

Abstract
Background
Dystonia and ataxia are manifestations of numerous disorders, and indeed, an ever‐expanding spectrum of genes causing diseases that encompass dystonia and ataxia are discovered with the advances of genetic techniques. In recent years, a pathophysiological link between both clinical features and the role of the cerebellum in the genesis of dystonia, in some cases, has been proposed. In clinical practice, the genetic diagnosis of dystonia‐ataxia syndromes is a major issue for genetic counseling, prognosis and, occasionally, specific treatment.
Methods
For this pragmatic and educational review, we conducted a comprehensive and structured literature search in Pubmed, OMIM, and GeneReviews using the key words “dystonia” and “ataxia” to identify those genetic diseases that may combine dystonia with ataxia.
Results
There are a plethora of genetic diseases causing dystonia and ataxia. We propose a series of clinico‐radiological algorithms to guide their differential diagnosis depending on the age of onset, additional neurological or systemic features, and imaging findings. We suggest a sequential diagnostic approach to dystonia‐ataxia syndromes. We briefly highlight the pathophysiological links between dystonia and ataxia and conclude with a review of specific treatment implications.
Conclusions
The clinical approach presented in this review is intended to improve the diagnostic success of clinicians when faced with patients with dystonia‐ataxia syndromes.
Keywords: ataxia, diagnosis, dystonia, genetics, movement disorders
Introduction
Dystonia is characterized by “sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both.”1 It can be the manifestation of a plethora of diseases, and with the advances of genetic techniques, we recognize an ever‐expanding spectrum of genes causing various dystonia syndromes.2 For clinical practice, the new classification of dystonia fosters a phenomenological approach with categorization according to recognizable common associations, which allows narrowing down the differential diagnosis.1 Such associations are, for example, combined dystonia syndromes such as dystonia‐myoclonus and dystonia‐parkinsonism, with well‐described differential diagnoses.3 In contrast, scarce information exists about the combination of dystonia and ataxia. Interestingly, there is also a pathophysiological link between both phenotypes, and the role of the cerebellum in the genesis of dystonia has been the focus of research lately.4, 5, 6 Based on a broad and exhaustive literature review, here we review the spectrum of disorders that can present with dystonia and ataxia, and propose a clinico‐radiological diagnostic algorithm. We examine existing evidence regarding different pathophysiological mechanisms and discuss specific treatment implications.
Methods
We conducted a comprehensive and structured search in PubMed, OMIM, and GeneReviews using the key words “dystonia” and “ataxia” to identify those genetic diseases that may combine dystonia with ataxia. Publications written in English and Spanish and published up to December 31, 2017, were reviewed.
Diagnostic Approach to Genetic Causes of Dystonia‐ataxia
There are a plethora of more than 100 genetic disorders giving rise to dystonia‐ataxia syndromes. In clinical practice, when evaluating patients with dystonia‐ataxia, the differential diagnosis can be largely guided by the age of onset, additional neurological or systemic features, and radiological findings. Clinical diagnostic algorithms that take into account the presence or absence of additional clinical clues and relevant complementary studies are illustrated in Fig. 1 (infancy/childhood onset) and Fig. 2 (adulthood onset). For didactic purposes, both figures include disorders that are most prevalent or relevant from a therapeutic perspective and therefore should be suspected first, according to the authors' opinion. Table 1 provides a more comprehensive list of differential diagnoses. Entities, where dystonia and ataxia are prominent and frequent, are considered first, before disorders where the combination of dystonia and ataxia is only found occasionally. A comprehensive summary of all diseases in which dystonia and ataxia were reported with their respective clinical features and relevant complementary studies are presented according to the mode of inheritance in Supporting Table 1. Sometimes, there are certain clinical clues (“red flags”) that can guide the differential diagnostic considerations (Table 2) and should therefore not be missed. Similarly, imaging findings are often helpful in directing further diagnostic evaluation (Fig. 3).
Figure 1.
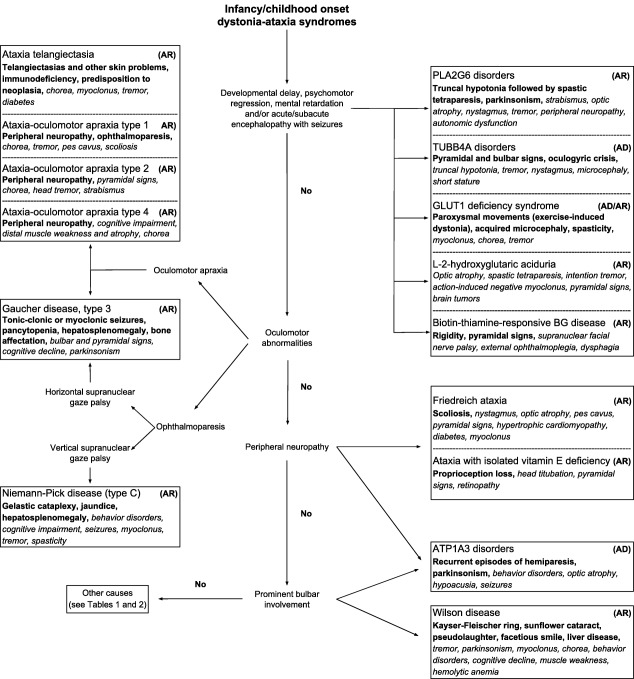
Clinical diagnostic algorithm for genetic dystonia‐ataxia syndromes with infancy or childhood onset.
Abbreviations: AD: autosomal dominant; AR: autosomal recessive; BG: basal ganglia; GLUT1: glucose transporter type 1.
Note: Red flags are shown in bold and possible symptoms are in italic type.
Figure 2.
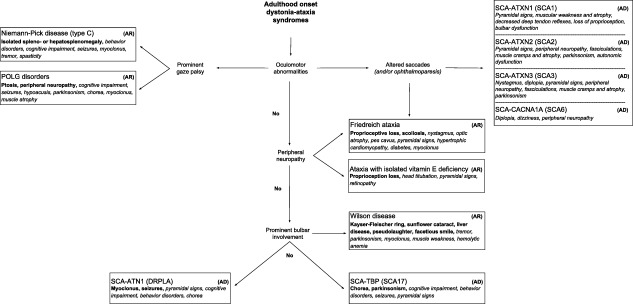
Clinical diagnostic algorithm for genetic dystonia‐ataxia syndromes with adulthood onset.
