

Abstract
Iron regulatory proteins 1 and 2 (IRP1 and IRP2) are RNA binding proteins that posttranscriptionally regulate the expression of mRNAs coding for proteins involved in the maintenance of iron and energy homeostasis. The RNA binding activities of the IRPs are regulated by changes in cellular iron. Thus, the IRPs are considered iron sensors and the principle regulators of cellular iron homeostasis. The mechanisms governing iron regulation of the IRPs are well described. Recently, however, much attention has focused on the regulation of IRPs by reactive nitrogen and oxygen species (RNS, ROS). Here we focus on summarizing the iron-regulated RNA binding activities of the IRPs, as well as the recent findings of IRP regulation by RNS and ROS. The recent observations that changes in oxygen tension regulate both IRP1 and IRP2 RNA binding activities will be addressed in light of ROS regulation of the IRPs.
Keywords: Iron regulatory proteins, Reactive nitrogen species, Reactive oxygen species
MAMMALIAN IRON HOMEOSTASIS
Iron is required by cells for survival and growth. This metal plays a role in a variety of biological processes and serves as a cofactor for many enzymes. Iron is also required for the synthesis of heme and, therefore, for the activity of all hemoproteins. Free iron is toxic due to its ability to catalyze the generation of free radicals that oxidize proteins and DNA and initiate peroxidation of lipid membranes. In humans, the accumulation of excess cellular iron can result in cirrhosis, arthritis, cardiomyopathy, diabetes mellitus, and increased risk of cancer and heart disease.
Cells balance their need for iron with the toxicity of iron by regulating both iron uptake and storage. Cells accumulate iron by transferrin (Tf)-dependent and Tf-independent mechanisms (2,3,17,79). The Tf-dependent process involves the binding of transferrin-Fe3+ to the transferrin receptor (TfR), which is then endocytosed into cells where iron is released. Tf-independent mechanisms involve the uptake of iron by the proton-coupled metal transporter, DMT1/ Nramp2/DCT1 (38), and by the iron transporter, SF1 (40). Iron taken up by cells enters a labile pool consisting of low molecular weight iron complexes (52). Although the exact nature of this pool is unknown, it appears that iron is chelated to ligands such as citrate, ascorbate, amino acids, and nucleotides. This pool is usually small due to iron’s ability to catalyze free radical formation. Increases in the iron pool result in the induction of ferritin, which stores iron in a form unavailable to catalyze free radical formation, and in the destabilization of the TfR mRNA, resulting in decreased iron uptake. The coordinate regulation of ferritin and TfR by iron provides a mechanism by which cells balance their requirement for iron with the toxicity of iron.
REGULATION OF IRON HOMEOSTASIS BY IRON REGULATORY PROTEINS
Iron homeostasis is regulated by the iron regulatory proteins (IRPs) 1 and 2. IRPs are cytosolic RNA binding proteins that posttranscriptionally regulate the expression of proteins involved in iron uptake, storage, and utilization (47,57,80,81). IRPs bind with high affinity to RNA stem-loops, known as iron-responsive elements (IREs). Structural studies have shown that IREs consist of a conserved six-membered loop and an unconserved base paired stem containing either a bulge-C or an internal loop/bulge UGC/C (1,34,88). IREs are located in the 5′-untranslated (UTR) regions of mRNAs encoding the iron storage protein, ferritin; the heme biosynthetic enzyme, erythroid-aminolevulinate synthase; and the Krebs cycle enzymes, mitochondrial (m)-aconitase and Drospophila succinate dehydrogenase. The binding of IRPs to the 5′ IREs inhibits translation of these mRNAs by preventing 43S ribosome binding (65). Five IREs are located in the 3′ UTR of the TfR mRNA, where IRP binding stabilizes this mRNA and protects it from endonuclease attack (6). One IRE is located in the 3′ UTR of the cation transporter mRNA, DMT1 (38).
Mammalian IRP1 shares ∼30% identity with the [4Fe-4S] containing enzyme, m-aconitase. Aconitase is a Krebs cycle enzyme that interconverts citrate and isocitrate via the intermediate cis-aconitate, and its activity depends on the presence of a [4Fe-4S] cluster. Aconitase contains four domains surrounding an active-site cleft where substrate is bound (4,36). Three iron atoms within the cluster are liganded to three cysteines within this cleft, whereas the fourth iron, Fea, is solvent exposed and has a free coordination site that binds substrate. The 18 active-site residues in m-aconitase, including the three cysteines that serve as ligands for the [4Fe-4S] cluster, are conserved in IRP1. IRP1 exhibits aconitase activity and is identical to a previously described c-aconitase (46).
IRP1 RNA binding activity is regulated by cellular iron levels. Iron posttranslationally converts the apo-RNA binding form into the active [4Fe-4S] c-aconitase form without changes in IRP1 protein or mRNA levels (Fig. 1). RNA binding and c-aconitase activities are mutually exclusive. RNA binding activity of IRP1 is enhanced by phosphorylation (84). The process of cluster assembly for the aconitases is not understood; however, a genetic screen in yeast revealed three proteins that are involved in assembly and maturation of mitochondrial Fe-S-containing proteins (90). The [4Fe-4S] cluster can be disassembled by nitric oxide (NO#) and ROS in vivo and in vitro (Fig. 1). Although the function of c-aconitase is unknown, the conservation of this activity suggests that it has a functional role in iron and/or energy homeostasis. IRP2 shares ∼60% identity with IRP1; however, unlike IRP1, IRP2 does not have detectable aconitase activity and does not appear to contain a [4Fe-4S] cluster. In addition, IRP2 is regulated by iron-induced proteolysis by the proteasome (39,51). Degradation is mediated by an iron-dependent oxidation mechanism that requires a unique 73-amino acid degradation domain containing three essential cysteines (39,50,51). Phosphorylation has also been shown to enhance IRP2 RNA binding activity (86).
FIG. 1.
