architecture of VTA Dopamine Neurons Revealed by Systematic Input-Output Mapping. Cell 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmaker RH, and Agam G (2008). Major depressive disorder. N Engl J Med 358, 55–68. [DOI] [PubMed] [Google Scholar]
- Calhoon GG, and Tye KM (2015). Resolving the neural circuits of anxiety. Nat Neurosci 18, 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calizo LH, Akanwa A, Ma X, Pan YZ, Lemos JC, Craige C, Heemstra LA, and Beck SG (2011). Raphe serotonin neurons are not homogenous: electrophysiological, morphological and neurochemical evidence. Neuropharmacology 61, 524–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JY, Amoroso MW, and Uchida N (2015). Serotonergic neurons signal reward and punishment on multiple timescales. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commons KG (2015). Two major network domains in the dorsal raphe nucleus. J Comp Neurol 523, 1488–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia PA, Lottem E, Banerjee D, Machado AS, Carey MR, and Mainen ZF (2017). Transient inhibition and long-term facilitation of locomotion by phasic optogenetic activation of serotonin neurons. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayan P, and Huys Q (2015). Serotonin’s many meanings elude simple theories. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do-Monte FH, Quinones-Laracuente K, and Quirk GJ (2015). A temporal shift in the circuits mediating retrieval of fear memory. Nature 519, 460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez SP, Cauli B, Cabezas C, Muzerelle A, Poncer JC, and Gaspar P (2016). Multiscale single-cell analysis reveals unique phenotypes of raphe 5-HT neurons projecting to the forebrain. Brain Struct Funct 221, 4007–4025. [DOI] [PubMed] [Google Scholar]
- Fonseca MS, Murakami M, and Mainen ZF (2015). Activation of Dorsal Raphe Serotonergic Neurons Promotes Waiting but Is Not Reinforcing. Curr Biol 25, 306–315. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, and Paxinos G (2013). Paxinos and Franklin’s The mouse brain in stereotaxic coordinates, Fourth edition edn (Amsterdam: Academic Press, an imprint of Elsevier; ). [Google Scholar]
- Gagnon D, and Parent M (2014). Distribution of VGLUT3 in highly collateralized axons from the rat dorsal raphe nucleus as revealed by single-neuron reconstructions. PLoS One 9, e87709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, Ellis RJ, Richie CT, Harvey BK, Dannals RF, et al. (2017). Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Doughty M, Harbaugh CR, Cummins A, Hatten ME, Heintz N, and Gerfen CR (2007). Targeting Cre recombinase to specific neuron populations with bacterial artificial chromosome constructs. J Neurosci 27, 9817–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, and El Mestikawy S (2002). A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci 22, 5442–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes WN, Seal RP, Oesch N, Edwards RH, and Diamond JS (2011). Genetic targeting and physiological features of VGLUT3+ amacrine cells. Vis Neurosci 28, 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, et al. (2014). Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura K, Takeuchi Y, Fujiwara K, Tominaga M, Yoshioka H, and Sawada T (1988). Quantitative analysis of the distribution of serotonin-immunoreactive cell bodies in the mouse brain. Neurosci Lett 91, 265–270. [DOI] [PubMed] [Google Scholar]