Abbreviations: AD: autosomal dominant; AR: autosomal recessive; DRPLA: dentatorubral‐pallidoluysian atrophy; SCA: spinocerebellar ataxia type.
Note: Red flags are shown in bold and possible symptoms are in italic type.
Table 1.
Genetic dystonia‐ataxia syndromes
| (A) Concomitant dystonia and ataxia |
| Spinocerebellar ataxias: SCA‐ATN1 * ; SCA‐ATXN2 * ; SCA‐ATXN3 * and SCA‐TBP * |
| Friedreich ataxia (FXN) * |
| Ataxia‐telangiectasia (ATM) * |
| Ataxia with isolated vitamin E deficiency (TTPA) * |
| Niemann‐Pick disease type C (NPC) * |
| Wilson disease (ATP7B) * |
| POLG‐disorders (POLG) * |
| L‐2‐hydroxyglutaric aciduria (L2HGDH) * |
| TUBB4A‐disorders (TUBB4A) * |
| ATP1A3‐disorders (ATP1A3) * |
| PLA2G6‐associated neurodegeneration (PLA2G6) * |
| Glucose transporter type 1 deficiency syndrome (SLC2A1) * |
| Ataxia‐oculomotor apraxia type 4 (PNKP) |
| Autosomal recessive spastic paraplegia type 48 (KIAA0415) |
| Autosomal recessive spastic ataxia type 3 (MARS2) |
| Autosomal recessive spastic ataxia with hypomyelinating leukodystrophy (NKX6‐2) |
| Cerebroretinal microangiopathy with calcifications and cysts or Coats plus syndrome (CTC1) |
| Childhood‐onset neurodegeneration with ataxia, dystonia, and gaze palsy (SQSTM1) |
| Birk‐Landau‐Perez syndrome (SLC30A9) |
| Pyruvate dehydrogenase E2 deficiency (DLAT) |
| Recessive dystonia‐ataxia syndrome due to mitochondrial complex IV deficiency (COX20) |
| Mitochondrial complex III deficiency, nuclear type 4 (UQCRQ) |
| B) Occasional dystonia‐ataxia combination |
| Spinocerebellar ataxias: SCA‐ATXN1; SCA‐SPTBN2; SCA‐CACNA1A; SCA‐ATXN7; SCA‐ATXN8OS; SCA‐ATXN10; SCA‐PPP2R2B; SCA‐KCNC3; SCA‐PRKCG; SCA‐KCND3; SCA‐AFG3L2; SCA‐TGM6; SCA‐NOP56 and SCA/HSP‐VAMP1 |
| Ataxia‐oculomotor apraxia type 1 (APTX) and type 2 (SETX) |
| Gaucher disease, type III (GBA) |
| Chediak‐Higashi syndrome (LYST) |
| Cockayne syndrome (ERCC) |
| Tay‐Sachs disease or GM2‐gangliosidosis type I (HEXA) |
| Biotin‐thiamine‐responsive basal ganglia disease (SLC19A3) |
| PRRT2‐associated disease spectrum (PRRT2) |
| Myoclonic epilepsy of Unverricht and Lundborg (CSTB) |
| Fatty acid hydroxylase‐associated neurodegeneration or autosomal recessive spastic paraplegia‐35 (FA2H) |
| Neurodegeneration with brain iron accumulation‐1 or pantothenate kinase‐associated neurodegeneration (PANK2) |
| Mitochondrial complex I deficiency (multiple genes) |
| Mitochondrial complex III deficiency, nuclear type 2 (TTC19) |
| Mitochondrial disorder due to genomic rearrangements affecting the ATAD3 gene |
| Mitochondrial DNA depletion syndrome type 13 (FBXL4) |
| Combined oxidative phosphorylation deficiency type 29 (TXN2) |
| Pyruvate dehydrogenase E1‐alpha deficiency (PDHA1) |
| Allan‐Herndon‐Dudley syndrome or monocarboxylate transporter type 8 deficiency (SLC16A2) |
| Sulfocysteinuria or sulfite oxidase deficiency (SUOX) |
| Primary coenzyme Q10 deficiency type 4 (ADCK3) |
| Autosomal recessive spinocerebellar ataxia type 5 or Galloway‐Mowat syndrome (WDR73) |
| Autosomal recessive spastic ataxia type 5 (AFG3L2) |
| Brain‐lung‐thyroid syndrome and benign hereditary chorea (NKX2‐1) |
| Congenital disorder of glycosylation type Ia (PMM2) |
| Ataxia‐telangiectasia‐like disorder‐1 (MRE11A) |
| Pelizaeus‐Merzbacher disease or hypomyelinating leukodystrophy type 1 (PLP1) |
| Progressive leukoencephalopathy with ovarian failure (AARS2) |
| Progressive encephalopathy with or without lipodystrophy (BSCL2) |
| Episodic encephalopathy due to thiamine pyrophosphokinase deficiency (TPK1) |
| Limb‐girdle muscular dystrophy type 2S (TRAPPC11) |
| Juvenile amyotrophic lateral sclerosis‐2 (ALS2) |
| X‐linked spinocerebellar ataxia type 1 (ATP2B3) |
| X‐linked syndromic mental retardation, Christianson type or Angelman‐like syndrome (SLC9A6) |
| Kallmann syndrome (KAL1) |
*Most frequent causes of concomitant dystonia and ataxia.
Table 2.
