

Abstract
The accumulation of therapeutic and imaging agents at sites of interest is critical to their efficacy. Similarly, off-target effects (especially toxicity) are a major liability for these entities. For this reason, the use of delivery vehicles to improve the distribution characteristics of bio-active agents has become ubiquitous in the field. However, the majority of traditionally employed, cargo-bearing platforms rely on passive accumulation. Even in cases where “targeting” functionalities are used, the agents must first reach the site in order for the ligand–receptor interaction to occur. The next stage of vehicle development is the use of “recruited” entities, which respond to biological signals produced in the tissues to be targeted, resulting in improved specificities. Recently, many advances have been made in the utilization of cells as delivery agents. They are biocompatible, exhibit excellent circulation lifetimes and tissue penetration capabilities, and respond to chemotactic signals. In this Minireview, we will explore various cell types, modifications, and applications where cell-based delivery agents are used.
Keywords: biological activity, bioorganic chemistry, cancer, drug delivery, nanotechnology
Graphical Abstract
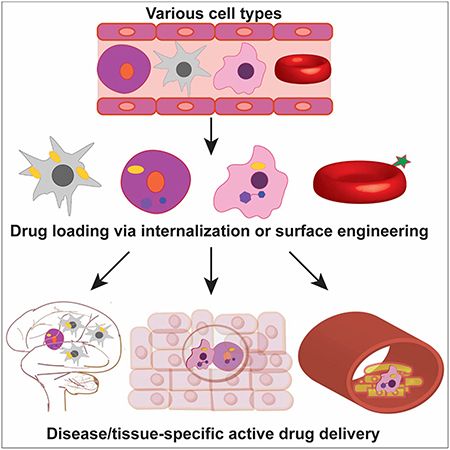
Introduction
A variety of drug-delivery systems are in development and in use clinically as aids for the transportation of therapeutic entities to diseased sites within the human body. The administration of biologic and/or small molecule drugs via encapsulation or conjugation to a delivery vehicle can result in improved therapeutic characteristics by increasing the drug quantity transported to and retained at the targeted site. Cargo-bearing vehicles based on nanoparticles,[1] liposomes,[2] polymeric micelles,[3] and hydrogels[4] have been employed to improve the net efficacy of drugs delivered. The circulation half-lives and toxicity profiles of these entities can vary widely, depending on their composition and cargo. However, these traditional strategies generally rely upon passive targeting of disease areas,[5] and often face various systemic and disease-specific biological barriers to delivery. For example, access to hypoxic areas, typically located within tumors,[6] and crossing of the blood–brain barrier (BBB) to reach brain cells and tissue[7] are both significant challenges. Even those entities that are “targeted” must first reach the site of interest in order for the targeting ligand (e.g. antibody, aptamer, peptide, or small molecule) to bind its receptor.[8]
While nanoparticles represent one of the most widely used vehicles, a recent statistical analysis of the literature revealed that only 0.7% (median) of administered doses reach solid tumors, indicating “a delivery problem.”[9] Independently transported functionalized nanoparticles (NPs) can accumulate in non-desired locations, leading to unwanted reactivity and toxicity, or be cleared from circulation without reaching the tissue of interest.[10] Depending on their size, systemically administered agents are generally cleared via the renal (kidney) or mononuclear phagocytic system (MPS, including the liver and spleen). These issues are generally the result of nanoparticles’ reliance on passive accumulation to reach their site of action, referred to as enhanced permeability and retention (EPR). Cell based delivery systems can mitigate these shortcomings via active recruitment of the cellular carrier to the diseased site. Hence, recruited (or “actively targeted”) vehicles are desirable for improved disease-specific treatment paradigms, and represent the next generation in drug delivery system development.
Cell-based therapeutic strategies have been used in the clinic and are being developed for the treatment of a variety of ailments, including Crohn’s and other inflammatory diseases,[11] stroke,[12] cancer,[13] and osteoarthritis.[14] Taking these approaches as inspiration, drug delivery systems where circulatory cells are used as biological vehicles are emerging as active therapeutic carriers. Cellular delivery systems have several distinct advantages over others in terms of circulation time, inherent biocompatibility, and excellent in vivo distribution characteristics. But perhaps most importantly, cells are actively recruited and able to accumulate in particular tissues and at targets of interest.
Cellular delivery vehicles respond to chemotactic markers and cytokines present in disease microenvironments, facilitating the design of optimal cell vehicle-drug combinations.[15] As a result, therapeutics carried by cells have a greater propensity to accumulate at the targeted tissues compared to those using no delivery or passively targeting vehicles. With this increased specificity, the biological efficacy of the cargo is enhanced, and there is a reduction in off-target toxicity. Various cell types, including erythrocytes (red blood cells, or RBCs),[16] stem cells (SCs),[17] and phagocytic leukocytes (monocytes and macrophages),[18] have been used for the specific delivery of genes,[19] nanoparticles,[20] proteins,[21] polymers,[22] and small molecules.[18] Similarly diverse are the applications they are used in, which include the imaging and/or treatment of cancer,[23] neurodegenerative,[21] and cardiovascular diseases[20] (Table 1). Some cellular delivery systems have also been fine-tuned for the controlled release of therapeutics via external stimulus (e.g. light),[24] or environmental cues (e.g. pH).[17] The unique properties of cell-based delivery systems have made them a focus of current research toward the development of novel drug delivery systems, with some already being tested in clinical trials.[25–27]
Table 1.
Cellular vehicles and their applications.
| Cell types | Loading methods | Target tissues | Cargo |
|---|---|---|---|
| leukocytes | phagocytosis, surface deposition/modification | plaques, various tumors | genes, nanoparticles, small molecules |
| RBCs | encapsulation, surface deposition/modification | lungs, various tumors | enzymes, nanoparticles, small molecules |
| SCs | internalization, surface deposition/modification | brain, various tumors | genes, nanoparticles, small molecules |
In this Minireview, we will discuss cell-based vehicles, with a particular focus on modification methods as applied to different cell types. In addition, we will describe their applications, including cargos and disease targets. We will also consider future work and remaining challenges toward the broad application of these actively targeted platforms.
Cell Types Used for Delivery
After briefly introducing the main cell types being used and studied for delivery applications (Figure 1), we will discuss the methods utilized to modify these cells, along with cargo types, and conditions where they are being employed.
Figure 1.

Cell types employed as delivery vehicles and locations where they are recruited/used to deliver agents to.