- Klein S, Staring M, Murphy K, Viergever MA, and Pluim JP (2010). elastix: a toolbox for intensity-based medical image registration. IEEE Trans Med Imaging 29, 196–205. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhong W, Wang D, Feng Q, Liu Z, Zhou J, Jia C, Hu F, Zeng J, Guo Q, et al. (2016). Serotonin neurons in the dorsal raphe nucleus encode reward signals. Nat Commun 7, 10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZX, Zhou JF, Li Y, Hu F, Lu Y, Ma M, Feng QR, Zhang JE, Wang DQ, Zeng JW, et al. (2014). Dorsal Raphe Neurons Signal Reward through 5-HT and Glutamate. Neuron 81, 1360–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry CA, Johnson PL, Hay-Schmidt A, Mikkelsen J, and Shekhar A (2005). Modulation of anxiety circuits by serotonergic systems. Stress 8, 233–246. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewcz CA, Mazzone CM, D’Agostino G, Halladay LR, Hardaway JA, DiBerto JF, Navarro M, Burnham N, Cristiano C, Dorrier CE, et al. (2016). Serotonin engages an anxiety and fear-promoting circuit in the extended amygdala. Nature 537, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Sorokin JM, Wong JC, Christian CA, Goldin AL, Escayg A, and Huguenard JR (2017). Regulation of Thalamic and Cortical Network Synchrony by Scn8a. Neuron 93, 1165–1179 e1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer HY (1994). An overview of the mechanism of action of clozapine. J Clin Psychiatry 55 Suppl B, 47–52. [PubMed] [Google Scholar]
- Miyazaki KW, Miyazaki K, and Doya K (2012). Activation of Dorsal Raphe Serotonin Neurons Is Necessary for Waiting for Delayed Rewards. J Neurosci 32, 10451–10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CP, and Jacobs BL (2010). Handbook of the Behavioral Neurobiology of Serotonin (Elsevier; ). [Google Scholar]
- Muzerelle A, Scotto-Lomassese S, Bernard JF, Soiza-Reilly M, and Gaspar P (2016). Conditional anterograde tracing reveals distinct targeting of individual serotonin cell groups (B5–B9) to the forebrain and brainstem. Brain Struct Funct 221, 535–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RL, Neve KA, Nestler EJ, and Carlezon WA Jr. (2005). Use of herpes virus amplicon vectors to study brain disorders. Biotechniques 39, 381–391. [DOI] [PubMed] [Google Scholar]
- Nicholson C, and Freeman JA (1975). Theory of current source-density analysis and determination of conductivity tensor for anuran cerebellum. J Neurophysiol 38, 356–368. [DOI] [PubMed] [Google Scholar]
- Niederkofler V, Asher TE, Okaty BW, Rood BD, Narayan A, Hwa LS, Beck SG, Miczek KA, and Dymecki SM (2016). Identification of Serotonergic Neuronal Modules that Affect Aggressive Behavior. Cell Reports 17, 1934–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa SK, Cohen JY, Hwang D, Uchida N, and Watabe-Uchida M (2014). Organization of monosynaptic inputs to the serotonin and dopamine neuromodulatory systems. Cell Rep 8, 1105–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, Wang Q, Lau C, Kuan L, Henry AM, et al. (2014). A mesoscale connectome of the mouse brain. Nature 508, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaty BW, Freret ME, Rood BD, Brust RD, Hennessy ML, deBairos D, Kim JC, Cook MN, and Dymecki SM (2015). Multi-Scale Molecular Deconstruction of the Serotonin Neuron System. Neuron 88, 774–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit-Demouliere B, Chenu F, and Bourin M (2005). Forced swimming test in mice: a review of antidepressant activity. Psychopharmacology (Berl) 177, 245–255. [DOI] [PubMed] [Google Scholar]
- Pollak Dorocic I, Furth D, Xuan Y, Johansson Y, Pozzi L, Silberberg G, Carlen M, and Meletis K (2014). A whole-brain atlas of inputs to serotonergic neurons of the dorsal and median raphe nuclei. Neuron 83, 663–678. [DOI] [PubMed] [Google Scholar]
