

Abstract
Stimuli-responsive nanoparticles hold great promise for drug delivery to improve the safety and efficacy of cancer therapy. One of the most investigated stimuli-responsive strategies is to induce drug release by heating with laser, ultrasound, or electromagnetic field. More recently, cryosurgery (also called cryotherapy and cryoablation), destruction of diseased tissues by first cooling/freezing and then warming back, has been used to treat various diseases including cancer in the clinic. Here we developed a cold-responsive nanoparticle for controlled drug release as a result of the irreversible disassembly of the nanoparticle when cooled to below ~10 °C. Furthermore, this nanoparticle can be used to generate localized heating under near infrared (NIR) laser irradiation, which can facilitate the warming process after cooling/freezing during cryosurgery. Indeed, the combination of this cold-responsive nanoparticle with ice cooling and NIR laser irradiation can greatly augment cancer destruction both in vitro and in vivo with no evident systemic toxicity.
Keywords: Cold-Responsive, NIPAM, PLGA, photothermal, combination therapy
1. Introduction
Stimuli-responsive drug release holds great promise to improve the safety and efficacy of chemotherapy for treating cancer [1–3]. In order to minimize drug release in normal tissue, both endogenous (e.g., ATP, glutathione, and pH) and external (e.g., laser, ultrasound, and electromagnetic field) stimuli have been explored to control the drug release from nanoparticles in tumor [3–8]. Thermally triggered drug release is one of the most investigated stimuli-responsive strategies, and has been widely explored for cancer therapy [9,10]. Existing thermally responsive strategies have been focused on heating to specifically increase the temperature in tumor using laser, microwave, or alternating magnetic field (for magnetic nanoparticles) [11–17]. However, in the clinic, cryosurgery (also called cryotherapy or cryoablation) that destroys diseased tissues by cooling/freezing, has been used to treat various diseases including cancer and cardiovascular malfunctions [18–22].
During cryosurgery, an iceball of the diseased tissue is created by using one or more cryosurgical probes and an intraoperative imaging technique (e.g., ultrasound) is often used to monitor the iceball size [18–22]. However, there is a significant temperature gradient between the cryoprobe (less than approximately −100 °C) and the iceball surface (approximately 0 °C) [23–26], and a freezing temperature of less than approximately −20 °C is required to achieve effective killing of diseased tissues/cells [27,28]. In other words, the diseased cells/tissues in an iceball (particularly in the peripheral region next to the iceball surface with a cold temperature of ~0 °C) visualized by the intraoperative imaging technique may not be completely killed, which results in uncertainty and ineffectiveness of the cryosurgical treatment. To address this issue, various chemical adjuvants including the small molecule anticancer drug have been combined with cryosurgery for improved therapy [29–32]. Moreover, two studies have explored the use of nanoparticles to deliver the chemical adjuvants for combining with cryosurgery [33,34]. However, none of the nanoparticles used is sensitive to cold temperature. Therefore, rational design of nanoparticles that are responsive to cold temperature and capable of cold-triggered drug release is in demand to minimize the systemic toxicity of the chemical adjuvants and improve the efficacy of cryosurgery against diseased cells/tissues at cold temperature (similar to that next to the surface of the frozen tissue iceball monitored intraoperatively). Because cold temperature can be achieved by cooling with ice readily available in a hospital setting without the need of any specialized instrument for surface cooling or by using cooling catheters for deep-tissue cooling [35–37], development of cold-responsive nanoparticles for drug delivery should facilitate the clinical application of both cryosurgery and nanomedicine.
Thermally responsive drug delivery systems have been made using poly(N-isopropyl acrylamide (PNIPAM) that exhibits a lower critical solution temperature (LCST) at ~32 °C in aqueous solution [38–41] or Pluronic F127 (PF127) with a LCST of ~25 °C on average in aqueous solution [42,43]. The polymers are hydrophobic at temperatures higher than their LCST, while they have high solubility in water at temperatures lower than their LCST. In previous studies, PNIPAM or PF127-based nanoparticles are used to achieve triggered drug release at temperatures higher than room temperature by heating to shrink/collapse the nanoparticles [40,44–46] or at temperatures lower than body temperature but higher than room temperature by cooling (also called cold shock, which is somewhat misleading because the LCST is higher than room temperature) to expand them [41–43]. For fabrication of polymeric nanoparticles, one of the widely used strategies is to utilize the hydrophobic and hydrophilic properties of different polymers to achieve self-assembly of the polymers by emulsification of water and organic solvents (e.g., double emulsion or emulsion solvent evaporation) [47–49]. Therefore, if the LCST of the thermo-responsive polymer is lower than room temperature, this polymer can be used as a hydrophobic polymer to prepare nanoparticles at room temperature. Once the temperature is lower than the LCST, the thermo-responsive polymer becomes hydrophilic (i.e., highly soluble in water) and can cause disassembly of the nanoparticles in aqueous solutions. Unlike previous studies, this nanoparticle is responsive to low temperature (or cold) and drug release can be triggered by the cold-induced disassembly of the nanoparticle rather than shrinkage in size.
Cancer stem-like cells (CSCs) have attracted a great deal of attention in the past several decades due to their high resistance to chemotherapy that is responsible for the many failures of chemotherapy [50]. Fortunately, hyaluronic acid (HA) is the natural ligand of the variant CD44 receptors overexpressed on many types of cancer cells and CSCs in tumor [51]. Therefore, we decorated the surface of the cold-responsive nanoparticles with HA for targeted delivery of theranostic agents to cancer cells and CSCs. Furthermore, the photothermal effect has been shown to be efficient for improving the antitumor capacity of chemotherapeutic drugs [52]. We therefore encapsulate a near infrared (NIR) dye (indocyanine green, ICG in short) in the nanoparticles, which could generate heat under NIR laser irradiation for the photothermal effect [53]. The goal of this study is to synthesize such cold-responsive nanoparticles for controlled drug delivery and enhanced combination therapy of orthotopic triple-negative human mammary cancer in vitro and in vivo.
2. Materials and methods
2.1. Materials
Pluronic F127 (PF127) and PNIPAM-B were purchased from Sigma (St. Louis, MO, USA). Hyaluronic acid (HA, Mw: 66–90 kDa) was purchased from Lifecore Biomedical (Chaska, MN, USA). Polyvinyl Alcohol (PVA, Mw: 100 kDa) was purchased from Fisher Scientific (Pittsburgh, PA, USA). Chitosan oligosaccharide of pharmaceutical grade (Mw: 1.2 kDa, 95% deacetylation) was purchased from Zhejiang Golden Shell Biochemical Co. Ltd (Yuhuan, Zhejiang, China). The CCK-8 cell proliferation reagent was purchased from Dojindo Molecular Technologies (Rockville, MD, USA). Fetal bovine serum (FBS) and penicillin/streptomycin were purchased from Invitrogen (Carlsbad, CA, USA). The Dulbecco Modified Eagle Medium (DMEM) was purchased from ATCC (Manassas, VA, USA). Irinotecan was purchased from Selleck Chemicals (Houston, TX, USA). All other chemicals were purchased from Sigma (St. Louis, MO, USA) unless specifically mentioned otherwise.
2.2. Preparation of chitosan-modified PF127 (chitosan-PF127)
The chitosan-PF127 was prepared using a previously reported procedure [54]. Briefly, a total of 30 ml of PF127 solution (26 mM in benzene) was added dropwise into 30 ml of 4-nitrophenyl chloroformate (4-NPC) solution (160 mM in benzene) and the mixture was stirred for 3 h in N2 atmosphere at room temperature to activate PF127. The activated polymer was then precipitated and filtered in excess (ice-cold) diethyl ether for three times and dried under vacuum overnight. To synthesize chitosan-PF127, 10 ml of chitosan solution (200 mg/ml) in deionized (DI) water was added dropwise into 10 ml of activated PF127 solution (400 mg/ml, in DI water). After stirring for 12 h, this mixture was dialyzed (molecular weight cut off or MWCO: 7 kDa) against DI water for 24 h. Lastly, the polymer was freeze-dried for 48 h to remove water for further use.
2.3. Preparation of nanoparticles
To prepare nanoparticles with PF127 or poly(N-isopropylacrylamide-co-butylacrylate) (PNIPAM-B) alone, a double emulsion (water in oil in water or W-in-O-in-W) method with slight modification was used [47]. Briefly, 10 mg of PF127 or PNIPAM-B was dissolved in 2 ml of dichloromethane and the solution together with 0.4 ml of DI water was transferred into a centrifuge tube where the two immiscible solutions were emulsified by sonication for 1 min using a Branson 450 sonifier. Afterward, this initial emulsion and 4 ml of 2% polyvinyl alcohol (PVA) solution (in deionized or DI water) were emulsified by sonication for 2 min to obtain the double emulsion that was further processed by slowly dropping into 6 ml of 0.6% PVA (in DI water) and stirred for 10 min at room temperature. After rotary evaporation of the double emulsion to remove organic solvent (i.e., oil), the samples prepared with PNIPAM-B alone were collected by centrifugation at 13,800 g for 10 min at room temperature and washed twice with DI water. The samples prepared with PF127 alone were collected by using freeze-dry.
To prepare PN and HCPN (H for HA, C for chitosan, P for PF127, N for PNIPAM-B, with or without drug) nanoparticles, 10 mg of PNIPAM-B and 20 mg of PF127 together with or without a desired amount irinotecan (CPT in short) were dissolved in 2 ml of dichloromethane. After adding 0.4 ml DI water either with or without indocyanine green (ICG in short), the immiscible solutions were emulsified by sonication for 1 min. Then, this first emulsion and 4 ml of the solution of either PVA (for PN nanoparticles) or the mixture of chitosan-PF127 and HA (for HCPN nanoparticles) were emulsified by sonication for 2 min. After rotary evaporation to remove organic solvent, the nanoparticles were collected by centrifugation at 13,800 g for 10 min at room temperature and washed twice with DI water.
2.4. Encapsulation of agents in the nanoparticles and in vitro drug release
The encapsulation efficiency (EE) of agents (CPT and ICG) using the nanoparticles was calculated with the following equation:
| (1) |
where Wencapsulated represents the amount (in weight) of agents encapsulated into nanoparticles and Wfed is the initial total amount of agents fed for encapsulation. The amount of CPT and ICG was determined spectrophotometrically using a Beckman Coulter (Indianapolis, IN, USA) DU 800 UV-Vis spectrophotometer based on their absorbance at 354 and 778 nm, respectively.
To determine the drug release in vitro, drug-laden nanoparticles (20–30 mg) were reconstituted in PBS (pH 7.4) and transferred into dialysis bags (MWCO: 20 kDa) that were placed in 30 ml of the same PBS solution at 37 °C and stirred at 110 rpm using a mini-stir bar. At various times, 100 μl of the dialysate was collected and the remaining dialysate replenished with the same amount of fresh PBS. The concentration of the released CPT in the removed dialysate was determined using UV-Vis spectrophotometry based on absorbance at 354 nm. For ice (incubated in ice for 5 min), pH-responsive (pH 5.0, 5 min), or near infrared (NIR) laser (1 W/cm2, 5 min) irradiation-triggered drug release, the nanoparticles solution in tube were simply incubated in ice, changed with pH 5.0 solution, or irradiated with NIR laser. The supernatant of the nanoparticle solutions was obtained by centrifuging at 13,800 g and analyzed in the same way using UV-Vis spectrophotometry.