Leukocytes (monocytes/macrophages)
Leukocytes represent the largest category of cells used as delivery vehicles. Also referred to as white blood cells, they are cells of the innate immune system that originate from bone marrow, and are involved in host defense. Monocytes are a class of leukocytes that circulate in the bloodstream and migrate to sites of infection or inflammation following chemotactic signals from cytokines. Once recruited, they differentiate into macrophages, which fulfill niche-specific functions.[28] Apart from their roles in the phagocytic engulfment of pathogens and clearance of apoptotic cell debris, macrophages are also involved in maintenance of homeostasis and tissue repair.[29]
Both monocytes and macrophages inherently home to hypoxic, ischemic, and necrotic environments, which are often associated with disease. The mechanisms for hypoxia-mediated accumulation of macrophages at tumors[30] and ischemic areas of atherosclerotic plaques[31] have been well studied.[32,33] Monocytes and macrophages are often recruited to tumor sites via directed chemotactic migrations induced by signaling proteins. Many cancer types secrete chemo-attractants, such as macrophage colony stimulating factor (referred to as M-CSF or CSF-1)[34] and chemokine (C–C motif) ligand 2 (CCL2).[35] These homing mechanisms have been taken advantage of for the delivery of small molecule drugs,[18] biological entities,[36] imaging agents,[37] and supramolecular constructs (NPs, polymers, etc.),[23] among others.
Stem cells (mesenchymal and neural)
Stem cells are pluripotent, plastic cells that can be differentiated in an inducible, tissue-specific manner.[38] As such, they have been broadly utilized in regenerative medicine.[39] Two specific types of SCs are most prevalently used in cell-based delivery—mesenchymal stem cells (MSCs) and neural stem cells (NSCs). MSCs, which are also referred to as multipotent stem cells, are present in almost all types of tissue. They can differentiate into a variety of cell types including adipocytes, osteocytes, and muscle cells.[40] NSCs are self-regenerating cells of the central nervous system that can differentiate into neurons and brain cells.[41]
Both types of stem cells have the ability to migrate to specific tissues, tracking various cytokines that act as signals.[42–46] For example, in liver injury, MSCs home toward the site via chemo-attraction mediated by stromal cell-derived factor 1 (SDF-1) and hepatocyte growth factor (HGF).[45] MSCs home to cancers via interactions with cytokines including interleukins IL-6 and IL-1b, and growth factors such as transforming factor-b1 (TGF-b1) and SDF-1, secreted by the tumor stroma.[46] NSCs also show tumor-tropic behavior on account of their chemo-taxis toward cytokines including IL-6 and VEGF, among others.[43] These characteristics have made SCs attractive carriers for specific delivery to tumors and CNS tissues. They are especially important in crossing the blood–brain barrier to access brain tissues, an application of which has progressed to clinical trials[25] (described in further detail below).
Red blood cells
Red blood cells (RBCs) are the most abundant non-nuclear blood cell type in vertebrates, and have a circulation lifetime of approximately four months. RBCs can reach most internal tissues of physiological systems; their vascular access and membrane permeation abilities make them ideal carriers of drug molecules in sustained release treatments.[16] RBCs bearing particles on their cell surfaces have been reported to shield their cargo from hepatic and spleen filtration,[47] and have been used as nanoparticle vehicles.[48] While RBCs are nonhagocytic cells, methods have been developed for their internalization of cargo, facilitating another means of delivery.[49] Red blood cell-based agents have also been used clinically[26,27] (described in further detail below).
Cellular Modifications and Cargo Loading for Delivery Applications
There are several critical factors in the selection of a cell type for use as a delivery vehicle. These include compatibility of the carrier with the target site, and desired cargo loading strategy and release mechanisms. In this section, we provide an overview of cellular modification methods, along with specific examples of payloads and cell types employed (Figure 2).
Figure 2.

Non-covalent cellular modifications for cargo delivery. (Above) Red blood cells (red), leukocytes (pink) and stem cells (purple) have been noncovalently modified at the cell surface to display various agent types shown. (Below) Stem cells and leukocytes have the innate ability to internalize cargo shown. Specialized methods must be used for RBCs (not shown) to uptake therapeutic entities.
Phagocytosis, internalization, and transfection
The two general means of converting cells into delivery agents are via internalization of cargo (via various methods) or exterior modification of cells to display therapeutic entities. Phagocytosis (engulfment by the carrier cell) is perhaps the most straight-forward method of loading cargo into cells. Phagocytosis is dependent both on the size[50] and geometry[51] of the entities being engulfed. Different strategies for efficient loading via this method have been explored, using nanoparticles,[52] and polymeric micelles[53] and bubbles,[22] in addition to the encasement of small molecules.[18,23] While examples exist where cells are used as delivery intermediates in situ, including uptake of single-walled carbon nanotubes by circulating monocytes,[54] and nano-therapeutic engulfment and release by tumor-associated macrophages,[55] our focus here is on systems that include cells as part of the dosing regimen.
The phagocytic loading of monocytes, macrophages, and stem cells has been extensively used to deliver nanoparticles and other entities (Figure 2). In what is commonly referred to as a “Trojan horse” strategy,[52] the imaging and/or therapeutic entity is encased within a biocompatible covering (in this case, a cell). Monocytes carrying gold nanoparticles have been used to image atherosclerosis using computed tomography (CT), with no effect on the viability or cytokine release by the cells.[20] Similarly, magnetic resonance imaging (MRI) has been employed to visualize macrophages bearing silica-coated, magnetic NPs in acute inflammation[56] and cancer[53] applications. However, the most widespread application of cellular vehicles is disease therapy.