Clinical clues associated with main genetic dystonia‐ataxia syndromes
| Clinical features | Disease (gene name) |
|---|---|
| Developmental delay/mental retardation | Glucose transporter type 1 deficiency syndrome (SLC2A1) |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| TUBB4A‐disorders (TUBB4A) | |
| L‐2‐hydroxyglutaric aciduria or academia (L2HGDH) | |
| Recessive dystonia‐ataxia syndrome due to mitochondrial complex IV deficiency (COX20) | |
| Pyruvate dehydrogenase E2 deficiency (DLAT) | |
| Cognitive decline | Wilson disease (ATP7B) |
| Niemann‐Pick disease (NPC) | |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| Glucose transporter type 1 deficiency syndrome (SLC2A1) | |
| Autosomal recessive spastic ataxia type 3 (MARS2) | |
| Cerebroretinal microangiopathy with calcifications and cysts syndrome (CTC1) | |
| Childhood‐onset neurodegeneration with ataxia, dystonia, and gaze palsy (SQSTM1) | |
| Ataxia‐oculomotor apraxia type 4 (PNKP) | |
| Dentatorubral‐pallidoluysian atrophy (ATN1) | |
| Spinocerebellar ataxia type 17 (TBP) | |
| Opthalmoparesis | Spinocerebellar ataxia type 1 (ATXN1) |
| Spinocerebellar ataxia type 2 (ATXN2) | |
| Spinocerebellar ataxia type 3 (ATXN3) | |
| Niemann‐Pick disease (NPC) | |
| Gaucher disease, type III (GBA) | |
| POLG‐disorders (POLG) | |
| Childhood‐onset neurodegeneration with ataxia, dystonia, and gaze palsy (SQSTM1) | |
| Oculomotor apraxia | Ataxia‐telangiectasia (ATM) |
| Ataxia‐oculomotor apraxia type 1 (APTX) | |
| Ataxia‐oculomotor apraxia type 2 (SETX) | |
| Ataxia‐oculomotor apraxia type 4 (PNKP) | |
| Gaucher disease, type III (GBA) | |
| Childhood‐onset neurodegeneration with ataxia, dystonia, and gaze palsy (SQSTM1) | |
| Retinopathy | Cerebroretinal microangiopathy with calcifications and cysts (CTC1) |
| Ataxia with isolated vitamin E deficiency (TTPA) | |
| Optic atrophy | Friedreich ataxia (FXN) |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| L‐2‐hydroxyglutaric aciduria or academia (L2HGDH) | |
| Cerebroretinal microangiopathy with calcifications and cysts (CTC1) | |
| ATP1A3‐disorders (ATP1A3) | |
| Abnormal eye movement saccades | Spinocerebellar ataxia type 1 (ATXN1) |
| Spinocerebellar ataxia type 2 (ATXN2) | |
| Spinocerebellar ataxia type 3 (ATXN3) | |
| Spinocerebellar ataxia type 6 (CACNA1A) | |
| Friedreich ataxia (FXN) | |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| Seizures | Niemann‐Pick disease (NPC) |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| Dentatorubral‐pallidoluysian atrophy (ATN1) | |
| Spinocerebellar ataxia type 17 (TBP) | |
| TUBB4A‐disorders (TUBB4A) | |
| Glucose transporter type 1 deficiency syndrome (SLC2A1) | |
| L‐2‐hydroxyglutaric aciduria or academia (L2HGDH) | |
| Peripheral neuropathy | Friedreich ataxia (FXN) |
| Ataxia with isolated vitamin E deficiency (TTPA) | |
| Ataxia‐telangiectasia (ATM) | |
| Ataxia‐oculomotor apraxia type 1 (APTX) | |
| Ataxia‐oculomotor apraxia type 2 (SETX) | |
| Ataxia‐oculomotor apraxia type 4 (PNKP) | |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| POLG‐disorders (POLG) | |
| ATP1A3‐disorders (ATP1A3) | |
| Spasticity | Spinocerebellar ataxia type 2 (ATXN2) |
| Spinocerebellar ataxia type 3 (ATXN3) | |
| TUBB4A‐disorders (TUBB4A) | |
| Autosomal recessive spastic ataxia type 3 (MARS2) | |
| Autosomal recessive spastic paraplegia type 48 (KIAA0415) | |
| Cerebroretinal microangiopathy with calcifications and cysts (CTC1) | |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| Glucose transporter type 1 deficiency syndrome (SLC2A1) | |
| Niemann‐Pick disease (NPC) | |
| Myoclonus | Wilson disease (ATP7B) |
| Ataxia‐telangiectasia (ATM) | |
| Niemann‐Pick disease (NPC) | |
| Dentatorubral‐pallidoluysian atrophy (ATN1) | |
| Glucose transporter type 1 deficiency syndrome (SLC2A1) | |
| Parkinsonism | Spinocerebellar ataxia type 2 (ATXN2) |
| Spinocerebellar ataxia type 3 (ATXN3) | |
| Spinocerebellar ataxia type 17 (TBP) | |
| Wilson disease (ATP7B) | |
| ATP1A3‐disorders (ATP1A3) | |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| Childhood‐onset neurodegeneration with ataxia, dystonia, and gaze palsy (SQSTM1) | |
| Chorea | Wilson disease (ATP7B) |
| PLA2G6‐associated neurodegeneration (PLA2G6) | |
| Ataxia‐telangiectasia (ATM) | |
| Dentatorubral‐pallidoluysian atrophy (ATN1) | |
| Spinocerebellar ataxia type 17 (TBP) | |
| Gelastic cataplexy | Niemann‐Pick disease (NPC) |
| Hypogonadism | Ataxia‐telangiectasia (ATM) |
| Congenital disorder of glycosylation type Ia (PMM2) | |
| Kallmann syndrome (KAL1) | |
| Telangiectasias | Ataxia‐telangiectasia (ATM) |
| Macrocephaly | Mitochondrial complex I deficiency (multiple genes) |
| Anosmia | Kallmann syndrome (KAL1) |
| Hepatic disease | Niemann‐Pick disease (NPC) |
| Wilson disease (ATP7B) | |
| Mitochondrial complex I deficiency (multiple genes) |
Figure 3.
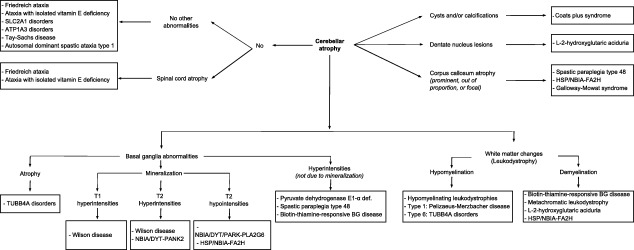
Predominant imaging findings in genetic dystonia‐ataxia syndromes.
Abbreviations: BG: basal ganglia; NBIA: neurodegeneration with brain iron accumulation. For didactic purposes we show only the predominant imaging findings of the diseases that can present with dystonia and ataxia that are most representative, either by its frequency or its ability to be treatable.