2.5. Characterization of HCPN-CG nanoparticles
The size of HCPN-CG (C for CPT and G for ICG) nanoparticles was assessed using a Brookhaven 90 Plus/BI-MAS dynamic light scattering (DLS) instrument by dispersing the nanoparticles (1 mg/ml) in DI water. The morphology of nanoparticles was characterized using both transmission (TEM) and scanning (SEM) electron microscopy. For TEM studies, the nanoparticles were examined after negatively stained with uranyl acetate solution (2%, w/w) using an FEI (Moorestown, NJ, USA) Tecnai G2 Spirit transmission electron microscope. The SEM experiments were conducted by depositing 10 μl of aqueous solutions of the nanoparticles on a freshly cleaved mica grid and allowing them to dry for 60 minutes in air. A thin film of Au was then sputtered onto the nanoparticles on the substrate. Samples were imaged with an FEI NOVA Nano400 scanning electron microscope.
2.6. Cell culture and in vitro cell viability
Triple negative human breast adenocarcinoma MDA-MB-231 cancer cells (ATCC) were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C in a humidified 5% CO2 incubator. To obtain 3D mammospheres enriched with CSCs, the well-established suspension culture was used [55]. Briefly, detached single cancer cells were cultured in 24-well ultralow attachment plates (Corning, Lowell, MA, USA) at a density of 20,000 cells/ml in 1 ml CSC medium consisting of serum-free DMEM/F12-K supplemented with 5 μg/ml insulin, 20 ng/ml epidermal growth factor (EGF), 20 ng/ml basic fibroblast growth factor (bFGF), 1 × B27 (Invitrogen), and 0.4% (w/v) bovine serum albumin (BSA). After 10 days, the mammospheres were collected for further experimental use. For 2D cultured cell viability, 10,000 cells were cultured in 96-well plates for 12 h, and then treated with various drug formulations. For quantifying the viability of 3D mammospheres, mammospheres cultured in 24-well plates as aforementioned were treated with various drug formulations directly. For further cooling and heating, cells ware incubated on ice for 5 min, cultured in incubator (37 °C) for 5 min, followed by either irradiating with laser (1 W/cm2 for 2 min) or incubating in waterbath (42 °C). The total cells in each sample were quantified using Cell Counting Kit-8 (CCK-8) assay according to the manufacturer’s instruction by measuring absorbance at 450 nm using a PerkinElmer VICTOR X4Multilabel plate reader.
2.7. In vitro imaging
Cells were seeded onto collagen-coated cover glasses (Nunc, Thermo Fisher Scientific Inc., Waltham, MA, USA) at a density of 2 × 105 cells/well in 6-well plate and grown at 37 °C for 12 h. The medium was then replaced with 2 ml of fresh medium containing different drug formulations. After incubation at 37 °C for 3 h, the cover glass attached with cells was mounted onto a glass slide with anti-fade mounting medium (Vector Laboratories Burlingame, CA, USA) for examination using an Olympus FluoView™ FV1000 confocal microscope.
2.8. TEM Imaging of Cells
Cells were seeded into Nunc™ Lab-Tek™ II Chamber Slide™ System (Thermo Fisher Scientific Inc., Waltham, USA) at a density of 1×106 cells/ml and were incubated with nanoparticles for 3 h. Ice treatment and laser irradiation are applied in the same way as that for cell viability studies. Samples were prepared for TEM according to standard procedures and examined using a FEI (Moorestown, NJ, USA) Tecnai G2 Spirit transmission electron microscope.
2.9. Flow cytometry analysis
MDA-MB-231 cells were first incubated in medium containing PN-CG and HCPN-CG for 3 h. After washing with PBS for 3 times, the cells were fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature. The cells were then washed with PBS twice and analyzed using a BD (Franklin Lakes, NJ, USA) LSR-II flow cytometer and Diva software.
2.10. Animals and xenograft Tumors
Athymic female NU/NU nude mice of 6-week old were purchased from Charles River (Wilmington, MA, USA) and maintained on a 16:8 h light-dark cycle. All procedures for animal use were approved by the Institutional Animal Care and Use Committee (IACUC) at The Ohio State University and all efforts were made to minimize animal suffering. To obtain xenograft of human breast tumor in the nude mice, detached mammosphere cells were suspended at 2 × 105 cells/ml in a mixture (1:1) of 1x PBS and matrigel. A total of 20,000 cells in 100 μl of the mixture were injected into the fat pad of mammary gland of nude mice of each 7-week-old mouse.
2.11. In vivo imaging and biodistribution
For in vivo imaging studies, after the tumor reached a volume of ~100 mm3, the mice were injected with 100 μl of saline, a simple mixture of 50 μg of ICG in 100 μl of saline, PN-CG nanoparticles (ICG: 50 μg and CPT: 200 μg), and HCPN-CG nanoparticles (ICG: 50 μg and CPT: 200 μg) in 100 μl of saline. In vivo fluorescence images were taken at 1, 3, 6, and 9 h after intravenous injection via the tail vein using a PerkinElmer (Waltham, MA, USA) IVIS instrument with excitation at 780 nm and an 830 nm filter to collect the fluorescence emission of ICG. After in vivo imaging, the mice were sacrificed and the tumor, liver, kidney, lung, spleen, and heart were removed and collected for further ex vivo fluorescence imaging of ICG using the same IVIS instrument.
2.12. In vivo antitumor efficacy and safety
After tumors reached a volume of ~100 mm3, mice were treated with 100 μl of saline or HCPN nanoparticles, CPT&ICG (with ice treatment for 5 min and NIR laser irradiation, 1 W/cm2 for 2 min, CPT: 3 mg/kg, ICG: 1.2 mg/kg body weight), HCPN-CG nanoparticles with NIR laser irradiation (1 W/cm2 for 2 min, CPT: 3 mg/kg, ICG: 1.2 mg/kg body weight), HCPN-CG nanoparticles with ice treatment (5 min, CPT: 3 mg/kg, ICG: 1.2 mg/kg body weight), PN-CG nanoparticles with both ice treatment (5 min) and NIR laser irradiation (~5 min after ice treatment, 1 W/cm2 for 2 min, CPT: 3 mg/kg, ICG: 1.2 mg/kg body weight), and HCPN-CG nanoparticles with ice treatment (5 min) and NIR laser irradiation (~5 min after ice treatment, 1 W/cm2 for 2 min, CPT: 3 mg/kg, ICG: 1.2 mg/kg body weight). A total of 100 μl of saline was used as the carrier for all the drug formulations. The ice treatment and NIR laser irradiation were conducted at 12 h after the intravenous drug injection. After ice treatment and/or laser irradiation, mice were also checked for the whole body temperature by using infrared thermographic maps. Tumor growth was monitored every 5 days. The tumor volume (V) was calculated as: V = (L × W2)/2, where L is long diameter and W is short diameter determined using a caliper. The mice were euthanized at day 30 after the drug injection. Tumors, livers, lungs, hearts, spleens, and kidneys were collected, formalin fixed, paraffin embedded, and haematoxylin&eosin (H&E) stained for further standard histological analysis.
2.13. Statistical analysis
All data are reported as mean ± standard deviation (SD) from at least three independent runs. The Kruskal-Wallis H test and the Mann-Whitney U test were used to assess the overall among-group and two-group differences, respectively. All statistical analyses were carried out with the IBM (Chicago, IL, USA) SPSS 22 software.
3. Results
3.1. Preparation and characterization of cold-responsive nanoparticles
In this study, we used a “green” approach to prepare cold-responsive nanoparticles without the need of chemical reaction to break or form covalent bonds. As shown in Fig. 1A, this was achieved by assembling Pluronic F127 (PF127), poly(N-isopropylacrylamide-co-butylacrylate) (PNIPAM-B instead of PNIPAM with no modification, NIPAM:B = 8:1, Mn = 30,000), chitosan-modified PF127 (PF127-chitosan), and hyaluronic acid (HA) using a double emulsion approach. The amphiphilic PF127 consists of both more hydrophilic polyethylene glycol (PEG) blocks and more hydrophobic polypropylene glycol (PPG) blocks and is soluble in both oil and water at room temperature. The PNIPAM-B is a thermally responsive polymer with an LCST of 14–16 °C, which is lower than the LCST of PNIPAM with no modification. This means it is hydrophobic at room temperature (~22 °C). Therefore, the cold-responsive nanoparticles were synthesized by first dissolving PF127 and PNIPAM-B in oil (O) to emulsify with water (W) at room temperature during the first emulsion (W-in-O) (Fig. 1A). At the same time, the hydrophobic irinotecan (CPT-11 or CPT in short) was dissolved in oil and incorporated into the hydrophobic shell of PNIPAM-B and PPG during the first emulsion as the chemotherapy drug. We further encapsulated the hydrophilic indocyanine green (ICG, dissolved in water) inside the hydrophilic core during the first emulsion (Fig. 1A), to generate heat in the nanoparticles under near infrared (NIR, ~800 nm) laser irradiation [56].
Fig. 1.
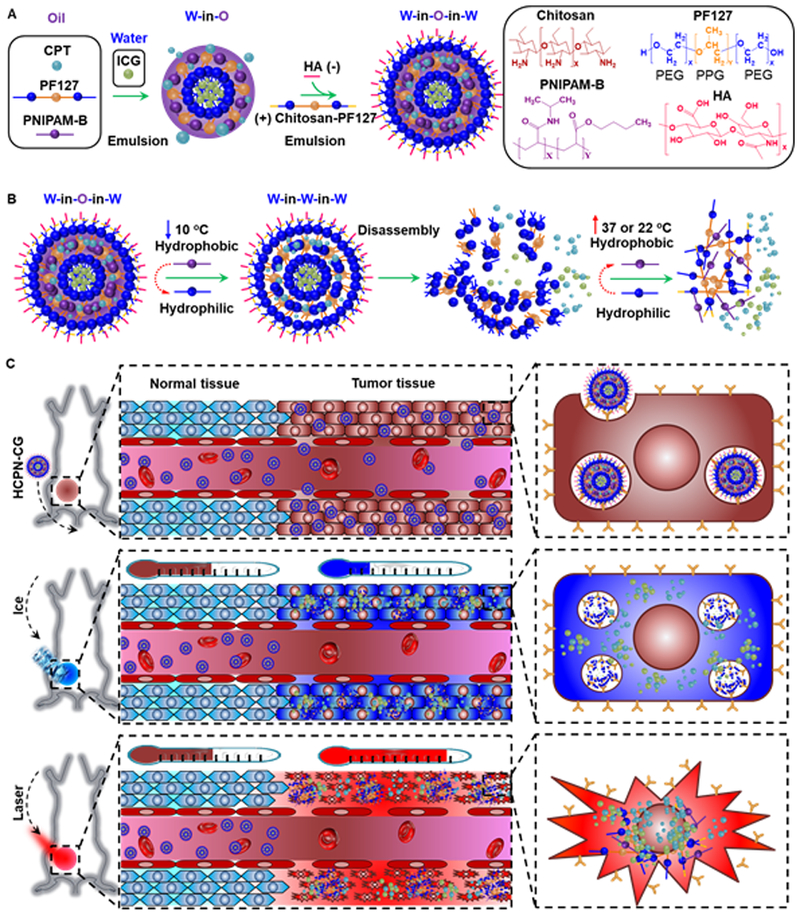
Synthesis and mechanism of cold-responsive nanoparticle for drug delivery to treat cancer. (A) Hyaluronic acid (HA or H), chitosan-modified Pluronic F127 (chitosan-PF127 or C), PF127 (P), and poly(N-isopropylacrylamide-co-butylacrylate) (PNIPAM-B or N) were used to prepare the irinotecan (CPT or C) and indocyanine green (ICG or G)-laden HCPN-CG nanoparticles using the double-emulsion method. (B) The thermal phase transition behavior of PNIPAM-B from being water-insoluble to highly water-soluble can cause disassembly of the HCPN-CG nanoparticles upon cooling to below room temperature, which can result in burst release of the encapsulated drug. (C) In vivo accumulation of HCPN-CG nanoparticles in tumor through the enhanced permeability and retention (EPR) effect of tumor vasculature, burst drug release upon cooling with ice, and enhanced antitumor efficacy with ice cooling followed by near infrared (NIR) laser irradiation.
