

Abstract
Inflammatory cytokines of the tumor necrosis factor (TNF) family mediate a large variety of cellular and organismal inflammatory responses and are important to the pathogenesis of a number of important disease states including arthritis, septic shock, inflammatory bowel disease, and, possibly, type II diabetes. Many of the responses to these cytokines require de novo gene expression mediated by the activator protein-1 (AP-1) heterodimeric transcription factor. This review will discuss what is known of how cytokines of the TNF family, acting at the cell surface, recruit two mitogen-activated protein kinase (MAPK) subfamilies, the stress-activated protein kinases (SAPKs, also called JNKs) and the p38s, to transduce signals to AP-1.
Keywords: AP-1, c-Jun, JNK, p38, SAPK, TNF, TRAF
TUMOR necrosis factor (TNF) was first identified over 20 years ago as the causative agent of bacterial lipopolysaccharide-induced sepsis and hemorrhagic tumor necrosis. Since then, a large variety of related cytokines, the TNF superfamily, has been identified. These include interleukin-1 (IL1), lymphotoxin-β (LT-β), CD154 (CD40L), CD70 (CD27L), FasL, receptor activator of NF-κB ligand [(RANKL), also called osteoprotegerin ligand (OPGL) or TNF-related activation-induced cytokine (TRANCE)], TNF-related apoptosis-inducing ligand (TRAIL), and CD153 (CD30L). These bind to a parallel family of polypeptides, the TNF receptor (TNFR) superfamily, that includes not only receptors for inflammatory mediators but a number of viral proteins such as the Epstein-Barr virus latent membrane protein-1 (LMP1) and A35R, a protein encoded in the genome of some vaccinia virus strains. Consistent with the structural conservation within the TNF superfamily, receptors of the TNFR family all possess homologous extracellular ligand binding domains with cysteine-rich repeats; however, the intracellular extensions of TNFR family receptors are significantly divergent. Cytokines of the TNF superfamily regulate a wide variety of proinflammatory responses and are critical to immune cell development, innate and acquired immunity, as well as the pathogenesis of a number of diseases such as inflammatory bowel disease, arthritis, and type II diabetes mellitus. Only recently, however, have the signal transduction mechanisms employed by these important proinflammatory mediators been identified (2,53,63).
Given the importance of the TNFR family to the inflammatory response, dissection and characterization of the signal transduction mechanisms by which these receptors exert their cellular effects has attracted wide interest. This review will focus on recent advances in our understanding of how the TNFR family signals to the activator protein-1 (AP-1) transcription factor.
EARLY EVENTS IN TNFR FAMILY SIGNALING
Receptors of the TNFR family do not possess intrinsic enzymatic activity. Instead, upon binding ligand, these receptors typically undergo homotrimerization of hetero oligomerization with receptor accessory proteins. This oligomerization, in turn, triggers the binding of adapter proteins that couple to downstream effectors (Fig. 1). Several TNFR family receptors (TNFR1 and Fas are notable examples) possess an 80–100 AA extension, the death domain, that was originally identified as a motif required for signaling apoptosis. It is now known that death domains mediate protein-protein homo- and heterodimerization and are required for nucleating receptor-effector complexes and for implementing several signaling programs, including gene expression (Fig. 1) (2,67,68).
FIG. 1.
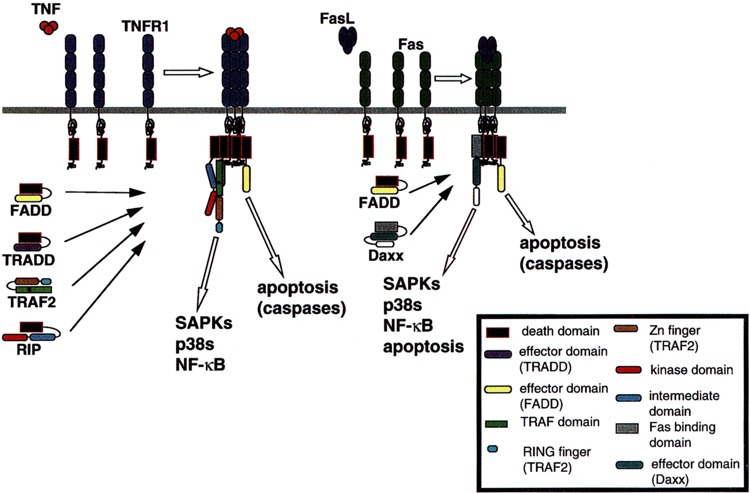
Recruitment of signaling adapter molecules to TNFR1 and Fas, two TNFR superfamily receptors. Domains responsible for the various interactions are discussed in the text. Adapter protein binding is dependent upon engagement and oligomerization of receptor proteins.
TNF binds to one of two receptors, the 55-kDa TNFR1 and the 75-kDa TNFR2 (67). Signal transduction by TNFR1 has been studied in some detail. Upon TNF-induced receptor trimerization, the death domain of TNFR 1 binds the death domain-containing adapter protein TNFR-associated death domain (TRADD). TRADD consists of a carboxyl-terminal death domain (AAs 195–305) and an amino-terminal effector binding domain (AAs 1–195) that binds proteins of the TNFR-associated factor (TRAF) family, including TRAF2 (24,25,48).
Receptor-interacting protein (RIP) is a second death domain-containing adapter protein that is recruited in a TNF-dependent manner to the TNFR1 complex (26,56,61). RIP consists of an amino-terminal protein Ser/Thr kinase domain (AAs 1–304), a carboxyl-terminal death domain (AAs 553–656), and an intermediate domain (AAs 305–552) that recruits downstream target proteins (56). The recruitment of RIP to the TNFR1 complex is indirect—the RIP death domain does not bind the death domain of TNFR1. Instead, the RIP death domain binds that of TRADD. RIP can also interact in vivo with TRAF2, and TNFR engagement is thought to foster the formation of a TRADD-RIP-TRAF2 complex (Fig. 1) (26,61).
TRAFs are an emerging family of at least six mammalian polypeptides (TRAFs 1–6; Table 1). All TRAF proteins possess two tandem carboxyl-terminal TRAF domains (TRAF-N and TRAF-C), preceded by a zinc finger domain and, with the exception of TRAF1, an amino-terminal RING finger domain. TRAF domains are necessary for binding both upstream activators and TRAF effectors (2). Thus, for example, the TRAF interaction motif of TRADD interacts with both the TRAF-C (AAs 356–501) and TRAF-N (AAs 272–355) domains of TRAF2, whereas the RIP kinase and intermediate domains interact with the TRAF2 TRAF-N domain (25,61). The RING and, possibly, the zinc finger motifs of TRAF proteins are necessary for activation of downstream targets, and deletion of the RING domain of TRAF2 (AAs 1–80) abrogates TRAF-mediated signal transduction (40,43,52).
TABLE 1.
TRAF REGULATION AND FUNCTION
| TRAF | Upstream TNFR Family Receptors (Selected) | Effector(s) That Couple(s) to SAPKs and p38s |
|---|---|---|
| TRAF1 | TNFR2, CD30, 4-1BB, Ox40, LMP-1 | does not activate SAPKs/p38s |
| TRAF2 | TNFR1, TNFR1—indirectly, CD27, CD30, CD40, 4-1BB, Ox40, LMP-1 | GCK, GCKR, ASK1 |
| TRAF3 | CD27, CD30, CD40, 4-1BB, Ox40, LT-βR, LMP-1 | does not activate SAPKs/p38s |
| TRAF4 | ? | does not activate SAPKs/p38s |
| TRAF5 | CD27, CD30, CD40, Ox40, LT-βR, LMP-1 | ASK1 |
| TRAF6 | CD40, IL-1R | GCK, ASK1 |
TRAF regulation and function. Receptors upstream of TRAFs are reviewed in Arch et al. (2). Recruitment of effector proteins is discussed in the text.