Dystonia and ataxia may develop during the disease course sequentially, or in rare cases simultaneously. Interestingly, some disorders that usually present with specific features, including ataxia in ataxia‐telangiectasia can also have a very different clinical picture, with isolated or predominant dystonia without ataxia.7, 8 In line with this, some complex phenotypic disorders may initially mimic isolated dystonia before other clinical characteristics become evident (e.g., Wilson's disease or several spinocerebellar ataxias).9, 10
Different types or forms of dystonia were described in the genetic diseases listed: focal, segmental, generalized, and hemidystonia (e.g., in autosomal dominant progressive external ophthalmoplegia type 1 due to POLG mutations11 and in Coats plus syndrome12) as well as task‐specific dystonia (e.g., in several spinocerebellar ataxias,10, 13 mitochondrial disorders,14 ataxia‐telangiectasia‐like disorder type 1,15 and in L‐2‐hydroxyglutaric aciduria16), paroxysmal, or episodic dystonia (paroxysmal kinesigenic dyskinesia due to PRRT2 mutations,17 episodic ataxia type 2,18 and biotin‐thiamine‐responsive basal ganglia disease due to SLC19A3 mutations19). Supporting Table 1 lists the various forms of dystonia described in genetic dystonia‐ataxia syndromes. In some cases, dystonia can spontaneously attenuate over time as occurs in ataxia‐oculomotor apraxia type 4,20, 21 or is induced by different triggers (e.g., exercise, infections, or emotional stress [PxMD‐SLC2A1 disorders,22, 23 pyruvate dehydrogenase E1‐alpha deficiency,24 biotin‐thiamine‐responsive basal ganglia disease25]). In some unusual cases, a phenotype of dystonia can replace or overshadow ataxia during the disease course (e.g., in ataxia with vitamin E deficiency26) or vice versa (SCA‐CACNA1A27). Last, the combination of dystonia and ataxia often correlates with disease course or severity. For example, the presence of dystonia was associated with greater severity of ataxia in the spinocerebellar ataxias SCA‐ATXN1, SCA‐ATXN2, and SCA‐ATXN3.28 Also, SCA‐ATXN2 and SCA‐ATXN3 patients with dystonia showed greater CAG repeat expansion.28, 29 In contrast, dystonia was associated a slower progression in SCA‐CACNA1A.29
Diagnostic Management
After careful clinical examination, MR imaging is essential as the presence or absence of cerebellar atrophy and other structural abnormalities will crucially guide differential diagnostic considerations. An initial screening of certain biochemical markers would depend on age of onset and clinical suspicion, but should always include serum copper and ceruloplasmin (plus 24 h urinary copper excretion, depending on the level of probability). Other parameters in peripheral blood are used to screen for more common conditions, with specific treatment implications that include vitamin E levels (ataxia with vitamin E deficiency), glucocerebrosidase enzyme activity in leukocytes (reduced in Gaucher disease), and triol levels (elevated in Niemann‐Pick disease type C). If the differential diagnosis includes ataxia telangiectasia (AT) and its lookalikes, serum testing should include alpha‐fetoprotein (elevated in AT and ataxia‐oculomotor apraxia type 2), immunoglobulin levels (often reduced in AT), albumin (reduced in ataxia‐oculomotor apraxia type 1), and cholesterol (increased in ataxia‐oculomotor apraxia type 1). In a case of suspected glucose transporter type 1 deficiency syndrome, serum and CSF should be tested for the glucose levels (CSF glucose concentration < 60 mg/dL) and its ratio (< 0.4). If these tests were negative, genetic testing of most frequent causes of dystonia‐ataxia syndromes, including Friedreich ataxia and several spinocerebellar ataxias (SCA‐ATN1, SCA‐ATXN2, SCA‐ATXN3, SCA‐CACNA1A, SCA‐TBP, and SCA‐ATXN1) should be explored first (again depending on clinical features and age). These are all disorders caused by repeat expansions and are therefore not detectable by next‐sequencing genetic tests. After these common causes are ruled out, genetic testing should be continued with whole exome sequencing or comprehensive dystonia and ataxia panels that include the dystonia‐ataxia syndromes here described.
The Role of the Cerebellar Dysfunction in Dystonia
Dystonia is usually associated with dysfunction of basal ganglia circuits, rather than alteration of the cerebellum.30 Many of the genetic diseases reviewed here encompass complex phenotypes due to neurodegeneration of multiple systems and structures, including the alteration of both basal ganglia and cerebellum, which may explain the occurrence of dystonia in the setting of ataxia and other neurological features. There is growing evidence of an important role of the cerebellar dysfunction in dystonia.4, 5, 6, 31, 32, 33, 34, 35, 36 Animal models of generalized dystonia showed abnormal cerebellar activity,37, 38, 39 and dystonia can be independent of the basal ganglia and can be alleviated or abolished by inactivation of the cerebellum.40, 41, 42 Moreover, a sophisticated network approach strongly suggested that the molecular pathways of ataxia and dystonia are closely related.43 Thus, the large numbers of disorders featuring both dystonia and ataxia are not too surprising. However, correlational studies, such as these network approaches cannot dissect cause and effect.
Treatment
Physical, occupational, and speech rehabilitation therapy are usually combined with pharmacological treatment where appropriate.44, 45 Unfortunately, there currently exists no medication that has been approved for the treatment of cerebellar ataxia or that can prevent or slow‐down neurodegenerative processes that are not related to metabolic diseases (with the exception of aminopyridines and acetazolamide for episodic ataxia in episodic ataxia type 2).44 Most drugs used to treat ataxia or other cerebellar features failed to achieve significant and sustained improvement.44, 45 For patients with Friedreich ataxia or spinocerebellar ataxia, riluzole showed modest improvement in ataxia at 12 months in a Class I study (Supporting Table 2).46 Careful attention should be paid to drugs that can exacerbate ataxia, such as alcohol, lithium, phenytoin, and phenobarbital47 or factors that can precipitate episodic ataxia in paroxysmal kinesigenic dyskinesia and episodic ataxia type 2, including physical or emotional stressful situations, alcohol, fatigue, or exertion. Likewise, drugs that are capable of exacerbating dystonic symptoms, including neuroleptics, piperazine derivatives with calcium antagonist properties (cinnarizine, flunarizine), or antiemetics (metoclopramide; among others) should be avoided.48
Disorders with Specific Management Implications
Some of the genetic dystonia‐ataxia syndromes are preventable by avoiding triggers or are treatable either by reduction of toxic products, dietary interventions, or vitamin supplements.49 Early diagnosis and therapy may slow or halt the clinical course, partially reverse symptoms, or prevent their development altogether (Supporting Table 2). In general, the response to treatment is more likely at early stages of the disease and in children rather than adults.
