

Abstract
Heat shock factor 1 (HSF1) is a stress-responsive transcription factor that regulates expression of protein chaperones and cell survival factors. In cancer, HSF1 plays a unique role, hijacking the normal stress response to drive a cancer-specific transcriptional program. These observations suggest that HSF1 inhibitors could be promising therapeutics. However, HSF1 is activated through a complex mechanism, which involves release of a negative regulatory domain, leucine zipper 4 (LZ4), from a masked oligomerization domain (LZ1-3), and subsequent binding of the oligomer to heat shock elements (HSEs) in HSF1-responsive genes. Recent crystal structures have suggested that HSF1 oligomers are held together by extensive, buried contact surfaces, making it unclear whether there are any possible binding sites for inhibitors. Here, we have rationally designed a series of peptide-based molecules based on the LZ4 and LZ1-3 motifs. Using a plate-based, fluorescence polarization (FP) assay, we identified a minimal region of LZ4 that suppresses binding of HSF1 to the HSE. Using this information, we converted this peptide into a tracer and used it to understand how binding of LZ4 to LZ1-3 suppresses HSF1 activation. Together, these results suggest a previously unexplored avenue in the development of HSF1 inhibitors. Furthermore, the findings also highlight how native interactions can inspire the design of inhibitors for even the most challenging protein-protein interactions (PPIs).
Graphical Abstract
Introduction
Heat shock factor 1 (HSF1) is a transcription factor that binds to heat shock elements (HSE) and transcriptionally regulates expression of heat shock proteins (HSPs) and other pro-survival targets [1, 2] [3–5]. Accordingly, active HSF1 is required for cancer cell survival, where stress and biosynthetic demands are higher than in normal cells [6]. In addition, HSF1−/− knock-out mice are strongly protected from chemical skin carcinogenesis [7], suggesting that HSF1 could be a potential target to prevent tumorigenesis. Compared to traditional chemotherapy or target-based drug discovery, inhibition of HSF1 might also provide higher selectivity and a lower chance of drug resistance due to its broad roles in a cancer cell’s transcriptional program [8].
These observations have motivated many groups to pursue chemical inhibitors of HSF1 [9, 10]. Although reported compounds suppress HSF1 transcriptional activity in cells, their molecular targets, binding sites and mechanisms remain uncertain because they have been discovered through phenotypic screens. A promising alternative would be to discover inhibitors through biochemical screens, using purified HSF1. However, HSF1 protein has been difficult to express until recently, when methods for purification of human HSF1 and its close paralog HSF2 have been reported [11]. In addition, it was found that HSF1 could be isolated as either a stable monomer or trimer. These advances have created an opportunity to revisit methods for HSF1 inhibition.
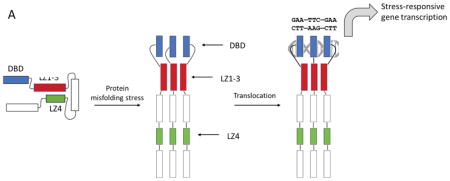
HSF1 is composed of a DNA-binding domain (DBD), an oligomerization motif (termed LZ1-3 or HR-A/B) [12, 13] an intrinsically disordered regulatory region, and a C-terminal coiled-coil, LZ4 (or HR-C). HSF1 is normally held in an inactive, monomeric state by the activity of chaperones and other proteins, which also seem to require an intramolecular interaction between LZ4 and LZ1-3 (Figure 1A) that maintains the monomer in a “paperclip” conformer [14, 15]. During activation, the LZ4 is released, so that LZ1-3 is allowed to form extensive coiled-coil interactions and align the DBDs for interactions with HSEs within the trimer. This oligomerization also facilitates the interaction of HSF1 with regulatory proteins important for transcription [2, 16].
Figure 1.

Structure and molecular mechanisms of HSF1. (A) HSF1 is held in a repressed state through interactions between LZ1-3 and LZ4. A stress response leads to oligomerization and transcriptional activation of heat shock responsive genes. (B) Proposed mechanisms by which LZ1-3 or LZ4 derived peptides might mimic intra-molecular interactions and suppress HSF1 activation. This activity might be detected by a fluorescence polarization (FP) experiment, in which changes in binding of HSF1 to fluorescent HSE is measured. (C) Homology model of human HSF1 LZ1-3 domain homotrimer. Coiled-coils are shown in cartoon form (left and middle), while the peptide template for LZ1-3 ligand design is shown as a yellow cartoon (right).