THE TNF FAMILY AND GENE EXPRESSION: ACTIVATION OF AP-1
General Considerations
A major function of TNF and cytokines of the TNF family is the activation of de novo gene expression mediated in large part by the recruitment of two important multimeric transcription factors: nuclear factor-κB (NF-κB) and activator protein-1 (AP-1). NF-κB is discussed by Gaynor et al. in this issue. AP-1 consists of bZIP transcription factors—typically c-Jun, JunD, along with members of the fos (usually c-Fos) and ATF (usually ATF2) families. All bZIP transcription factors contain leucine zippers that enable homo- and heterodimerization; and, accordingly, AP-1 components are organized into Jun-Jun, Jun-Fos, or Jun-ATF dimers. Upon activation, AP-1 binds and trans-activates genes containing the TPA response element (TRE—consensus sequence: TGAC/GTCA). AP-1 heterodimers containing ATFs can also bind and trans-activate genes containing the cAMP response element (CRE—consensus sequence: TGACGTCA). Activation of AP-1 involves the direct phosphorylation/dephosphorylation of AP-1 components as well as the phosphorylation and activation of transcription factors that induce elevated expression of c-jun or c-fos. These events can be activated independently by several different signaling pathways. TNF activation of AP-1 is important to key elements of the inflammatory response including the expression of proinflammatory cell adhesion molecules such as E-selectin, and the expression of tissue remodeling proteases, such as collagenase [reviewed in (30)].
Activation of AP-1 by Members of the MAPK Family
Members of the mitogen-activated protein kinase (MAPK) family are critical to most aspects of AP-1 regulation (30,35,36,64). Three mammalian MAPK groups have been studied in detail: the extracellular signal-regulated kinases (ERKs)-l and -2 are effectors of the Ras proto-oncoprotein and are activated in response to mitogenic stimuli (3). By contrast, the stress-activated protein kinases [(SAPKs), also called c-Jun-NH2-terminal kinases (JNKs)] and the p38 MAPKs (Table 2) are activated preferentially by environmental stresses and inflammatory cytokines of the TNF family (35,36). Phosphorylation of c-Jun or ATF2 within their trans-activation domains correlates well with enhanced trans-activating activity (17,30). The SAPKs can phosphorylate the c-Jun trans-activating domain at Ser63 and Ser73. These residues are phosphorylated in vivo under conditions wherein the SAPKs are activated; and depletion of SAPK from cell extracts removes all stress-activated c-Jun kinase. Thus, the SAPKs appear to be the dominant kinases responsible for stress-induced c-Jun phosphorylation in vivo (7,34–36). The SAPKs and p38s can phosphorylate ATF2 at Thr69 and Ser71 in the trans-activation domain. Again, these residues are phosphorylated under circumstances when the SAPKs/JNKs and p38s are activated. Phosphorylation of ATF2 activates its trans-activating activity (17).
TABLE 2.
MAMMALIAN MAP KINASE PATHWAY NOMENCLATURE
| Name | Alternate Names | Substrate(s) (Transcription Factors Only) | Substrate(s) | Effectors |
|---|---|---|---|---|
| MAPKs | ||||
| ERK1 | p44-MAPK | Elkl | ||
| ERK2 | p42-MAPK | Elkl | ||
| SAPK-α | JNK2, SAPKla | c-Jun, JunD, ATF2, Elkl | ||
| SAPK-β | JNK3, SAPKlb | c-Jun, JunD, ATF2, Elkl | ||
| SAPK-γ | JNK1, SAPKlc | c-Jun, Jun D, ATF2, Elkl | ||
| p38α | SAPK2a, CSBP1 | ATF2, Elkl, MEF2C | ||
| p38β | SAPK2b | ATF2 | ||
| p38γ | SAPK3 | ATF2 | ||
| p38δ | SAPK4 | ATF2 | ||
| MEKs | ||||
| MEK1 | MAPKK1, MKK1 | ERK1, ERK2 | ||
| MEK2 | MAPKK2, MKK2 | ERK1, ERK2 | ||
| SEK1 | MKK4, JNK kinase (JNKK)-l, MEK4, SAPK-kinase (SKK)-l | SAPKs | ||
| MKK7 | JNKK2, MEK7, SKK4 | SAPKs | ||
| MKK3 | MEK3, SKK2 | p38s | ||
| MKK6 | MEK6, SKK3 | p38s | ||
| MAP3Ks | ||||
| A,B-Raf, Raf-1 | MEK1, MEK2 | |||
| MEKK1 | SEK1, MKK7 | |||
| MEKK2 | SEK1, MEK1 | |||
| MEKK3 | SEK1, MEK1 SEK1, MKK3, | |||
| MEKK4 | MTK1 | MKK6 SEK1, MKK3, | ||
| ASK1 | MKK6 SEK1, MKK3, | |||
| TAK1 | MKK6 | |||
| Tpl-2 | Cot | MEK1, SEK1 | ||
| MLK2 | MST | SEK1, MKK7 | ||
| MLK3 | SPRK, PTK1 | SEK1 | ||
| DLK | MUK, ZPK | ? | ||
| TAOl/2 | MKK3 | |||
| GCKs | ||||
| GCK1 | GCK | MEKK1, ?MLK3 | ||
| GCKR | kinase homologous to Ste20 (KHS) | MEKK1 | ||
| GLK1 | ? | |||
| HPK1 | MEKK1, MLK3 | |||
| NIK | HPKl/GCK-4ke kinase (HGK) | MEKK1 |
Nomenclature for mammalian MAPK pathway components. Included are commonly accepted nomenclatures found in the primary literature. Not all of these names are included in the text; only SAPK and p38 regulation and function are discussed in the text.
The SAPKs and p38s also contribute to AP-1 activation by stimulating the transcription of genes encoding AP-1 components. One of the earliest transcriptional events known to occur in response to mitogen is the induction of c-fos expression. The fos promoter contains a cis-acting element, the serum response element (SRE), which mediates the recruitment of transcription factors that induce c-fos expression. The SRE binds a heterodimeric transcription factor containing two polypeptides, the serum response factor (SRF) and the ternary complex factor (TCF). The TCFs are a family of Ets domain transcription factors that includes Elk-1 [reviewed in (64)]. The SAPKs and p38s (as well as the ERKs) can phosphorylate two critical residues in the Elk1 C-terminus (Ser383, Ser389). This enhances the binding of Elk1 to the SRF and thereby elevates trans-activation at the SRE. Accordingly, the TCFs represent a point of signal integration whereby MAPKs activated by a variety of stimuli contribute to c-fos induction (64,69).
The p38s can also phosphorylate and activate the trans-activating activity of the transcription factor myocyte enhancer factor-2C (MEF2C), a member of the MEF subgroup of the MCM1-agamous and deficiens-SRF (MADS) box transcription factor family. MEF2C was originally identified as a transcription factor that bound to AT-rich sequences and trans-activated a number of genes involved in myoblast differentiation; however, MEF2C is widely expressed and may mediate numerous transcriptional regulatory events. p38s phosphorylate Thr293 and Thr300 of MEF2C; and Thr293/Thr300 phosphorylation is sufficient to activate MEF2C trans-activating function. A cis element for MEF2C resides within the c-jun promoter; thus, p38 activation can contribute to the induction of c-jun expression (18).
REGULATION OF THE SAPKs AND p38s
MAPK Pathway Regulation: The Core Signaling Module
All MAPKs are regulated as part of three-tiered core signaling modules wherein the MAPKs are activated upon concomitant Tyr and Thr phosphorylation of the consensus sequence Thr-X-Tyr (X is Glu for the ERKs, Pro for the SAPKs, and Gly for the p38s) within the P-loop of subdomain VIII of the kinase domain. This phosphorylation is catalyzed by members of the MAPK/ERK-kinase (MEK) family. MEKs, in turn, are regulated by Ser/Thr phosphorylation, also in the subdomain VIII P-loop, catalyzed by any of several protein kinases collectively referred to as MAPK-kinase-kinases (MAP3Ks). Consistent with the diversity of upstream agonists that can recruit different MAPK pathways, MAP3Ks themselves are thought to be regulated by a somewhat daunting array of potential upstream activators ranging from Ras superfamily GTPases and polypeptides induced by DNA damage to other protein kinases and adapter proteins coupled to TNFR family receptors (19,35–37,60,66).