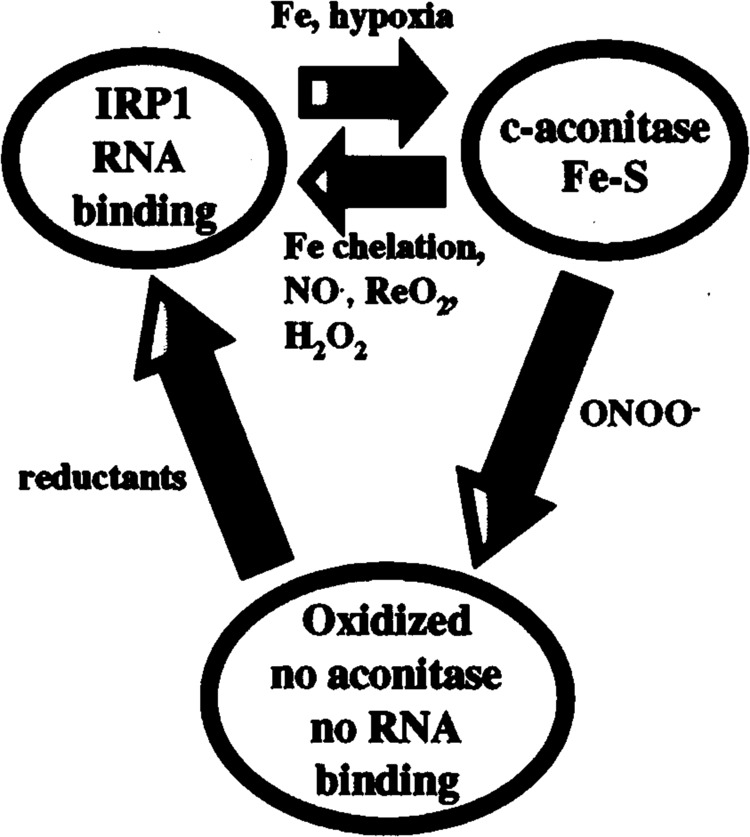
Model for the regulation of IRP1 by iron, ROS, and RNS. IRP1 interconverts between a RNA binding form and a [4Fe-4S] c-aconitase form. Iron or hypoxia converts the apo-RNA binding form into the [4Fe-4S] aconitase form. NO•, H2O2, iron chelation, or reoxygenation (ReO2) results in the formation of the IRP1 RNA binding form. ONNO– results in the disassembly of the [4Fe-4S] cluster and oxidation of cysteines required for RNA binding. This results in the formation of an oxidized protein, lacking RNA binding and c-aconitase activities. Addition of reductants to oxidized IRP1 restores RNA binding activity.
The functional role of two IRPs in the regulation of iron homeostasis is not known. IRP1 and IRP2 can specifically bind to unique subsets of synthetic IREs (12,45), and appropriately regulate RNAs containing these IREs in vivo (64). IRP1 and IRP2 bind naturally occurring IREs with different affinities. For example, IRP2 binds IREs containing the UGC/C-bulge (ferritin IRE) with higher affinity than C-bulge IREs (TfR-B, aconitase, and aminolevulinate synthase IREs) (56). These data suggest that IRP1 and IRP2 may bind and regulate different IRE mRNAs in vivo. The distinctive roles of IRP1 and IRP2 in iron homeostasis is further questioned by the finding of a cell line lacking IRP1, indicating the importance of IRP2 in IRE regulation (85). Identification of novel IRE mRNAs with specificity for IRP1 or IRP2 will expand our knowledge of the repertoire of the IRP network.
REGULATION OF IRPs BY NO•
Nitric oxide (NO•) is a reactive nitrogen species that is synthesized by nitric oxide synthases (NOSs) (67). NO• serves as a ubiquitous second messenger and modulates a myriad of physiological functions, including neuronal signaling, vascular tone, and antibacterial activity of macrophages. NO• could be cytotoxic due to its ability to react with transition metals and sulfhydryls of proteins, leading to their inactivation (89). NO• produced by activated macrophages can kill tumor cells and combat invading microorganisms (68). When activated macrophages are co-cultured with tumor cells, the production of macrophage NO• results in the inactivation of mitochondrial Fe-S-containing enzymes, leading to inhibition of respiration and subsequent tumor cell death (20,21).
The affinity of NO• for Fe-S clusters, and the inactivation of m-aconitase by NO• led to the notion that NO• might also modulate IRP1 activity (18,19). When macrophages (22,77,97), fibroblasts (70,73), hepatoma cells (75), and erythroleukemia cells (78,97) are stimulated with cytokines to produce NO• or treated with NO•-generating chemicals, c-aconitase was inactivated and converted into its IRP1 RNA binding form. These data supported a model whereby NO• modulated IRP1/c-aconitase activity; however, questions arose concerning whether inactivation in vivo was due to a direct effect by NO• or by a NO•-derived species such as peroxynitrite (ONOO−). ONOO− is the reaction product of superoxide anion (O2 •–) and NO•. Two studies reported that ONOO−, but not NO•, inactivated aconitase (14,44). Recent studies by Gardner and colleagues refuted these data, demonstrating that the NO•-mediated inactivation of Escherichia coli aconitase in vivo was due to S-nitrosylation of the [4Fe-4S] center, which occurred independently of ONOO− (28). These data were supported by an electron paramagnetic resonance study showing that NO• reacts directly with the [4Fe-4S] cluster of m-aconitase and c-aconitase, resulting in cluster disassembly and the formation of a dintrosyl-iron-dithiol complex (58). ONOO− can inactivate c-aconitase in cell extracts; however, in contrast to NO•, ONOO− does not activate IRP1 RNA binding (8). In addition to disrupting the [4Fe-4S] cluster, ONOO− oxidizes critical cysteines required for RNA binding (8). The physiological role of ONOO− in IRP1 regulation in vivo is unclear. Whether NO• modulates IRP1 activity by S-nitrosylation of thiol groups in IRP1 as suggested by Ponka and colleagues remains to be determined (78).
Conflicting data regarding IRP2 regulation by NO• have been reported. IRP2 RNA binding was activated in J774 macrophages (96,97), fibroblasts (73), and in the liver during inflammation (13), but not in rat hepatoma cells (75). In contrast, other studies using J774 (77) and RAW264.7 macrophages (7) stimulated with cytokines to produce NO•, inactivation of IRP2 RNA binding was observed. In J774 macrophages, the addition of the inducible NOS inhibitor, N G-mono-methyl-L-arginine monoacetate or the iron chelator, deferioxamine, prevented the decrease in IRP2 RNA binding activity, indicating roles for both NO• and iron in IRP2 inactivation (77). Iron can be released from activated macrophages co-cultured with tumor cells, suggesting that NO• may alter iron levels at least in some cell types (48). In RAW264.7 macrophages, the decrease in IRP2 RNA binding activity was independent of NO•, suggesting that cytokines may modulate IRP2 activity by other pathways (7).