A recent homology model of the human LZ1-3 trimer attracted our attention [12, 13, 17]. We reasoned that pharmacologically targeting this oligomerization domain with mimetics of the LZ1-3 or LZ4 motifs might disrupt HSF1 activity and potentially its oligomerization (Figure 1B). However, one challenge is that the observed HSF1 coiled-coils are long, only about 20 residues shorter than the entire LZ1-3 domain, and they feature three α-helices intertwined from N to C-terminus to form a tight bundle (Figure 1C). More specifically, the protein-protein interactions (PPIs) in this region appear to be driven by four leucine zippers (LZs) distributed within the 50-residued α–helix, giving a large interface that lacks well-defined binding pockets for small molecules. However, Navitoclax [18, 19], the first marketed drug targeting intracellular PPIs, was built by mimicking a >20-residue α–helix, suggesting that such a strategy might potentially work for HSF1. Also, there have been recent successes in either stabilizing helical conformation, or converting short peptides into mimetics, yielding potent pharmacological tools or even clinical candidates [20–23], further motivating this approach.
To enable inhibitor discovery for HSF1, we first needed to tackle the formidable challenge of understanding which regions of LZ1-3 and/or LZ4 might be the best starting points. Accordingly, we report here the design of peptides that mimic discrete stretches of LZ1-3 and LZ4. Using a new, plate-based fluorescence polarization (FP) assay, we screened the peptides for potential inhibitors of HSF1-HSE interactions. Strikingly, none of the peptides derived from LZ1-3 were found to be inhibitors, while the shorter peptides from LZ4 were relatively potent (EC50 ~ 13 μM). Leveraging this knowledge, we created an LZ4-based FP tracer and used it to measure direct interactions between the inhibitory region and HSF1. These results are an important first step in mapping the PPI surfaces of HSF1 for potentially “druggable” sites. Specifically, we suggest that mimetics of the natural regulatory domain, LZ4, might prove to be an unexpectedly promising lead towards the development of the first rationally designed HSF1 inhibitors. In addition, the FP tracer described here could be a useful chemical probe for better understanding HSF1 activation and/or discovering small molecules.
Materials and Methods
HSF1 protein expression
HSF1 monomer, homotrimer and ΔLZ1-3 were expressed in Escherichia coli and the monomeric and trimeric fractions were separated through Size Exclusion Chromatography (SEC) using a previously described procedure [11]. Briefly, pET15b-6xHis-HSF1 was transformed in BL21 E. coli. A single colony was transferred to an overnight stationary culture, which was diluted 1:100 and grown to an Optical Density 600 of 0.5 at 37 °C. Cultures were transferred to 15 °C and induced with addition of 1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) for 16 hours. Cells were harvested with a 10 min spin at 10,000×g, and pellets were re-suspended in Nickel Lysis Buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 20 mM Imidazole) and lysed with sonication (three, 30 second bursts with ten minutes between each burst. Cell lysate was cleared at 4 °C by 30 min centrifugation at 20,000×g and soluble lysate was incubated with 2 mL of equilibrated Ni NTA agarose resin (QIAGEN), washed three times with Nickel Wash Buffer (Buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 40 mM Imidazole), and washed twice with Nickel Wash Buffer supplemented with 5 mM ATP and 20 mM MgCl2. HSF1 was eluted with Nickel Elution Buffer (Buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 250 mM Imidazole). Nickel elution was further separated by SEC using an ÄKTA system with a Sephacryl S-400 (GE Healthcare). SEC buffer (25 mM pH 7.5 HEPES and 150 mM NaCl) was used at a flow rate of 1.0 mL/min and fractions containing HSF1 trimer (Ve~160 mL) and monomer (Ve~210 mL) were collected. Concentration was assessed using both Bradford and bicinchoninic acid assay (BCA) and resulting HSF1 monomer or trimer samples were flash frozen in liquid nitrogen and stored at −80 degrees C in SEC buffer. The protein purity was estimated to be greater than 95% by SDS-PAGE. Proteins were stored frozen at −80 °C in 25 mM HEPES, pH 7.5, and 150 mM NaCl buffer.