- Prouty EW, Chandler DJ, and Waterhouse BD (2017). Neurochemical differences between target-specific populations of rat dorsal raphe projection neurons. Brain Res 1675, 28–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prut L, and Belzung C (2003). The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol 463, 3–33. [DOI] [PubMed] [Google Scholar]
- Ravindran LN, and Stein MB (2010). The pharmacologic treatment of anxiety disorders: a review of progress. J Clin Psychiatry 71, 839–854. [DOI] [PubMed] [Google Scholar]
- Ren J, Qin C, Hu F, Tan J, Qiu L, Zhao S, Feng G, and Luo M (2011). Habenula “cholinergic” neurons co-release glutamate and acetylcholine and activate postsynaptic neurons via distinct transmission modes. Neuron 69, 445–452. [DOI] [PubMed] [Google Scholar]
- Renier N, Adams EL, Kirst C, Wu Z, Azevedo R, Kohl J, Autry AE, Kadiri L, Umadevi Venkataraju K, Zhou Y, et al. (2016). Mapping of Brain Activity by Automated Volume Analysis of Immediate Early Genes. Cell 165, 1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, et al. (2015). Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A, Bocchio M, Bannerman DM, Sharp T, and Capogna M (2017). Control of Amygdala Circuits by 5-HT Neurons via 5-HT and Glutamate Cotransmission. J Neurosci 37, 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamonin DP, Bron EE, Lelieveldt BP, Smits M, Klein S, Staring M, and Alzheimer’s Disease Neuroimaging I (2013). Fast parallel image registration on CPU and GPU for diagnostic classification of Alzheimer’s disease. Front Neuroinform 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbusch HW (1981). Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience 6, 557–618. [DOI] [PubMed] [Google Scholar]
- Teissier A, Chemiakine A, Inbar B, Bagchi S, Ray RS, Palmiter RD, Dymecki SM, Moore H, and Ansorge MS (2015). Activity of Raphe Serotonergic Neurons Controls Emotional Behaviors. Cell Rep 13, 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban DJ, Zhu H, Marcinkiewcz CA, Michaelides M, Oshibuchi H, Rhea D, Aryal DK, Farrell MS, Lowery-Gionta E, Olsen RHJ, et al. (2016). Elucidation of The Behavioral Program and Neuronal Network Encoded by Dorsal Raphe Serotonergic Neurons. Neuropsychopharmacol 41, 1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP (1991). A PHA-L analysis of ascending projections of the dorsal raphe nucleus in the rat. J Comp Neurol 313, 643–668. [DOI] [PubMed] [Google Scholar]
- Walf AA, and Frye CA (2007). The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc 2, 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, Adhikari A, Tye KM, Frank LM, and Deisseroth K (2012). A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waselus M, Valentino RJ, and Van Bockstaele EJ (2011). Collateralized dorsal raphe nucleus projections: a mechanism for the integration of diverse functions during stress. J Chem Neuroanat 41, 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbourd B, Ren J, DeLoach KE, Guenthner CJ, Miyamichi K, and Luo L (2014). Presynaptic partners of dorsal raphe serotonergic and GABAergic neurons. Neuron 83, 645–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney MS, Shemery AM, Yaw AM, Donovan LJ, Glass JD, and Deneris ES (2016). Adult Brain Serotonin Deficiency Causes Hyperactivity, Circadian Disruption, and Elimination of Siestas. J Neurosci 36, 9828–9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Finke S, Conzelmann KK, and Callaway EM (2007). Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat Methods 4, 47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2017). Depression.