Various gold-based agents have been internalized and used in photodynamic therapies, including MSCs with hollow gold nanoparticles,[57] macrophages with nanoshells,[52,58] and NSCs with gold nanorods.[57,59,60] All of these entities have been shown to target tumor cells without significant undesired toxicity, and where assessed, were more effective than analogous agents utilizing only the EPR effect. In a multi-modality treatment, monocytes bearing super-paramagnetic iron oxide nano-particles, photosensitizers, and oxygen-loaded polymer bubbles were used to treat hypoxic areas of tumors.[61]
Cells have also been used to deliver chemotherapeutics, including macrophages directly loaded with doxorubicin (DOX)[18] or paclitaxel (PAX),[23] or via internalization of nanoparticles or other entities bearing small molecules.[23,53,62] In one example, following loading of 100 mg of DOX into 1 million macrophages, >80% of the cells remained viable up to 72 h; cell-based delivery resulted in increased rates of survival in cancer models versus free DOX treatment.[18] Similarly, liposomal-DOX delivered via macrophages out-performed DOX-liposomes alone.[53]
While few examples exist, responsive systems have been developed for the specific release of drugs. Echogenic polymer bubbles and DOX-loaded vesicles susceptible to focused ultra-sound liberation, co-delivered in macrophages, resulted in substantial tumor uptake, while free NP-DOX largely accumulated in the liver; the cell-based agents penetrated 150 mm from the nearest blood vessel, however NPs were limited to 10–15 mm.[22] Photo-release of drugs has been used with red, farred, and near-IR light in erythrocytes,[24,63] which can uptake entities as described in further detail below. Iron oxide nano-particles phagocytosed by macrophages can also assist in “directing” cellular delivery via magnetic resonance imaging[64] or electromagnetic actuation.[65]
Cargo-bearing cells loaded in this manner have been utilized to cross the blood–brain-barrier. In the delivery of drug-laden nanoparticles for the treatment of glioma, macrophage viability was not affected at doxorubicin concentrations up to 25 μg mL−1 and drug-loaded nanoparticles exhibited better tumor targeting efficiency and penetration.[62] For a nano-formulated version of the therapeutic anti-oxidant enzyme catalase for Parkinson’s disease, the unidirectional influx rate of monocyte-loaded nanozyme was 0.026 versus 0.014 μLg−1 x min for free nanozyme, indicating improved BBB penetration, in addition to superior half-life (3.3 versus 2.5 h).[21]
In some instances, phagocytic cell-based delivery systems have been compared to one another. In a direct assessment, doxorubicin- or paclitaxel-nanoparticle conjugates phagocytosed by macrophages were found to have greater efficacy than the small molecules internalized alone, or NP- or free-drugs independently dosed.[23] NSCs used to deliver platinumloaded silica nanoparticles, either loaded into or conjugated to cells were shown to result in higher levels of Pt in ovarian tumors compared to free drug or nanoparticle-drug conjugates, with substantially deeper tissue penetration.[66] Furthermore, no evidence of localization to normal tissue was observed. Choi and co-workers similarly showed that gold NPs engulfed by macrophages reach tumor sites more efficiently than NPs administered using other methods that access tumor interiors via leaky vasculature-reliant EPR.[52] In the discussion section below, we describe the necessity to perform additional comparative analyses for the field to move forward.
Since RBCs are inherently a non-phagocytic cell type, specialized strategies must be employed for encapsulation of cargo. In a hypotonic buffer of osmolarity less than 160 m-osm/L, RBCs undergo reversible membrane expansion, creating pores large enough for macromolecules to permeate through.[49] This passive diffusion-mediated drug loading and subsequent release in isotonic environments is referred to as hypotonic dialysis (HD). HD has been employed to load therapeutic enzymes,[27,67] nanoparticles,[48] and drugs.[63,68] These same (“blood-banking”) conditions are also used for blood storage and transfusion. An apparatus, termed a “Red Cell Loader” (Sorin Group, Italy) has been developed to automate the process, and used to encapsulate a variety of drugs.[68] RBCs loaded with therapeutic enzymes and small molecule drugs for treatment of leukemia[27] and neurodegenerative diseases,[26] respectively, are in clinical trials. RBCs bearing l-asparaginase (GRASPA) are in Phase II for the treatment of Philadelphia chromosome-negative acute lymphoblastic leukemia, and have been shown to result in a 70% complete remission rate with limited toxicity.[27] Similarly, intra-erythrocyte administration of Dexamethasone (EryDex) was well-tolerated, and alleviated neurological symptoms of ataxia teleangiectasia, a rare neurodegenerative disease, in patients.[26] While most (including clinical) studies have utilized the HD method of cell loading, others exist, including chloropromazine (cpz) treatment and liposomal fusion, which have been shown to be superior to HD in terms of loading efficiency, induction of hemolysis, and cellular deformation post-loading.[69]
As living, actively targeted vehicles, cells have been used as generators of biological entities at targeted locations (referred to here as gene delivery). Because nucleic acid-based material cannot permeate the cell membrane and DNA cannot result in protein production independently, specialized methods for transfection are typically employed. These often require lentior adeno-viruses, although new techniques are being developed.[70] Stem cells and macrophages are the two cell types that have been most commonly used in these approaches.
Macrophages, which home to atherosclerotic plaques, have been used to generate Apolipoprotein E (ApoE), in order to reduce the size and presence of lesions.[71] They have also been used to kill cancer cells via production of the therapeutic human protein cytochrome P4502B6 under hypoxic conditions.[19] MSCs have been used to express anti-proliferative, an giogenesis-inhibiting matrix metalloproteinase C-terminal hemopexin-like domain fragments (PEX)[70] and immunoproapoptotic molecules[72,73] in a site-specific manner, while retaining cellular viability. Significant toxicities were not observed and improved anti-cancer effects were shown versus purified proteins alone and delivered via other means. Tumor-tropic NSCs have been genetically modified to secrete the anti-HER2 immunoglobulin Herceptin (trastuzumab) to inhibit the growth of HER2-expressing cancers.[74] The antibody produced in situ was shown to be functionally equivalent to the standard drug. While independently injected trastuzumab distribution was heterogeneous and localized near tumor vasculature, the NSC-produced antibody was distributed throughout the tumor.
Cells expressing an enzyme that converts a pro-drug into its active form for treatment of glioma, have recently undergone a first-in-human study.[25,75] The NSCs generated cytosine deaminase to produce 5-fluorouracil from 5-fluorocytosine in the brain and were able to migrate to distant tumor sites. No dose-dependent toxicity or immunologic response was observed. Also prevalent in the development of this strategy toward cancer treatment is the use of cells to deliver several types of oncoviruses, including macrophages used to deliver/produce adenoviruses in prostate cancers,[36,76] and MSCs with herpes simplex virus for breast cancer[77] and metastases,[78] measles virus for ovarian cancer,[79] and adenovirus for pancreatic cancer,[80] all of which have been shown to significantly affect tumor burden with few side-effects.
Covalent and Non-Covalent Cell Surface Modifications
Surface modification (also referred to as surface engineering) of carrier cells is emerging as a more frequently employed cargo loading method due to various factors (Table 2). By comparison with cargo internalization methods, it facilitates greater control over cellular loading and cargo release. There is also a diminished likelihood of the intra-cellular environment affecting and/or trapping the cargo itself. Surface modifications are amenable for use with any cell type, including non-phagocytic cells, which otherwise must undergo rigorous and sometimes toxic methods (e.g. electroporation) for membrane permeabilization to load cargo.[81] Modifications where an entity is adhered to cell surface physically (via electrostatic forces or lithographic deposition) are here referred to as non-covalent surface modifications or adherence (Figure 2). While these methods are facile, there is little control over payload release. On the other hand, when cargo is covalently conjugated to the cellular vehicle, it is robustly attached,[82] but these methods may require more involved chemical manipulation, including design of linkers for therapeutic release. While few direct comparisons have been made, surface functionalization does not generally result in dramatic changes to the modified cells, leaving their intrinsic properties, including homing capability, largely intact.