Plate-based fluorescence polarization (FP) assay
Preparation of the 5-fluorescein labeled HSE oligonucleotides followed a previous approach [11]. FP experiments were performed in HEPES buffer: 25 mM HEPES, 75 mM NaCl, pH 7.5, with addition of 10 mM MgCl2 and 0.01% Triton-X to improve DNA stability and prevent nonspecific aggregations. The concentration of labeled HSE for the screen was determined by serial dilution of monomeric HSF1 protein (18 μM) into either 15 nM or 30 nM of the tracer. This mixture was co-incubated for 15 mins in Costar 96-well, black, flat bottom plates before measurement in a SpectraMax M5 plate reader (Molecular Devices) using an excitation wavelength of 485 nm and an emission wavelength of 530 nm. These mP values were plotted and fit using a one-site binding model in GraphPad Prism 5 (Figure S1). A similar procedure was used to obtain saturation curves for HSF1 trimer. In those experiments, we also varied the concentration of DMSO and co-incubation time to evaluate assay stability. The final conditions for peptide screening were selected as: 250 nM HSF1 trimer (75% of saturation), 15 nM of HSE tracer and 5% DMSO, using an incubation time of 15 min and 100 μL volume. A similar procedure was employed to get saturation curves of the 14F tracer (10 nM) binding to wild-type HSF1 monomer (8 μM) or ΔLZ1-3 recombinant HSF1 (8 μM).
Peptide synthesis
HSF1-based peptides were either purchased from commercial source or synthesized manually using microwave-assisted, solid-phase peptide synthesis in an Fmoc protecting group strategy. Synthesized peptides were cleaved using 87.5% trifluoroacetic acid, 5% dithiothreitol, 5% water and 2.5% triisopropylsilane, precipitated with ethyl ether and purified with reverse phase, semi-preparative HPLC. The peptide-based tracer was made manually by attaching 5-fluorescein to the N-terminus of 14 using 4-equivalent NHS-fluorescein 8% acetic anhydride and 8% pyridine in dry DMF, two β-alanine was introduced as a spacer. Peptide purities were determined to be > 90% by HPLC. Peptides were stored as dry powders at −80 °C and dissolved in DMSO prior to use.
Competitive FP assay for peptide screening
Competitive FP binding assays were designed to evaluate the activity of peptides to inhibit HSF1 from binding to DNA. Peptides dissolved in DMSO were co-incubated with HSF1 trimer protein in a 96 well plate at room temperature for 1 hour with gentle shaking. The tracer was then added into the reaction to make an assay condition with 400 μM peptide, 250 nM HSF1 trimer and 15 nM DNA tracer, which was shaken for additional 15 mins before reading. Percentage inhibition values were calculated by an equation including factors of maximum signal from DMSO control, baseline signal from tracer-only group and signals given by each tested peptide: %inhibition= (mPDMSO - mPPeptides)/(mPDMSO- mPtracer) %. For dose-dependent inhibition, serial dilutions of peptides were performed from 800 μM before co-incubating with the protein. In the trimer de-activation study, the peptide was added into a pre-formed protein (250 nM) and tracer (15 nM) complex and co-incubated for additional 1 hour before reading. IC50 values were determined by nonlinear regression fitting of the competition curves.
Tracer binding assay
Serial dilutions (from 400 μM) of peptide solutions were co-incubated with 15 nM tracer for 15 mins, 1 hour, 3 hours and 24 hours before reading. FP readings were stable for >120 minutes, but mP values at 15 mins were used to plot the curves.
Circular dichroism
Circular dichroism experiments were performed using Jasco J-720 spectropolarimeter. Peptide powders were dissolved in 10 mM phosphate buffer (pH = 7.5) to produce a 100 μM solution. A spectrum was generated using the method described previously [24].
Size exclusion chromatography experiments
Homotrimeric full-length human HSF1 was collected, concentrated, and re-purified again by Size Exclusion Chromatography (SEC) using the GE S400 column. As expected, trimeric HSF1 eluted at ~165 mL as a monodispersed peak. Fractions containing trimeric HSF1 were pooled with a final concentration of 2 μM HSF1; this HSF1 trimer was divided and used to determine the oligomeric state of HSF1 incubated with peptide or DMSO by SEC. 20 μM peptide (10-fold molar excess) or DMSO (2% v/v) was incubated with HSF1 at 4°C for 30 minutes. After Incubating, the samples were subjected SEC and fractions were collected and analyzed by immunoblot.