- Wu Q, Clark MS, and Palmiter RD (2012). Deciphering a neuronal circuit that mediates appetite. Nature 483, 594–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, Ren J, Luo L, Horowitz AM (2018). Mapping Mouse Brain Slice Sequence to a Reference Brain Without 3D Reconstruction. bioRxiv doi: 10.1101/357475 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Spatial Organization of DR Serotonin Neurons with Respect to Axonal Projections, Related to Figure 1. (A) Examples of labeled neurons at eight sites of injecting HSV-Cre into Ai14 mice (yellow arrows). Scale, 500 μm. (B) Schematic of data collection for 3D reconstruction of DR serotonin neurons. After Tph2 staining, the positions for all the positive neurons were recorded. The line connecting the highest and lowest points of the aqueduct (AQ; dashed ovals) was defined as the midline, and a mirror image was created for each cell across the midline (reflecting the bilateral symmetry of DR serotonin neurons), and then the two mirror images were merged (left four panels). The “zero” along the dorsal-ventral axis refers to the lowest point of AQ for individual slices during data collection. In registered brains, “zero” refers to the brain surface (right panel). (C) Coronal (left) and sagittal (right) view of individual cells’ location (cyan) from four subcortical projecting DR serotonin neuron groups. Yellow represent the surface of the cluster that include all Tph2+ cells. (D) Coronal (left) and sagittal (right) view of individual cells’ location (red) from OB and three cortical projecting DR serotonin neuron groups. (E) Quantification of the vertical distance between cells from either DRTph2→SC group (cyan) or DRTph2→AC group (red) and a plane 3742 μm ventral from the brain surface. (Unpaired twotailed test, t=4.43, df=2286). ****p<0.01. Error bars, SEM. (F) Coronal (left) and sagittal (right) view of individual cells’ location from a combination of PVH-, CeA-, LHb-, and dLGN-projecting DR serotonin groups (cyan) and a combination of OB-, PIR-, OFC- and ENT- projecting DR serotonin groups (red). Dashed line, 3752 μm below the brain surface. (G) Quantification of cell density along the dorsal-ventral axis. Dashed line shows where the two clusters share the same line density at 3752 μm ventral to the brain surface. The probability for cyan and red cells being dorsal to the 3752-μm deep plane is 75.08% and 15.67%, respectively. (H) Quantification of the vertical distance between cells from either of the two clusters in F and 3752-μm deep plane. (Unpaired two tailed test, t=30.9, df=2958). ****p<0.01. Error bars, SEM. (I) Quantification of dual-site retrograde tracing. HSV-Cre was injected into OFC of the three groups of Ai14 mice (n=3), and AAVretro-CAG-GFP was injected into OFC, OB, or CeA for individual group. Anti-Tph2 staining was performed on consecutive coronal sections containing DR. The proportion of Tph2, GFP, and tdTomato triple positive neurons in GFP+/Tph2+ or tdTomata+/Tph2+ neurons for the three co-injection experiments are listed on the y-axis.
Figure S2. Select Target Regions Highlight the Complementary Nature of Axonal Projections from DRSert→CeA and DRSert→OFC Neurons, Related to Figure 2. (A) DRSert→OFC axons heavily innervate OFC, OB, and the cortical amygdala (CoA) (within the yellow outlines). DRSert→CeA axons largely avoid these regions. (B) DRSert→CeA axons target the CeA as expected, but are also found in PVH and BNST while DRSert→OFC axons are largely absent from these regions. Images of axons are aligned to each brain’s own autofluorescence and both channels are matched to the Allen Common Coordinate Framework. All sections are sagittal with 20 μm in z; scale, 400 μm.
Figure S3. Control Experiments and Additional Images for cTRIO, Related to Figure 3. (A) Schematic of control groups for cTRIO experiments applied on DR. Wild type (WT) instead of Sert-Cre mice were used in No Cre Ctrl, and AAV8-FLExFRT-G was omitted in No G Ctrl. (B) Schematic of brain regions quantified for input neurons. Regions approximately 1 mm anterior and posterior to the center of the DR were excluded from analysis due to local background labeling from rabies virus (see Weissbourd et al., 2014), including median raphe (MR), ventral tegmental area (VTA), substantia nigra pars compacta (SNc), interpeduncular nucleus (IPN), periaqueductal gray (PAG), locus coeruleus (LC) and several medulla nuclei. (C) Quantification of long-distance background infection in control groups. Y-axis presents total input cell numbers, and X-axis lists group names. n=9, 8, 4, 4, 4, 4. Error bars, SEM. (D) Representatives of GFP labeled ipsilateral input cells in PVH (at four coronal section planes from anterior to posterior; left panels) and in medulla nuclei and DCN (right panels) to OFC- and CeA-projecting DR serotonin neurons. Scale, 250 μm.