The discrepancy among these studies is unclear. Recent studies have shown that in addition to iron-mediated oxidative damage (50), IRP2 activity is sensitive to oxidants. For example, IRP2 RNA binding is inactivated by 5′5′-dithiobis(2-nitrobenzoic) (DTNB) (75), ONOO− (7,8), and cobalt chloride (42). In contrast, under hypoxic conditions where ROS are expected to be altered, IRP2 RNA binding is activated (42). It is possible that cell growth conditions (density, medium, and serum) vary among the studies, resulting in subtle differences in the relative concentrations of iron and/or ROS, which can modulate IRP2 RNA binding activity. One recent study showed that in macrophages grown in l-arginine-depleted medium, inducible NOS generates both NO• and O2 •–, which react to form ONOO−, revealing the importance of culture conditions on ROS/RNS formation (99).
What effect does NO• regulation of IRP1 and IRP2 have on target IRE mRNAs? Activation of IRP1 and IRP2 RNA binding correlated with decreased ferritin synthesis in macrophages (96,97), hepatoma cells (75), and fibroblasts (73), and increased TfR mRNA levels in erthroleukemia cells (70,78). In experiments where IRP2 activity decreased, ferritin synthesis increased and TfR mRNA levels decreased (77). The contributions of IRP1 and IRP2 in the regulation of ferritin, TfR, and other IRE mRNAs remain to be determined.
The physiological significance of NO• regulation of IRPs is unclear. Cairo and colleagues proposed that the increase in ferritin synthesis in J774 macrophages producing NO• is consistent with in vivo models of inflammation where iron is retained in reticuloendothelial cells (77). Regulation of IRP1 and IRP2 by NO• may be important in the anemia of chronic disease where the sequestration of iron by macrophages limits iron for hematopoiesis (18,77).
REACTIVE OXYGEN SPECIES
The O2 •– anion is a ubiquitous and naturally produced reactive oxygen metabolite (25). There are several sources leading to the production of cytosolic 0r including the NADP(H) oxidase (41) and xanthine oxidase systems (63). A significant amount of cellular O2 •– production is derived from the incomplete reduction of O2 by the mitochondrial electron transport machinery (15,23,93). It has been estimated that approximately 1–2% of the total cellular uptake of O2 is not fully reduced during respiration (9,10). The potential toxicity of mitochondrial- and cytosolic-derived CV is countered by the superoxide dismutases (SODs) that dismutates O2 •– to the less reactive and more membrane diffusible H2O2 molecule (25,26). The concerted activities of the SODs and the H2O2 enzymatic decomposing systems, catalase and glutathione peroxidase, result in a steady-state level of ∼10−10 M for O2 •– and 10−8 M for H2O2 (9).
ROS levels increase during aging and pathophysiological conditions that can lead to significant cellular oxidative damage (5). Furthermore, ROS have important roles in regulating cellular gene expression. For instance, H2O2 has gained recognition as an important signaling molecule involved in the regulation of MAP kinase pathways, as well as mediating transcriptional regulation of the NF-kB and AP1 transcription factors (55,66,91). The IRPs represent a posttranscriptional gene regulatory system that also responds to ROS. Regulation of IRPs by ROS allows IRPs to sense signals from oxygen-derived species in addition to sensing iron. Although not yet fully elucidated, the convergence of these signals on IRPs presumably coordinates communication between iron, oxidative stress, and oxygen homeostasis. Understanding the respective contributions that these signals have on IRP regulation in vivo is an important and formidable task.
REGULATION OF IRPs BY ROS
Regulation of IRP1/c-Aconitase by H2O2
IRP1/c-aconitase activities are affected by H2O2 (8,62,69,71–73) and O2 •– (33,71). A central feature that these species share is the ability to modulate the aconitase [4Fe-4S] cluster. The best understood example of ROS regulation of IRP1/c-aconitase is the pathway initiated by extracellular H2O2. Martins et al. (62) and Pantopoulos et al. (71) were the first to report that IRP1 RNA binding activity is regulated by oxidative stress when they demonstrated rapid activation of IRP1 RNA binding activity by H2O2. De novo protein synthesis is not required for activation, demonstrating that H2O2 operates by a posttranslational mechanism (71). Furthermore, the continuous presence of H2O2 for IRP1 activation is not required. Rather, brief exposure to H2O2 is sufficient to initiate a signaling pathway resulting in IRP1 activation (62,71). As would be predicted, IRP1 activation by H2O2 results in the repression of ferritin protein synthesis and the upregulation of TfR mRNA (71). The observation that H2O2 activates IRP1 has since been reproduced by other laboratories [(64); E. S. Hanson and E. A. Leibold, unpublished results].
The regulation of IRP1 by H2O2 is mediated by an unknown extracellular event that initiates a signaling cascade (71–73). Using cytosolic extracts, H2O2 was not able to activate RNA binding (62,71), indicating that H2O2 is not directly acting on IRP1. Recently, an in vitro assay has recapitulated H2O2 activation of IRP1 similar to that seen using intact cells. Both cytosolic and membrane fractions are required for H2O2 activation of IRP1, demonstrating activation requires a multicomponent system (69). Because H2O2 activation of IRP1 is concomitant with decreased c-aconitase activity, the proximal signal in IRP1 activation may involve [4Fe-4S] cluster disassembly (53,71). The signaling pathway and the precise mechanism for IRP1 activation by extracellular H2O2 have yet to be defined.
Regulation of Aconitase by O2 •–
Aconitase activity is dependent on the presence of a cubane [4Fe-4S] cluster. Oxidative destruction of the [4Fe-4S] cluster leads to enzyme inactivation. This property is not unique to aconitases because other [4Fe-4S]-containing enzymes undergo oxidatively induced inactivation as well (24). It is well established that aconitases from E. coli (24,30,31,44) and mammalian (32,33) sources are sensitive to inactivation by O2 •–. Both m- and c-aconitase activities are inactivated by O2 •– (33,44). Superoxide-induced inactivation of aconitase results from the oxidation of the [4Fe-4S] cluster due to the loss of a labile, solvent-exposed iron atom (Fea) yielding a [3Fe-4S] cluster ([4Fe-4S]+2 → [3Fe-4S]+1). Due to the continuous attack of the [4Fe-4S] cluster by steady-state levels of O2 •–, it has been estimated that ∼15% of total cellular aconitase activity is inactivated at any given time under normal respiratory conditions (33). Inactivation is inversely proportional to O2 •– concentration (29,30,32). Accordingly, overexpression of the mitochondrial Mn-SOD (33) or the cytosolic CuZn-SOD (92) results in increased aconitase activity. Furthermore, Mn-SOD+/– mice have a ∼30% decrease in m-aconitase activity, but no change in c-aconitase activity (60,98). Therefore, it is predicted that pathophysiological situations resulting in increased O2 •– levels will be associated with an increased rate of cluster disintegration. Conversely, decreased O2 •– should stabilize the Fe-S cluster leading to increased aconitase activity. The fate of O2 •–-inactivated aconitase is not unidirectional, because rapid reactivation was demonstrated in a lung cell line (33) and in E. coli (30). Thus, reintroduction of the fourth iron into the [3Fe-4S] form results in a cyclic process of inactivation–reactivation (27,32,33).