Results
Design of a plate-based fluorescence polarization assay to screen HSF1 inhibitors
Recent work established a recombinant expression system for producing human HSF1 and separating samples of either HSF1 monomer or trimer [11]. Further, a cuvette-based FP assay was developed to measure binding of these proteins to fluorophore-labeled DNA containing HSEs. Due to the scope of the intended study and the challenge of producing human HSF1 is sufficient quantities, we first needed to miniaturize the FP assay, reducing the volume for use in 96-well plates. Briefly, we performed a matrix optimization of tracer concentration and tracer:protein ratio (Fig S1) for purified samples of either HSF1 monomer or trimer, ultimately reducing overall volume by 10-fold (100 μL), without compromising signal:noise (> 200 ΔmP). In addition, we added 0.01% Triton to the FP buffer to reduce nonspecific interactions or aggregation. Using the modified assay conditions, we obtained a robust saturation-binding isotherm, with suitably low protein concentration (250 nM) to detect nanomolar range IC50 values. Finally, we found that the assay signal was stable for at least 2 hours (Fig 2A) and was not sensitive to DMSO up to ~5% (Fig 2B).
Figure 2.

Development of assay conditions for the fluorescence polarization-based detection of HSF1 binding to HSE. Labeled HSE was incubated with purified HSF1 in 96-well plates, as discussed in the Methods. (A) FP signal is stable for at least 120 minutes. Results are the average of experiments performed in triplicate and error bars represent standard error of the mean (SEM). Many of the error bars are smaller than the symbols. (B) The FP signal is not sensitive to DMSO concentrations to ~ 5%. The results are the average of duplicates and error bars represent standard deviation (SD). Many of the error bars are smaller than the symbols.
Rational design of a peptide library to inhibit HSF1 activation
Our approach was to leverage the FP assay and the available HSF1 structures to design peptidomimetics, focused on the natural sites of PPIs: the leucine zippers of LZ1-3 and LZ4. We envisioned that this experiment might be the first step towards creating orthosteric inhibitors, based on precedent in other PPI systems [25]. Towards that goal, we first examined the primary sequences and the homology models of the human HSF1 trimer, revealing that both the intermolecular interactions of LZ1-3 with other LZ1-3 regions and the binding of LZ4 to LZ1-3 all seemed to involve leucine zippers. Therefore, we designed the first target peptides to encompass the six, leucine zipper repeats in LZ1-3, and the two consecutive zippers in the middle of LZ4 (Table 1). Our design started with peptides of 30 residues in length, comprising 4 heptad units and the intervening, non-leucine zipper regions. In addition, we included one extra residue on both the N- and C-termini (1–3, 11). A truncation was also made close to the middle of LZ1-3, with the goal of disclosing the contributions between N-terminal and C-terminal leucine zippers, respectively (4 and 5).
Table 1.
Sequence of HSF1-based peptides and their activity against HSF1-HSE

|

|
All of the peptides were capped with acetyl on the N-terminus and amide at the C-terminus.
Each Heptad Repeat is shown in colors, while hydrophobic residues on leucine zippers are underlined.
%inhibition= (mPDMSO − mPPeptides)/(mPDMSO − mPtracer) %. Results are the average of octuplicates and error bars represent SD.
In addition to an analysis of leucine zippers, we also relied on data generated from a hydrogen deuterium exchange (HDXE) study of the inactive HSF1 monomer [26]. That study showed that HR-A/B (LZ1-3) and HR-C (LZ4) were protected from deuterium exchange, and therefore not solvent-exposed. Specifically, regions around Leu141-Leu158, Arg176-Leu189 and Asp389-Leu395 showed high protection, suggesting that they might contribute to intramolecular binding affinity that maintains HSF1 in its monomeric state. We took advantage of this information and designed 6–9, 12–13 to cover all six fragments showed in the HDXE study, and combined some short sequences to make 10 and 14. Together, this peptide library contained 14 rationally designed sequences from both oligomerization (LZ1-3) and auto-inhibitory (LZ4) domains. These peptides were synthesized by standard Fmoc coupling and purified by HPLC (see Methods).