Figure S4. Summary of Optical Fiber Placement and Representative Traces for Fiber Photometry Recording, Related to Figure 4. Fiber photometry recording was performed on DRSert (A), DRSert→OFC (B), and DRSert→CeA (C) neurons expressing GCaMP6m, and on DRSert neurons expressing eGFP (D). (A1, B1, C1) Schematic DR diagram showing the end of optic fiber placement (oval) for each mouse. AQ, aqueduct. Note that we adjusted the fiber implant positions to maximize DRSert→OFC and DRSert→CeA neurons based on data from Figure 1. (A2, B2, C2) Mean responses of individual mice to lever press. Time 0 is aligned to lever press (vertical dashed line). Time window of −0.117s to −0.65s is in shaded grey (a), and time window of −0.65s to −0.13s is in shaded pink (b). (A3, B3, C3) Group data from all the trials of individual mice showing the quantification of the mean ΔF/F during “window a” compared to the mean ΔF/F during “window b”. A3, Signal of three mice from DRSert group was significantly increased (p<0.0001). B3, Signal of four mice from DRSert→OFC group was significantly increased (p<0.01), and one significantly decreased (p<0.05). C4, Signal of one DRSert→CeA group was significantly decreased (p<0.05). (two-tail paired t test). See Table S3 for detailed data. (A4, B4, C4) Example responses to sucrose consumption observed in one mouse from the DRSert (A4), DRSert→OFC (B4), or DRSert→CeA (C4) group. Time 0 is aligned to the initiation of sucrose water licking. (A5, B5, C5) Example of responses to foot-shock observed in one mouse each from the DRSert (A5), DRSert→OFC (B5), or DRSert→CeA (C5) group. Time 0 is aligned to the onset of foot-shock (yellow shadow covered the foot-shock duration). Solid and dotted lines represent mean ± SEM. (D1) Schematic of viral injection and optical fiber implantation of the eGFP control group. (D2) Confocal images of coronal sections showing fiber optic placement (dotted rectangle) and the expression of eGFP (green) in the DR. Vertical dashed lines represent the midline. Scale, 100 μm. (D3) Mean responses of individual mice (n=6, each with a different color) to sucrose consumption after lever press. Time 0 is aligned to lick initiation (vertical dashed line). (D4) Mean responses of individual mice from to electrical shock. Time 0 is aligned to onset of 1-sec electric shock delivery.
Figure S5. Functional Validation of hM3Dq and AAVretro-Cre in Serotonin Neurons. Related to Figure 5–7. (A) Sample traces showing that the addition of CNO to two acute brain slices from Sert-Cre mice transduced with AAV-FLExloxP-hM3Dq increased the firing rate of DR serotonin neurons (top trace, example from 7/9 slices) or transformed tonic firing to phasic firing (lower trace, example from 2/9 slices). (B) Left, grouped data from the experiments performed in A (two-tail paired t-test, **p<0.005, t=3.95, df=8. n=9, from 9 slices of 4 mice); right, grouped data from experiments performed in control Sert-Cre;Ai14 mice (two-tail paired t-test, n.s. not significant, t=0.050, df=6. n=7, from 6 slices of 2 mice). (C) Representative images of OFC, CeA, and hippocampus showing serotonin staining at terminals after AAVretro-Cre injection into either OFC or CeA of Tph2flox/flox mice. Scale, 50 μm. (D) Quantification of the fluorescence intensity in OFC and CeA slices, normalized to the staining intensity in hippocampal sections from each brain. n=3, 3, **p<0.01; ***p<0.001. (E) Quantification of OFC- (upper panel) and CeA- (lower panel) projecting serotonin neurons located in either DR or MR. AAVretro-Cre-2A-GFP was unilaterally injected into either OFC or CeA of Ai14 mice. tdTomato+/Tph2+ dual-labeled neurons in DR and MR were counted. n=3. Error bars, SEM.