Regulation of IRP1 RNA Binding Activity During Hypoxia
One situation that may represent O2 •– regulation of IRP1/c-aconitase is hypoxia. Hypoxia is an important regulator of gene expression (11). The best understood example of mammalian oxygen-regulated gene expression is that mediated by the transcription factor hypoxia-inducible factor-1 (HIF-1) (11,37,76,87), whereas relatively little information is available regarding posttranscriptional mechanisms (16,59). Because IRP1 is regulated by ROS, and because ROS production is ultimately dictated by O2 concentration, we speculated that IRP1 may be regulated by changes in O2 concentration. To investigate a potentially novel pathway for gene regulation during hypoxia, a study on the regulation of the IRPs by changes in O2 concentration was undertaken.
We have recently reported that hypoxia (1–3% O2) posttranslationally downregulates IRP1 while upregulating IRP2 RNA binding activity in a variety of cell types (Fig. 2) (42,43). Iron is required for IRP1 hypoxic inactivation implicating the Fe-S cluster in “sensing” changes in O2. Decreased IRP1 RNA binding activity during hypoxia is accompanied by ∼40% increase in c-aconitase activity (E. S. Hanson and E. A. Leibold, unpublished results). Because there are no detectable changes in IRP1 protein levels (43), and because inactivation is cycloheximide insensitive (E. S. Hanson and E. A. Leibold, unpublished data), it appears that the [4Fe-4S] cluster is stabilized during hypoxia. This indicates that decreased O2 concentration promotes a posttranslational conversion from IRP1 RNA binding to its [4Fe-4S] aconitase form. How does the Fe-S cluster of IRP1 “sense” changes in O2? At least two possibilities arise that are not mutually exclusive (Fig. 3). First, hypoxia may increase iron leaching from mitochondrial Fe-S clusters due to hypoxia-induced increases in mitochondrial O2 •– production (23,61,93). Iron liberated in this manner could encourage c-aconitase [4Fe-4S] cluster formation at the expense of RNA binding activity. If this were the case, IRP2 activity would be predicted to decrease when in fact hypoxia increases IRP2 activity (Fig. 2). Thus, it appears that any increase in cellular iron during hypoxia cannot be the only factor in the hypoxic regulation of IRPs. A second possibility for the hypoxic stabilization of c-aconitase may involve decreased cytosolic O2 •– production during hypoxia. A decrease in cytosolic O2 •– would slow the rate of O2 •–-mediated cluster disassembly. Because aconitase inactivation has been used as a marker for O2 •– (25,30,74), this latter scenario would suggest that the increase in mitochondrial generated O2 •– that accompanies hypoxia is not accessible to the cytosol (23,93). This result indicates that O2 •– is compartmentalized, which is consistent with data demonstrating that Mn-SOD+/– mice display a decrease in m-aconitase but not c-aconitase (98). It should be noted, however, that there are contradicting reports regarding cellular compartmentalization of O2 •– (15,33,93).
FIG. 2.

Hypoxic regulation of IRP1 and IRP2 Hepa-1 cells. Mouse Hepa-1 clc4 were exposed to normoxia (N) or hypoxia (1% O2) for the indicated times. Bandshift analysis was performed by incubating cytosolic extracts (12 μg) with a 32P-labeled iron responsive element RNA probe. The RNA–protein complexes were resolved on a 5% nondenaturing polyacrylamide gel, and the gel was exposed to film. IRP1–RNA and IRP2–RNA complexes are indicated.
FIG. 3.
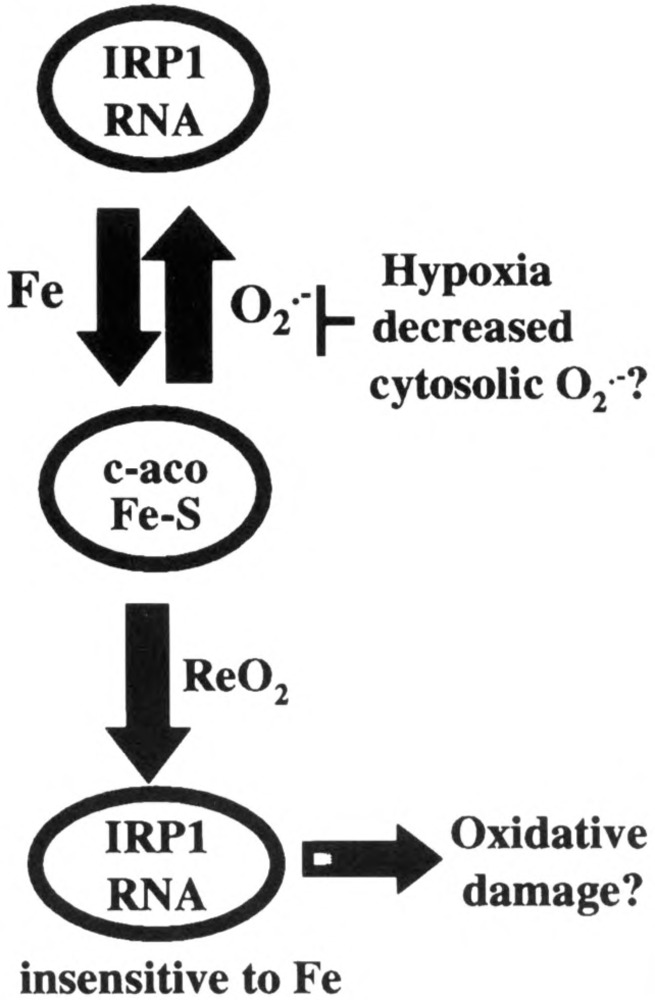
Model depicting oxygen regulation of IRP1. During normoxia IRP1 interconverts between its RNA binding form and its [4Fe-4S] c-aconitase form. This interconversion is dependent on the relative levels of iron and O2 •–. Hypoxia decreases RNA binding activity (see Fig. 2) and increases c-aconitase activity. The model suggests that hypoxic regulation of IRP1 could be due to increased iron and/or decreased cytosolic O2 •–, either of which would lead to stabilization of the [4Fe-4S] cluster at the expense of RNA binding activity. Reoxygenation (ReO2) activates IRP1 to a constitutively active RNA binding form. The dysregulated form of IRP1 (shaded) is refractory to iron downregulation. By adversely affecting iron levels, it is possible that this form of IRP1 may contribute to ReO2-induced oxidative damage.