MEKs and MAP3Ks Upstream of the SAPKs andp38s
The SAPKs are regulated by at least two MEKs: SAPK/ERK-kinase-1 [(SEK1), also called MAPK-kinase-4 (MKK4) and JNK-kinase-1 (JNKK1)] and MKK7 (also called JNKK2) (Table 2). SEK1 is more strongly activated by environmental stresses whereas MKK7 is more strongly recruited by inflammatory cytokines. However, full SAPK activation may require the concerted activity of both SEK1 and MKK7 inasmuch as SEK1, in vitro, acts primarily to phosphorylate the SAPKs at Tyr whereas MKK7 is preferentially a SAPK Thr kinase (8,22,39,51,62). Two p38-specific MEKs are known: MKK3 and MKK6. MKK6 is robustly activated by all known stimuli that recruit the p38s. By contrast, MKK3 is selectively activated by environmental stresses (Table 2) (6,8,46). It has not yet been established whether or not MKK3 or MKK6 preferentially phosphorylates Thr or Tyr.
The SAPKs and p38s are regulated by a very large number of MAP3Ks. These fall into several protein kinase families. The MEK kinases (MEKKs) bear structural homology, within their kinase domains, to the kinase domain of S. cerevisiae STE11, a MAP3K in the mating pheromone response pathway. Mammalian MEKKs include MEKKs 1–4 TGF-β-activated kinase-1 (TAK1), apoptosis signal-regulated kinase-1 (ASK1), NF-κB inducing kinase, a selective activator of the NF-κB pathway (see Gaynor et al. in this issue), and tumor progression locus-2 (Tpl-2). Tpl-2 is the rat homologue of the product of the human proto-oncogene cot. MEKK1 is predominantly specific for the SAPK pathway and is a potent activator of SEK1. MEKK1 can also strongly activate MKK7. MEKK2 and-3 can activate both the SAPK pathway (via SEK1) and the ERK pathway (via MEK1). Transient expression of Tpl-2 also recruits the SAPK and ERK pathways with equal potency; and in vivo and in vitro, Tpl-2 is a MAP3K that can activate MEK1 and SEK1. MEKK4, TAK1, and ASK1 can activate both the SAPK pathway (via SEK1) and the p38 pathway (via MKK3 and MKK6; Table 2) (4,14,29,38,41,42,50,59,70–72).
Mixed lineage kinases (MLKs) are also MAP3Ks. MLKs are a small family of protein Ser/Thr kinases that share a general structural configuration wherein an amino-terminal kinase domain is followed by one to two leucine zippers, a Cdc42/Rac interaction and binding (CRIB) domain, and a carboxyl-terminal proline-rich domain with several consensus SH3 binding motifs. MLK2 [also called MKN28 cell-derived Ser/Thr kinase (MST)] and MLK3 [also called SH3 domain-containing proline-rich kinase (SPRK), or protein Tyr kinase-1 (PTK1)] contain, in addition to these features, an amino-terminal SH3 domain. The name mixed lineage kinase comes from the fact that the kinase domains of the MLKs bear structural similarities to both Ser/Thr and Tyr kinases (10,13,23,66). Dual leucine zipper bearing kinase [(DLK), also called MAPK upstream kinase (MUK) or zipper-containing protein kinase (ZPK)] MLK2, and MLK3 are MAP3Ks that selectively recruit the SAPKs via direct activation of SEK1 and MKK7 (11,20,21,47).
Thousand-and-one (TAO) kinases (TAO1 and TAO2) are a novel family of 1001 amino acid Ser/Thr kinases. TAOs consist of amino-terminal kinase domains and extensive (700 AA) carboxyl-terminal extensions of unknown function. The kinase domains of TAOs are significantly homologous to S. cerevisiae STE20 (40% identity) and the germinal center kinases (43% identity with GCK) (see below). However, there is also appreciable identity with MLK2 (33% overall). Notably, most of the identity with MLK2 resides within the substrate binding motifs of the kinase domain. In vitro, the purified kinase domain of TAO1 will directly phosphorylate and activate SEK1, MKK3, and MKK6; however, in vivo only MKK3 is activated. Thus, TAO1 appears to be a MAP3K selective for the p38s. The ability of TAO1 to catalyze directly the activation of MEKs may be due to the kinase domain homology with MLK2 (Table 2) (28).
Given this remarkable heterogeneity of upstream activators, how, then, do receptors of the TNFR family couple to the SAPKs and p38s? Transient expression of TRAF2, -5, and -6, as well as RIP, but not TRAF1, -3, or -4 activates the SAPKs (12,40,43,54,76). In addition, thus far, it is known that expression of TRAF2 and RIP also activates the p38s (76). Gene disruption studies indicate that deletion of traf2 abrogates TNF activation of the SAPKs, but not NF-κB, indicating a pivotal role for TRAF2 in TNF signaling to SAPK (74). Deletion of rip has no effect on TNF activation of SAPK, but prevents TNF activation of NF-κB (32); insofar as RIP interacts strongly with TRAF2, these gene deletion results indicate that RIP activation of SAPK may represent one of several redundant mechanisms of SAPK activation by TNF, all of which emanate from TRAF2.
Inasmuch as TRAF2 plays an essential role in TNF signaling to the SAPKs, activation of the SAPKs by TRAF5 and -6 may occur as a consequence of engagement of other receptors of the TNF family. In support of this, activation of the SAPKs by RANKL proceeds in large part through TRAF6, whereas activation of the SAPKs by CD27 may be mediated by both TRAF2 and TRAF5 (1,12). However, until recently, aside from these observations, there has been little insight into how TRAFs couple to the MAP3K → MEK → MAPK core signaling modules that regulate the SAPKs and p38s.
COUPLING STRESS-REGULATED MAP3Ks TO TRAFs
General Considerations
Four broad classes of regulators are thought to lie upstream of stress-regulated MAP3Ks: (i) Ras superfamily GTPases [reviewed in (66)], (ii) polypeptides transcriptionally induced by stress (60), (iii) protein kinases homologous to germinal center kinase (9,27,33,45,52,57,58,65), and (iv) adapter proteins coupled to receptors of the TNFR family (1,2,12,40,43,76).The latter two mechanisms are most germane to TNFR signaling.
The Germinal Center Kinase Family Includes Potent and Selective Activators of the SAPKs
Germinal center kinase-1 [(GCK1), also called germinal center kinase (GCK)] was cloned by Kehrl and colleagues as part of a subtractive screen to identify novel polypeptides selectively expressed in B follicular germinal centers (GCs) (31). Subsequently, 11 mammalian protein Ser/Thr kinases related to GCK1 have been identified. In addition, there are Drosophila, C. elegans, Dictyostelium, and S. cerevisiae homologues. All members of the GCK family possess amino-terminal protein kinase domains and extensive carboxyl-terminal regulatory domains referred to as CTDs. The kinase domains of GCKs are distantly related to those of S. cerevisiae STE20, and, accordingly, GCKs and Ste20s have often been grouped into a single family. However, unlike the Ste20s, GCKs do not possess amino-terminal regulatory regions. Nor do the GCKs possess CRIB motifs. Thus, GCKs cannot bind Rho subfamily GTPases. Therefore, GCKs should be considered a separate protein kinase family [reviewed in (37)].
The GCK family can be subdivided into two groups (group-I and group-II) based on structural and functional properties. Group-I GCKs are most closely related to GCK1. These include GCK1, GCK-related (GCKR), GCK-like kinase (GLK), hematopoietic progenitor kinase-1 (HPK1), and Nck-interacting kinase (NIK). (NIK is also, somewhat confusingly, an abbreviation for NF-κB-inducing kinase. In this review, NIK refers to the GCK homologue.) Drosophila Misshapen and C.elegans Mig-15 are also group-I GCKs (Table 2) (9,27,33,37,52,57,58,65). Group-II GCKs are poorly understood. These enzymes are more distantly related to GCK1 and more closely homologous to S. cerevisiae SPS1, a gene required for encapsulation of haploid nuclei during sporulation [reviewed in (37)].