Table 2.
Cell surface modification methods.
| Method | Modified Sites | Advantages | Disadvantages |
|---|---|---|---|
| cellular hitchhiking | non-specific | mild attachment conditions | weak adherence |
| cellular backpacks | local patches on cell surface | more stable adherence | complexity of deposition methods |
| biotin-avidin | variable | strong linkage, modular | large protein conjugation |
| native amino acids | thiols, primary amines | single step modification | internalization by cells, synthetic cargo modification |
| click chemistry | incorporated azido-sugars | chemically orthogonal | long incubation times, multi-step |
Non-covalent surface modifications
Among non-covalent surface engineering strategies, one straight-forward method is via “cellular hitchhiking”—particle adhesion to cell surfaces via electrostatic, van der Waals, hydrogen bonding, and hydrophobic interactions.[83] Cationic entities can be immobilized on the hydrophobic cell surfaces mainly through interactions between these opposite charges.[84] Other weaker interactions, like van der Waals and hydrogen bonding assist this adherence, and displacement of water molecules at the interface gives an entropic advantage for NP assembly and stability at the surface.[85] Spherical polystyrene NPs adhered to red blood cells via this method have not been shown to affect RBC bio distribution, avoiding splenic and hepatic filtration.[47] At the same time, the RBC-nanoparticles showed a threefold increase in blood circulation time, and sevenfold increased accumulation in the lungs versus free nanoparticles. Polymeric nanoparticles similarly attached possessed extended circulatory lifetimes of almost 24 h compared to non-adhered NPs, greater than 95% of which were cleared in less than 30 min. As long as the particles remained attached to the cells, they stayed in circulation, however sheer forces and cell-cell interactions result in passive removal followed by clearance via the liver and spleen.[82,83]
In another non-covalent surface modification method, the cargo is confined to a local vesicle (a “cellular backpack”) on the cell membrane.[86] These backpacks can be fabricated using layer-by-layer spray deposition of films consisting of alternating hydrogen bond donor-acceptor pairs, for example, PLGA/FITC-BSA [(PLGA =polylactic-co-glycolic acid), BSA=bovine serum albumin]. Particles delivered in this manner were shown to accumulate in inflamed tissues, while avoiding clearance organs; free particles showed threefold increased levels in the liver and spleen, and were almost undetectable in areas of inflammation.[86] Similarly, backpacks bearing the antioxidant enzyme catalase on macrophages were shown to facilitate blood brain barrier crossing in targeting brain inflammation in vivo.[87] While the presence of backpacks slowed cellular trafficking across the BBB, neither catalase alone nor unconjugated backpacks reached the brain. Cellular backpacks have also been employed by Rubner and co-workers to load monocytes with doxorubicin, which were viable for over 72 h, in order to target cancer.[88] Cells can also be non-covalently modified via small-molecule lipid association. For example, macrophages have been labeled with DiR (1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyanine iodide) for tracking via fluorescence-mediated tomography (FMT) in the visualization and quantification of inflammatory responses in vivo.[37] Cell viability and function (phagocytosis, nitric oxide production, and adherence) were not affected.
Use of biotin–streptavidin linkage
Several different methods have been used to modify cell surfaces in a covalent manner for the conjugation of traceable and/or therapeutic entities for imaging and delivery applications (Figure 3). Biotin-streptavidin-based cargo loading methods take advantage of one of the strongest non-covalent interactions known in biology,[89] and are the most commonly employed. Biotin is a water-soluble, small biomolecule with high affinity for streptavidin-related proteins (dissociation constant (Kd) ≈10−15M).[90] It can be conjugated to cell surfaces via various chemical reactions, resulting in display of streptavidin-linked entities following association. This method is biocompatible in the sense that it does not appear to affect cell viability, tissue tropism, or other characteristics. N-hydroxysuccinimide (NHS)-ester-biotin is a common, commercially available biotinylation reagent that can introduce biotin on membranes containing primary amines by forming a stable amide bond within hours at room temperature.[91] This surface functionalization has found varied applications from cell targeting[92] to loading drugs or therapeutic particles.[93]
Figure 3.

Strategies for covalent cell surface modifications. Click chemistry methods involve coupling of metabolically incorporated azides to phosphines or alkynes for cargo delivery. NHS esters and maleimides can be used for conjugations to amines and thiols, respectively. Biotin can be linked to the cell membrane via several means, and then associate with streptavidin-related proteins linked to chemical moieties.