Hypoxic inactivation of IRP1 is reversible because upon reexposure to normoxia RNA binding ensues (Fig. 3) (43). Importantly, reactivated IRP1 is modified to a dysregulated form since iron does not down-regulate its RNA binding activity. The mechanism for IRP1 dysregulation is not known; however, one possibility may involve some form of oxidative protein damage that precludes [4Fe-4S] cluster formation. Interestingly, the prooxidant condition associated with aged house flies is responsible for oxidative damage and inactivation of m-aconitase (100). Whether this bears any relationship to reoxygenation-induced IRP1 activation is not known. Furthermore, it will be of interest to determine if mammalian IRP1/c-aconitase is altered during aging in mammalian cells. A second possibility is that IRP1 may be phosphorylated during reoxygenation, which could preclude Fe-S assembly (84). Finally, ROS produced during reoxygenation may disassemble the Fe-S cluster faster than cluster assembly. Regardless of the mechanism, the inability of IRP1 to sense iron during reoxygenation is predicted to result in aberrant iron homeostasis, and may be an important contributor to reoxygenation-induced cell injury, an event known to be exacerbated by increased iron.
Hypoxic Activation of IRP2
IRP2 RNA binding activity is significantly upregulated during hypoxia (Fig. 2). The increase in RNA binding activity parallels an equal increase in IRP2 protein levels (42). Hypoxic activation of IRP2 has several intriguing similarities to the well-studied HIF-1α subunit of HIF-1. HIF-1 is a heterodimeric transcription factor (HIF-1α/β) composed of two helix–loop–helix PAS family proteins (37,76,94). HIF-1 activates the transcription of many genes during hypoxia and therefore is believed to be an important mediator of cellar adaptation to hypoxia. Whereas HIF-1 p levels do not change during hypoxia, both HIF-1 α and IRP2 are activated during hypoxia by a mechanism involving protein stabilization (42,49,82). Iron chelation and CoCl2 mimic hypoxia by increasing HIF-1 α (54,95) and IRP2 protein levels (39,42,83). Although CoCl2 increases IRP2 levels, IRP2 is unable to bind RNA in bandshift analysis unless first reduced with DTT. Thus, CoCl2 has the dual effect of inducing IRP2 accumulation while at the same time reversibly inactivating RNA binding. Whether redox regulation of IRP2 occurs in vivo under more physiological conditions is not known.
It has been suggested that CoCl2 and iron chelation mimic hypoxia by inactivation of a cytosolic hemoprotein that functions as an O2 sensor (11,35,37). Such an O2 sensor could modulate a signal during low O2 conditions that could result in IRP2 and HIF-1α stabilization. However, such a cytosolic O2 sensing protein has not yet been definitively identified. Mechanistic studies have demonstrated that IRP2 is oxidized by metal-catalyzed oxidation mechanism requiring Fe2+ and O2. In turn, oxidized IRP2 results in a good substrate for ubiquitination and proteasomal degradation (Fig. 4) (50). Based on this finding, we suggest a model whereby altering the rate of IRP2 metal-catalyzed oxidation by hypoxia and CoCl2 activates IRP2 by a mechanism involving protein stabilization (Fig. 4). CoCl2 could also compete for iron binding at the degradation domain, thus blocking IRP2 iron sensing.
FIG. 4.

Model for the oxygen regulation of IRP2. Normoxic degradation of IRP2 by the proteasome is dependent on the 73-amino acid degradation domain that contains three essential cysteines required to sense Fe2+ (hashed) (51). In this model, IRP2 stability is dependent on Fe2+ and H2O2, which are required for oxidative modification leading to ubiquitination (Ub) and proteasomal degradation (50). Hypoxia, iron chelation, and CoCl2 increase IRP2 protein levels by stabilization. Hypoxia may act through an unknown O2 sensor that lowers cytosolic H2O2 resulting in decreased IRP2 oxidation and degradation. CoCl2 mimics hypoxia by possibly altering the activity of an O2 sensor or by competing with iron at an iron binding site on the degradation domain.
It should be noted that recent data indicate that the mitochondria may function as an O2 sensor that initiates signaling to HIF-1α by increasing rather than decreasing H2O2 levels (15,23,93). Elucidating the source(s) and defining the precise signaling pathway(s) for IRP2 and HIF-1α hypoxic stabilization is an important next step in understanding hypoxic signaling and gene regulation.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grants GM45201 (to E.A.L.) and T32 DK07115 (to E.S.H.), and a grant from the Huntsman Cancer Institute (to E.A.L.). We thank Dr. Prem Ponka for critical reading of this manuscript.