As was mentioned above, although GCK1 is widely expressed, its distribution in B lymphocyte follicular tissues is restricted largely to the GCs and not the surrounding mantle zone (31). GCs are regions of B cell selection and differentiation, processes that require in part agonists of the TNF family including CD40 and TNF itself (53,63). Consistent with this, the three most closely related GCKs— GCK1, GCKR, and GLK—are all activated in vivo by TNF. Given the ability of the TNF family to recruit the SAPKs, it was not surprising that the group-I GCKs, the members of the GCK family most closely related to GCK1, are, upon transient expression, also strong SAPK activators. Expression of group-I GCKs does not result in activation of the ERKs or p38s.
The CTDs of group-I GCKs all contain at least two PEST motifs and at least two consensus binding sites for polypeptides with SH3 domains (37). Most strikingly, however, all group-I GCKs share a strongly conserved ∼350-AA domain at far carboxyl-terminal end of the CTD that contains a leucine-rich region and a short carboxyl-terminal extension, the CT motif. One of the SH3 binding motifs is present at the amino-terminal end of the Leu-rich region of four of the mammalian group-I GCKs: GCK1, GCKR, GLK, and HPK1. The Leu-rich and CT motifs have been implicated in the binding of MAP3Ks and TRAFs (Fig. 2) (37,58,76).
FIG. 2.
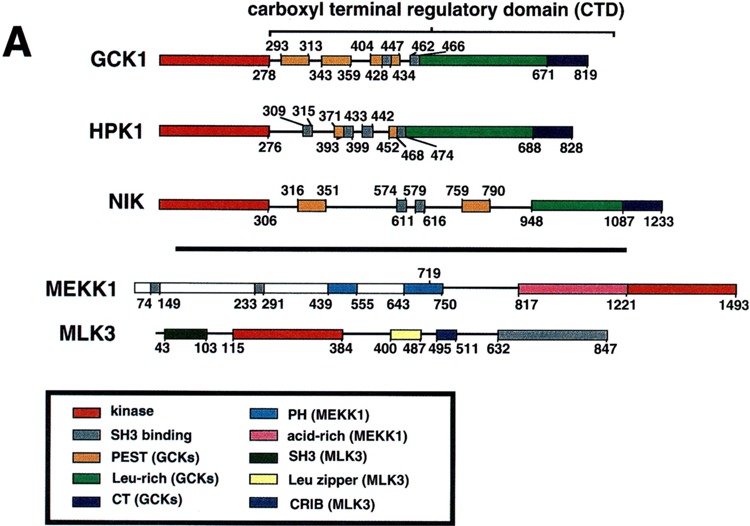

Structure of GCK family kinases. (A) Schematic diagram of three representative group-I GCKs: GCK1, HPK1 and NIK [reviewed in (37)]. Structures of MEKK1 and MLK3, MAP3Ks known to bind these GCKs, are also indicated. (B) Sequence alignment of the Leu-rich and CT motifs of the five known mammalian group-I GCKs. Blue shading illustrates residues shared by all five enzymes. Green shading illustrates residues shared by four of the five enzymes. Gray shading indicates residues shared by the three group-I enzymes known to be activated by TNF: GCK1, GCKR and GLK. Red printing delineates the known TRAF2 binding regions (the CT motifs) of GCK and GCKR. A consensus sequence is shown. Green printing indicates the conserved amino-terminal extension of the Leu-rich domain present in GCK1, GCKR, GLK, and HPK1 and absent in NIK. See text for details. Alignment was achieved using the PILEUP and CLUSTAL-W programs and further optimized by eye.
Binding of MAP3Ks to Group-I GCKs
The mechanism(s) by which group-I GCKs recruit the SAPKs was initially obscure until it was observed that these kinases could directly interact in vivo and in vitro with MAP3Ks. Thus, a yeast two-hybrid screen that employed HPK1 as bait identified MLK3 as an HPK1 interactor (33). The SH3 domain of MLK3 interacts in vivo and in vitro with the carboxyl-terminal two polyproline SH3 binding sites on the HPK1 CTD (33). HPK1 can also interact with MEKK1 in vivo. This interaction has been mapped to the HPK1 CTD; however, it is not yet known which region(s) of the HPK1 CTD bind MEKK1 (27). It is likely that MLK3 and MEKK1 are HPK1 effectors inasmuch as kinase-dead forms of either MAP3K can effectively block HPK1 recruitment of the SAPKs (27,33).
NIK can interact in vivo with MEKK1. In this instance, the binding has been mapped to the Leu-rich and CT motifs of the NIK CTD, and AAs 1–719 of MEKK1 (Fig. 2A). Deletion of either region abrogates binding; and the two domains, as free, truncated polypeptides, can interact in vivo. Consistent with the possibility that MEKK1 is a NIK effector, kinase-in-active mutants of MEKK1 can effectively block NIK activation of the SAPKs (58).
GCK1 can interact in vivo with either endogenous or recombinant MEKK1 (Figs. 2A and 3). This interaction can be replicated in vitro. Truncation studies indicate that MEKK1 interacts with GCK1 through an acid-rich region of the MEKK1 amino-terminal regulatory domain, AAs 817–1221, which is clearly distinct from the domain, AAs 1–719, that interacts with NIK (Figs. 2A and 3) (58,76). MEKK1 is a likely GCK1 effector inasmuch as expression of AAs 817–1221 of MEKK1, a region devoid of substrate binding motifs, completely inhibits GCK1 signaling to the SAPKs (76). The domains on the GCK1 CTD that bind MEKK1 also differ from those of NIK that interact with MEKK1. Deletion of the GCK1 CT motif abrogates MEKK1 binding whereas deletion of the Leu-rich motif restores binding. Subsequent deletion of the C-terminal PEST motif (PEST3) again prevents binding (76).
FIG. 3.
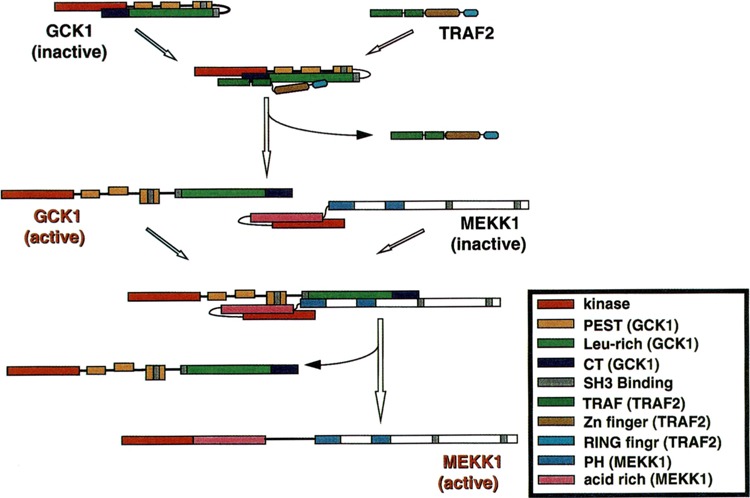
Signaling from TRAF2 to GCK and MEKK1. GCK is activated as a consequence of binding TRAF2; although the mechanism has not been characterized completely, the kinase activity of GCK is likely ultimately enhanced by TRAF2. Activated GCK then binds MEKK1. Results (76) indicate that the kinase activity of GCK permits binding of MEKK1 and efficient turnover of activated MEKK1. The exact mechanism of MEKK1 activation is still largely unclear.