Streptavidin-like proteins conjugated to biotinylated NPs on NSCs and MSCs have been shown to facilitate tumor-selective distribution in intracranial[94] and tumor tropic[93] drug delivery, respectively. In both cases, cell viability was not affected, and NSC-NP conjugates studied in the glioma model had improved distribution and retention versus NPs alone.[94] RBCs can also be conveniently modified at the surface via biotin–streptavidin conjugation. They have been modified in this manner with insulin and IgG to facilitate targeting for directed delivery of albumin to endothelial cells,[95] and plasminogen-activating NPs as prophylactic measures for fibrinolysis, which were shown to persist in the bloodstream for tenfold longer than non-conjugated entities.[92] In a stimuli-responsive example by Berlin et al., cell surfaces were biotinylated via acid-labile hydrazine linkages, and connected to pH-responsive nanoparticles through an avidin protein.[17] NSC viability and tumor tropism were not affected, and the efficacy of docetaxel-NPs was improved when incorporated with the cell-based vehicle. It has been noted that in addition to streptavidin-biotin interactions, contributions from streptavidin-integrin and passive adsorption of nanoparticles to the cell surface may occur.[94] However, non-specifically adsorbed NPs have not been shown to be retained following cellular migration.[17]
Direct conjugation to native amino acids
As an alternative to the display of a large protein on the cell surface, as in biotin–streptavidin methods (avidin is 66–69 kDa), direct linking of cargo to membranes has been employed. In limited cases, endogenous cell surface protein–ligand combinations are used. For example, monoclonal anti bodies for CD73 and CD90 have been used to link doxorubicin-anchored silica nano-rattles to MSCs for tumor tropic migration.[96] Cell-based delivery here resulted in better tumor distribution and retention, and improved induction of apoptosis versus free doxorubicin and non-attached nano-rattles. MSC viability was 85% or greater up to 50 mg mL@1 drug. Strategies to modify exposed amines and thiols on cell membranes are more prevalent. N-hydroxysuccinimide (NHS) ester-based cross-linkers are the most frequently employed to link therapeutic payloads to accessible amines in this approach. In proof of concept work, Rossi and co-workers attached polyglcerol-NHS moieties to the surfaces of RBCs, showing that the modification resulted in significant reduction in Rh antigen immunogenicity,[97] with good cell viability up to 4 mm. Maleimide based cross-linkers have been utilized to conjugate bio-active molecules to accessible thiols present on carrier cell surfaces. Cytokines (IL-15Rα and IL-21) bearing maleimide-functionalized polymeric NPs have been conjugated to stem cells, and were found to be more stable than particles adsorbed to the cell surface.[98] The cell-conjugated NP-drugs were almost sixfold more effective than free drug, and the NPs did not result in an immune reaction or alter the tumor-tropic movements of the cells. In another case, covalent surface modifications of RBCs were shown to have no detrimental effects on their structure, function, and in vivo circulation and survival, which were akin to non-modified cells.[99]
Other cellular conjugation strategies
Additional chemical reactions are also amenable for use as cell modification methods, although these have not yet been broadly employed in the generation of delivery vehicles. While copper-catalyzed Huisgen cycloadditions between azides and alkynes can be used to label cells,[100] they often result in residual toxicity. This can be mitigated via use of tris(hydroxypropyltriazolyl)methylamine (THPTA), yielding improved cell viability after labelling. Other click-type reactions that use metabolically installed azides to link functionalities to cells include the Staudinger ligation with phosphines[101] and strain-promoted copper-free cycloadditions[102] developed by Bertozzi and coworkers, neither of which result in appreciable cellular toxicity. As an example, paclitaxel-loaded diarylcyclooctyne-functionalized nanoparticles linked to glyco-modified MSCs have been shown to result in significant tumor growth inhibition in an orthotopic metastatic ovarian cancer model.[103] The viability of glyco-engineered cells was unaffected up to a week, and the system resulted in significant reduction in tumor size and improved survival versus nanoparticles alone. Tetrazines and alkenes can also be ligated together in a biorthogonal manner,[104] which can be applied to cells by appending either an alkene[105] or tetrazine[106] moiety prior to conjugation.
Summary and Future Perspectives
Cellular vehicles have been used for the delivery of various nanoparticles and molecules via direct attachment or internalization of cargo. These systems have several advantages over their non-cellular counterparts in terms of circulation, site-specific recruitment and delivery, and bio-compatibility. However, the use of a biological system as a vehicle is also accompanied by several complexities. Specialized laboratory equipment and protocols are required, and unlike super-molecular entities (e.g. nanoparticles), cells cannot simply be “pulled off of the shelf.” Furthermore, issues of consistency and generalizability have not yet been addressed. Likely due to the platform’s nascence, quantitative studies involving the direct comparison of delivery efficiency, specificity, bio-compatibility, and other characteristics are generally lacking. Experiments assessing the impact of cell type and modification method would enrich the existing knowledge in the field, and contribute towards optimization of cell-based delivery vehicles. For the successful translation of these approaches toward use as clinically viable tools, several additional issues need to be overcome.
Except for red blood cells, vector cells are generally scarce on account of the difficulty in their harvest and low natural abundance. Following FDA approval of cellular immunotherapy for cancer treatment,[107] there has been much industrial investment in the commercialization of cell-based products, which should lead to developments in methods for producing and harvesting these cells at higher yields.
A general concern for using cell-based vehicles is potential immunogenicity. Most studies described here do not evaluate the immune responses that may be generated by the cells themselves, and it is likely that internalized cargo will result in different profiles from those displayed on the surface. Additional modifications, including use of PEG, may be required. PEGylation of RBCs[99] and pancreatic islets[108] has been shown to be compatible with cells, non-toxic in vivo, and can temper immunogenicity.
Another limitation associated with cell-based delivery vehicles is that many current loading strategies do not facilitate control over cargo release. Cleavable linkers between therapeutics and carriers can facilitate specific delivery to target tissues, leading to even greater treatment efficacy, and decreased risk of off-target effects. A potential strategy for development of such a system could involve the use of a moiety between the cell and therapeutic, labile under conditions present in the tissue or disease microenvironment, or a cellular “kill switch” resulting in release of payload from within the cell interior. Polymers and linkers that respond to subtle changes in pH and temperature[109,110] and undergo degradation-based controlled release[111] are being developed for drug delivery. Similarly, disulfide bonds have also been used as linkers,[112] as have peptides that are cleaved by enzymes present in specific disease and/or tissue microenvironments.
With increasing research and clinical applications of both cellular therapies[12–14] and vehicles,[25–27] the foundation has been laid for further development of cell-based delivery agents. It is likely that any of the issues described here will be addressed in the near future. The field of cell-based delivery is one that continues to expand, with new modification strategies, cargo types, and health applications being explored. This platform has the potential to improve efficacy and reduce undesirable effects of many existing therapies, and lead to a new generation of therapeutics and imaging agents.
Acknowledgements
The authors wish to thank Hui-Hsien Lin for helpful discussions, and Javier A. Mas and L. D. Sujeewa Sampath for assistance in generating illustrations. B.P.J. and J.H. were supported by NIH R21 EB023369.
Biographies
Bishnu Prasad Joshi is a native of Nepal and received his B.S. and M.S. degrees in chemistry from Sri Sathya Sai Institute of Higher Learning in India. Following his graduation in 2015, he joined the Ph.D. program in chemistry at the University of Massachusetts Amherst. He is currently working on the development of macrophages as cell-based imaging and drug delivery tools, and the concomitant effects of functionalization on macrophage biology.

Joseph M. Hardie received his B.S. degree in chemistry from the College of New Jersey in 2013. Afterwards, he started his Ph.D. studies in chemistry at the University of Massachusetts Amherst under the joint supervision of Professors Michelle E. Farkas and Vincent M. Rotello. His research currently focuses on immunomodulation of macrophage behavior using nanomaterials for disease therapy.

Michelle E. Farkas received her B.A. in chemistry from Wellesley College in 2001, her Ph.D. in chemistry from the California Institute of Technology (under supervision of Peter B. Dervan) in 2010, and performed postdoctoral work in the College of Chemistry at the University of California, Berkeley (under supervision of Matthew B. Francis). In 2013, she began her independent career at the University of Massachusetts Amherst, where her group is interested in developing new tools for imaging, studying, and treating cancer.

Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Parveen S, Misra R, Sahoo SK, Nanomed. Nanotechnol. Biol. Med 2012, 8, 147–166. [DOI] [PubMed] [Google Scholar]
- [2].Allen TM, Cullis PR, Adv. Drug Delivery Rev 2013, 65, 36–48. [DOI] [PubMed] [Google Scholar]
- [3].Matsumura Y, Kataoka K, Cancer Sci 2009, 100, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ashley GW, Henise J, Reid R, Santi DV, Proc. Natl. Acad. Sci. USA 2013, 110, 2318–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brannon-Peppas L, Blanchette JO, Adv. Drug Delivery Rev 2004, 56, 1649–1659. [DOI] [PubMed] [Google Scholar]
- [6].Bottaro DP, Liotta LA, Nature 2003, 423, 593–595. [DOI] [PubMed] [Google Scholar]
- [7].Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, Engelhardt B, Grammas P, Nedergaard M, Nutt J, Pardridge W, Rosenberg GA, Smith Q, Drewes LR, Lancet Neurol 2008, 7, 84–96. [DOI] [PubMed] [Google Scholar]
- [8].Muro S, J. Controlled Release 2012, 164, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW, Nat. Rev. Mater 2016, 1, 16014. [Google Scholar]
- [10].De Jong WH, Borm PJA, Int. J. Nanomed 2008, 3, 133. [Google Scholar]
- [11].Duran NE, Hommes DW, Ther. Adv. Gastroenterol 2016, 9, 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Coburn ML, Billigen JB, Kim AS, Johnson JN, Bates D, King B, Stroke 2016, 47, 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Newick K, O’Brien S, Moon E, Albelda SM, Annu. Rev. Med 2017, 68, 139–152. [DOI] [PubMed] [Google Scholar]
- [14].Burke J, Hunter M, Kolhe R, Isales C, Hamrick M, Fulzele S, Clin. Transl. Med 2016, 5, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Su Y, Xie Z, Kim GB, Dong C, Yang J, ACS Biomater. Sci. Eng 2015, 1, 201–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Muzykantov VR, Expert Opin. Drug Delivery 2010, 7, 403–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mooney R, Weng Y, Garcia E, Bhojane S, Smith-Powell L, Kim SU, Annala AJ, Aboody KS, Berlin JM, J. Controlled Release 2014, 191, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fu J, Wang D, Mei D, Zhang H, Wang Z, He B, Dai W, Zhang H, Wang X, Zhang Q, J. Controlled Release 2015, 204, 11–19. [DOI] [PubMed] [Google Scholar]
- [19].Griffiths L, Binley K, Iqball S, Kan O, Maxwell P, Ratcliffe P, Lewis C, Harris A, Kingsman S, Naylor S, Gene Ther 2000, 7, 255. [DOI] [PubMed] [Google Scholar]
- [20].Chhour P, Naha PC, O’Neill SM, Litt HI, Reilly MP, Ferrari VA, Cormode DP, Biomaterials 2016, 87, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhao Y, Haney MJ, Mahajan V, Reiner BC, Dunaevsky A, Mosley RL, V Kabanov A, Gendelman HE, V Batrakova E, J. Nanomed. Nanotechnol 2011, Suppl 4, 003; 10.4172/2157-7439.S4-00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang W-C, Chiang W-H, Cheng Y-H, Lin W-C, Yu C-F, Yen C-Y, Yeh C-K, Chern C-S, Chiang C-S, Chiu H-C, Biomaterials 2015, 71, 71–83. [DOI] [PubMed] [Google Scholar]
- [23].Li S, Feng S, Ding L, Liu Y, Zhu Q, Qian Z, Gu Y, Int. J. Nanomed 2016, 11, 4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gao M, Hu A, Sun X, Wang C, Dong Z, Feng L, Liu Z, ACS Appl. Mater. Interfaces 2017, 9, 5855–5863. [DOI] [PubMed] [Google Scholar]
- [25].Portnow J, Synold TW, Badie B, Tirughana R, Lacey SF, D’Apuzzo M, Metz MZ, Najbauer J, Bedell V, Vo T, Clin. Cancer Res 2017, 23, 2951–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chessa L, Leuzzi V, Plebani A, Soresina A, Micheli R, D’Agnano D, Venturi T, Molinaro A, Fazzi E, Marini M, Orphanet J Rare Dis 2014, 9,5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hunault-Berger M, Leguay T, Huguet F, LeprÞtre S, Deconinck E, Ojeda-Uribe M, Bonmati C, Escoffre-Barbe M, Bories P, Himberlin C, Am. J. Hematol 2015, 90, 811–818. [DOI] [PubMed] [Google Scholar]
- [28].Ginhoux F, Jung S, Nat. Rev. Immunol 2014, 14, 392–404. [DOI] [PubMed] [Google Scholar]
- [29].Eming SA, Krieg T, Davidson JM, J. Invest. Dermatol 2007, 127, 514–525. [DOI] [PubMed] [Google Scholar]
- [30].Vaupel P, Kallinowski F, Okunieff P, Cancer Res 1989, 49, 6449–6465. [PubMed] [Google Scholar]
- [31].Bjçrnheden T, Levin M, Evaldsson M, Wiklund O, Arterioscler. Thromb. Vasc. Biol 1999, 19, 870–876. [DOI] [PubMed] [Google Scholar]
- [32].Murdoch C, Giannoudis A, Lewis CE, Blood 2004, 104, 2224–2234. [DOI] [PubMed] [Google Scholar]
- [33].Brown JM, Mol. Med. Today 2000, 6, 157–162. [DOI] [PubMed] [Google Scholar]
- [34].Pixley FJ, Stanley ER, Trends Cell Biol 2004, 14, 628–638. [DOI] [PubMed] [Google Scholar]
- [35].Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser E, Snyder L, Pollard JW, Nature 2011, 475, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Muthana M, Giannoudis A, Scott SD, Fang H-Y, Coffelt SB, Morrow FJ, Murdoch C, Burton J, Cross N, Burke B, Cancer Res 2011, 71, 1805–1815. [DOI] [PubMed] [Google Scholar]
- [37].Eisenbl-tter M, Ehrchen J, Varga G, Sunderkçtter C, Heindel W, Roth J, Bremer C, Wall A, J. Nucl. Med 2009, 50, 1676–1682. [DOI] [PubMed] [Google Scholar]
- [38].Wagers AJ, Weissman IL, Cell 2004, 116, 639–648. [DOI] [PubMed] [Google Scholar]
- [39].Bianco P, Robey PG, Nature 2001, 414, 118–121. [DOI] [PubMed] [Google Scholar]
- [40].Chamberlain G, Fox J, Ashton B, Middleton J, Stem Cells 2007, 25, 2739–2749. [DOI] [PubMed] [Google Scholar]