HA is decorated on the nanoparticle surface because it is a natural ligand of the variant CD44 commonly overexpressed on many types of cancer cells and particularly CSCs [54]. The cold-responsive nanoparticles were formed by further emulsifying the first emulsion (W-in-O) in water dissolved with HA and PF127-chitosan for the second emulsion (W-in-O-in-W, Fig. 1A). The PPG of PF127-chitosan is integrated into the hydrophobic shell formed during the first emulsion while HA and chitosan-PEG stabilize and decorate the surface of the resultant nanoparticles through the electrostatic interactions between HA (negatively charged) and chitosan (positively charged) during the second emulsion [17,51]. The nanoparticles can be collected by centrifugation after removing oil by rotary evaporation under vacuum. The encapsulation efficiency of CPT and ICG in the resultant nanoparticles is 78.5 ± 4.9% and 45.2 ± 3.7%, respectively. We anticipate that the nanoparticles should disassemble when the temperature is lower than the LCST of PNIPAM-B to trigger quick release of the encapsulated drug (Fig. 1B). After injected into mice, nanoparticles of 20–150 nm can preferentially accumulate in tumor as a result of the enhanced permeability and retention (EPR) effect of tumor that has a leaky vasculature with no lymphatic drainage (Fig. 1C) [57,58]. The tumor will be cooled with ice to trigger the drug release first and then irradiated with NIR laser to increase the temperature to enhance the antitumor ability (Fig. 1C) [56].
Typical transmission electron microscopy (TEM) image of the resultant nanoparticles (HCPN-CG, H for HA, C for chitosan, P for PF127, N for PNIPAM-B, C for CPT, and G for ICG) is shown in Fig. 2A. The nanoparticles have a spherical morphology and core-shell structure. In contrast, no nanoparticles could be collected by centrifugation if PF127 was used as the only polymer for nanoparticle synthesis with the procedure shown in Fig. 1A (Fig. S1A). After freeze-drying the supernatant, no nanoparticles and only polymer aggregates could be seen in the freeze-dried product (Fig. S1B). Although some materials could be collected if PNIPAM-B was used as the only polymer to prepare the nanoparticles (Fig. S1A), no nanoparticles could be seen in the SEM image of the collected materials either (Fig. S1C). This is not surprising as PNIPAM-B has poor solubility in water at room temperature, and easily forms aggregates that can be spun down by centrifugation. This is further confirmed by shining a red laser beam through the aqueous PF127, PNIPAM-B, and HCPN (made without CPT and ICG using the same procedure as that shown in Fig. 1A) samples. A light track is discernible only in the HCPN sample (Fig. S2, the Tyndall effect) as a result of light scattering by the HCPN nanoparticles. The absence of the Tyndall effect indicates a negligible amount of nanoparticles in the other two samples.
Fig. 2.
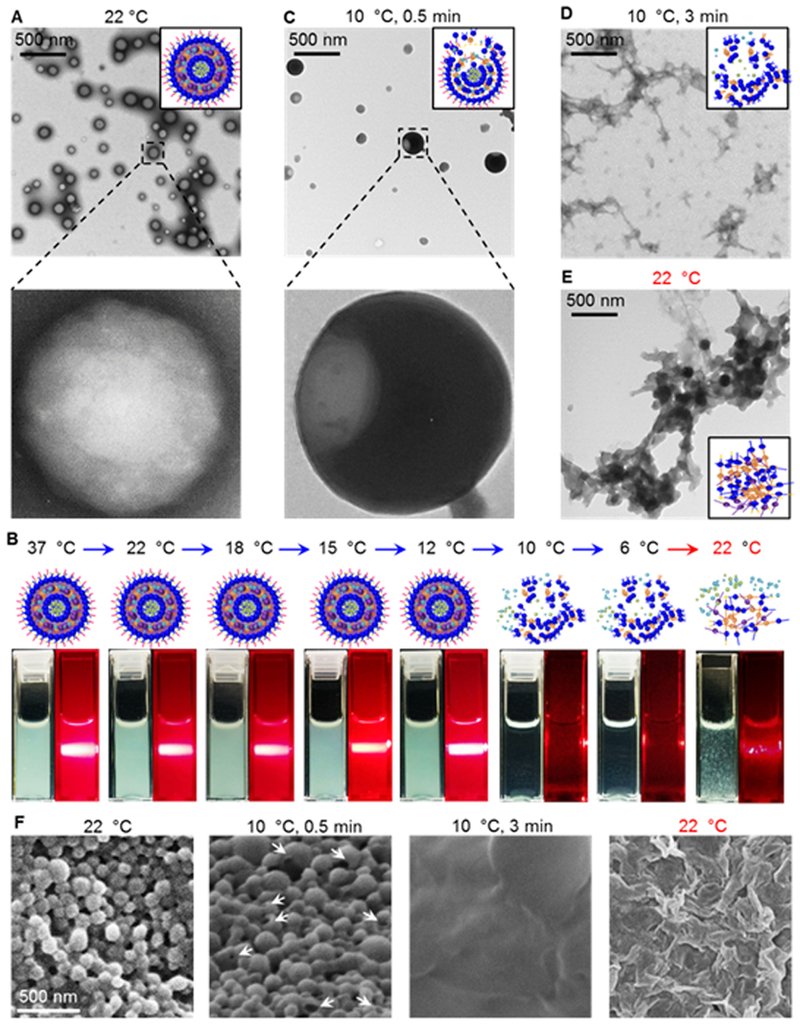
Characterization of HCPN-CG nanoparticles and their cold responsiveness. (A) TEM images showing the HCPN-CG nanoparticles are spherical with a core-shell configuration. (B) Typical schemes and photographs of the aqueous samples of HCPN-CG nanoparticles at various temperatures before and after shining a red laser beam through them in the dark. As a result of the Tyndall effect (i.e., scattering of laser beam by nanoparticles in solution), a bright white track of light is visible in the dark in the solutions of HCPN-CG nanoparticles above 10 °C. However, it is not clearly observable at or below 10 °C and after warming back to room temperature (i.e., 22 °C), indicating the HCPN-CG nanoparticles disassemble upon cooling to 10 °C (or a lower temperature) and the disassembling process is not reversible. (C) TEM images showing the partially dissembled HCPN-CG nanoparticles after incubating at 10 °C for 0.5 min. (D) The nanoparticles become completely disassembled after 3 min incubation at 10 °C. (E) No nanoparticles are clearly observable after warming back to 22 °C. (F) SEM images of HCPN-CG nanoparticles showing the same cold responsiveness observed with the TEM studies. Arrows indicate partially dissembled HCPN-CG nanoparticles after incubating at 10 °C for 0.5 min.
3.2. Cold-responsive drug release of HCPN-CG nanoparticles
Next, we investigated the cold-responsive property of the HCPN-CG nanoparticles. As shown in Fig. 2B, the solution of HCPN-CG nanoparticles has the Tyndall effect at or above 12 °C, confirming the presentence of the nanoparticles in the solution. Interestingly, when the temperature decreases to 10 or 6 °C, the solution of HCPN-CG nanoparticles becomes clear and transparent and no evident Tyndall effect could be observed, suggesting disassembly of the nanoparticles. Furthermore, the disassembly appears to be irreversible because the Tyndall effect is not evident when the temperature is increased back to room temperature (22 °C, Fig. 2B). We further confirmed the cold-responsiveness of HCPN-CG nanoparticles with TEM. When incubated at 10 °C for 0.5 min, the HCPN-CG nanoparticles become partially dissembled (Fig. 2C). Moreover, no nanoparticles are clearly observable if the incubation time at 10 °C is 3 min (Fig. 2D). Extensive polymer aggregates formed when the temperature was increased back to room temperature (22 °C, Fig. 2E). These observations at the different temperatures and times are further supported by SEM studies (Fig. 2F). The extensive polymer aggregates may be responsible for the vague Tyndall effect observed in the sample after warming back to 22 °C, as shown in Fig. 2B.
The size distribution of HCPN-CG nanoparticles in aqueous solutions at different temperatures was determined by dynamic light scattering (DLS) analyses. As shown in Fig. S3A, the HCPN-CG nanoparticles in aqueous solution have a narrow size distribution and their size is similar (~90 nm on average) at all the temperature above 12 °C. However, no stable measurement could be obtained at 10 and 6 °C, probably because nearly all the nanoparticles are dissembled at the cold temperatures. Similarly, the zeta potential of HCPN-CG nanoparticles is also stable at all the temperature above 12 °C, and could not be detected at 10 and 6 °C (Fig. S3B). More importantly, the HCPN-CG nanoparticles are stable at room temperature for at least 56 days according to the DLS size analyses (Fig. S4). For long-time storage of the HCPN-CG nanoparticles without significant drug release, one convenient way is to remove most of the supernatant after centrifugation during the preparation of the HCPN-CG nanoparticles. As shown in Fig. S5A–B, after kept at room temperature for 56 days, the HCPN-CG nanoparticles can still be homogenously dispersed in DI water and the size of the HCPN-CG nanoparticles is similar to that on the day 0 (Fig. S5B) with minimal drug release (⁓2%). This may allow convenient handling (e.g., washing by centrifuging and re-suspending) of the drug-laden nanoparticles at room temperature without significantly losing the payload, unlike a previously reported nanoparticle that is responsive to room temperature [41–43]. The stability of HCPN-CG nanoparticles under physiological condition is also investigated by dispersing the HCPN-CG nanoparticles in fetal bovine serum (FBS). As shown in Fig. S6, the size of the HCPN-CG nanoparticles in FBS solution is stable and did not form any large aggregates for at least 3 days. In order to confirm the cold-responsive property of the HCPN nanoparticles is due to PNIPAM-B, poly (lactic-co-glycolic acid) (PLGA, a polymer widely used for preparing nanoparticles) was used to replace PNIPAM-B for preparing nanoparticles. As shown in Fig. S7, the PLGA-based nanoparticles are not cold responsive. We further confirmed the cold-responsive property of HCPN nanoparticles is due to PNIPAM-B polymer by using PVA instead of HA and PF127-chitosan to prepare the nanoparticles (PN-CG). As shown in Fig. S8A, the size of the PN-CG nanoparticles is 103.9 ± 1.5 nm and zeta potential is −43.7 ± 2.7 at room temperature. The PN-CG nanoparticles have a narrow size distribution at the temperature at 18, 15, and 12 °C (Fig. S8B), but no stable measurement could be obtained at 10 and 6 °C. This is further confirmed by taking photograph of the PN-CG nanoparticles solution at 22 and 10 °C. As shown in Fig. S8C, the solution of PN-CG nanoparticles has the Tyndall effect at 22 °C, but no evident Tyndall effect could be observed at 10 °C and the solution becomes clear and transparent. This suggests disassembly of the nanoparticles at the cold temperature.