Given the striking conservation of group-I GCK Leu-rich and CT extensions (Fig. 2B), what could account for the different mechanisms by which NIK and GCK1 interact with MEKK1? An explanation may lie in the choice of MEKK1 constructs used in the mapping studies, and in nonconserved regions of the Leu-rich domains of group-I GCKs; and, in fact, MEKK1 may interact with upstream group-I GCKs at multiple contact points. Thus, although GCK1 can bind full-length MEKK1, the mapping studies of the GCK1-MEKK1 interaction, unlike those of the NIK-MEKK1 interaction, employed a truncated MEKK1 construct consisting of residues 817–1493 (76). Conversely, the effect on MEKK1 binding of deletion of the NIK CT motif was not examined (58). Finally, NIK is the most distantly related of the group-I GCKs, and the Leu-rich regions of GCK1, GCKR, GLK, and HPK1 contain a highly conserved ∼60–70-AA amino-terminal extension not present in the NIK Leu-rich domain (Fig. 2B) (37,58). In the resting cell, this domain could serve to inhibit the binding of MEKK1. GCK1 (or GCKR, GLK, or HPK1) activation, perhaps initiated by regulatory proteins binding (or dissociating) from the CT motif, could reverse this inhibition (37).
Binding and Regulation of Group-I GCKs by TRAFs
Ectopic expression of TRAF2, -5, and -6 results in robust activation of the SAPKs and p38s (1,2,12,40,43,54,76). Gene disruption studies indicate that TRAF2 is required for TNF activation of the SAPKs (74); TRAF5 and -6 could couple the SAPKs to other receptors of the TNFR family. GCK1, GCKR, and GLK are all activated in vivo by TNF, and there is substantial evidence that GCK1 and GCKR are effectors for TRAFs. Thus, expression of TRAF2 activates GCKR; and expression of GCKR antisense mRNA blocks TNF and TRAF2 activation of SAPK in 293 cells (52). Furthermore, both GCK1 and GCKR can physically associate in vivo with TRAF2. GCK1 can also bind TRAF6 (37,52,76).
The binding of TRAF2 to both GCK1 and GCKR requires the CT motifs of both kinases. This motif is 55% identical, 81% conserved at the amino acid level. The CT motifs interact with the TRAF domains of TRAF2, whereas the TRAF domains bind GCK1 and GCKR. However, it is likely that the RING domains mediate any regulation of GCK1 and GCKR inasmuch as a truncated TRAF2 construct wherein the RING domain is deleted binds GCK1 and GCKR, but cannot activate in vivo coexpressed GCKR (37,52,76).
The mechanism of GCK1/GCKR activation by TRAFs remains to be determined. The relative SAPK activating activity and MEKK1 binding affinity of wild-type and truncated or kinase-dead GCK1 suggest that upstream components may activate GCK1’s kinase activity, an event that then gates both the binding and efficient subsequent activation of MEKK1 (Fig. 3). Thus, full-length GCK1, kinase-dead GCK1 (Lys44 → Met), and the free GCK1 CTD can all activate coexpressed SAPK to varying degrees. Wild-type GCK1 is the strongest SAPK activator, whereas activation by K44M GCK1 and the GCK1 CTD is comparatively modest. This result is consistent with the idea that the GCK1 CTD either titers out GCK1 inhibitors or serves as a domain for homo oligomerization-dependent activation (45,76). All three GCK1 constructs can interact in vivo with MEKK1; however, in apparent contrast to the relative ability of these GCK constructs to recruit the SAPKs, the free GCK1 CTD interacts most strongly with MEKK1, whereas the interaction between wild-type GCK1 and MEKK1 is weaker, and that between K44M GCK1 and MEKK1 is barely detectable (76). Taken together, these findings suggest that activation of GCK1’s kinase activity both permits MEKK1 binding and efficient activation/turnover of activated MEKK1. TRAF2 might provide an initiating step in GCK1 activation, by triggering disinhibition of MEKK1 binding (Figs. 3 and 4). In this regard, it is noteworthy that the CT motif of the GCK1 CTD is required for both MEKK1 and TRAF2 binding (76). Actual activation of MEKK1, subsequent to GCK1 binding, might involve direct phosphorylation. Indeed, GCK1 can phosphorylate MEKK1 in vitro (76). In addition, GCK1 binding may translocate MEKK1 to regions of the cell that contain additional regulatory components such as inositol phospholipids, which could bind to the PH domains on the MEKK1 polypeptide, or Ras superfamily GTPases, which are known to bind to MEKK1 in a GTP-dependent manner. GCK1 binding could also promote oligomerization-dependent activation of MEKK1.
FIG. 4.
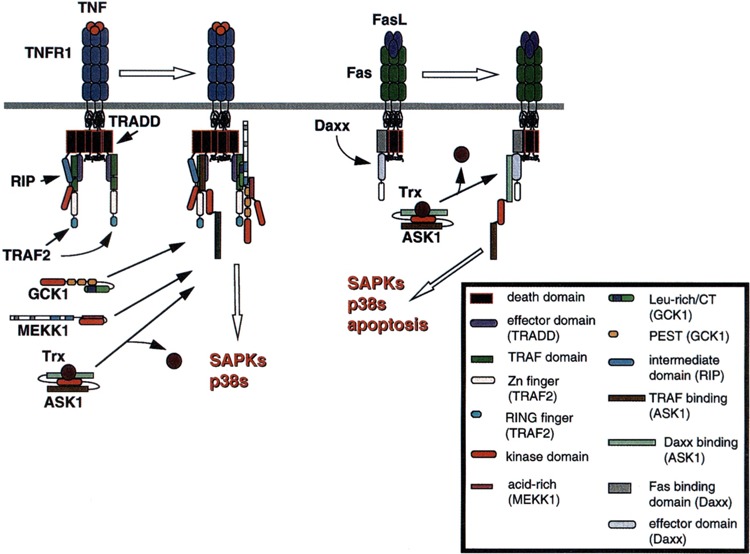
Recruitment of elements upstream of the SAPKs and p38s by the TNFR complex. See Fig. 1 and text for details on formation of the TNFR complex. The various domains involved in the molecular interactions shown are illustrated in the key.
Binding and Regulation of ASK 1 by TRAFs and Daxx
GCK1 and GCKR do not represent the only mechanism by which TRAFs recruit the SAPKs. Most notably, TRAF2 expression activates both the SAPKs and p38s (40,43,76); by contrast, GCK1, GCKR, and MEKK1 are selective activators of the SAPKs (45,52,72). ASK1 is a MAP3K that can activate both the SAPKs and p38s (29). Moreover, the MAP3K activity of ASK1 itself is activated by TNFR1 and Fas engagement, and ASK1 is emerging as a parallel effector for TNFR family signaling to the SAPKs and p38s (5,29,44).
Two recent studies indicate that ASK1 binds and is regulated by adapter proteins coupled to TNFR family receptors. Fas is a widely expressed cell death receptor that is crucial to immune cell regulation where it governs in part the ablation of autoreactive T lymphocytes [reviewed in (53,75)]. Engagement of Fas by FasL triggers apoptosis through at least two mechanisms. The first of these to be discovered involves the adapter protein Fas-associated death domain protein (FADD), the death domain of which interacts with that of activated Fas to trigger the apoptotic caspase cascade [reviewed in (75)]. Daxx is a novel adapter protein that also interacts with Fas and couples Fas to both the SAPKs and a second, parallel proapoptotic pathway. Daxx does not possess a death domain; instead, the carboxyl-terminal end of Daxx (AAs 625–739) binds directly to the Fas death domain, but not to FADD (Figs. 1 and 4). AAs 500–625 of Daxx appear necessary for signaling apoptosis, and the remainder of the Daxx polypeptide (AAs 626–739 and 1–500) is thought to perform an autoinhibitory function that is relieved upon the interaction of Daxx with Fas. Ectopic expression of Daxx activates the SAPKs—a reaction also apparently mediated by AAs 500–625 (Figs. 1 and 4). Expression of a construct consisting solely of the Fas binding region of Daxx (AAs 625–739) blocks both Fas-induced SAPK activation and apoptosis. The SAPKs are required for Daxx-induced cell death, but only in a subset of the cell lines tested (293 and L929 cells, but not HeLa cells) (73).
Coexpression of Daxx and ASK1 results in ASK1 activation, and either endogenous or recombinant Daxx can associate directly in vivo with ASK1, a reaction that is Fas dependent and requires the amino-terminal 648 AAs of ASK1. Kinase-inactive ASK1 effectively blocks both Daxx-induced apoptosis and SAPK activation. From these results, it is clear that ASK1 is a downstream target of Daxx (5).