- [41].Rakic P, Nat. Rev. Neurosci 2009, 10, 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liu S, Ginestier C, Ou SJ, Clouthier SG, Patel SH, Monville F, Korkaya H, Heath A, Dutcher J, Kleer CG, Cancer Res 2011, 71, 614–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhao D, Najbauer J, Annala AJ, Garcia E, Metz MZ, Gutova M, Polewski MD, Gilchrist M, Glackin CA, Kim SU, Stem Cells 2012, 30, 314–325. [DOI] [PubMed] [Google Scholar]
- [44].Perrigue PM, Silva ME, Warden CD, Feng NL, Reid MA, Mota DJ, Joseph LP, Tian YI, Glackin CA, Gutova M, Mol. Cancer Res 2015, 13, 636–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen Y, Xiang L, Shao J, Pan R, Wang Y, Dong X, Zhang G, J. Cell. Mol. Med 2010, 14, 1494–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Guan J, Chen J, Biomed. Rep 2013, 1, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Anselmo AC, Gupta V, Zern BJ, Pan D, Zakrewsky M, Muzykantov V, Mitragotri S, ACS Nano 2013, 7, 11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang C, Sun X, Cheng L, Yin S, Yang G, Li Y, Liu Z, Adv. Mater 2014, 26, 4794–4802. [DOI] [PubMed] [Google Scholar]
- [49].Hoffman JF in Use Resealed Erythrocytes as Carriers Bioreactors, Springer, Amsterdam, 1992, pp. 1–15. [Google Scholar]
- [50].Champion JA, Walker A, Mitragotri S, Pharm. Res 2008, 25, 1815–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Champion JA, Mitragotri S, Proc. Natl. Acad. Sci. USA 2006, 103, 4930–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Choi M-R, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, Halas NJ, Clare SE, Nano Lett 2007, 7, 3759–3765. [DOI] [PubMed] [Google Scholar]
- [53].Choi J, Kim H-Y, Ju EJ, Jung J, Park J, Chung H-K, Lee JS, Lee JS, Park HJ, Song SY, Biomaterials 2012, 33, 4195–4203. [DOI] [PubMed] [Google Scholar]
- [54].Smith BR, Ghosn EEB, Rallapalli H, Prescher JA, Larson T, Herzenberg LA, Gambhir SS, Nat. Nanotechnol 2014, 9, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Miller MA, Zheng Y, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC, Weissleder R, Nat. Commun 2015, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kang S, Lee HW, Jeon YH, Singh TD, Choi YJ, Park JY, Kim JS, Lee H, Hong KS, Lee I, Mol. Imaging Biol 2015, 17, 643–651. [DOI] [PubMed] [Google Scholar]
- [57].Encabo-Berzosa MM, Gimeno M, Lujan L, Sancho-Albero M, Gomez L, Sebastian V, Quintanilla M, Arruebo M, Santamaria J, Martin-Duque P, RSC Adv 2016, 6, 58723–58732. [Google Scholar]
- [58].Baek S-K, Makkouk AR, Krasieva T, Sun C-H, Madsen SJ, Hirsch-berg H, J. Neuro-Oncol 2011, 104, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Schnarr K, Mooney R, Weng Y, Zhao D, Garcia E, Armstrong B, Annala AJ, Kim SU, Aboody KS, Berlin JM, Adv. Healthcare Mater 2013, 2, 976–982. [DOI] [PubMed] [Google Scholar]
- [60].Mooney R, Roma L, Zhao D, Van Haute D, Garcia E, Kim SU, Annala AJ, Aboody KS, Berlin JM, ACS Nano 2014, 8, 12450–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Huang W-C, Shen M-Y, Chen H-H, Lin S-C, Chiang W-H, Wu P-H, Chang C-W, Chiang C-S, Chiu H-C, J. Controlled Release 2015, 220, 738–750. [DOI] [PubMed] [Google Scholar]
- [62].Pang L, Qin J, Han L, Zhao W, Liang J, Xie Z, Yang P, Wang J, Onco-target 2016, 7, 37081–37091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hughes RM, Marvin CM, Rodgers ZL, Ding S, Oien NP, Smith WJ, Lawrence DS, Angew. Chem. Int. Ed 2016, 55, 16080–16083; Angew. Chem. 2016, 128, 16314–16317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Muthana M, Kennerley AJ, Hughes R, Fagnano E, Richardson J, Paul M, Murdoch C, Wright F, Payne C, Lythgoe MF, Nat. Commun 2015, 6, 8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Han J, Zhen J, Van Du Nguyen GG, Choi Y, Ko SY, Park J-O, Park S, Sci. Rep 2016, 6, 28717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cao P, Mooney R, Tirughana R, Abidi W, Aramburo S, Flores L, Gilchrist M, Nwokafor U, Haber T, Tiet P, Annala AJ, Han E, Dellinger T, Aboody KS, Berlin JM, Bioconjugate Chem 2017, 28, 1767–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ihler GM, Glew RH, Schnure FW, Proc. Natl. Acad. Sci. USA 1973, 70, 2663–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Magnani M, Rossi L, D’ascenzo M, Panzani I, Bigi L, Zanella A, Biotechnol. Appl. Biochem 1998, 28, 1–6. [PubMed] [Google Scholar]
- [69].Favretto ME, Cluitmans JCA, Bosman G, Brock R, J. Controlled Release 2013, 170, 343–351. [DOI] [PubMed] [Google Scholar]
- [70].Haber T, Baruch L, Machluf M, Sci. Rep 2017, 7, 42046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].He W, Qiang M, Ma W, Valente AJ, Quinones MP, Wang W, Reddick RL, Xiao Q, Ahuja SS, Clark RA, Hum. Gene Ther 2006, 17, 949–959. [DOI] [PubMed] [Google Scholar]
- [72].Yan F, Li X, Li N, Zhang R, Wang Q, Ru Y, Hao X, Ni J, Wang H, Wu G, Cancer Lett 2017, 402, 32–42. [DOI] [PubMed] [Google Scholar]
- [73].Cai Y, Xi Y, Cao Z, Xiang G, Ni Q, Zhang R, Chang J, Du X, Yang A,Yan B, Cancer Lett 2016, 381, 104–112. [DOI] [PubMed] [Google Scholar]