We further checked the ultraviolet-visible (UV-Vis) absorbance of the HCPN nanoparticles at different temperatures. As shown in Fig. 3A, the absorbance of the HCPN nanoparticles decreases significantly when temperature is lowered to 10 or 6 °C, suggesting disassembly of the nanoparticles into soluble polymers in the solution. After encapsulated with CPT and ICG, although the absorbance peak (arrowhead) of ICG at 778 nm is evident at all temperatures, the absorbance peak (arrow) of CPT at 354 nm is barely identifiable at or above 12 °C (Fig. 3A, bottom) due to the strong absorbance of the HCPN nanoparticles at the same wavelength (Fig. 3A, top). Moreover, it shows up clearly at 10 or 6 °C when the nanoparticles are dissembled because of dissolution of all polymers in the sample. Next, we investigated the fluorescence intensity of CPT in HCPN-CG nanoparticles at different temperatures (Fig. 3B). The strongest fluorescence intensity is observed at 10 or 6 °C, suggesting CPT is released from the nanoparticles at the cold temperatures with minimal fluorescence quenching that could occur when CPT is encapsulated in the nanoparticles. To confirm this, the drug release profile of HCPN-CG nanoparticles was conducted by using ice to cool the samples. Although the drug release from HCPN nanoparticles is slow at 37 °C (~2% in 5 h), more than 80% of CPT can be released from the nanoparticles after cooled on ice for 5 min (Fig. 3C). In contrast, the drug release is less than 5% if pH is lowered to 5 for 5 min. As aforementioned, ICG encapsulated inside the nanoparticles can absorb NIR to generate heat. Indeed, under NIR laser irradiation (1 W/cm2), the temperature of HCPN-CG nanoparticle solution increases depending on the ICG concentration and time (Fig. S9). Therefore, we checked the high temperature triggered drug release. As shown in Fig. 3C, ~20% of the CPT is released from the nanoparticles during laser irradiation (1 W/cm2 for 5 min). We further checked the drug release profile when the stimuli are applied sequentially. After the cold-triggered drug release, the impact of NIR irradiation on the drug release is negligible (probably because most of the drug is already released after the cold treatment). In contrast, after NIR irradiation to induce ~20% drug release, the cold-triggered drug release is still apparent leading to a total of ~90% of drug release. Similarly, after acidic (pH 5) treatment with minimal drug release, the cold treatment can still induce significant drug release (~80%) from the nanoparticles. These data indicate that the cold triggered drug release is much more efficient than that induced by heating or low pH, and the latter two treatments do not significantly affect the efficiency of the cold-triggered drug release.
Fig. 3.
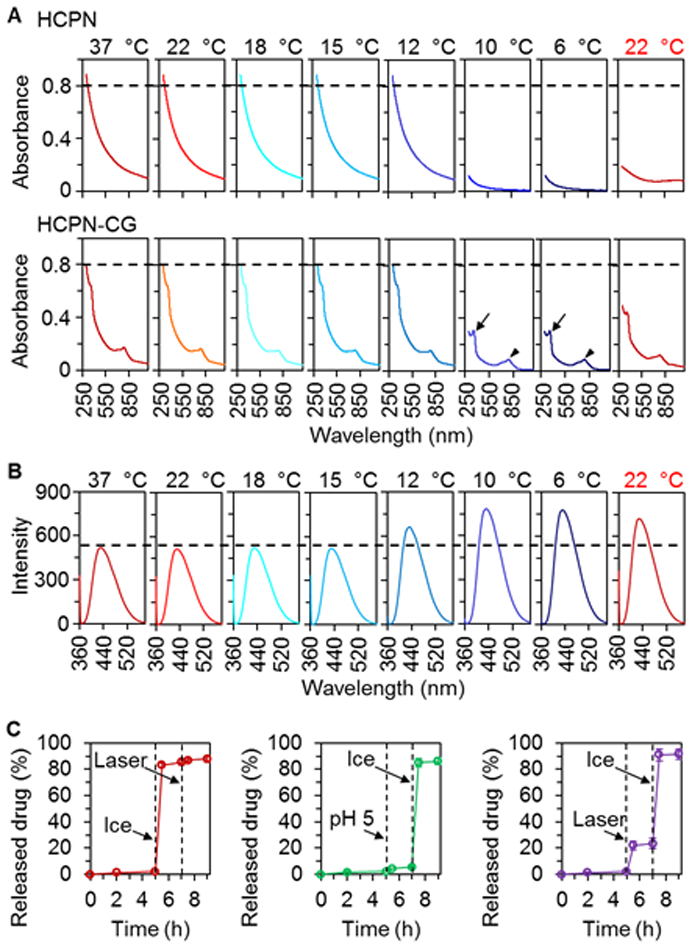
Characterization of cold-responsive drug release from HCPN-CG nanoparticles. (A) UV-Vis absorbance of HCPN (top) and HCPN-CG (bottom) nanoparticles at different temperature showing the cold-responsive ability of HCPN-CG nanoparticles. Arrow and arrowhead indicate the absorbance peaks of CPT and ICG, respectively. (B) Fluorescence emission spectra of HCPN-CG nanoparticles at various temperatures showing cold-triggered release of CPT. (C) A comparison of the triggered release of CPT from HCPN-CG nanoparticles by ice cooling (5 min), acidic pH (5.0, 5 min), and NIR laser irradiation (1 W/cm2, 5 min), showing the cold-triggered drug release from HCPN-CG nanoparticles is much more effective than the other two treatments. Moreover, the efficient drug release triggered by cold is not affected by pre-treatment with the NIR laser or low pH. Error bars represent ± S.D. (n = 3).
3.3. In vitro targeting and antitumor capacity of HCPN-CG nanoparticles
Both 2D-cultured MDA-MB-231 cancer cells and 3D-cultured MDA-MB-231 mammospheres enriched with CSCs were used to examine the capability of HCPN-CG nanoparticles for targeted drug delivery. For cellular uptake, we treated the 2D cells and 3D mammospheres with the mixture of the two free drugs (CPT&ICG) and HCPN-CG nanoparticles for 3 h. As shown in Fig. S10 (for free drug treated cells) and Fig. 4A–B (for nanoparticles treated cells), there was almost no free CPT in the 2D cells and 3D mammospheres, probably due to its hydrophobic nature (poorly soluble in water) with low bioavailability to the cells. In contrast, after encapsulated in HCPN-CG nanoparticles, CPT could be observed (i.e., taken up) in the 2D cells and 3D mammospheres after 3 h of incubation. We further checked the drug release inside the cells by treating them with laser (L) or ice (I). Stronger fluorescence of CPT could be seen in the cells after ice treatment than NIR laser irradiation, suggesting the cold-responsiveness of the HCPN-CG nanoparticles can be utilized to better control drug release inside cells than laser heating. The targeting capability of HCPN-CG nanoparticles was confirmed by treating cancer cells using nanoparticles without HA modification on their surface (PN-CG, Fig. S11). More CPT and ICG could be delivered inside cells by using HCPN than PN nanoparticles due to the targeting capability of HA on the HCPN nanoparticles [17,51]. The targeting capability of HCPN-CG nanoparticles was further supported by flow cytometry studies on cell uptake of CPT encapsulated in the nanoparticles. As shown in Fig. S12, the HA decoration could significantly increase the intracellular delivery of CPT using nanoparticles. More importantly, with further cooling by ice and then heating by laser irradiation, the morphology of the 2D cells treated with HCPN-CG was completely changed and became shrunken and spiky compared with other groups (last row of Fig. 4A). Similarly, cells in the 3D mammospheres treated with HCPN-CG also shrunk after the further ice and laser treatments, which is significant according to the quantitative data of the cell area shown in Fig. S13.
Fig. 4.
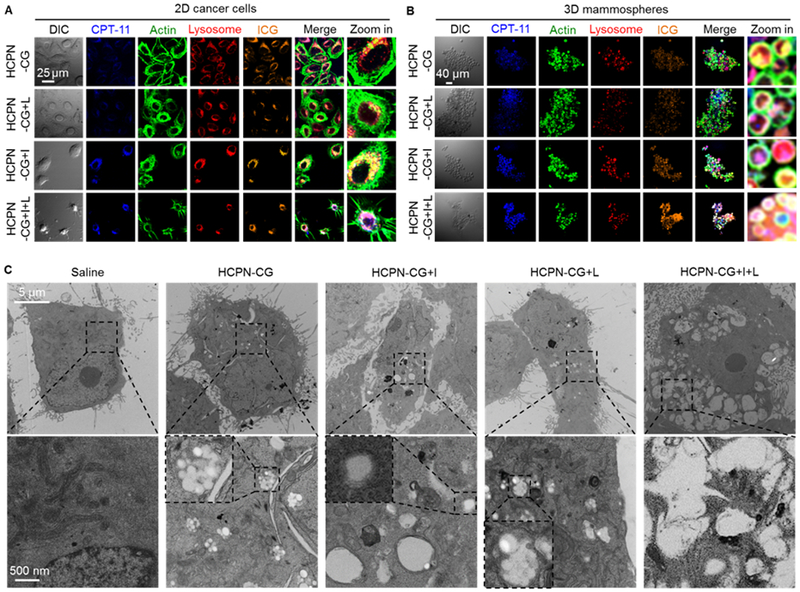
In vitro cell uptake of nanoparticles and their cold responsiveness inside cells. Confocal micrographs of (A) 2D cancer cells and (B) 3D mammosphere cells after incubating with HCPN-CG nanoparticles for 3 h at 37 °C, followed by cooling on ice (+I, 5 min), laser irradiation (+L, 1 W/cm2, 2 min), or both (+I+L). The cells incubated with HCPN-CG nanoparticles and further treated with ice cooling and laser irradiation (HCPN-CG+I+L) appear shrunken and/or spiky. (C) TEM images of cancer cells treated with saline, HCPN-CG nanoparticles alone, and HCPN-CG nanoparticles with ice cooling for 5 min (+I, 5 min), NIR laser irradiation for 2 min at 1 W/cm2 (+L, 1 W/cm2, 2 min), or both (+I+L). Endo/lysosomes can be easily observed as white dots in the low-magnification images (top row) in HCPN-CG treated cells (no such white dots in control cells treated with saline), due to the uptake of multiple HCPN-CG nanoparticles with a core-shell structure (Figure 2A) in the endo/lysosomes of the cells (bottom row). Importantly, almost all the nanoparticles disappeared/disassembled in the endo/lysosomes after ice cooling, but not NIR irradiation alone. The insets in the high-magnification images in the bottom row show endo/lysosomes either with intact HCPN-CG nanoparticles or without discernable nanoparticles (due to their disassembly upon ice cooling, +I). DIC: differential interference contrast.
In order to confirm the temperature changes in the cells, FLIR (Wilsonville, Oregon, USA) near infrared thermography were used to determine the temperature in samples. As illustrated in Fig. S14A and shown in the last column of Fig. S14B, the temperature of cells treated with HCPN-CG nanoparticle is ~37 °C in the incubator, decreases to ~4 °C after incubating on ice for 5 min. Then, the temperature could recover to ~37 °C by warming in incubator for 5 min and increases to ~42 °C after further treated with laser for 2 min (1 W/cm2). For cells treated with saline, free CPT&ICG, and PN-CG nanoparticles, the temperature increase is not as much as that of the HCPN-CG nanoparticle-treated cells, suggesting more ICG could be delivered into the cells with HCPN-CG nanoparticles (Fig. S14B). In addition to heating with laser, the HCPN-CG nanoparticle-treated cells, after cooling on ice and incubating in incubator at 37 °C, were further heated to 42 °C in waterbath at 42 °C for 30 min (Fig. S15). As shown in Fig. S16, the 2D-cultured cells were shrunken but round, which is very different from the spiky morphology shown in Fig. 4A for the cells with laser heating. There is no obvious change of 3D mammospheres before and after waterbath treatment. These results indicate that although the maximum bulk temperature is all ~42 °C, the fast and localized laser heating for 2 min from inside cells via ICG is more damaging to the cells than the non-localized waterbath heating for ~30 min from outside the cells.