Figure S6. DRSert→OFC fibers release serotonin and glutamate to modulate OFC network activity, related to Figures 1, 5–7. To examine how activation of DR fibers modulates neural activity in the OFC, we utilized mice expressing ChR2 in DRSert neurons for in vitro electrophysiology. Light-evoked local field potentials (LFPs) were recorded from OFC slices in an interface-recording chamber. Current source density (CSD) analysis was performed on LFP data to determine the location, magnitude, and direction of synaptic currents evoked by ChR2 activation in the OFC circuit in the absence or presence of cocktail of serotonin or ionotropic glutamate receptor blockers. (A) Upper panel: schematic of the OFC circuit including pyramidal neurons (Pyr), interneurons (Int), as well as the potential locations of serotonergic innervation by DR fibers (brown). Bottom panel: Experimental design of LFP recordings. A silicon multichannel probe was placed in the OFC (grey area outlined in the right schematic), while an optical fiber was situated medial to the recording array above layer II/III. Additionally, a bipolar tungsten electrode was placed in layer V to deliver electrical pulses (0.1 ms, 100 μA) to evoke synaptic responses and confirm the efficacy of drugs altering neurotransmission. (B) Representative LFP recording in response to a 10-ms/25-mW blue light pulse. (C) Representative CSD derived from the LFP recording. In response to the blue light pulse, a consistent response was observed involving prominent current sinks (net positive current flowing into the cell) in layer (L) II (orange star) and LV (green star), and a smaller source in LIII (net negative current flowing inside neurons; purple star). A 100-ms time window ~160 ms following the end of the light pulse (between dashed vertical lines) was used to quantify CSD and the effect of neurotransmitter blockers in D−G below. This window was chosen to avoid potential contamination from the light artifact, while ensuring that we could measure significant current sinks and sources (STAR Methods). To confirm that the responses we quantified were direct rather than via polysynaptic circuit activation, we applied 1 μM tetrodotoxin to block voltagegated Na+ channels with, and 100 μM 4-aminopyridine to partially block K+ channels (n=6 slices), and found that the responses persisted. (D) Representative responses of L2 sink, L3 source, and L5 sink in the absence (baseline [BL], solid trace) or presence (drug, dashed trace) of cocktail of serotonin receptor (HTR) antagonists GR 113808 (10 μM), Metergoline (10 μM), and Ondansetron (1 **M). (E) Representative responses of L2 sink, L3 source, and L5 sink in presence and absence of ionotropic glutamate receptor (iGluR) antagonists 6,7-dinitroquinoxaline-2,3-dione (DNQX, 25 μM) and 3-((+-)-2-carboxypiperazin-4-yl)-propyl-1- phosphonic acid (CPP, 1 μM). (F) Quantification of the CSD at baseline (BL, ChR2 stimulation alone) and in the presence of HTR antagonists (from 4 mice). While the L2 sink and L3 source were not affected, the L5 sink was significantly reduced (less negative, p = 0.008), suggesting serotonin release from DR fibers in the OFC contributes to activation of L5 OFC neurons. (G) Same as F, but comparing CSDs and in baseline and the presence of iGluR antagonists (from 5 mice). The L2 Sink shows a weak trend of a reduction (p = 0.073), but both the L3 Source and L5 Sink are significantly reduced in response to iGluR antagonists (p = 0.003 and p = 0.015 respectively). Given that ChR2-evoked responses were reduced both by serotonin receptor and iGluR antagonists, our data suggest that DR neurons co-release serotonin and glutamate in the OFC.