- [74].Frank RT, Edmiston M, Kendall SE, Najbauer J, Cheung C-W, Kassa T, Metz MZ, Kim SU, Glackin CA, Wu AM, PLoS One 2009, 4, e8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Aboody KS, Najbauer J, Metz MZ, D’Apuzzo M, Gutova M, Annala AJ, Synold TW, Couture LA, Blanchard S, Moats RA, Garcia E, Aramburo S, Valenzuela VV, Frank RT, Barish ME, Brown CE, Kim SU, Badie B, Portnow J, Sci. Transl. Med 2013, 5, 184ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Muthana M, Rodrigues S, Chen Y-Y, Welford A, Hughes R, Tazzyman S, Essand M, Morrow F, Lewis CE, Cancer Res 2013, 73, 490–495. [DOI] [PubMed] [Google Scholar]
- [77].Leng L, Wang Y, He N, Wang D, Zhao Q, Feng G, Su W, Xu Y, Han Z, Kong D, Biomaterials 2014, 35, 5162–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Leoni V, Gatta V, Palladini A, Nicoletti G, Ranieri D, Dall’Ora M, Grosso V, Rossi M, Alviano F, Bonsi L, Oncotarget 2015, 6, 34774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mader EK, Butler G, Dowdy SC, Mariani A, Knutson KL, Federspiel MJ, Russell SJ, Galanis E, Dietz AB, Peng K-W, J. Transl. Med 2013, 11, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kaczorowski A, Hammer K, Liu L, Villhauer S, Nwaeburu C, Fan P, Zhao Z, Gladkich J, Groß W, Nettelbeck DM, Oncotarget 2016, 7, 9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Baron S, Poast J, Rizzo D, McFarland E, Kieff E, Immunol J. Methods 2000, 242, 115–126. [DOI] [PubMed] [Google Scholar]
- [82].Chambers E, Mitragotri S, Exp. Biol. Med 2007, 232, 958–966. [PubMed] [Google Scholar]
- [83].Chambers E, Mitragotri S, J. Controlled Release 2004, 100, 111–119. [DOI] [PubMed] [Google Scholar]
- [84].Gribova V, Auzely-Velty R, Picart C, Chem. Mater 2012, 24, 854–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kinge S, Crego-Calama M, Reinhoudt DN, ChemPhysChem 2008, 9, 20–42. [DOI] [PubMed] [Google Scholar]
- [86].Anselmo AC, Gilbert JB, Kumar S, Gupta V, Cohen RE, Rubner MF, Mitragotri S, J. Controlled Release 2015, 199, 29–36. [DOI] [PubMed] [Google Scholar]
- [87].Klyachko NL, Polak R, Haney MJ, Zhao Y, Neto RJG, Hill MC, Kabanov AV, Cohen RE, Rubner MF, Batrakova EV, Biomaterials 2017, 140, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Polak R, Lim RM, Beppu MM, Pitombo RNM, Cohen RE, Rubner MF, Adv. Healthcare Mater 2015, 4, 2832–2841. [DOI] [PubMed] [Google Scholar]
- [89].Livnah O, Bayer EA, Wilchek M, Sussman JL, Proc. Natl. Acad. Sci. USA 1993, 90, 5076–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wilchek M, Bayer EA, Methods Enzymol 1990, 184, 5–13. [DOI] [PubMed] [Google Scholar]
- [91].Abbina S, Siren EMJ, Moon H, Kizhakkedathu JN, ACS Biomater. Sci. Eng 2017; DOI: 10.1021/acsbiomaterials.7b00514. [DOI] [PubMed] [Google Scholar]
- [92].Murciano J-C, Medinilla S, Eslin D, Atochina E, Cines DB, Muzykantov VR, Nat. Biotechnol 2003, 21, 891–896. [DOI] [PubMed] [Google Scholar]
- [93].Cheng H, Kastrup CJ, Ramanathan R, Siegwart DJ, Ma M, Bogatyrev SR, Xu Q, Whitehead KA, Langer R, Anderson DG, ACS Nano 2010, 4, 625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mooney R, Weng Y, Tirughana-Sambandan R, Valenzuela V, Aramburo S, Garcia E, Li Z, Gutova M, Annala AJ, Berlin JM, Future Oncol 2014, 10, 401–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sternberg N, Georgieva R, Duft K, B-umler H, J. Microencapsulation 2012, 29, 9–20. [DOI] [PubMed] [Google Scholar]
- [96].Li L, Guan Y, Liu H, Hao N, Liu T, Meng X, Fu C, Li Y, Qu Q, Zhang Y, ACS Nano 2011, 5, 7462–7470. [DOI] [PubMed] [Google Scholar]
- [97].Rossi NAA, Constantinescu I, Kainthan RK, Brooks DE, Scott MD, Kizhakkedathu JN, Biomaterials 2010, 31, 4167–4178. [DOI] [PubMed] [Google Scholar]
- [98].Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ, Nat. Med 2010, 16, 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Murad KL, Mahany KL, Brugnara C, Kuypers FA, Eaton JW, Scott MD, Blood 1999, 93, 2121–2127. [PubMed] [Google Scholar]
- [100].Hong V, Steinmetz NF, Manchester M, Finn MG, Bioconjugate Chem 2010, 21, 1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Laughlin ST, Bertozzi CR, Nat. Protoc 2007, 2, 2930–2944. [DOI] [PubMed] [Google Scholar]
- [102].Jewett JC, Sletten EM, Bertozzi CR, J. Am. Chem. Soc 2010, 132, 3688–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Layek B, Sadhukha T, Prabha S, Biomaterials 2016, 88, 97–109. [DOI] [PubMed] [Google Scholar]
- [104].Blackman ML, Royzen M, Fox JM, J. Am. Chem. Soc 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Devaraj NK, Weissleder R, Hilderbrand SA, Bioconjugate Chem 2008, 19, 2297–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ni Z, Zhou L, Li X, Zhang J, Dong S, PLoS One 2015, 10, e0141918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Salgaller ML, Tjoa BA, Lodge PA, Ragde H, Kenny G, Boynton A, Murphy GP, Crit. Rev. Immunol 1998, 18, 109–119. [DOI] [PubMed] [Google Scholar]
- [108].Scott MD, Chen AM, Transfus. Clin. Biol 2004, 11, 40–46. [DOI] [PubMed] [Google Scholar]
- [109].Schmaljohann D, Adv. Drug Delivery Rev 2006, 58, 1655–1670. [DOI] [PubMed] [Google Scholar]
- [110].Lu J, Jiang F, Lu A, Zhang G, Int. J. Mol. Sci 2016, 17, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Xie Z, Zhang Y, Liu L, Weng H, Mason RP, Tang L, Nguyen KT, Hsieh J, Yang J, Adv. Mater 2014, 26, 4491–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Patil US, Qu H, Caruntu D, O’Connor CJ, Sharma A, Cai Y, Tarr MA, Bioconjugate Chem 2013, 24, 1562–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]