REFERENCES
- 1. Addess K. J.; Basilion J. P.; Klausner R. D.; Rou-ault T. A.; Pardi A. Structure and dynamics of the iron responsive element RNA: Implications for binding of the RNA by iron regulatory binding proteins. J. Mol. Biol. 274:72–83; 1997. [DOI] [PubMed] [Google Scholar]
- 2. Aisen P.; Wessling-Resnick M.; Leibold E. A. Iron metabolism. Curr. Opin. Chem. Biol. 3:200–206; 1999. [DOI] [PubMed] [Google Scholar]
- 3. Andrews N. C. ; Levy J. E. Iron is hot: An update on the pathophysiology of hemochromatosis. Blood 92: 1845–1851; 1998. [PubMed] [Google Scholar]
- 4. Beinert H.; Kennedy M. C. Aconitase, a two-faced protein: Enzyme and iron regulatory factor. FASEB J. 7:1442–1449; 1993. [DOI] [PubMed] [Google Scholar]
- 5. Berlett B. S.; Stadtman E. R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272:20313–20316; 1997. [DOI] [PubMed] [Google Scholar]
- 6. Binder R.; Horowitz J. A.; Basilion J. P.; Koeller D. M.; Klausner R. D.; Harford J. B. Evidence that the pathway of transferrin receptor mRNA degradation involves an endonucleolytic cleavage within the 3′UTR and does not involve poly(A) tail shortening. EMBOJ. 13:1969–1980; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouton C.; Oliveira L.; Drapier J. Converse modulation of IRP1 and IRP2 by immunological stimuli in murine RAW 264.7 macrophages. J. Biol. Chem. 273:9403–9408; 1998. [DOI] [PubMed] [Google Scholar]
- 8. Bouton C.; Hirling H.; Drapier J. Redox modulation of iron regulatory proteins by peroxynitrite. J. Biol. Chem. 272:199969–199975; 1997. [DOI] [PubMed] [Google Scholar]
- 9. Boveris A.; Cadenas E. Cellular sources and steady-state levels of reactive oxygen species. In: Biadasz Clerch L.; Massaro D. J., eds. Oxygen gene expression and cellular function. New York: Marcel Dekker, Inc.; 1997:1–25. [Google Scholar]
- 10. Boveris A.; Oshino N.; Chance B. The cellular production of hydrogen peroxide. Biochem. J. 128:617–630; 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bunn H. F.; Poyton R. O. Oxygen sensing and molecular adaptation to hypoxia. Physiol. Rev. 76:839–885; 1996. [DOI] [PubMed] [Google Scholar]
- 12. Butt J.; Kim H.; Basilion J.; Cohen S.; Iwai K.; Philpott C.; Altschul S.; Klausner R.; Rouault T. Differences in the RNA binding sites of iron regulatory proteins and potential target diversity. Proc. Natl. Acad. Sci. USA 93:4345–4349; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cairo G.; Pietrangelo A. Nitric-oxide-mediated activation of iron-regulatory protein controls hepatic iron metabolism during acute inflammation. Eur. J. Biochem. 232:358–363; 1995. [DOI] [PubMed] [Google Scholar]
- 14. Castro L.; Rodriguez M.; Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J. Biol. Chem. 269:29409–29415; 1994. [PubMed] [Google Scholar]
- 15. Chandel N.; Maltepe E.; Goldwasser E.; Mathieu C.; Simon M.; Schumacker P. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 95:11715–11720; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Czyzyk-Krzeska M. F.; Paulding W. R.; Beresh J. E.; Kroll S. L. Post-transcriptional regulation of tyrosine hydroxylase gene expression by oxygen in PC 12 cells. Kidney Int. 51:585–590; 1997. [DOI] [PubMed] [Google Scholar]
- 17. De Silva D.; Askwith C. C.; Kaplan J. Molecular mechanisms of iron uptake in eukaryotes. Physiol. Rev. 76:31–47; 1996. [DOI] [PubMed] [Google Scholar]
- 18. Domachowske J. B. The role of nitric oxide in the regulation of cellular iron metabolism. Biochem. Mol. Med. 60:1–7; 1997. [DOI] [PubMed] [Google Scholar]
- 19. Drapier J. C.; Bouton C. Modulation by.nitric oxide metalloprotein regulatory activities. Bioessays 18: 549–556; 1996. [DOI] [PubMed] [Google Scholar]
- 20. Drapier J. C.; Hibbs J. B. Differentiation of murine macrophages to express nonspecific cytotoxicity for tumor cells results in L-arginine-dependent inhibition of mitochondrial iron-sulfur enzymes in the macro-phage effector cells. J. Immunol. 140:2829–2838; 1988. [PubMed] [Google Scholar]
- 21. Drapier J. C.; Hibbs J. B. J. Murine cytotoxic activated macrophages inhibit aconitase in tumor cells. Inhibition involves the iron-sulfur prosthetic group and is reversible. J. Clin. Invest. 78:790–797; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Drapier J. C.; Hirling H.; Wietzerbin J.; Kaldy P.; Kühn L. C. Biosynthesis of nitric oxide activates iron regulatory factor in macrophages. EMBO J. 12:3643–3649; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Duranteau J.; Chandel N. S.; Kulisz A.; Shao Z.; Schumacker P. T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 273:11619–11624; 1998. [DOI] [PubMed] [Google Scholar]
- 24. Flint D. H.; Tuminello J. F.; Emptage M. H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 268:22369–22376; 1993. [PubMed] [Google Scholar]
- 25. Fridovich I. Superoxide anion radical (O2 •-), superoxide dismutases, and related matters. J. Biol. Chem. 272:18515–18517; 1997. [DOI] [PubMed] [Google Scholar]
- 26. Fridovich I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 64:97–112; 1995. [DOI] [PubMed] [Google Scholar]
- 27. Gardner P. R. Superoxide-driven aconitase FE-S center cycling. Biosci. Rep. 17:33–42; 1997. [DOI] [PubMed] [Google Scholar]
- 28. Gardner P. R.; Costantino G.; Szabo C.; Salzman A. L. Nitric oxide sensitivity of the aconitases. J. Biol. Chem. 272:25071–25076; 1997. [DOI] [PubMed] [Google Scholar]
- 29. Gardner P. R.; Fridovich I. Effect of glutathione on aconitase in Escherichia coli . Arch. Biochem. Bio-phys. 301:98–102; 1993. [DOI] [PubMed] [Google Scholar]
- 30. Gardner P. R.; Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli . J. Biol. Chem. 267:8757–8763; 1992. [PubMed] [Google Scholar]
- 31. Gardner P. R.; Fridovich I. Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 266:19328–19333; 1991. [PubMed] [Google Scholar]
- 32. Gardner P. R.; Nguyen D. H.; White C. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. USA 91:12248–12252; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gardner P. R.; Rained I.; Epstein L. B.; White C. W. Superoxide radical and iron modulate aconitase activity in mammalian cells. J. Biol. Chem. 270: 13399–133405; 1995. [DOI] [PubMed] [Google Scholar]
- 34. Gdaniec Z.; Sierzputowska-Gracz H.; Theil E. C. Iron regulatory element and internal loop/bulge structure for ferritin mRNA studied by cobalt(III) hexamine binding, molecular modeling, and NMR spectroscopy. Biochemistry 37:1505–1512; 1998. [DOI] [PubMed] [Google Scholar]