Figure S7. Functional Analysis of CeA- and OFC-projecting DR Serotonin Neurons in Fear Learning and Memory, Related to Figure 6, 7. (A) Chemogenetic activation of DRSert→OFC neurons does not affect freezing time to conditioned tone (CS+) during fear conditioning (one-way ANOVA, F[2, 31] = 1.28. n= 9, 11, 14). (B) Chemogenetic activation of DRSert→OFC neurons does not affect freezing time to CS+ during 1-day recall (one-way ANOVA, F[2, 31] = 1.195. n= 9, 11, 14). (C) Chemogenetic activation of DRSert→CeA neurons increases freezing time to CS+ during fear conditioning. ***p<0.001, ****p<0.0001 (one-way ANOVA followed by multiple t-tests; F[2, 29] = 18.0; t= 5.55, 4.70. n= 10, 11, 12). (D) Chemogenetic activation of DRSert→CeA neurons increases freezing time to CS+ during 1-day recall. **p<0.01, ***p<0.001 (one-way ANOVA followed by multiple t-tests, F[2, 29] = 9.61; t=4.58, 3.429. n= 10, 11, 12). (E) Conditionally knocking out Tph2 from DRSert→OFC neurons does not affect freezing time to CS+ during fear conditioning (two-tail unpaired t-test; t=1.47, df=17. n=9, 10). (F) Conditionally knocking out Tph2 from DRSert→OFC neurons does not affect freezing time to CS+ during 1-day recall (two-tail unpaired t-test; t=0.0624, df=17. n=9, 10). (G) Conditionally knocking out Tph2 from DRSert→CeA neurons does not affect freezing time to CS+ during fear conditioning (two-tail unpaired t-test; t=0.990, df=14. n=9, 10). (H) Conditionally knocking out Tph2 from DRSert→CeA neurons does not affect freezing time to CS+ during 1-day recall (two-tail unpaired t-test; t=0.685, df=14. n=9, 10). (I) Activation of DRSert→OFC neurons does not affect the immobility time in forced-swim test on day-1 training session (one-way ANOVA, F[2, 31] = 1.44, n= 9, 11, 14).
Movie S1: 3D Representation of DR Serotonin Neurons’ Spatial Organization with Respect to Axonal Projections and Vglut3 Co-Expression, Related to Figure 1.
Movie S2: 3D Rendering of DRSert→OFC and DRSert→CeA Axon Collateralization Patterns, Related to Figure 2B–D. Projecting rostrally passing through the lateral hypothalamus, the DRSert→OFC axons split into a medial branch that travels dorsally to medial frontal cortex; the rest fans out laterally to the cortex, with some projecting posteriorly to the ENT and others innervating the PIR and insular cortex anteriorly, eventually combining with the medial branch to jointly innervate other prefrontal areas and the OB. By contrast, the DRSert→CeA axons intensely innervated most amygdala nuclei and several hypothalamic nuclei, notably the ventrolateral preoptic nucleus, PVH, and the ventral BNST. Consistent with our retrograde tracing experiments, DRSert→OFC axons did not innervate most forebrain subcortical regions, whereas DRSert→CeA axons rarely invaded any cortical areas.
Movie S3: Fly-through Coronal Density Maps of DRSert→OFC (left) and DRSert→CeA (middle) Projections, and A P-value Map (right), Related to Figure 2F.
Table S1: Allen Brain Atlas IDs and Their Corresponding Names as Identified by the 2017 Common Coordinate Framework, Related to Figure 2. Regions were selected prior to analysis such that areas defined by individual layers (e.g., cortical layers I−VI), cell identity, and anatomical cardinal directions are collapsed into their parent region. Individual normalized regional densities for each brain are aligned to the heat maps from Figure 2G.
Table S2: Quantification of cTRIO: Number of Cells in Region (Proportion of Total), Related to Figure 3. For each sample, the raw cell count for each brain region is displayed in the left column, the fraction of total input for that brain region is displayed in the right column, and the total number of inputs is displayed at the bottom. Medulla nuclei were first calculated individually and then all collapsed into other medulla nuclei expect for the nucleus of the solitary tract (NST).
Table S3: Details of Statistical Analyses of Fiber Photometry Recording Data. Related to Figure 4 and Figure S4.
Table S4: Summary of Number of Cells Labeled by Retrograde Tracing. Related to Figure 1, Figure 5.