ASK1 can also interact with TRAF2, -5, and -6 in vivo (Table 1), and appears to be a parallel component by which TNF recruits the SAPKs. Thus, endogenous TRAF2 and ASK1 interact in vivo in a TNF-dependent manner; and this interaction can be replicated in vivo using recombinant proteins. The interaction between ASK1 and TRAF2 requires the TRAF domains of TRAF2 and a carboxyl-terminal regulatory domain of the ASK1 polypeptide (AAs 940–1375, Fig. 4). Coexpression of ASK1 and TRAF2 also results in activation of ASK1, and kinase-inactive ASK1 can block TRAF2 activation of the SAPKs. Thus, it is plausible to conclude that ASK1 is a TRAF2 effector. Inasmuch as ASK1 is a potent activator of p38, the TRAF2-ASK1 interaction might also mediate the activation of p38 by TNF (44).
How do Daxx and TRAF2 activate ASK1? Coimmunoprecipitation experiments indicate that GCK1 and ASK1 do not reliably interact in vivo. Moreover, GCK1 is a selective SAPK activator. Thus, it appears unlikely that TRAF2 (or Daxx) activation of ASK1 proceeds through GCK1. Yeast two-hybrid screening has revealed that the redox sensing enzyme thioredoxin (trx) is an endogenous inhibitor of ASK1. This inhibition requires that Trx be in a reduced state. Thus, treatment of cells with oxidant stresses (H2O2) triggers the dissociation of Trx from ASK1 and activation of Ask1 in vivo (Fig. 4) (49). Furthermore, TNF treatment is known to generate a pulse of reactive oxygen intermediates (ROIs) with slow kinetics (20 min-1 h) that parallel TNF/Fas activation of ASK1 (15,16,49,55). Indeed, TNF fosters the dissociation of ASK1 from Trx by a process that can be blocked with free radical scavengers (49). Thus, TNF-induced and, possibly, Fas-induced ROI formation could trigger release of Trx from ASK1 and ASK1 activation. Although TRAF2 is clearly necessary for TNF activation of ASK1, whether or not Trx dissociation from ASK1 precedes or is followed by TRAF2/Daxx binding remains to be determined.
A consequence of Trx dissociation from ASK1 is likely to be dimerization-dependent activation of ASK1. Upon overexpression, ASK1 spontaneously dimerizes in vivo, and TNF promotes the dimerization of indegenous ASK1 by a mechanism that requires ROIs and can be reversed with free redical scavengers. Moreover, expressed fusion protiens of ASK1 and DNA gyrase can be forced to dimerize in vivo upon administration of the binary DNA gyrase-binding drug coumermycin. This coumermycin-induced dimerization results in substantial activation of coexpressed SAPK (16); and subsequent to Trx dissociation from ASK1, one to be foster the dimerization/activation of associated ASK1.
Activation of SAPK and p38 by RIP
RIP, like TRAF2 and ASK1, is a potent activator of both SAPK and p38 (40,76). The RIP intermediate domain is both necessary and sufficient for this function. A dominant inhibitory construct of RIP wherein the intermediate domain is deleted (RIP-ΔID) effectively blocks TNF activation of both SAPK and p38. By contrast, although RIP-ΔID blocks TRAF2 activation of p38, activation of SAPK is unaffected (76). RIP and TRAF2 can interact directly in vivo; however, gene disruption studies indicate that deletion of rip does not affect TNF activation of SAPK, but instead abrogates activation of NF-κB (32,61). Taken together, these results support the contention that multiple, parallel mechanisms couple TRAF2 to the SAPKs. Although a similar situation may exist for p38, RIP appears necessary for TRAF2 activation of p38 (76). Immunoprecipitates of the RIP intermediate domain contain significant amounts of an endogenous MAP3K activity that can activate MKK6 (and p38) in a coupled assay in vitro (76). Given the known TNF-dependent association of TRAF2 and ASK1, as well as TRAF2 and RIP (44,61), it will be important to determine if this RIP-associated MAP3K is, in fact, a complex of TRAF2 and ASK1.
CONCLUDING REMARKS
This review has focused on recent studies of the mechanisms by which TNFR1, and related receptors that signal through TRAFs, recruit the SAPKs and p38s. The more comprehensive studies of SAPK activation that have been published to date indicate that there are several signaling components that emanate from TRAFs to recruit the SAPKs: GCK1/GCKR-MEKK1, ASK1 through RIP. The RIP mechanism (plus the ASK1 mechanism) represents mechanisms that may also recruit p38. What is the reason for this apparent redundancy? TNF is a pleiotrophic cytokine that affects many different cells. Different cell types may preferentially utilize different mechanisms. Multiple pathways may also allow for differential kinetics of SAPK and p38 activation. Thus, activation of GCKR (and GCK1) by TNF is comparatively rapid whereas activation of ASK1 requires more time, due to the obligate generation of ROIs necessary for ASK1 activation. These differential kinetics could allow a cell to select immediate and sustained versus more transient SAPK activation. In addition, many studies of SAPK regulation by the TNFR family have used overexpression strategies that may mask subtle differences in the recruitment of effectors by different TRAFs. Thus, the subset of effectors recruited by TRAF2 may differ from those recruited by TRAF5 or-6. Finally, GCK1, GCKR, ASK1, and RIP likely have additional functions outside activation of the SAPKs and p38s. Thus, ASK1 can stimulate apoptosis whereas RIP is required for TNF activation of NF-κB. Recruitment of these different pathways may foster different groups of responses. Mammalian gene knockout studies and the exploitation of model organisms amenable to genetic manipulation will be important in dissecting these complex regulatory pathways.
REFERENCES
- 1. Akiba H.; Nakano H.; Nishinaka S.; Shindo M.; Kobata T.; Atsuta M.; Morimoto C.; Ware C. F.; Malinin N.; Wallach D.; Yagita H.; Okumura K. CD27, a member of the tumor necrosis factor receptor super-family, activates NF-κB and stress-activated protein kinase/c-Jun N-terminal kinase via TRAF2, TRAF5 and NF-κB-inducing kinase. J. Biol. Chem. 273: 13353–13358; 1998. [DOI] [PubMed] [Google Scholar]
- 2. Arch R. H.; Gedrich R. W.; Thompson C. B. Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev. 12:2821–2830; 1998. [DOI] [PubMed] [Google Scholar]
- 3. Avruch J.; Zhang X.-f.; Kyriakis J. M. Raf meets Ras: Completing the framework of a signal transduction pathway. Trends Biochem. Sci. 19:279–283; 1994. [DOI] [PubMed] [Google Scholar]
- 4. Blank J. L.; Gerwins P.; Elliot E. M.; Sather S.; Johnson G. L. Molecular cloning of mitogen activated protein/ERK kinase kinases (MEKK) 2 and 3. J. Biol. Chem. 271:5361–5368; 1996. [DOI] [PubMed] [Google Scholar]
- 5. Chang H. Y.; Nishitoh H.; Yang X.; Ichijo H.; Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 281:1860–1863; 1998. [DOI] [PubMed] [Google Scholar]
- 6. Cuenda A.; Alonso G.; Morrice N.; Jones M.; Meier R.; Cohen P.; Nebreda A. Purification and cDNA cloning of SAPKK3, the major activator of RK/p38 in stress- and cytokine-stimulated monocytes and epithelial cells. EMBO J. 15:4156–4164; 1996. [PMC free article] [PubMed] [Google Scholar]
- 7. Dérijard B.; Hibi M.; Wu I.-H.; Barrett T.; Su B.; Deng T.; Karin M.; Davis R. J. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun transactivation domain. Cell 76:1025–1037; 1994. [DOI] [PubMed] [Google Scholar]