In order to further confirm the cold-responsive property of the HCPN-CG nanoparticles inside cells and the severe damage to the nanoparticles laden cells treated with ice cooling and laser irradiation, we further checked the cellular uptake of the HCPN-CG nanoparticles and cell morphology by using TEM. As shown in Fig. 4C, endo/lysosomes can be easily observed as white dots in the low-magnification images (top row) in HCPN-CG treated cells (no such white dots in control cells treated with saline), due to the uptake of multiple HCPN-CG nanoparticles with a core-shell structure (Fig. 2A) in the endo/lysosomes of the cells (bottom row). Importantly, almost all the nanoparticles disappeared/disassembled in the endo/lysosomes after ice cooling, suggesting the cold-responsive property of HCPN-CG nanoparticles retains after they are taken up inside cells. In contrast, the PLGA-based nanoparticles are stable in cells either kept at 37 °C or with ice treatment (Fig. S17). Similarly, the HCPN-CG nanoparticles are stable after NIR laser irradiation alone. More importantly, severe cell damage can be observed only in the TEM image of cells treated by HCPN-CG nanoparticles together with ice cooling and laser irradiation, which is in accordance with the confocal data (Fig. 4A).
To investigate the anticancer capacity of HCPN-CG nanoparticles, both the 2D cancer cells and 3D mammospheres were treated with HCPN nanoparticles, free CPT, simple mixture of free CPT&ICG, PN-CG nanoparticles, and HCPN-CG nanoparticles at various concentrations without or with ice (+I), laser (+L), and both (+I+L, for which cells were treated with ice for 5 min first and then laser for 2 min at 1 W/cm2). According to the viability data of both 2D-cultured cancer cells (Fig. 5A) and 3D mammospheres (Fig. 5B and Fig. S18), blank HCPN nanoparticles were not harmful to the cells. Interestingly, HCPN-CG nanoparticles with ice (HCPN-CG+I) treatment induced higher cytotoxicity than the treatment of HCPN-CG nanoparticles alone (p < 0.05 for 2D cultured cells and 3D mammospheres at drug concentrations of 50 and 10 μg/ml), suggesting the cold-responsive drug release could enhance the anticancer activity because blank HCPN nanoparticles are not harmful to cells after ice, laser, or combined treatment (Fig. S19). This is further supported by the data showing no significant difference in viability of the cells treated with free drugs with or without ice cooling (i.e., CPT&ICG versus CPT&ICG+I). Similarly, the toxicity of HCPN-CG nanoparticles to the 2D cells was also increased when combined with laser. More importantly, the HCPN-CG nanoparticles combined with both ice and laser (HCPN-CG+I+L) show the best antitumor capacity compared with all other groups (Fig. 5B). The cells were also treated with HCPN-CG nanoparticles in combination with ice and waterbath heating (Fig. S15). As shown in Fig. S20, the waterbath heating could enhance the in vitro antitumor ability of HCPN-CG nanoparticles, but it is not as effective as laser irradiation. This is consistent with the morphological change observed for waterbath versus laser heating (Fig. 4A–B versus Fig. S16).
Fig. 5.
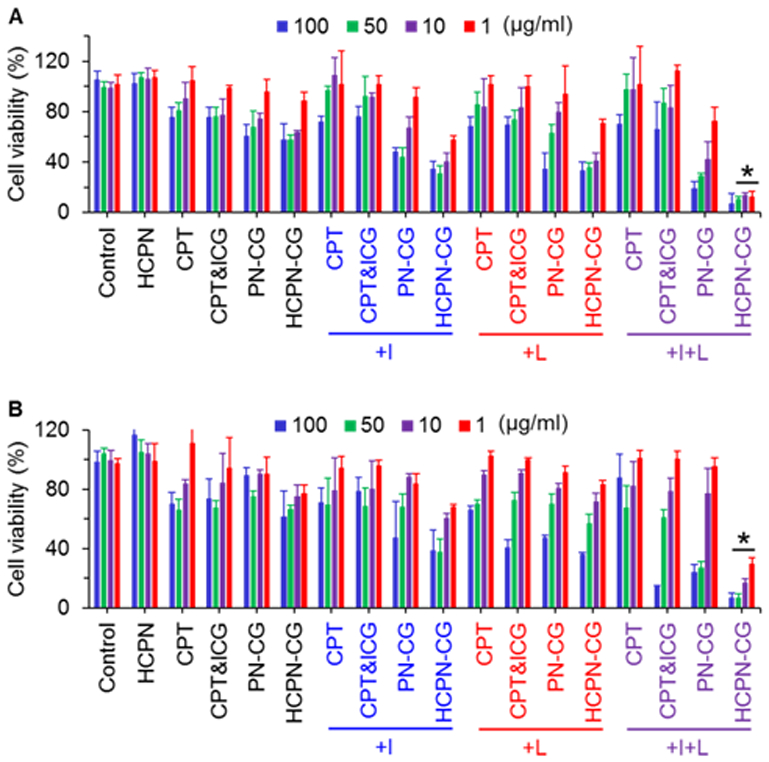
In vitro anticancer capacity. Viability of (A) 2D cancer cells and (B) 3D mammosphere cells after treated with blank nanoparticles (HPCN), free CPT, simple mixture of free CPT&ICG, PN-CG nanoparticles, and HCPN-CG nanoparticles without or with ice cooling for 5 min (+I), NIR laser irradiation for 2 min (+L), or both (+I+L). Control cells were cultured in pure medium without any treatment. Error bars represent S.D. (n = 3). *: p < 0.05 (Kruskal-Wallis H test).
3.4. In vivo targeting and antitumor capacity of HCPN-CG nanoparticles
We next investigated the bio-distribution of the HCPN-CG nanoparticles in orthotopic MDA-MB-231 (triple-negative) human mammary tumor-bearing mice by utilizing the fluorescence of ICG. Tumors were produced by injecting 20,000 MDA-MB-231 mammosphere cells per mouse into the fat pad of 6–8 week-old female nude mice. As shown in Fig. 6A, the ICG fluorescence was detectable over almost the whole animal body for PN-CG nanoparticles, HCPN-CG nanoparticles, and free ICG at 1 h after intravenous injection. More importantly, stronger fluorescence is observable only in the tumors of mice treated with the PN-CG and HCPN-CG nanoparticles, suggesting preferential accumulation in tumor (i.e., tumor targeting) of the PN-CG and HCPN-CG nanoparticles. The fluorescence in tumor in mice treated with PN-CG and HCPN-CG nanoparticles further increases at 3, 6, and 9 h. By contrast, the fluorescence in tumor in mouse treated with free ICG is weak and decreases after 6 h post injection (Fig. 6A). Moreover, the fluorescence in tumor treated with HCPN-CG nanoparticles appears stronger than that treated with PN-CG nanoparticles at 6 and 9 h although no obvious difference is discernable at 1 and 3 h. To confirm the observations from whole animal imaging based on ICG fluorescence, various organs were harvested for ex vivo imaging to check the distribution of ICG after sacrificing the mice at 9 h. As shown in Fig. 6B, the tumor from mouse treated with HCPN-CG nanoparticles show stronger ICG fluorescence than that from mouse treated with PN-CG nanoparticles, which is consistent with the whole-animal imaging data. Interestingly, the fluorescence in liver and lung from the HCPN-CG nanoparticles treated mouse is weaker than that from the PN-CG nanoparticles treated mouse, suggesting that the HA modification on the HCPN-CG nanoparticles could reduce their accumulation in the two critical organs. This is possibly because HA is a polymer naturally present in various tissues as well as body fluids [59]. Therefore, the HA modification may help to reduce the chance of HCPN-CG nanoparticles being captured by Kupffer cells or macrophages in liver, which is a main organ of the mononuclear phagocyte system (MPS) to remove foreign materials [60]. In contrast, the surface of PN-CG nanoparticles consists of polymers that are not naturally present in the body (e.g., PF127). Therefore, they may be more easily recognized by Kupffer cells or macrophages in liver, resulting in the accumulation of more PN-CG nanoparticles in liver.
Fig. 6.
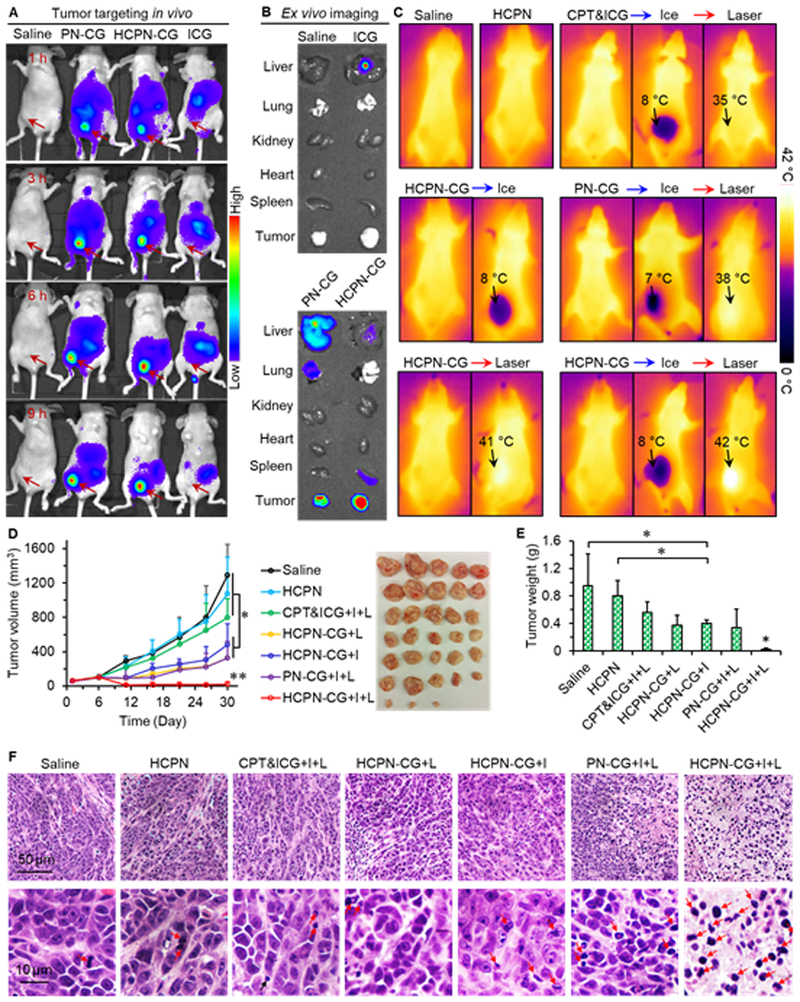
In vivo biodistribution and antitumor capacity. (A) In vivo whole animal imaging of ICG fluorescence at different times after intravenous injection of free ICG, ICG-laden PN-CG nanoparticles, and HCPN-CG nanoparticles via the tail vein. The arrows indicate the locations of tumors in mice. (B) Ex vivo imaging of ICG fluorescence in tumor and five critical organs collected after sacrificing the mice at 9 h. (C) Infrared thermographic maps of whole animal after ice treatment (5 min) and near infrared laser irradiation (at ~5 min after ice cooling, 1 W/cm2, 2 min) on tumor. (D) Tumor growth curves and tumor image for seven different treatments. Error bars represent S.D. (n = 5). *: p < 0.05, **: p < 0.01 (Kruskal-Wallis H test). (E) Weight of the tumors collected after sacrificing the mice on day 30. Error bars represent S.D. (n = 5). *: p < 0.05 (Kruskal-Wallis H test). (F) Representative histology (H&E) images of the tumors collected on day 30. Most of the cells in HCPN-CG+I+L treated tumor are necrotic. I: ice cooling for 5 min and L: laser irradiation at 1 W/cm2 for 2 min. The red arrows indicate necrotic cells.