Identification of an HSF1 inhibitory region
Peptides were first evaluated by FP at a single concentration (400 μM) and % inhibition values were calculated (Table 1, Fig S2). In this experiment, we pre-incubated peptide with HSF1 trimer for 15 minutes prior to addition of labeled HSE tracer. We specifically chose the protein concentration to enable potential identification of both inhibitors and activators of HSF1-HSE interactions. This decision was made because there is also interest in activation of HSF1 in neurodegenerative diseases [2, 27]. From this screen, peptides 4, 11 and 14 showed >60% reduction of binding, while 5 and 10 significantly increased the FP signal. Most of the peptides used in the screen were inactive, suggesting that the tractable inhibitory regions of HSF1 are, as might be expected, quite limited.
To confirm the screening results, we performed a series of validation experiments. First, we performed FP experiments to measure any nonspecific interactions between the peptides and the HSE tracer. When HSF1 was absent, 5 and 10 still increased the FP signals, suggesting that they bind non-specifically to the tracer DNA (Fig 3A). Then, we performed circular dichroism (CD) studies to explore the secondary structure of the peptides. We found that peptides 4, 11 and 14 had different structures by CD: 11 was α-helical, 14 behaved as an unstructured peptide, and 4 was likely a small, soluble aggregate (Fig 3B). Together, these studies focused our attention on 11 and 14 for further confirmation.
Figure 3.

Peptides from the screen are dose-dependent and reproducible inhibitors of HSF1 binding to HSE. (A) Control experiments revealed non-specific binding of peptides 5 and 10 to the HSE tracer. Results are the average of triplicates and error bars represent SD. Some of the error bars are smaller than the symbols. (B) Circular dichroism revealed the predominant secondary structures in peptides 4, 11 and 14. (C) Dose-dependent inhibition of HSF1 trimer binding to DNA, with DNA tracer added before peptide-protein co-incubation. The results from both of the duplicate experiments are shown. (D) Dose-dependent inhibition of HSF1 trimer binding to DNA, with peptide 14 added after tracer-protein complex was formed. The results from both of the duplicate experiments are shown.
Dose-dependence experiments confirmed that 14, the sequence from LZ4, had the most potent IC50 value (13 μM), while 11 had an IC50 of 150 μM (Fig 3C). Based on this result, we hypothesized that 14 might be acting like the natural inhibitory motif to suppress HSF1 oligomerization. To further probe this question, we added 14 to a pre-formed mixture of HSF1 and DNA tracer. Strikingly, peptide 14 was able to disassemble the preformed HSF1-DNA complex with an IC50 value of 14 μM (Fig 3D), suggesting that it is capable of disrupting interactions of the HSF1 trimer with DNA.
Creating a new probe for HSF1
To leverage these findings, we created a new tracer by coupling a fluorescein derivative (FAM) to the N-terminus of 14 (14F, Fig 4A and Fig S3). Using this tracer, we asked whether the LZ4 region binds directly to the the LZ1-3 region. Specifically, we measured binding of 14F to purified HSF1 monomer or a truncated version lacking LZ1-3 (HSF1 ΔLZ1-3; Fig 4B). We found that 14F binds to wild-type HSF1 monomer (EC50 of 4.5 μM), whereas there was no binding to ΔLZ1-3 (EC50 ≫ 8 μM). These results suggest that LZ4 might bind directly to LZ1-3.
Figure 4.

New fluorescence polarization assay using 14-based tracer. (A) Structure and sequence of 14-based tracer (14F). (B) 14F dose-dependently binds to wild-type HSF1 monomer, but fails to bind HSF1 lacking the LZ1-3 domain. Results of duplicate experiments are shown.
LZ4 peptides disrupt HSF1-HSE binding without disrupting the trimer
We considered two possible mechanisms for how 14 could disrupt HSF1 trimer binding to HSE. One possibility is that its interaction with LZ1-3 could disrupt the trimer and release inactive monomer. The other possibility is that the interaction could prevent tight HSE binding through allosteric mechanisms. To test these ideas, we performed size exclusion chromatography (SEC) experiments. It is known [11] that purified human HSF1 can be separated into trimer and monomer peaks by SEC (Fig 5A; black line) and that trimer samples can be isolated as stable solutions (blue line; 160 mL). When we treated an HSF1 trimer with peptide 14 (20 μM; 10-fold excess), there was little change in its position (Fig 5B) and SDS-PAGE confirmed that HSF1 remained largely in the trimer peak (Fig 5C). Thus, binding to LZ4 may allosterically disrupt HSE binding without dramatically altering HSF1 trimer structure.