- 8. Dérijard B.; Raingeaud J.; Barrett T.; Wu L.-H.; Han J.; Ulevitch R. J.; Davis R. J. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267:682–685; 1995. [DOI] [PubMed] [Google Scholar]
- 9. Diener K.; Wang X. S.; Chen C.; Meyer C. F.; Keesler G.; Zukowski M.; Tan T.-H.; Yao Z. Activation of the c-Jun N-terminal kinase pathway by a novel protein kinase related to human germinal center kinase. Proc. Natl. Acad. Sci. USA 94:9687–9692; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dorow D. S.; Devereux L.; Dietzsch E.; De Kretser T. Identification of a new family of hyman epithelial protein kinases containing two leucine/isoleucine-zipper domains. Eur. J. Biochem. 213:701–710; 1993. [DOI] [PubMed] [Google Scholar]
- 11. Fan G.; Merritt S. E.; Kortenjann M.; Shaw P. E.; Holzman L. B. Dual leucine zipper-bearing kinase (DLK) activates p46SAPK and p38mapk but not ERK2. J. Biol. Chem. 271:24788–24793; 1996. [DOI] [PubMed] [Google Scholar]
- 12. Galibert L.; Tometsko M. E.; Anderson D. M.; Cosman D.; Dougall W. C. The involvement of multiple tumor necrosis factor receptor (TNFR)-associated factors in the signaling mechanisms of receptor activator of NF-κB, a member of the TNFR superfamily. J. Biol. Chem. 273: 34120–34127; 1998. [DOI] [PubMed] [Google Scholar]
- 13. Gallo K.; Mark M. R.; Scadden D. T.; Wang Z.; Gu Q.; Godowski P. Identification and characterization of SPRK, a novel src-homology 3 domain-containing proline-rich kinase with serine/threonine kinase activity. J. Biol. Chem. 269:15092–15100; 1994. [PubMed] [Google Scholar]
- 14. Gerwins P.; Blank J. L.; Johnson G. L. Cloning of a novel mitogen-activated protein kinase-kinase-kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J. Biol. Chem. 272:8288–8295; 1997. [DOI] [PubMed] [Google Scholar]
- 15. Goossens V.; Grooten J.; De Vos K.; Fiers W. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc. Natl. Acad. Sci. USA 92:8115–8119; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gotoh Y.; Cooper J. A. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-α signal transduction. J. Biol. Chem. 273:17477–17482; 1998. [DOI] [PubMed] [Google Scholar]
- 17. Gupta S.; Campbell D.; Dörijard B.; Davis R. J. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 267:389–393; 1995. [DOI] [PubMed] [Google Scholar]
- 18. Han J.; Jiang Y.; Li Z.; Kravchenko V. V.; Ulevitch R. J. MEF2C participates in inflammatory responses via p38-mediated activation. Nature 386:563–566; 1997. [DOI] [PubMed] [Google Scholar]
- 19. Herskowitz I. MAP kinase pathways in yeast: For mating and more. Cell 80:187–197; 1995. [DOI] [PubMed] [Google Scholar]
- 20. Hirai S.-i.; Katoh M.; Terada M.; Kyriakis J. M.; Zon L. I.; Rana A.; Avruch J.; Ohno S. MST/MLK2, a member of the miked lineage kinase family, directly phosphorylates and activates SEK1, an activator of c-Jun N-terminal kinase/stress-activated protein kinase. J. Biol. Chem. 272:15167-15173; 1997. [DOI] [PubMed] [Google Scholar]
- 21. Hirai S.-i.; Noda K.; Moriguchi T.; Nishida E.; Yamashita A.; Deyama T.; Fukuyama K.; Ohno S. Differential activation of two JNK activators, MKK7 and SEK1, by MKN28-derived nonreceptor serine/threonine kinase/mixed lineage kinase 2. J. Biol. Chem. 273:7406–7412; 1998. [DOI] [PubMed] [Google Scholar]
- 22. Holland P. M.; Suzanne M.; Campbell J. S.; Noselli S.; Cooper J. A. MKK7 is a stress-activated mitogen-activated protein kinase kinase functionally related to hemopterous. J. Biol. Chem. 272:24994–24998; 1997. [DOI] [PubMed] [Google Scholar]
- 23. Holtzman L. B.; Merritt S. E.; Fan G. Identification, molecular cloning and characterization of dual leucine zipper bearing kinase. J. Biol. Chem. 269:30808–30817; 1994. [PubMed] [Google Scholar]
- 24. Hsu H.; Xiong J.; Goeddel D. V. The TNF receptor-1-associated protein TRADD signals cell death and NF-κB activation. Cell 81:495–504; 1995. [DOI] [PubMed] [Google Scholar]
- 25. Hsu H.; Shu H.-B.; Pan M.-G.; Goeddel D. V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84:299–308; 1996. [DOI] [PubMed] [Google Scholar]
- 26. Hsu H.; Huang J.; Shu H.-B.; Baichwal V.; Goeddel D. V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4:387–396; 1996. [DOI] [PubMed] [Google Scholar]
- 27. Hu M. C.-T.; Qiu W. R.; Wang X.; Meyer C. F.; Tan T.-H. Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes Dev. 10:2251–2264; 1996. [DOI] [PubMed] [Google Scholar]
- 28. Hutchison M.; Berman K. S.; Cobb M. H. Isolation of TAO1, a protein kinase that activates MEKs in stress-activated protein kinase cascades. J. Biol. Chem. 273:28625–28632; 1998. [DOI] [PubMed] [Google Scholar]
- 29. Ichijo H.; Nishida E.; Irie K.; ten Dijke P.; Saitoh M.; Moriguchi T.; Takagi M.; Matsumoto K.; Miyazono K.; Gotoh Y. (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275:90–94; 1997. [DOI] [PubMed] [Google Scholar]
- 30. Karin M.; Liu Z.-g.; Zandi E. AP-1 function and regulation. Curr. Opin. Cell Biol. 9:240–246; 1997. [DOI] [PubMed] [Google Scholar]
- 31. Katz P.; Whalen G.; Kehrl J. H. Differential expression of a novel protein kinase in human B lymphocytes: Preferential localization in the germinal center. J. Biol. Chem. 269:16802–16809; 1994. [PubMed] [Google Scholar]
- 32. Kelliher M. A.; Grimm S.; Ishida Y.; Kuo F.; Stanger B. Z.; Leder P. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8: 297–303; 1998. [DOI] [PubMed] [Google Scholar]
- 33. Kiefer F.; Tibbies L. A.; Anafi M.; Janssen A.; Zanke B. W.; Lassam N.; Pawson T.; Woodgett J. R.; Iscove N. R. HPK1, a hematopoietic protein kinase activating the SAPK/JNK pathway. EMBO J. 15: 7013–7025; 1996. [PMC free article] [PubMed] [Google Scholar]
- 34. Kyriakis J. M.; Banerjee P.; Nikolakaki E.; Dai T.; Rubie E. A.; Ahmad M. F.; Avruch J.; Woodgett J. R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369:156–160; 1994. [DOI] [PubMed] [Google Scholar]
- 35. Kyriakis J. M.; Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays 18:567–577; 1996. [DOI] [PubMed] [Google Scholar]
- 36. Kyriakis J. M.; Avruch J. Sounding the alarm: Protein kinase cascades activated by stress and inflammation. J. Biol. Chem. 271:24313–24316; 1996. [DOI] [PubMed] [Google Scholar]
- 37. Kyriakis J. M. Signaling by the germinal center kinase family of protein kinases. J. Biol. Chem. 274:5259–5262; 1999. [DOI] [PubMed] [Google Scholar]
- 38. Lange-Carter C. A.; Pleiman C. M.; Gardner A. M.; Blumer K. J.; Johnson G. L. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science 260:315–319; 1993. [DOI] [PubMed] [Google Scholar]
- 39. Lawler S.; Fleming Y.; Goedert M.; Cohen P. Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr. Biol. 8:1387–1390; 1998. [DOI] [PubMed] [Google Scholar]
- 40. Liu Z.-g.; Hsu H.; Goeddel D. V.; Karin M. Dissection of TNF receptor-1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 87:565–576; 1996. [DOI] [PubMed] [Google Scholar]
- 41. Malinin N. L.; Boldin M. P.; Kovalenko A. V.; Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD-95 and IL-1. Nature 385:540–544; 1997. [DOI] [PubMed] [Google Scholar]