Lastly, we treated the orthotopic tumor-bearing mice with different drug formulations to understand the safety and efficacy of the HCPN-CG nanoparticles for cancer therapy. To assess the cooling and warming effects in vivo, ice and NIR laser were applied through the skin over the tumor area. The tumor-bearing mice were randomly divided into 7 groups: saline, blank nanoparticles (HCPN), free CPT&ICG with ice and laser treatments (CPT&ICG+I+L), HCPN-CG nanoparticles with laser irradiation (HCPN-CG+L), HCPN-CG nanoparticles with ice treatment (HCPN-CG+I), PN-CG nanoparticles with ice and laser treatments (PN-CG+I+L), and HCPN-CG nanoparticles with ice and laser treatments (HCPN-CG+I+L). Mice were treated with the various formations at a total CPT dose of 3 mg/kg body weight (for formulations with CPT) via intravenous injection when the tumor reached a volume of ~100 mm3 on day 6. No mice died during the course of the one-month treatment and observation.
To confirm the ice cooling and NIR laser heating effects in vivo, FLIR near infrared thermography was used to detect the temperature under ice cooling and/or laser irradiation (Fig. 6C). Without treatment, the temperature is ~35 °C for all the groups. After treating with ice for 5 min, the temperature in the tumor area decreases to 7–8 °C, which is low enough to induce disassembly of the nanoparticles. Then, NIR laser was applied over the tumor area. Unlike ice treatment that cools anything in touch, the increase in temperature with laser treatment is dependent on the amount of ICG (Fig. S9). Indeed, the increase in temperature in the tumor area of mice injected with free CPT&ICG from the baseline (~35 °C) is negligible after laser irradiation. This further suggests free ICG cannot accumulate efficiently in tumor after intravenous injection, consistent with the biodistribution data shown in Fig. 6A–B. In contrast, the temperature in the tumor area of mice treated with both PN-CG and HCPN-CG nanoparticles could be higher than the baseline after laser treatment (Fig. 6C). More importantly, the temperature in the tumor area of mice treated with HCPN-CG nanoparticles is higher than that of mice treated with PN-CG nanoparticles (38 versus 42 °C), indicating the HCPN-CG nanoparticles have better in vivo tumor targeting capability than the PN-CG nanoparticles. This is in accordance with in in vivo and ex vivo imaging data.
Probably due to aforementioned in vivo tumor targeting capability, cold-responsive drug release, and localized heating via ICG with laser irradiation, the HCPN-CG nanoparticles with ice and laser (HCPN-CG+I+L) exhibit the best anti-tumor capacity (Fig. 6D–E). The tumor size (Fig. 6D) and weight (Fig. 6E) of the HCPN-CG+I+L group are significantly less than that of all the other treatment groups. Tumor growth for treatments with blank nanoparticles (HCPN) or free CPT&ICG+I+L is similar to that of saline control. For the HCPN-CG+I, HCPN-CG+L, and PN-CG+I+L treatments, tumor volume is also significantly less than that of the saline control while they are not as effective as the HCPN-CG+I+L treatment. Especially, by comparing HCPN-CG+L and HCPN-CG+I+L groups, the antitumor capacity of HCPN-CG nanoparticles can be significantly improved when combined with ice cooling. This confirms the importance of using the cold-responsive property of the HCNPN-CG nanoparticles in destroying tumor. Further histological examination (hematoxylin and eosin or H&E stain) reveals extensive necrosis in the tumors from HCPN-CG+I+L group while tumors from the other groups appear more viable (Fig. 6F and Fig. S21). Collectively, these data demonstrate the remarkable in vivo antitumor efficacy of the HCPN-CG+I+L treatment. Equally important, we did not notice any obvious sign of side effects for the HCPN-CG+I+L treatment. Neither death nor significant drop of body weight was noted for all the seven different treatments (Fig. S22). Major organs from the HCPN-CG+I+L treated mice were collected at 30 days after the treatment for histology analysis. No obvious damage to the critical organs was observable in the H&E stained tissue slices (Fig. S23). These results indicate the excellent safety of the HCPN-CG nanoparticles for delivering therapeutic agents in vivo. This is probably because the four polymers used to synthesize the nanoparticles are biocompatible [61–64]. Moreover, the body temperature (~37 °C) is always higher than the disassembly/dissolution temperature (~10 °C) of the HCPN-CG nanoparticles. As a result, the nanoparticles are stable when circulating in the body with minimal release of drug to reduce any potential side effects. These results indicate that HCPN-CG+I+L treatment can greatly augment cancer destruction in vivo with no evident systemic toxicity.
It is worth noting that although it could be further reduced by replacing the butyl group in the PNIPAM-B polymer with a more hydrophobic group (e.g., pentyl group) [65], a transition temperature of ~10 °C is desired in this study. This is because the ice is applied on the skin surface above the tumor for cooling in this study and the temperature in tumor should be higher than that of ice. If the transition temperature is further lowered, it may be inefficient to induce triggered drug release with ice cooling from the skin surface. For future potential application of the HCPN-CG nanoparticles to treat tumors in deep organs, cold temperature can be achieved by using a cooling catheter to generate local hypothermia guided by minimally invasive surgical approaches such as endoscopy, thoracoscopy, and laparoscopy [35–37]. These minimally invasive surgical technologies have also been studied for delivering NIR laser into deep organs [66–69], and it is possible to combine the delivery of NIR laser and cold in one catheter. In fact, catheter or probes that can deliver both microwave/heat and cold has been reported in the literature before [70,71]. Therefore, the requirement of cooling together with NIR irradiation or heating is not an obstacle for the future clinical applications of our cold responsive nanoparticle system. Although the various drug formulations can be locally delivered into tumor using the aforementioned method, we chose intravenous injection in this study to demonstrate the preferential accumulation of the HCPN-CG nanoparticles in tumor via both passive (due to the EPR effect of tumor vasculature) and active (due to HA on the nanoparticle surface) targeting (Fig. 6A–B). Furthermore, systemic administration of chemotherapeutic drugs is widely used in the clinic to inhibit not only the growth of primary tumor but also the potential metastatic tumor formation [72,73].
4. Discussion
The resistance of CSCs to conventional chemotherapy is multifaceted including overexpressed drug efflux pumps, enhanced DNA repair, overexpression of anti-apoptotic proteins, and dormancy [51], In this study, HA is decorated on the surface of the HCPN-CG nanoparticles for targeted delivery of CPT to the CSCs. This may help to deliver more drugs into the CSCs to enhance the anti-tumor/CSCs capability of the nanoparticles. However, as a result of the slow/sustained drug release from the nanoparticles, the concentration of the released drug in CSCs may be still not high enough to efficiently kill the CSCs. We take advantage of the cold-responsive property of the HCPN-CG nanoparticles to achieve efficient burst-release of CPT from nanoparticles (more than 80% within minutes, Fig. 3C). To further improve the anti-CSCs capability of CPT, we co-encapsulated ICG into the nanoparticles for photothermal warming under NIR laser irradiation. This is because the photothermal effect could enhance the cytotoxicity of chemotherapeutic drugs [15]. Indeed, both in vitro and in vivo data suggest that HA-mediated CPT targeted delivery, ice cooling, or laser irradiation (photothermal warming) could enhance the antitumor capability of CPT. Importantly, the best antitumor can be achieved only by combining them together.
It is worth noting that the temperature is not constant (i.e., decreases with time from 37 °C to ~4–0 °C) during ice cooling for 5 min in this study, as shown in Fig. S14–15. The nanoparticles solution in centrifuge tube was put in ice for 5 min to determine the cold-triggered drug release (Fig. 3C). According to both the near infrared thermal images and the thermal history quantified using thermocouples (Fig. S14–15), the temperature of the samples is close to 0 °C after cooling in ice for 5 minutes and the drug can still be efficiently released from HCPN-CG nanoparticles (Fig. 3C). Similarly, the therapeutic effect of the HCPN-CG nanoparticles was conducted by cooling the MDA-MB-231 cells or tumor using ice for 5 min. Both the in vitro cytotoxicity and in vivo antitumor data show that the anticancer capability of HCPN-CG nanoparticles is augmented by ice cooling.
In this study, ICG was used as a photothermal agent to enhance the antitumor efficacy of CPT rather than thermal ablation (or destroy tumors at a temperature usually more than 43 °C, [23,24]). Therefore, the concentration of ICG was optimized to generate mild hyperthermia (up to ~42 °C) for both in vitro and in vivo studies. As shown in Fig. S14 for in vitro studies, the infrared thermographic maps indicate that the temperature of cells treated with HCPN-CG nanoparticles (ICG: 3 μg/ml) could be increased to ~42 °C after irradiated with NIR laser for 2 minutes. Therefore, the ICG concentration of 3 μg/ml was used for all the in vitro studies. Similarly, the concentration of ICG for in vivo studies was optimized to be 3 mg/kg to increase the temperature of tumor to ~42 °C after laser irradiation (Fig. 6C). The concentration of CPT was varied from 1 to 100 μg/ml for testing the cytotoxicity in vitro and the CPT dose of 3 mg/kg was used for in vivo studies based on previous research [74]. Mice (for in vivo studies) were irradiated with NIR laser at 12 h after treated with HCPN-CG nanoparticles, also based on previous studies shown that the nanoparticles could well accumulate in tumor and be cleared from normal organs at ~12 h after intravenous injection of nanoparticles [75]. For consistency, cells (for in vitro studies) were treated with NIR laser irradiation after incubated with HCPN-CG nanoparticles for 12 h.
ICG is a clinically approved NIR dye with low cytotoxicity [76,77], especially at the low concentration (3 μg/ml) used in this study. To confirm this, we investigated the cytotoxicity of free ICG and HCPN nanoparticles encapsulated ICG (HCPN-G, without CPT). As shown in Fig. S24A, neither free ICG nor HCPN-G nanoparticles is toxic to the MDA-MB-231 cells. The HCPN-G nanoparticles under NIR laser irradiation (HCPN-G+L) are still not toxic to the cells. This is probably because the concentration of ICG was optimized to generate only mild hyperthermia (up to ~42 °C) in this study. It is worth noting that the HCPN-G nanoparticles with both ice cooling and laser irradiation (HCPN-G+I+L) could inhibit the growth of the cancer cells (Fig. S24A), which may be due to the structural damage during the cooling and heating process as shown in Fig. S24B. However, the cytotoxicity of HCPN-G+I+L is much lower than HCPN-CG+I+L (Fig. 5A), suggesting that the contribution of ICG is limited to the generation of mild hyperthermia during NIR laser irradiation. The anticancer contribution of CPT was investigated by encapsulating CPT alone in the HCPN nanoparticles (HCPN-C). As shown in Fig. S24C, free CPT can inhibit the growth of MDA-MB-231 cells, which can be improved by encapsulating the drug inside the HCPN nanoparticles. The cytotoxicity of HCPN-C nanoparticles was further enhanced when combined with ice cooling, confirming the therapeutic benefit of burst drug release. In contrast, NIR laser irradiation does not cause significant difference in the viability of the HCPN-C nanoparticles-treated cancer cells (Fig. S24C). This is probably because no ICG is encapsulated to generate mild hyperthermia for enhancing the cytotoxicity of CPT. With ice cooling and/or NIR laser irradiation, the cytotoxicity of HCPN-C+I+L is similar to HCPN-C+I and lower than HCPN-CG+I+L (see Fig. 5A). All these results suggest that the contribution of ICG used in this study is to induce mild hyperthermia (or photothermal warming instead of photothermal ablation to kill tumor at more than 43 °C [23,24]), and the cytotoxicity of CPT can be improved by the combination of the mild hyperthermia and cold-triggered burst drug release.