Symptomatic Therapy of Dystonia
Treatment of dystonic features remains difficult. In very rare cases, they can resolve spontaneously and completely over time (e.g., in ataxia‐oculomotor apraxia type 4 and SCA‐CACNA1A)21, 27 Without counting on these exceptions, dystonic symptoms are always persistent and hardly alleviated with drugs such as levodopa, anticholinergics, or botulinum toxin.13, 26, 50, 51, 52, 53 It is important to emphasize that botulinum toxin use can be dangerous in the treatment of cervical or oromandibular dystonia in patients with spinocerebellar ataxias; this is because dysphagia is very common in this group of neurodegenerative diseases.54 In some of the genetic dystonia‐ataxia syndromes, including SCA‐ATXN2, SCA‐ATXN3, and ataxia‐telangiectasia, dystonic features can show a marked response to levodopa8, 55, 56 and illustrate the rationale of a trial with this drug.10, 11, 13, 57, 58, 59 Indeed, dopamine replacement therapy was established in 16 of 140 (11%) patients with spinocerebellar ataxia, with a partial response in 75% of the cases.13 Pharmacological treatment with trihexyphenidyl, baclofen, benzhexol, diazepam, or clonazepam may also produce variable relief of dystonic postures.26, 60, 61, 62 Currently, there is lack of evidence for the efficacy of non‐invasive brain stimulation techniques, like transcranial magnetic stimulation and transcranial direct/alternating current stimulation in patients with dystonia.63 In contrast, surgical interventions for dystonia such as GPi‐DBS have been found effective to some extent in patients with Wilson disease,64, 65 ataxia‐telangiectasia and its variant64, 66 spinocerebellar ataxias,64, 67, 68, 69 episodic ataxia type 2,70 Cockayne syndrome,71, 72 PLA2G6‐associated neurodegeneration,73 and neurodegeneration with brain iron accumulation type 1 due to mutations in the PANK2 gene.74, 75, 76, 77 Responses are often transient or partial, usually in the range of 10 to 30% of improvement, which is far less than the benefit reported in patients with primary generalized dystonia,64, 76 but greater improvements of approximately 70% during the first years after surgery were also found.74, 75 Patients with dystonia combined with parkinsonism showed a greater response than those with dystonia‐ataxia syndromes.64 Surgical outcome seems to be independent from age at surgery, duration of disease, dystonic features at surgery, or dystonia severity.64
Conclusions
The dystonia‐ataxia syndromes are a clinically and genetically heterogeneous group of disorders that hold a major diagnostic challenge for neurologists. In clinical practice, the etiological diagnosis of dystonia‐ataxia syndromes is key in guiding genetic counseling, prognosis, and in some cases49 specific treatment. Clinico‐radiological algorithms serve to narrow down the differential diagnosis for genetic testing and are crucial to avoid unnecessary complementary studies in scenarios where next‐generation techniques are inaccessible to physicians or unaffordable to patients.2, 9, 78, 79, 80 In addition, algorithms and lists of genes associated with dystonia‐ataxia syndromes may direct genetic research and are also important to assist molecular geneticists in the interpretation of whole exome or genome sequencing data to achieve high diagnostic accuracy.80 For example, as more than 100 genes and genetic diseases need to be screened to diagnose dystonia‐ataxia syndromes, clinico‐radiological algorithms may facilitate the implementation of disease‐focused gene panels or dedicated exome strategies to prioritize those genes that overlap between dystonia and ataxia.43 Most important useful handles in guiding diagnosis with the herein proposed clinical algorithms for dystonia‐ataxia syndromes include the age of disease onset, some associated clinical clues and particular imaging findings. However, major criticism to clinical algorithms arise on the clinical and genetic heterogeneity81 (e.g., many entities or combined syndromes are not distinct conditions), but may represent a continuum between different phenotypes, as is the case for ATP1A3‐, or PLA2G6‐related disorders.82, 83, 84, 85, 86 In addition, clinical algorithms may not fit for atypical phenotypes or be useless if they are too simplistic or rigid, as they may delay or obstruct the identification of the underlying genetic cause in a determined patient.80, 81 In our opinion, clinical algorithms should be used as tools that can orient to specific disorders and they may also be useful to validate genetic findings. The modern next‐sequencing genetic tests do not eradicate the need for an exhaustive clinical assessment and are not extent of limitations, for example, the inability to detect copy number variations or repeat expansions that cause ataxias (e.g., Friedreich ataxia and several spinocerebellar ataxias).79, 80
The list of genetic diseases that display either dystonia or ataxia or both in combination as dystonia‐ataxia syndromes will continue to increase with the widespread access to next‐generation sequencing techniques. Continuous updating of dystonia‐ataxia syndromes will be possible with online resources, such as GeneReviews (available at https://www.ncbi.nlm.nih.gov/books/NBK1116/) and the International Parkinson and Movement Disorder Society Genetic Mutation Online Database (available at http://www.mdsgene.org/).87
In conclusion, the clinical approach presented here is intended to improve diagnostic success of clinicians when facing with patients with dystonia‐ataxia syndromes. Future perspectives resides in research that would provide a better understanding of the role of the cerebellum in dystonia that may in turn result in targeted treatment approaches to help both dystonia and ataxia features.
Author Roles
1. Research project: A. Conception, B. Organization, C. Execution;
2. Manuscript Preparation: A. Writing of the first draft, B. Review and Critique;
M.R.: 1A, 1B, 1C, 2A
B.B.: 1A, 1B, 1C, 2A, 2B
P.M.: 1A, 1B, 1C, 2A
K.B.: 1A, 1B, 1C, 2B
M.M.: 1A, 1B, 1C, 2B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The authors confirm that the approval of an institutional review board was not required for this work.
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: The authors declare that there are no disclosures to report.
Supporting information
Supporting information may be found in the online version of this article.