- 35. Goldberg M. A.; Dunning S. P.; Bunn H. F. Regulation of the erythropoietin gene: Evidence that the oxygen sensor is a heme protein. Science 242:1412–1415; 1988. [DOI] [PubMed] [Google Scholar]
- 36. Gruer M. J.; Artymiuk P. J.; Guest J. R. The aconitase family: Three structural variations on a common theme. Trends Biochem. Sci. 22:3–6; 1997. [DOI] [PubMed] [Google Scholar]
- 37. Guillemin K.; Krasnow M. A. The hypoxic response: Huffing and HIFFing. Cell 89:9–12; 1997. [DOI] [PubMed] [Google Scholar]
- 38. Gunshin H.; Mackenzie B.; Berger U. V.; Gunshin Y.; Romero M. F.; Boron W. F.; Nussberger S.; Gollan J. L.; Hediger M. A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388:482–488; 1997. [DOI] [PubMed] [Google Scholar]
- 39. Guo B.; Phillips J. D.; Yu Y.; Leibold E. A. Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J. Biol. Chem. 270: 21645–21651; 1995. [DOI] [PubMed] [Google Scholar]
- 40. Gutierrez J. A.; Yu J.; Rivera S.; Wessling-Resnick M. Functional expression cloning and characterization of SFT, a stimulator of Fe transport. J. Cell Biol. 139:895–905; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hampton M. B.; Kettle A. J.; Winterbourn C. C. Inside the neutrophil phagosome: Oxidants, myeloperoxidase, and bacterial killing. Blood 92:3007–3017; 1998. [PubMed] [Google Scholar]
- 42. Hanson E. S.; Foot L. M.; Leibold E. A. Hypoxia post-translationally activates iron regulatory protein 2. J. Biol. Chem. 274:5047–5052; 1999. [DOI] [PubMed] [Google Scholar]
- 43. Hanson E. S.; Leibold E. A. Regulation of iron regulatory protein 1 during hypoxia and hypoxia/reoxygenation. J. Biol. Chem. 273:7588–7593; 1998. [DOI] [PubMed] [Google Scholar]
- 44. Hausladen A.; Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 269:29405–29408; 1994. [PubMed] [Google Scholar]
- 45. Henderson B. R.; Menotti E.; Bonnard C.; Kühn L. C. Optimal sequence and structure of iron-responsive elements. J. Biol. Chem. 269:17481–17489; 1994. [PubMed] [Google Scholar]
- 46. Henson C. P.; Cleland W. W. Purification and kinetic studies of beef liver cytoplasmic aconitase. J. Biol. Chem. 242:3833–3838; 1967. [PubMed] [Google Scholar]
- 47. Hentze M. W.; Kühn L. C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. USA 93:8175–8182; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hibbs J. B.; Taintor R. R.; Vavrin Z. Iron depletion: Possible cause of tumor cell cytotoxicity induced by activated macrophages. Biochem. Biophys. Res. Commun. 123:716–723; 1984. [DOI] [PubMed] [Google Scholar]
- 49. Huang L.; Arany Z.; Livingston D.; Bunn H. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 271:32253–32259; 1996. [DOI] [PubMed] [Google Scholar]
- 50. Iwai K.; Drake S. K.; Wehr N. B.; Weissman A. M.; LaVaute T.; Minato N.; Klausner R. D.; Levine R. L.; Rouault T. A. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: Implications for degradation of oxidized proteins. Proc. Natl. Acad. Sci. USA 95:4924–2928; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iwai K.; Klausner R. D.; Rouault T. A. Requirements for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J. 14:5350–5357; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jacobs A. Low molecular weight intracellular iron transport compounds. Blood 50:433–439; 1977. [PubMed] [Google Scholar]
- 53. Janero D. R.; Hreniuk D. Suppression of TCA cycle activity in the cardiac muscle cell by hydroperoxide-induced oxidant stress. Am. J. Physiol. 270:C1735–1742; 1996. [DOI] [PubMed] [Google Scholar]
- 54. Kallio P. J.; Pongratz I.; Gradin K.; McGuire J.; Poellinger L. Activation of hypoxia-inducible factor 1 alpha: Posttranscriptional regulation and conformational change by recruitment of the Amt transcription factor. Proc. Natl. Acad. Sci. USA 94:5667–5672; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karin M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann. NY Acad. Sci. 851:139–146; 1998. [DOI] [PubMed] [Google Scholar]
- 56. Ke Y.; Wu J.; Leibold E. A.; Walden W. E.; Theil E. C. Loops and bulge/loops in iron-responsive element isoforms influence iron regulatory protein binding. Fine-tuning of mRNA regulation? J. Biol. Chem. 273:23637–23640; 1998. [DOI] [PubMed] [Google Scholar]
- 57. Kennedy M. C. Role of iron-sulfur proteins in gene regualtion. Met. Ions Biol. Syst. 32:579–602; 1996. [PubMed] [Google Scholar]
- 58. Kennedy M. C.; Antholine W. E.; Beinert H. An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J. Biol. Chem. 272:20340–20347; 1997. [DOI] [PubMed] [Google Scholar]
- 59. Levy N. S.; Chung S.; Furneaux H.; Levy A. P. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J. Biol. Chem. 273:6417–6423; 1998. [DOI] [PubMed] [Google Scholar]
- 60. Li Y.; Huang T. T.; Carlson E. J.; Melov S.; Ursell P. C. Olson J. L.; Noble L. J.; Yoshimura M. P.; Berger C.; Chan P. H. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 11:376–381; 1995. [DOI] [PubMed] [Google Scholar]
- 61. Liochev S. I.; Fridovich I. The role of O2 •- in the production of HO•: In vitro and in vivo. Free Radic. Biol. Med. 16:29–33; 1994. [DOI] [PubMed] [Google Scholar]
- 62. Martins E. A.; Robalinho R. L.; Meneghini R. Oxidative stress induces activation of a cytosolic protein responsible for control of iron uptake. Arch. Biochem. Biophys. 316:128–134; 1995. [DOI] [PubMed] [Google Scholar]
- 63. McCord J. M.; Fridovich I. The reduction of cytochrome c by milk xanthine oxidase. J. Biol. Chem. 243:5753–5760; 1968. [PubMed] [Google Scholar]
- 64. Menotti E.; Henderson B. R.; Kühn L. C. Translational regulation of mRNAs with distinct IRE sequences by iron regulatory proteins 1 and 2. J. Biol. Chem. 273:1821–1824; 1998. [DOI] [PubMed] [Google Scholar]
- 65. Muckenthaler M.; Gray N.; Hentze M. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell. 2:383–388; 1998. [DOI] [PubMed] [Google Scholar]
- 66. Muller J. M.; Rupec R. A.; Baeuerle P. A. Study of gene regulation by NF-kappa B and AP-1 in response to reactive oxygen intermediates. Methods 11:301–312; 1997. [DOI] [PubMed] [Google Scholar]
- 67. Nathan C.; Xie Q. Nitric oxide synthases: Roles, tolls, and controls. Cell 78:915–918; 1994. [DOI] [PubMed] [Google Scholar]
- 68. Nathan C. F.; Hibbs J. B. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 3:65–70; 1991. [DOI] [PubMed] [Google Scholar]