In order to show the advantage of encapsulating both CPT and ICG in the HCPN nanoparticles (Fig. 5), we further investigated the cytotoxicity of HCPN-C nanoparticles with different concentrations of CPT after treated with both ice cooling and laser irradiation. As shown in Fig. S25A–B, under the same concentration of CPT, the HCPN-CG+I+L treatment is significantly more cytotoxic than HCPN-C+I+L treatment for both 2D cultured MDA-MB-231 cells and 3D microscale tumors (i.e., mammospheres) enriched with CSCs. This is not surprising as the ICG is necessary for photothermal warming to enhance the cytotoxicity of CPT. Without chemotherapeutic drug encapsulation, the cytotoxicity of HCPN-G+I+L treatment is significantly lower than HCPN-CG+I+L treatment for all different concentrations of CPT. We did not include the animal groups treated with the nanoparticles encapsulated with CPT or ICG alone. This is because the purpose of the in vivo study is to understand the benefit of combining ice cooling and NIR laser irradiation for tumor destruction.
Cryotherapy has been used to destroy tumors in various organs including liver, kidney, lung, prostate, breast, skin, bone, and other tissues [18,21,22,78,79]. However, tumor cells in an iceball (particularly in the peripheral region next to the iceball surface with a cold temperature of ~0 °C) visualized by intraoperative imaging techniques may not be completely killed, which may result in tumor recurrence [23–25]. This issue could be addressed by combining the cold-responsive nanoparticles developed in this study with cryotherapy. This is because the nanoparticle is capable of burst-release of most encapsulated drug at cold temperature to enhance cancer destruction in the peripheral region of the iceball. For future potential clinical application, patients may be treated with the cold-responsive nanoparticles first and then cryotherapy at a few hours later to allow the preferential accumulation of nanoparticles in tumor (Fig. 6A–B). As a result, cancer cells that are not killed by cryotherapy in the peripheral region of an icerball may be killed by the chemotherapeutic drug released from the cold-responsive nanoparticles in response to the cold temperature in the peripheral region of the iceball during cryotherapy.
5. Conclusions
In summary, we have developed novel cold-responsive HCPN-CG nanoparticles composed of HA, chitosan, PNIPAM-B, and PF127 for targeted co-delivery of chemotherapeutics (CPT) and photothermal agent (ICG) into orthotopic human mammary tumor. Due to the dissolution of PNIPAM-B, the nanoparticles could disassemble quickly at temperatures below 12 °C. This cold-responsiveness can induce much more efficient drug release from the nanoparticles than acidic pH or NIR-induced photothermal effect. Interestingly, ice cooling followed by short NIR irradiation could cause severe structural damage/shrinkage to the HCPN-CG nanoparticle-laden cells. Our extensive in vitro studies with both 2D-cultured cancer cells and 3D microscale tumors (i.e., mammospheres) enriched with CSCs and in vivo studies using orthotopic triple negative human breast tumors grown in mice demonstrate the great potential of the HCPN nanoparticles with ice cooling and laser irradiation for enhancing drug delivery to combat cancer.
Supplementary Material
Acknowledgments
This work was partially supported by American Cancer Society (#120936-RSG-11-109-01-CDD), NIH (R01CA206366), and Pelotonia (Postdoctoral Fellowship to HW).
Footnotes
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.biomaterials.2018.07.021
Data Availability
All data supporting the findings of this study are available from the corresponding authors upon request.
References
- [1].Stuart MAC, Huck WT, Genzer J, Müller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Emerging applications of stimuli-responsive polymer materials, Nat Mater 9(2) (2010) 101–113. [DOI] [PubMed] [Google Scholar]
- [2].Lu Y, Aimetti AA, Langer R, Gu Z, Bioresponsive materials, Nat. Rev. Mater 2 (2016) 16075. [Google Scholar]
- [3].Mura S, Nicolas J, Couvreur P, Stimuli-responsive nanocarriers for drug delivery, Nat Mater 12(11) (2013) 991–1003. [DOI] [PubMed] [Google Scholar]
- [4].Kelley EG, Albert JN, Sullivan MO, Epps TH III, Stimuli-responsive copolymer solution and surface assemblies for biomedical applications, Chem. Soc. Rev 42(17) (2013) 7057–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Arruebo M, Fernández-Pacheco R, Ibarra MR, Santamaría J, Magnetic nanoparticles for drug delivery, Nano Today 2(3) (2007) 22–32. [Google Scholar]
- [6].Rapoport N, Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery, Prog. Polym. Sci 32(8) (2007) 962–990. [Google Scholar]
- [7].Muhammad F, Guo M, Qi W, Sun F, Wang A, Guo Y, Zhu G, pH-triggered controlled drug release from mesoporous silica nanoparticles via intracelluar dissolution of ZnO nanolids, J. Am. Chem. Soc 133(23) (2011) 8778–8781. [DOI] [PubMed] [Google Scholar]
- [8].Cassano D, Santi M, Cappello V, Luin S, Signore G, Voliani V, Biodegradable passion fruit-like nano-architectures as carriers for cisplatin prodrug, Part. Part. Syst. Charact 33 (2016) 818–824. [Google Scholar]
- [9].Zhang L, Guo R, Yang M, Jiang X, Liu B, Thermo and pH dual-responsive nanoparticles for anti-cancer drug delivery, Adv. Mater 19(19) (2007) 2988–2992. [Google Scholar]
- [10].Cammas S, Suzuki K, Sone C, Sakurai Y, Kataoka K, Okano T, Thermo-responsive polymer nanoparticles with a core-shell micelle structure as site-specific drug carriers, J. Control. Release 48(2) (1997) 157–164. [Google Scholar]
- [11].Zhang Z, Wang L, Wang J, Jiang X, Li X, Hu Z, Ji Y, Wu X, Chen C, Mesoporous silica-coated gold nanorods as a light-mediated multifunctional theranostic platform for cancer treatment, Adv. Mater 24(11) (2012) 1418–1423. [DOI] [PubMed] [Google Scholar]
- [12].Zheng M, Yue C, Ma Y, Gong P, Zhao P, Zheng C, Sheng Z, Zhang P, Wang Z, Cai L, Single-step assembly of DOX/ICG loaded lipid–polymer nanoparticles for highly effective chemo-photothermal combination therapy, ACS Nano 7(3) (2013) 2056–2067. [DOI] [PubMed] [Google Scholar]
- [13].Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z, Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy, Nat. Commun 7 (2016) 13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu Y, Ai K, Liu J, Deng M, He Y, Lu L, Dopamine-melanin colloidal nanospheres: an efficient near-infrared photothermal therapeutic agent for in vivo cancer therapy, Adv. Mater 25(9) (2013) 1353–1359. [DOI] [PubMed] [Google Scholar]
- [15].Wang H, Agarwal P, Zhao S, Yu J, Lu X, He X, A biomimetic hybrid nanoplatform for encapsulation and precisely controlled delivery of therasnostic agents, Nat. Commun 6 (2015) 10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Han G, Xie J, Light-mediated deep-tissue theranostics, Theranostics 6(13) (2016) 2292–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang H, Agarwal P, Zhao S, Yu J, Lu X, He X, A near-infrared laser-activated “nanobomb” for breaking the barriers to microRNA delivery, Adv. Mater 28(2) (2016) 347–355. [DOI] [PubMed] [Google Scholar]
- [18].Gage AA, Cryosurgery in the treatment of cancer, Surg. Gynecol. Obstet 174(1) (1992) 73–92. [PubMed] [Google Scholar]
- [19].Avitall B, Kalinski A, Cryotherapy of cardiac arrhythmia: From basic science to the bedside, Heart Rhythm 12(10) (2015) 2195–203. [DOI] [PubMed] [Google Scholar]
- [20].Villablanca PA, Rao G, Briceno DF, Lombardo M, Ramakrishna H, Bortnick A, Garcia M, Menegus M, Sims D, Makkiya M, Mookadam F, Therapeutic hypothermia in ST elevation myocardial infarction: a systematic review and meta-analysis of randomised control trials, Heart 102(9) (2016) 712–9. [DOI] [PubMed] [Google Scholar]
- [21].Gage AA, Baust JG, Cryosurgery for tumors, J. Am. Coll. Surg 205(2) (2007) 342–356. [DOI] [PubMed] [Google Scholar]
- [22].Onik GM, Cohen JK, Reyes GD, Rubinsky B, Chang Z, Baust J, Transrectal ultrasound-guided percutaneous radical cryosurgical ablation of the prostate, Cancer 72(4) (1993) 1291–1299. [DOI] [PubMed] [Google Scholar]
- [23].He X, Bischof JC, Quantification of temperature and injury response in thermal therapy and cryosurgery, Crit. Rev. Biomed. Eng 31(5–6) (2003) 355–422. [DOI] [PubMed] [Google Scholar]
- [24].He X, Thermostability of biological systems: fundamentals, challenges, and quantification, Open Biomed. Eng. J 5 (2011) 47–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gilstrap K, Hu X, Lu X, He X, Nanotechnology for energy-based cancer therapies, Am. J. Cancer Res 1(4) (2011) 508–520. [PMC free article] [PubMed] [Google Scholar]
- [26].He X, Bischof JC, Analysis of thermal stress in cryosurgery of kidneys, J. Biomech. Eng 127(4) (2005) 656–661. [DOI] [PubMed] [Google Scholar]
- [27].Hoffmann NE, Bischof JC, The cryobiology of cryosurgical injury, Urology 60(2) (2002) 40–49. [DOI] [PubMed] [Google Scholar]
- [28].Gage AA, Baust J, Mechanisms of tissue injury in cryosurgery, Cryobiology 37(3) (1998) 171–186. [DOI] [PubMed] [Google Scholar]
- [29].Clarke DM, Baust JM, Van Buskirk RG, Baust JG, Chemo-cryo combination therapy: an adjunctive model for the treatment of prostate cancer, Cryobiology 42(4) (2001) 274–285. [DOI] [PubMed] [Google Scholar]
- [30].Jiang J, Goel R, Iftekhar MA, Visaria R, Belcher JD, Vercellotti GM, Bischof JC, Tumor necrosis factor-alpha-induced accentuation in cryoinjury: mechanisms in vitro and in vivo, Mol. Cancer Ther 7(8) (2008) 2547–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang CL, Teo KY, Han B, An amino acidic adjuvant to augment cryoinjury of MCF-7 breast cancer cells, Cryobiology 57(1) (2008) 52–59. [DOI] [PubMed] [Google Scholar]
- [32].Chao BH, He X, Bischof JC, Pre-treatment inflammation induced by TNF-alpha augments cryosurgical injury on human prostate cancer, Cryobiology 49(1) (2004) 10–27. [DOI] [PubMed] [Google Scholar]
- [33].Rao W, Bellotti A, Littrup PJ, Yu J, Lu X, He X, Nanoparticle-encapsulated doxorubicin enhances cryoablation of cancer stem-like cells, Technology 2 (2014) 28–35. [Google Scholar]
- [34].Goel R, Swanlund D, Coad J, Paciotti GF, Bischof JC, TNF-alpha-based accentuation in cryoinjury--dose, delivery, and response, Mol. Cancer Ther 6(7) (2007) 2039–2047. [DOI] [PubMed] [Google Scholar]