Table S1. Genetic diseases that may combine dystonia with ataxia
Table S2. Specific treatment implications
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balint B, Bhatia KP. Isolated and combined dystonia syndromes ‐ an update on new genes and their phenotypes. Eur J Neurol 2015;22:610–617. [DOI] [PubMed] [Google Scholar]
- 3. Balint B, Bhatia KP. Dystonia: an update on phenomenology, classification, pathogenesis and treatment. Curr Opin Neurol 2014;27:468–476. [DOI] [PubMed] [Google Scholar]
- 4. Sadnicka A, Hoffland BS, Bhatia KP, van de Warrenburg BP, Edwards MJ. The cerebellum in dystonia ‐ help or hindrance? Clin Neurophysiol 2012;123:65–70. [DOI] [PubMed] [Google Scholar]
- 5. Shakkottai VG, Batla A, Bhatia K, et al. Current Opinions and Areas of Consensus on the Role of the Cerebellum in Dystonia. Cerebellum 2017;16:577–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Teo JT, van de Warrenburg BP, Schneider SA, Rothwell JC, Bhatia KP. Neurophysiological evidence for cerebellar dysfunction in primary focal dystonia. J Neurol Neurosurg Psychiatry 2009;80:80–83. [DOI] [PubMed] [Google Scholar]
- 7. Carrillo F, Schneider SA, Taylor AM, Srinivasan V, Kapoor R, Bhatia KP. Prominent oromandibular dystonia and pharyngeal telangiectasia in atypical ataxia telangiectasia. Cerebellum 2009;8:22–27. [DOI] [PubMed] [Google Scholar]
- 8. Charlesworth G, Mohire MD, Schneider SA, Stamelou M, Wood NW, Bhatia KP. Ataxia telangiectasia presenting as dopa‐responsive cervical dystonia. Neurology 2013;81:1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rossi M, Perez‐Lloret S, Doldan L, et al. Autosomal dominant cerebellar ataxias: a systematic review of clinical features. Eur J Neurol 2014;21:607–615. [DOI] [PubMed] [Google Scholar]
- 10. van Gaalen J, Giunti P, van de Warrenburg BP. Movement disorders in spinocerebellar ataxias. Mov Disord 2011;26:792–800. [DOI] [PubMed] [Google Scholar]
- 11. Rossi M, Medina Escobar A, Radrizzani M, Tenembaum S, Perandones C, Merello M. Dystonia in a patient with autosomal‐dominant progressive external ophtalmoplegia type 1 caused by mutation in the POLG gene. Mov Disord Clin Pract 2015;4:266–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Briggs TA, Abdel‐Salam GM, Balicki M, et al. Cerebroretinal microangiopathy with calcifications and cysts (CRMCC). Am J Med Genet A 2008;146A:182–190. [DOI] [PubMed] [Google Scholar]
- 13. Rossi M, Perez Lloret S, Cerquetti D, Merello M. Movement Disorders in Autosomal Dominant Cerebellar Ataxias: A Systematic Review. Mov Disord Clin Pract 2014;1:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sudarsky L, Plotkin GM, Logigian EL, Johns DR. Dystonia as a presenting feature of the 3243 mitochondrial DNA mutation. Mov Disord 1999;14:488–491. [DOI] [PubMed] [Google Scholar]
- 15. Yoshida T, Awaya T, Shibata M, et al. Hypergonadotropic hypogonadism and hypersegmented neutrophils in a patient with ataxia‐telangiectasia‐like disorder: potential diagnostic clues? Am J Med Genet A 2014;164A:1830–1834. [DOI] [PubMed] [Google Scholar]
- 16. Termsarasab P, Frucht SJ. Writer's cramp as a presentation of L‐2‐hydroxyglutaric aciduria. J Clin Mov Disord 2014;1:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delcourt M, Riant F, Mancini J, et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J Neurol Neurosurg Psychiatry 2015;86:782–785. [DOI] [PubMed] [Google Scholar]
- 18. Spacey SD, Materek LA, Szczygielski BI, Bird TD. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol 2005;62:314–316. [DOI] [PubMed] [Google Scholar]
- 19. Serrano M, Rebollo M, Depienne C, et al. Reversible generalized dystonia and encephalopathy from thiamine transporter 2 deficiency. Mov Disord 2012;27:1295–1298. [DOI] [PubMed] [Google Scholar]
- 20. Bras J, Alonso I, Barbot C, et al. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet 2015;96:474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paucar M, Malmgren H, Taylor M, et al. Expanding the ataxia with oculomotor apraxia type 4 phenotype. Neurol Genet 2016;2:e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zorzi G, Castellotti B, Zibordi F, Gellera C, Nardocci N. Paroxysmal movement disorders in GLUT1 deficiency syndrome. Neurology 2008;71:146–148. [DOI] [PubMed] [Google Scholar]
- 23. Suls A, Dedeken P, Goffin K, et al. Paroxysmal exercise‐induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain 2008;131:1831–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castiglioni C, Verrigni D, Okuma C, et al. Pyruvate dehydrogenase deficiency presenting as isolated paroxysmal exercise induced dystonia successfully reversed with thiamine supplementation. Case report and mini‐review. Eur J Paediatr Neurol 2015;19:497–503. [DOI] [PubMed] [Google Scholar]
- 25. Tabarki B, Al‐Shafi S, Al‐Shahwan S, et al. Biotin‐responsive basal ganglia disease revisited: clinical, radiologic, and genetic findings. Neurology 2013;80:261–267. [DOI] [PubMed] [Google Scholar]
- 26. Roubertie A, Biolsi B, Rivier F, Humbertclaude V, Cheminal R, Echenne B. Ataxia with vitamin E deficiency and severe dystonia: report of a case. Brain Dev 2003;25:442–445. [DOI] [PubMed] [Google Scholar]
- 27. Muzaimi MB, Wiles CM, Robertson NP, Ravine D, Compston DA. Task specific focal dystonia: a presentation of spinocerebellar ataxia type 6. J Neurol Neurosurg Psychiatry 2003;74:1444–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuo PH, Gan SR, Wang J, et al. Dystonia and ataxia progression in spinocerebellar ataxias. Parkinsonism Relat Disord 2017;45:75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Monte TL, Pereira FS, Reckziegel EDR, et al. Neurological phenotypes in spinocerebellar ataxia type 2: Role of mitochondrial polymorphism A10398G and other risk factors. Parkinsonism Relat Disord 2017;42:54–60. [DOI] [PubMed] [Google Scholar]