Figure 5.

Peptide 14 does not disrupt pre-formed HSF1 trimers. (A) The SEC profile of wild-type, purified human HSF1 (black line) shows trimer (~160 mL) and monomer (~210 mL) peaks, as previously reported. The isolated trimer was then re-subjected to SEC, confirming it remained a stable sample (blue line). (B) Neither vehicle (DMSO) nor peptide 14 disrupted the HSF1 trimer. (C) SDS-PAGE and Western blots for HSF1 confirm that the protein stays in the trimer fraction after treatment with vehicle (DMSO) or peptide 14.
Discussion and Conclusions
In this study, we designed and screened for inhibitors of the transcription factor, HSF1. HSF1 is a promising target for anti-cancer therapy, but it has proven difficult to discover drug-like, small molecules that bind directly to this target. Indeed, this task seems quite difficult, as HSF1 has many features of an “undruggable” target, such as a large, flat buried surface area [28]. Towards this goal, we first developed a lower volume FP assay format, which enabled testing of fourteen putative mimics of the LZ1-3 and LZ4 motifs. The results suggested that the LZ4 motif might be a promising starting point for inhibitors. This finding was quite interesting because this region is normally involved in auto-inhibition of HSF1 monomers. Indeed, we found that peptide 14 could suppress HSF1 binding to DNA (EC50 ~ 13 μM) but, interestingly, it did not seem to disrupt the HSF1 trimer, as measured by SEC. Based on these results, we hypothesize that peptides, peptoids or even small molecules that mimic the activity of LZ4 might represent a promising path towards chemical inhibitors of HSF1. Such a strategy might take a similar path to that used in other systems, such as SMAC-IAP interactions [29], in which a native inhibitory mechanism is used to inspire drug-like compounds.
What are the next steps? The screening hit 14 has relatively modest potency (EC50 ~ 13 μM), so one of the next important steps is to optimize its affinity through a broader search of natural and non-natural amino acids. The peptide is comparatively short and our CD studies suggested that it was unable to retain a helical structure when dissolved in aqueous buffer (see Fig 3B). Thus, it might be fruitful to apply one of the α-helix stabilizing strategies [20, 30]. For similar reasons, it might also be worth pursuing continued optimization of 11, another LZ4 derived peptide, even though it seemed to be a weaker starting point. Overall, we envision that one objective of these optimization strategies would be to create a peptide-based FP tracer, similar to 14F, for use in screening diverse chemical libraries of drug-like molecules.
Supplementary Material
Acknowledgments
The authors thank Professor William DeGrado and Dr. Haifan Wu for help with peptide synthesis and characterization. We also thank Alex Jaeger and Leah Makley for helpful comments. E.T.B. is supported by a pre-doctoral fellowship from the US National Institutes of Health (F31 GM119375-02). This work was further supported by the NIH (R01NS059690) and the Tau Consortium (to J.E.G.).