- [35].Cattaneo G, Schumacher M, Maurer C, Wolfertz J, Jost T, Büchert M, Keuler A, Boos L, Shah M, Foerster K, Endovascular cooling catheter for selective brain hypothermia: an animal feasibility study of cooling performance, Am. J. Neuroradiol 37(5) (2016) 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Inoue S, Mori A, Shimizu H, Yoshitake A, Tashiro R, Kabei N, Yozu R, Combined use of an epidural cooling catheter and systemic moderate hypothermia enhances spinal cord protection against ischemic injury in rabbits, J. Thorac. Cardiovasc. Surg 146(3) (2013) 696–701. [DOI] [PubMed] [Google Scholar]
- [37].Crain DS, Spencer CR, Favata MA, Amling CL, Transureteral saline perfusion to obtain renal hypothermia: potential application in laparoscopic partial nephrectomy, JSLS 8(3) (2004) 217. [PMC free article] [PubMed] [Google Scholar]
- [38].Schmaljohann D, Thermo-and pH-responsive polymers in drug delivery, Adv. Drug. Deliv. Rev 58(15) (2006) 1655–1670. [DOI] [PubMed] [Google Scholar]
- [39].Chung J, Yokoyama M, Yamato M, Aoyagi T, Sakurai Y, Okano T, Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly (N-isopropylacrylamide) and poly (butylmethacrylate), J. Control. Release 62(1) (1999) 115–127. [DOI] [PubMed] [Google Scholar]
- [40].Sershen SR, Westcott SL, Halas NJ, West JL, Temperature-sensitive polymer-nanoshell composites for photothermally modulated drug delivery, J. Biomed. Mater. Res 51(3) (2000) 293–298. [DOI] [PubMed] [Google Scholar]
- [41].Yang Z, Cheng Q, Jiang Q, Deng L, Liang Z, Dong A, Thermo-sensitive nanoparticles for triggered release of siRNA, J. Biomater. Sci. Polym. Ed 26(4) (2015) 264–276. [DOI] [PubMed] [Google Scholar]
- [42].Zhang W, Gilstrap K, Wu L, K CR, Moss MA, Wang Q, Lu X, He X, Synthesis and characterization of thermally responsive Pluronic F127-chitosan nanocapsules for controlled release and intracellular delivery of small molecules, ACS Nano 4(11) (2010) 6747–6759. [DOI] [PubMed] [Google Scholar]
- [43].Lee SH, Choi SH, Kim SH, Park TG, Thermally sensitive cationic polymer nanocapsules for specific cytosolic delivery and efficient gene silencing of siRNA: swelling induced physical disruption of endosome by cold shock, J. Control. Release 125(1) (2008) 25–32. [DOI] [PubMed] [Google Scholar]
- [44].Wilks TR, Bath J, de Vries JW, Raymond JE, Herrmann A, Turberfield AJ, O’Reilly RK, “Giant surfactants” created by the fast and efficient functionalization of a DNA tetrahedron with a temperature-responsive polymer, ACS Nano 7(10) (2013) 8561–8572. [DOI] [PubMed] [Google Scholar]
- [45].Zhu A, Miao K, Deng Y, Ke H, He H, Yang T, Guo M, Li Y, Guo Z, Wang Y, Dually pH/reduction-responsive vesicles for ultrahigh-contrast fluorescence imaging and thermo-chemotherapy-synergized tumor ablation, ACS Nano 9(8) (2015) 7874–7885. [DOI] [PubMed] [Google Scholar]
- [46].Su S, Wang H, Liu X, Wu Y, Nie G, iRGD-coupled responsive fluorescent nanogel for targeted drug delivery, Biomaterials 34(13) (2013) 3523–3533. [DOI] [PubMed] [Google Scholar]
- [47].Wang H, Zhao Y, Wu Y, Hu Y.-l., Nan K, Nie G, Chen H, Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles, Biomaterials 32(32) (2011) 8281–8290. [DOI] [PubMed] [Google Scholar]
- [48].Zambaux M, Bonneaux F, Gref R, Maincent P, Dellacherie E, Alonso M, Labrude P, Vigneron C, Influence of experimental parameters on the characteristics of poly (lactic acid) nanoparticles prepared by a double emulsion method, J. Control. Release 50(1) (1998) 31–40. [DOI] [PubMed] [Google Scholar]
- [49].Kim B, Hwang S, Park J, Park HJ, Preparation and characterization of drug-loaded polymethacrylate microspheres by an emulsion solvent evaporation method, J. Microencapsul 19(6) (2002) 811–822. [DOI] [PubMed] [Google Scholar]
- [50].Ho MM, Ng AV, Lam S, Hung JY, Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells, Cancer Res 67(10) (2007) 4827–4833. [DOI] [PubMed] [Google Scholar]
- [51].Wang H, Agarwal P, Zhao S, Xu RX, Yu J, Lu X, He X, Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan for targeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells, Biomaterials 72 (2015) 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang W, Guo Z, Huang D, Liu Z, Guo X, Zhong H, Synergistic effect of chemo-photothermal therapy using PEGylated graphene oxide, Biomaterials 32(33) (2011) 8555–8561. [DOI] [PubMed] [Google Scholar]
- [53].Wang H, Gao Z, Liu X, Agarwal P, Zhao S, Conroy DW, Ji G, Yu J, Jaroniec CP, Liu Z, Targeted production of reactive oxygen species in mitochondria to overcome cancer drug resistance, Nat. Commun 9(1) (2018) 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rao W, Wang H, Han J, Zhao S, Dumbleton J, Agarwal P, Zhang W, Zhao G, Yu J, Zynger DL, Chitosan-decorated doxorubicin-encapsulated nanoparticle targets and eliminates tumor reinitiating cancer stem-like cells, ACS Nano 9(6) (2015) 5725–5740. [DOI] [PubMed] [Google Scholar]
- [55].Rao W, Zhao S, Yu J, Lu X, Zynger DL, He X, Enhanced enrichment of prostate cancer stem-like cells with miniaturized 3D culture in liquid core-hydrogel shell microcapsules, Biomaterials 35(27) (2014) 7762–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi S, Liu Y, Chen Y, Zhang Z, Ding Y, Wu Z, Yin J, Nie L, Versatile pH-response micelles with high cell-penetrating helical diblock copolymers for photoacoustic imaging guided synergistic chemo-photothermal therapy, Theranostics 6(12) (2016) 2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Maruyama K, Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects, Adv. Drug. Deliv. Rev 63(3) (2011) 161–169. [DOI] [PubMed] [Google Scholar]
- [58].Brigger I, Dubernet C, Couvreur P, Nanoparticles in cancer therapy and diagnosis, Adv. Drug. Deliv. Rev 54(5) (2002) 631–651. [DOI] [PubMed] [Google Scholar]
- [59].Fakhari A, Berkland C, Applications and emerging trends of hyaluronic acid in tissue engineering, as a dermal filler and in osteoarthritis treatment, Acta Biomate 9(7) (2013) 7081–7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Van Furth R, Cohn Z, Hirsch J, Humphrey J, Spector W, Langevoort H, The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells, Bull. World Health Organ 46(6) (1972) 845–852. [PMC free article] [PubMed] [Google Scholar]
- [61].Naha PC, Bhattacharya K, Tenuta T, Dawson KA, Lynch I, Gracia A, Lyng FM, Byrne HJ, Intracellular localisation, geno-and cytotoxic response of polyN-isopropylacrylamide (PNIPAM) nanoparticles to human keratinocyte (HaCaT) and colon cells (SW 480), Toxicol. Lett 198(2) (2010) 134–143. [DOI] [PubMed] [Google Scholar]
- [62].Mi F-L, Tan Y-C, Liang H-F, Sung H-W, In vivo biocompatibility and degradability of a novel injectable-chitosan-based implant, Biomaterials 23(1) (2002) 181–191. [DOI] [PubMed] [Google Scholar]
- [63].Duranti F, Salti G, Bovani B, Calandra M, Rosati ML, Injectable hyaluronic acid gel for soft tissue augmentation, Dermatol. Surg 24(12) (1998) 1317–1325. [DOI] [PubMed] [Google Scholar]
- [64].Shachaf Y, Gonen-Wadmany M, Seliktar D, The biocompatibility of Pluronic® F127 fibrinogen-based hydrogels, Biomaterials 31(10) (2010) 2836–2847. [DOI] [PubMed] [Google Scholar]
- [65].Speidel AT, Stuckey DJ, Chow LW, Jackson LH, Noseda M, Abreu Paiva M, Schneider MD, Stevens MM, Multimodal hydrogel-based platform to deliver and monitor cardiac progenitor/stem cell engraftment, ACS Cent. Sci 3(4) (2017) 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hyun JH, Kim SK, Kim KG, Kim HR, Lee HM, Park S, Kim SC, Choi Y, Sohn DK, A novel endoscopic fluorescent band ligation method for tumor localization, Surg. Endosc 30(10) (2016) 4659–4663. [DOI] [PubMed] [Google Scholar]
- [67].Zako T, Ito M, Hyodo H, Yoshimoto M, Watanabe M, Takemura H, Kishimoto H, Kaneko K, Soga K, Maeda M, Extra-luminal detection of assumed colonic tumor site by near-infrared laparoscopy, Surg. Endosc 30(9) (2016) 4153–4159. [DOI] [PubMed] [Google Scholar]
- [68].Anayama T, Qiu J, Chan H, Nakajima T, Weersink R, Daly M, McConnell J, Waddell T, Keshavjee S, Jaffray D, Irish JC, Hirohashi K, Wada H, Orihashi K, Yasufuku K, Localization of pulmonary nodules using navigation bronchoscope and a near-infrared fluorescence thoracoscope, Ann. Thorac. Surg 99(1) (2015) 224–230. [DOI] [PubMed] [Google Scholar]
- [69].Li H, Zhou J, Chi C, Mao Y, Yang F, Tian J, Wang J, Clinical application of near-infrared thoracoscope with indocyanine green in video-assisted thoracoscopic bullectomy, J. Thorac. Dis 8(7) (2016) 1841–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].He X, Mcgee S, Coad JE, Schmidlin F, Iaizzo PA, Swanlund DJ, Kluge S, Rudie E, Bischof JC, Investigation of the thermal and tissue injury behaviour in microwave thermal therapy using a porcine kidney model, Int. J. Hyperthermia 20(6) (2004) 567–593. [DOI] [PubMed] [Google Scholar]
- [71].Cai Z, Song M, Zhang A, Sun J, Xu LX, Numerical Simulation of a New Probe for the Alternate Cooling and Heating of a Subcutaneous Mouse Tumor Model, Numer. Heat Tr. A-Appl 63(7) (2013) 534–548. [Google Scholar]
- [72].Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2, N. Engl. J. Med 344(11) (2001) 783–792. [DOI] [PubMed] [Google Scholar]
- [73].Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer, N. Engl. J. Med 360(6) (2009) 563–572. [DOI] [PubMed] [Google Scholar]
- [74].Li F, Zhao X, Wang H, Zhao R, Ji T, Ren H, Anderson GJ, Nie G, Hao J, Multiple layer-by-layer lipid-polymer hybrid nanoparticles for improved FOLFIRINOX chemotherapy in pancreatic tumor models, Adv. Funct. Mater 25(5) (2015) 788–798. [Google Scholar]
- [75].Wang Z, Li S, Zhang M, Ma Y, Liu Y, Gao W, Zhang J, Gu Y, Laser-Triggered Small Interfering RNA Releasing Gold Nanoshells against Heat Shock Protein for Sensitized Photothermal Therapy, Adv. Sci 4(2) (2017) 1600327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Alander JT, Kaartinen I, Laakso A, Pätilä T, Spillmann T, Tuchin VV, Venermo M, Välisuo P, A review of indocyanine green fluorescent imaging in surgery, Int. J. Biomed. Imaging 2012 (2012) 940585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jackson TL, Vote B, Knight BC, El-Amir A, Stanford MR, Marshall J, Safety testing of infracyanine green using retinal pigment epithelium and glial cell cultures, Invest. Ophthalmol. Vis. Sci 45(10) (2004) 3697–3703. [DOI] [PubMed] [Google Scholar]
- [78].Wang H, Agarwal P, Zhao G, Ji G, Jewell CM, Fisher JP, Lu X, He X, Overcoming Ovarian Cancer Drug Resistance with a Cold Responsive Nanomaterial, ACS Cent. Sci 4(5) (2018) 567–581. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [79].Niu L, Zhou L, Xu K, Mu F, The role of cryosurgery in palliative care for cancer, Ann. Palliat. Med 2(1) (2013) 26–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.