- 30. Wichmann T, DeLong MR. Models of basal ganglia function and pathophysiology of movement disorders. Neurosurg Clin N Am 1998;9:223–236. [PubMed] [Google Scholar]
- 31. Lehericy S, Tijssen MA, Vidailhet M, Kaji R, Meunier S. The anatomical basis of dystonia: current view using neuroimaging. Mov Disord 2013;28:944–957. [DOI] [PubMed] [Google Scholar]
- 32. Batla A, Sanchez MC, Erro R, et al. The role of cerebellum in patients with late onset cervical/segmental dystonia?‐‐evidence from the clinic. Parkinsonism Relat Disord 2015;21:1317–1322. [DOI] [PubMed] [Google Scholar]
- 33. Filip P, Gallea C, Lehericy S, et al. Disruption in cerebellar and basal ganglia networks during a visuospatial task in cervical dystonia. Mov Disord 2017;32:757–768. [DOI] [PubMed] [Google Scholar]
- 34. Prudente CN, Hess EJ, Jinnah HA. Dystonia as a network disorder: what is the role of the cerebellum? Neuroscience 2014;260:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sadnicka A, Teo JT, Kojovic M, et al. All in the blink of an eye: new insight into cerebellar and brainstem function in DYT1 and DYT6 dystonia. Eur J Neurol 2015;22:762–767. [DOI] [PubMed] [Google Scholar]
- 36. Tewari A, Fremont R, Khodakhah K. It's not just the basal ganglia: Cerebellum as a target for dystonia therapeutics. Mov Disord 2017;32:1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ulug AM, Vo A, Argyelan M, et al. Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock‐in mice. Proc Natl Acad Sci U S A 2011;108:6638–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neychev VK, Fan X, Mitev VI, Hess EJ, Jinnah HA. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 2008;131:2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. LeDoux MS, Hurst DC, Lorden JF. Single‐unit activity of cerebellar nuclear cells in the awake genetically dystonic rat. Neuroscience 1998;86:533–545. [DOI] [PubMed] [Google Scholar]
- 40. Hisatsune C, Miyamoto H, Hirono M, et al. IP3R1 deficiency in the cerebellum/brainstem causes basal ganglia‐independent dystonia by triggering tonic Purkinje cell firings in mice. Front Neural Circuits 2013;7:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. LeDoux MS, Lorden JF, Ervin JM. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol 1993;120:302–310. [DOI] [PubMed] [Google Scholar]
- 42. Campbell DB, Hess EJ. L‐type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol 1999;55:23–31. [DOI] [PubMed] [Google Scholar]
- 43. Nibbeling EA, Delnooz CC, de Koning TJ, et al. Using the shared genetics of dystonia and ataxia to unravel their pathogenesis. Neurosci Biobehav Rev 2017;75:22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ilg W, Bastian AJ, Boesch S, et al. Consensus paper: management of degenerative cerebellar disorders. Cerebellum 2014;13:248–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sarva H, Shanker V. Treatment options in degenerative cerebellar ataxia: a systematic review. Mov Disord Clin Pract 2014;1:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol 2015;14:985–991. [DOI] [PubMed] [Google Scholar]
- 47. van Gaalen J, Kerstens FG, Maas RP, Harmark L, van de Warrenburg BP. Drug‐induced cerebellar ataxia: a systematic review. CNS Drugs 2014;28:1139–1153. [DOI] [PubMed] [Google Scholar]
- 48. van Harten PN, Hoek HW, Kahn RS. Acute dystonia induced by drug treatment. BMJ 1999;319:623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jinnah HA, Albanese A, Bhatia KP, et al. Treatable inherited rare movement disorders. Mov Disord 2018;33:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rossi M, Medina Escobar A, Ameghino L, Merello M. Expanding the phenotype of phosphomannomutase‐2 gene congenital disorder of glycosylation: Cervical dystonia. J Neurol Sci 2017;378:52–54. [DOI] [PubMed] [Google Scholar]
- 51. Wakusawa K, Haginoya K, Kitamura T, et al. Effective treatment with levodopa and carbidopa for hypomyelination with atrophy of the basal ganglia and cerebellum. Tohoku J Exp Med 2006;209:163–167. [DOI] [PubMed] [Google Scholar]
- 52. Feyma T, Ramsey K, Group CRR, et al. Dystonia in ATP2B3‐associated X‐linked spinocerebellar ataxia. Mov Disord 2016;31:1752–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Head RA, de Goede CG, Newton RW, et al. Pyruvate dehydrogenase deficiency presenting as dystonia in childhood. Dev Med Child Neurol 2004;46:710–712. [DOI] [PubMed] [Google Scholar]
- 54. Tuite PJ, Lang AE. Severe and prolonged dysphagia complicating botulinum toxin A injections for dystonia in Machado‐Joseph disease. Neurology 1996;46:846. [PubMed] [Google Scholar]
- 55. Wilder‐Smith E, Tan EK, Law HY, Zhao Y, Ng I, Wong MC. Spinocerebellar ataxia type 3 presenting as an L‐DOPA responsive dystonia phenotype in a Chinese family. J Neurol Sci 2003;213:25–28. [DOI] [PubMed] [Google Scholar]
- 56. Nandagopal R, Moorthy SG. Dramatic levodopa responsiveness of dystonia in a sporadic case of spinocerebellar ataxia type 3. Postgrad Med J 2004;80:363–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Aggarwal A, Bhatt M. The Pragmatic Treatment of Wilson's Disease. Mov Disord Clin Pract 2014;1:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kitahara M, Shimohata T, Tokunaga J, Nishizawa M. Cervical dystonia associated with spinocerebellar ataxia type 2 successfully treated with levodopa: a case report. Mov Disord 2009;24:2163–2164. [DOI] [PubMed] [Google Scholar]
- 59. Munchau A, Dressler D, Bhatia KP, Vogel P, Zuhlke C. Machado‐Joseph disease presenting as severe generalised dystonia in a German patient. J Neurol 1999;246:840–842. [DOI] [PubMed] [Google Scholar]
- 60. Koepp M, Schelosky L, Cordes I, Cordes M, Poewe W. Dystonia in ataxia telangiectasia: report of a case with putaminal lesions and decreased striatal [123I]iodobenzamide binding. Mov Disord 1994;9:455–459. [DOI] [PubMed] [Google Scholar]