Footnotes
Conflict of Interest Statement
The authors have no conflicts to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–555. doi: 10.1038/nrm2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomez-Pastor R, Burchfiel ET, Thiele DJ. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:4–19. doi: 10.1038/nrm.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matz JM, Blake MJ, Tatelman HM, Lavoi KP, Holbrook NJ. Characterization and regulation of cold-induced heat shock protein expression in mouse brown adipose tissue. Am J Physiol. 1995;269:R38–47. doi: 10.1152/ajpregu.1995.269.1.R38. [DOI] [PubMed] [Google Scholar]
- 4.Cao Y, Ohwatari N, Matsumoto T, Kosaka M, Ohtsuru A, Yamashita S. TGF-beta 1 mediates 70-kDa heat shock protein induction due to ultraviolet irradiation in human skin fibroblasts. Pflug Arch Eur J Phy. 1999;438:239–244. doi: 10.1007/s004240050905. [DOI] [PubMed] [Google Scholar]
- 5.Laplante AF, Moulin V, Auger FA, Landry J, Li H, Morrow G, Tanguay RM, Germain L. Expression of heat shock proteins in mouse skin during wound healing. J Histochem Cytochem. 1998;46:1291–1301. doi: 10.1177/002215549804601109. [DOI] [PubMed] [Google Scholar]
- 6.Dai C, Sampson SB. HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 2016;26:17–28. doi: 10.1016/j.tcb.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dobbelstein M, Moll U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov. 2014;13:179–196. doi: 10.1038/nrd4201. [DOI] [PubMed] [Google Scholar]
- 9.Dayalan Naidu S, Dinkova-Kostova AT. Regulation of the mammalian heat shock factor 1. FEBS J. 2017;284:1606–1627. doi: 10.1111/febs.13999. [DOI] [PubMed] [Google Scholar]
- 10.Yoon YJ, Kim JA, Shin KD, Shin DS, Han YM, Lee YJ, Lee JS, Kwon BM, Han DC. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J Biol Chem. 2011;286:1737–1747. doi: 10.1074/jbc.M110.179440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaeger AM, Makley LN, Gestwicki JE, Thiele DJ. Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J Biol Chem. 2014;289:30459–30469. doi: 10.1074/jbc.M114.591578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neudegger T, Verghese J, Hayer-Hartl M, Hartl FU, Bracher A. Structure of human heat-shock transcription factor 1 in complex with DNA. Nat Struct Mol Biol. 2016;23:140–146. doi: 10.1038/nsmb.3149. [DOI] [PubMed] [Google Scholar]
- 13.Jaeger AM, Pemble CWt, Sistonen L, Thiele DJ. Structures of HSF2 reveal mechanisms for differential regulation of human heat-shock factors. Nat Struct Mol Biol. 2016;23:147–154. doi: 10.1038/nsmb.3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabindran SK, Haroun RI, Clos J, Wisniewski J, Wu C. Regulation of Heat-Shock Factor Trimer Formation - Role of a Conserved Leucine Zipper. Science. 1993;259:230–234. doi: 10.1126/science.8421783. [DOI] [PubMed] [Google Scholar]
- 15.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Labbadia J, Morimoto RI. Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol. 2017;27:895–905. doi: 10.1016/j.tcb.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peteranderl R, Nelson HCM. Trimerization of the Heat-Shock Transcription Factor by a Triple-Stranded Alpha-Helical Coiled-Coil. Biochemistry-Us. 1992;31:12272–12276. doi: 10.1021/bi00163a042. [DOI] [PubMed] [Google Scholar]
- 18.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 20.Ran X, Liu L, Yang CY, Lu J, Chen Y, Lei M, Wang S. Design of High-Affinity Stapled Peptides To Target the Repressor Activator Protein 1 (RAP1)/Telomeric Repeat-Binding Factor 2 (TRF2) Protein-Protein Interaction in the Shelterin Complex. J Med Chem. 2016;59:328–334. doi: 10.1021/acs.jmedchem.5b01465. [DOI] [PubMed] [Google Scholar]
- 21.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karatas H, Townsend EC, Bernard D, Dou Y, Wang S. Analysis of the binding of mixed lineage leukemia 1 (MLL1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J Med Chem. 2010;53:5179–5185. doi: 10.1021/jm100139b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karatas H, Li Y, Liu L, Ji J, Lee S, Chen Y, Yang J, Huang L, Bernard D, Xu J, Townsend EC, Cao F, Ran X, Li X, Wen B, Sun D, Stuckey JA, Lei M, Dou Y, Wang S. Discovery of a Highly Potent, Cell-Permeable Macrocyclic Peptidomimetic (MM-589) Targeting the WD Repeat Domain 5 Protein (WDR5)-Mixed Lineage Leukemia (MLL) Protein-Protein Interaction. J Med Chem. 2017;60:4818–4839. doi: 10.1021/acs.jmedchem.6b01796. [DOI] [PubMed] [Google Scholar]
- 24.Kawamoto SA, Coleska A, Ran X, Yi H, Yang CY, Wang S. Design of triazole-stapled BCL9 alpha-helical peptides to target the beta-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J Med Chem. 2012;55:1137–1146. doi: 10.1021/jm201125d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arkin MR, Tang Y, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2014;21:1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hentze N, Le Breton L, Wiesner J, Kempf G, Mayer MP. Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. Elife. 2016;5 doi: 10.7554/eLife.11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14:248–262. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- 29.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 30.Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat Chem. 2013;5:161–173. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.