- 42. Moriguchi T.; Kuroyanagi N.; Yamaguchi K.; Gotoh Y.; Irie K.; Kano T.; Shirakabe K.; Muro Y.; Shibuya H.; Matsumoto K.; Nishida E.; Hagiwara M. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J. Biol. Chem. 271:13675–13679; 1996. [DOI] [PubMed] [Google Scholar]
- 43. Natoli G.; Costanzo A.; Ianni A.; Templeton D. J.; Woodgett J. R.; Balsano C.; Levrero M. Activation of SAPK/JNK by TNF receptor-1 through a noncyto-toxic TRAF2-dependent pathway. Science 275:200–203; 1997. [DOI] [PubMed] [Google Scholar]
- 44. Nishitoh H.; Saitoh M.; Mochida Y.; Takeda K.; Nakano H.; Rothe M.; Miyazono K.; Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 2:389–395; 1998. [DOI] [PubMed] [Google Scholar]
- 45. Pombo C. M.; Kehrl J. H.; Sánchez I.; Katz P.; Avruch J.; Zon L. I.; Woodgett J. R.; Force T.; Kyriakis J. M. Activation of the SAPK pathway by the human STE20 homologue germinal center kinase. Nature 377:750–754; 1995. [DOI] [PubMed] [Google Scholar]
- 46. Raingeaud J.; Whitmarsh A. J.; Barett T.; Dérijard B.; Davis R. J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell. Biol. 16:1247–1255; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rana A.; Gallo K.; Godowski P.; Hirai S.-i.; Ohno S.; Zon L. I.; Kyriakis J. M.; Avruch J. The mixed lineage protein kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK1. J. Biol. Chem. 271:19025–19028; 1996. [DOI] [PubMed] [Google Scholar]
- 48. Rome M.; Wong S. C.; Henzel W. J.; Goeddel D. V. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75-kDa tumor necrosis factor receptor. Cell 78:681–692; 1994. [DOI] [PubMed] [Google Scholar]
- 49. Saitoh M.; Nishitoh H.; Fujii M.; Takeda K.; Tobiume K.; Sawada Y.; Kawabata M.; Miyazono K.; Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17:2596–2606; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Salmeron A.; Ahmad T. B.; Carlile G. W.; Pappin D.; Narsimhan R. P.; Ley S. C. Activation of MEK-1 and SEK-1 by Tpl-2 proto oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 15:817–826; 1996. [PMC free article] [PubMed] [Google Scholar]
- 51. Sanchez I.; Hughes R. T.; Mayer B. J.; Yee K.; Woodgett J. R.; Avruch J.; Kyriakis J. M.; Zon L. I. Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature 372:794–798; 1994. [DOI] [PubMed] [Google Scholar]
- 52. Shi C.-S.; Kehrl J. H. Activation of stress-activated protein kinase/c-Jun N-terminal kinase, but not NF-κB, by the tumor necrosis factor (TNF) receptor 1 through a TNF receptor-associated factor 2- and germinal center kinase related-dependent pathway. J. Biol. Chem. 272:32102–32107; 1997. [DOI] [PubMed] [Google Scholar]
- 53. Smith C. A.; Farrah T.; Goodwin R. G. The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation and death. Cell 76:959–962; 1994. [DOI] [PubMed] [Google Scholar]
- 54. Song H. Y.; Régnier C. H.; Kirschning C. J.; Goeddel D. V.; Rothe M. Tumor necrosis factor (TNF)-mediated kinase cascades: Bifurcation of nuclear factor-B and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2 Proc. Natl. Acad. Sci. USA 94:9792–9796; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Staal F. J. T.; Roederer M.; Herzenberg L. A.; Herzenberg L. A. Intracellular thiols regulate activation of nuclear factor κB and transcription of human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 87:9943–9947; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stanger B. Z.; Leder P.; Lee T.-H.; Kim E.; Seed B. RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81:513–523; 1995. [DOI] [PubMed] [Google Scholar]
- 57. Su Y.-C.; Han J.; Xu S.; Cobb M.; Skolnik E. Y. NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 16:1279–1290; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Su Y.-C.; Treisman J. E.; Skolnik E. Y. The Drosophila Ste20-related kinase misshapen is required for embryonic dorsal closure and acts through a JNK MAPK module on an evolutionarily conserved signaling pathway. Genes Dev. 12:2371–2380; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Takekawa M.; Posas F.; Saito H. A human homolog of the yeast Ssk2/Ssk22 MAP kinase kinase kinases, MTK1, mediates stress-induced activation of the p38 and JNK pathways. EMBO J. 16:4973–4982; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takekawa M.; Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell 95:521–530; 1998. [DOI] [PubMed] [Google Scholar]
- 61. Takeuchi M.; Rothe M.; Goeddel D. V. Anatomy of TRAF2. J. Biol. Chem. 271:19935–19942; 1996. [DOI] [PubMed] [Google Scholar]
- 62. Tournier C.; Whitmarsh A. J.; Cavanagh J.; Barrett T.; Davis R. J. Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc. Natl. Acad. Sci. USA 94:7337–7342; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tracey K. J.; Cerami A. Tumor necrosis factor, other cytokines and disease. Annu. Rev. Cell Biol. 9:317–343; 1993. [DOI] [PubMed] [Google Scholar]
- 64. Treisman R. Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell. Biol. 8:205–215; 1996. [DOI] [PubMed] [Google Scholar]
- 65. Tung R. M.; Blenis J. A novel human SPS1/STE20 homologue, KHS, activates Jun N-terminal kinase. Oncogene 14:653–659; 1997. [DOI] [PubMed] [Google Scholar]
- 66. Van Aelst L.; D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 11:2295–2322; 1997. [DOI] [PubMed] [Google Scholar]
- 67. Vandenabeele P.; Declercq W.; Beyaert R.; Fiers W. Two tumour necrosis factor receptors: Structure and function. Trends Cell Biol. 5:392–399; 1995. [DOI] [PubMed] [Google Scholar]
- 68. Wallach D. Cell death induction by TNF: A matter of self control. Trends Biochem. Sci. 22:107–109; 1997. [DOI] [PubMed] [Google Scholar]
- 69. Whitmarsh A. J.; Shore P.; Sharrocks A. D.; Davis R. J. Integration of MAP kinase signal transduction pathways at the serum response element. Science 269:403–407; 1995. [DOI] [PubMed] [Google Scholar]
- 70. Xu S.; Robbins D. J.; Christerson L. B.; English J. M.; Vanderbilt G.; Cobb M. H. Cloning of Rat MEK kinase 1 cDNA reveals an endogenous membrane-associated 195-kDa protein with a large regulatory domain. Proc. Natl. Acad. Sci. USA 93:5291–5295; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yamaguchi K.; Shirakabi K.; Shibuya H.; Irie K.; Oishi I.; Ueno N.; Taniguchi T.; Nishida E.; Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-β signal transduction. Science 270:2008–2011; 1995. [DOI] [PubMed] [Google Scholar]
- 72. Yan M.; Dai T.; Deak J. C.; Kyriakis J. M.; Zon L. I.; Woodgett J. R.; Templeton D. J. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature 372:798–800; 1994. [DOI] [PubMed] [Google Scholar]
- 73. Yang X.; Khosravi-Far R.; Chang H. Y.; Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89:1067–1076; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yeh W.-C.; Shahinian A.; Speiser D.; Kraunus J.; Billia F.; Wakeham A.; de la Pompa J. L.; Ferrick D.; Hum B.; Iscove N.; Ohashi P.; Rothe M.; Goeddel D. V.; Mak T. W. Early lethality, functional NF-κB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7:715–725; 1997. [DOI] [PubMed] [Google Scholar]
- 75. Yuan J. Transducing signals of life and death. Curr. Opin. Cell Biol. 9:247–251; 1997. [DOI] [PubMed] [Google Scholar]
- 76. Yuasa T.; Ohno S.; Kehrl J. H.; Kyriakis J. M. Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. J. Biol. Chem. 273:22681–22692; 1998. [DOI] [PubMed] [Google Scholar]