- 61. Patterson MC, Hendriksz CJ, Walterfang M, et al. Recommendations for the diagnosis and management of Niemann‐Pick disease type C: an update. Mol Genet Metab 2012;106:330–344. [DOI] [PubMed] [Google Scholar]
- 62. Nardocci N, Bertagnolio B, Rumi V, Angelini L. Progressive dystonia symptomatic of juvenile GM2 gangliosidosis. Mov Disord 1992;7:64–67. [DOI] [PubMed] [Google Scholar]
- 63. Erro R, Tinazzi M, Morgante F, Bhatia KP. Non‐invasive brain stimulation for dystonia: therapeutic implications. Eur J Neurol 2017;24:1228–e1264. [DOI] [PubMed] [Google Scholar]
- 64. Beaulieu‐Boire I, Aquino CC, Fasano A, et al. Deep Brain Stimulation in Rare Inherited Dystonias. Brain Stimul 2016;9:905–910. [DOI] [PubMed] [Google Scholar]
- 65. Sidiropoulos C, Hutchison W, Mestre T, et al. Bilateral pallidal stimulation for Wilson's disease. Mov Disord 2013;28:1292–1295. [DOI] [PubMed] [Google Scholar]
- 66. Georgiev D, Mehta D, Zacharia A, et al. Bilateral Deep Brain Stimulation of the Globus Pallidus Pars Interna in a Patient with Variant Ataxia‐Telangiectasia. Mov Disord Clin Pract 2016;3:405–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oyama G, Thompson A, Foote KD, et al. Deep brain stimulation for tremor associated with underlying ataxia syndromes: a case series and discussion of issues. Tremor Other Hyperkinet Mov (N Y) 2014;4:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nam TM, Cho KR, Youn J, Cho JW, Lee JI. Deep brain stimulation in a dentatorubral‐pallidoluyisian atrophy patient with myoclonic dystonia. J Clin Neurosci 2015;22:1976–1978. [DOI] [PubMed] [Google Scholar]
- 69. Copeland BJ, Fenoy A, Ellmore TM, Liang Q, Ephron V, Schiess M. Deep brain stimulation of the internal globus pallidus for generalized dystonia associated with spinocerebellar ataxia type 1: a case report. Neuromodulation 2014;17:389–392. [DOI] [PubMed] [Google Scholar]
- 70. Harries AM, Sandhu M, Spacey SD, Aly MM, Honey CR. Unilateral pallidal deep brain stimulation in a patient with dystonia secondary to episodic ataxia type 2. Stereotact Funct Neurosurg 2013;91:233–235. [DOI] [PubMed] [Google Scholar]
- 71. Hebb MO, Gaudet P, Mendez I. Deep brain stimulation to treat hyperkinetic symptoms of Cockayne syndrome. Mov Disord 2006;21:112–115. [DOI] [PubMed] [Google Scholar]
- 72. Hamasaki K, Yamada K, Hamasaki T, Kuratsu J. GPi‐pallidal stimulation to treat generalized dystonia in Cockayne syndrome. Mov Disord 2010;25:656–658. [DOI] [PubMed] [Google Scholar]
- 73. Cif L, Kurian M, Gonzalez V, et al. Atypical PLA2G6‐Associated Neurodegeneration: Social Communication Impairment, Dystonia and Response to Deep Brain Stimulation. Mov Disord Clin Pract 2014;1:128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Krause M, Fogel W, Tronnier V, et al. Long‐term benefit to pallidal deep brain stimulation in a case of dystonia secondary to pantothenate kinase‐associated neurodegeneration. Mov Disord 2006;21:2255–2257. [DOI] [PubMed] [Google Scholar]
- 75. Castelnau P, Cif L, Valente EM, et al. Pallidal stimulation improves pantothenate kinase‐associated neurodegeneration. Ann Neurol 2005;57:738–741. [DOI] [PubMed] [Google Scholar]
- 76. Timmermann L, Pauls KA, Wieland K, et al. Dystonia in neurodegeneration with brain iron accumulation: outcome of bilateral pallidal stimulation. Brain 2010;133:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mikati MA, Yehya A, Darwish H, Karam P, Comair Y. Deep brain stimulation as a mode of treatment of early onset pantothenate kinase‐associated neurodegeneration. Eur J Paediatr Neurol 2009;13:61–64. [DOI] [PubMed] [Google Scholar]
- 78. Renaud M, Tranchant C, Martin JVT, et al. A recessive ataxia diagnosis algorithm for the next generation sequencing era. Ann Neurol 2017;82:892–899. [DOI] [PubMed] [Google Scholar]
- 79. Tranchant C, Koob M, Anheim M. Parkinsonian‐Pyramidal syndromes: A systematic review. Parkinsonism Relat Disord 2017;39:4–16. [DOI] [PubMed] [Google Scholar]
- 80. Sethi K, Lang AE. Will New Genetic Techniques Like Exome Sequencing Obviate the Need for Clinical Expertise? Mov Disord Clin Pract 2017;4:39–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Synofzik M, Schule R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov Disord 2017;32:332–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Synofzik M, Gasser T. Moving Beyond Syndromic Classifications in Neurodegenerative Disease: The Example of PLA2G6. Mov Disord Clin Pract 2017;4:8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Erro R, Balint B, Kurian M, et al. Early ataxia and subsequent parkinsonism: PLA2G6 mutations cause a continuum rather than three discrete phenotypes. Mov Disord Clin Pract 2017;4:125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Termsarasab P, Yang AC, Frucht SJ. Intermediate Phenotypes of ATP1A3 Mutations: Phenotype‐Genotype Correlations. Tremor Other Hyperkinet Mov (N Y) 2015;5:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nicita F, Travaglini L, Sabatini S, et al. Childhood‐onset ATP1A3‐related conditions: Report of two new cases of phenotypic spectrum. Parkinsonism Relat Disord 2016;30:81–82. [DOI] [PubMed] [Google Scholar]
- 86. Perez‐Torre P, Escobar Villalba A, Martinez Ulloa P, et al. PLA2G6‐associated neurodegeneration: report of a novel mutation in 2 siblings with striking different clinical presentation. Mov Disord Clin Pract 2017;4:129–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lill CM, Mashychev A, Hartmann C, et al. Launching the movement disorders society genetic mutation database (MDSGene). Mov Disord 2016;31:607–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information may be found in the online version of this article.
Table S1. Genetic diseases that may combine dystonia with ataxia
Table S2. Specific treatment implications