- 69. Pantopoulos K.; Hentze M. W. Activation of iron regulatory protein-1 by oxidative stress in vitro. Proc. Natl. Acad. Sci. USA 95:10559–10563; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pantopoulos K.; Hentze M. W. Nitric oxide signaling to iron regulatory protein: Direct control of ferritin mRNA translation and transferrin receptor mRNA stability in transfected fibroblasts. Proc. Natl. Acad. Sci. USA 92:1267–1271; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pantopoulos K.; Hentze M. W. Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J. 14:2917-2924; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pantopoulos K.; Mueller S.; Atzberger A.; Ansorge W.; Stremmel W.; Hentze M. W. Differences in the regulation of iron regulatory protein-1 (IRP-1) by extra- and intracellular oxidative stress. J. Biol. Chem. 272:9802–9808; 1997. [DOI] [PubMed] [Google Scholar]
- 73. Pantopoulos K.; Weiss G.; Hentze M. W. Nitric oxide and oxidative stress (H2O2) control mammalian iron metabolism by different pathways. Mol. Cell. Biol. 16:3781–3788; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Patel M.; Day B. J.; Crapo J. D.; Fridovich I.; Mc-Namara J. O. W. Requirement for superoxide in excitotoxic cell death. Neuron 16:345–355; 1996. [DOI] [PubMed] [Google Scholar]
- 75. Phillips J. D.; Guo B.; Yu Y.; Brown F. M.; Leibold E. A. Differential regulation of iron-regulatory proteins 1 and 2 by nitric oxide in hepatoma cells. Blood 50:1–10; 1996. [Google Scholar]
- 76. Ratcliffe P. J.; O’Rourke J. F.; Maxwell P. H.; Pugh C. W. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J. Exp. Biol. 201(Pt. 8):1153–1162; 1998. [DOI] [PubMed] [Google Scholar]
- 77. Recalcati S.; Taramelli D.; Conte D.; Cairo G. Nitric oxide-mediated induction of ferritin synthesis in J774 macrophages by inflammatory cytokines: Role of selective iron regulatory protein-2 downregulation. Blood 91:1059-1066: 1998. [PubMed] [Google Scholar]
- 78. Richardson D. R.; Neumannova V.; Nagy E.; Ponka P. The effect of redox-related species of nitrogen monoxide on transferrin and iron uptake and cellular proliferation of erythroleukemia (K562) cells. Blood 86:3211–3219. 1995. [PubMed] [Google Scholar]
- 79. Richardson D. R.; Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim. Biophys. Acta 1331:1–40; 1997. [DOI] [PubMed] [Google Scholar]
- 80. Rouault T. A.; Klausner R. D. The impact of oxidative stress on eukaryotic iron metabolism. EXS 77: 183–197; 1996. [DOI] [PubMed] [Google Scholar]
- 81. Rouault T. A.; Klausner R. D. Iron-sulfur clusters as biosensors of oxidants and iron. Trends Biochem. Sci. 121:174–177; 1996. [PubMed] [Google Scholar]
- 82. Salceda S.; Caro J. Hypoxia-inducible factor 1alpha (HIF-1 alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 272:22642–22647; 1997. [DOI] [PubMed] [Google Scholar]
- 83. Samaniego F.; Chin J.; Iwai K.; Rouault T. A.; Klausner R. D. Molecular characterization of a second iron-responsive element binding protein, iron regulatory protein 2. J. Biol. Chem. 49:30904–30910; 1994. [PubMed] [Google Scholar]
- 84. Schalinske K. L.; Anderson S. A.; Tuazon P. T.; Chen O. S.; Kennedy M. C. ; Eisenstein R. S. The iron-sulfur cluster of iron regulatory protein 1 modulates the accessibilty of RNA binding and phosphorylation sites. Biochemistry 36:3950–3958; 1997. [DOI] [PubMed] [Google Scholar]
- 85. Schalinske K. L.; Blemings K. P.; Steffen D. W.; Chen O. S.; Eisenstein R. S. Iron regulatory protein 1 is not required for the modulation of ferritin and transferrin receptor expression by iron in a murine pro-B lymphocyte cell line. Proc. Natl. Acad. Sci. USA 30:10681–10686; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Schalinske K. L.; Eisenstein R. S. Phosphorylation and activation of both iron regulatory proteins 1 and 2 in HL-60 cells. J. Biol. Chem. 271:7168–7176; 1996. [DOI] [PubMed] [Google Scholar]
- 87. Semenza G. L. Hypoxia-inducible factor 1 and the molecular physiology of oxygen homeostasis. J. Lab. Clin. Med. 131:207–214; 1998: [DOI] [PubMed] [Google Scholar]
- 88. Sierzputowska-Gracz H.; Theil E. C. 15N NMR and CD studies of the IRE (iron regulatory element) in ferritin mRNA with single base substitution in the hairpin loop. Nucleic Acids Symp. Ser. 33:203–206; 1995. [PubMed] [Google Scholar]
- 89. Stamler J. S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 78:931–936; 1994. [DOI] [PubMed] [Google Scholar]
- 90. Strain J.; Lorenz C. R.; Bode J.; Garland S.; Smolen G. A.; Ta D. T.; Vickery L. E.; Culotta V. C. Suppressors of superoxide dismutase (SOD1) deficiency in Saccharomyces cerevisiae. Identification of proteins predicted to mediate iron-sulfur cluster assembly. J. Biol. Chem. 273:31138–31144; 1998. [DOI] [PubMed] [Google Scholar]
- 91. Suzuki Y. J.; Forman H. J.; Sevanian A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med. 22:269–285; 1997. [DOI] [PubMed] [Google Scholar]
- 92. Teixeira H.; Schumacher R.; Meneghini R. Lower intracellular hydrogen peroxide levels in cells overexpressing CuZn-superoxide dismutase. Proc. Natl. Acad. Sci. USA 95:7872–7875; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vanden-Hoek T. L.; Becker L. B.; Shao Z.; Li C.; Schumacker P. T. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 273: 18092-18098; 1998. [DOI] [PubMed] [Google Scholar]
- 94. Wang G. L.; Jiang B. H.; Rue E. A.; Semenza G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad Sci. USA 92:5510–5514; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Wang G. L.; Semenza G. L. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood 82: 3610–3615; 1993. [PubMed] [Google Scholar]
- 96. Weiss G.; Bogdan C.; Hentze M. W. Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL-13. J. Immunol. 158:420–425; 1997. [PubMed] [Google Scholar]
- 97. Weiss G.; Goossen B.; Doppler W.; Fuchs D.; Pantopoulos K.; Werner-Felmayer G.; Wachter H.; Hentze M. W. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J. 12:3651–3657; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Williams M. D.; Van Remmen H.; Conrad C. C. ; Huang T. T.; Epstein C. J.; Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J. Biol. Chem. 273:28510–28515; 1998. [DOI] [PubMed] [Google Scholar]
- 99. Xia Y.; Zweier J. L. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. USA 94:6954–6958; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yan L.-J.; Levine R. L.; Sohal R. S. Oxidative damage during aging targets mitochondrial aconitase. Proc. Natl. Acad. Sci. USA 94:11168–11172; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]