

Abstract
Non‐small‐cell lung cancer (NSCLC) has been recognized as a highly heterogeneous disease with phenotypic and genotypic diversity in each subgroup. While never‐smoker patients with NSCLC have been well studied through next generation sequencing, we have yet to recognize the potentially unique molecular features of young never‐smoker patients with NSCLC. In this study, we conducted whole genome sequencing (WGS) to characterize the genomic alterations of 36 never‐smoker Chinese patients, who were diagnosed with lung adenocarcinoma (LUAD) at 45 years or younger. Besides the well‐known gene mutations (e.g., TP53 and EGFR), our study identified several potential lung cancer‐associated gene mutations that were rarely reported (e.g., HOXA4 and MST1). The lung cancer‐related copy number variations (e.g., EGFR and CDKN2A) were enriched in our cohort (41.7%, 15/36) and the lung cancer‐related structural variations (e.g., EML4‐ALK and KIF5B‐RET) were commonly observed (22.2%, 8/36). Notably, new fusion partners of ALK (SMG6‐ALK) and RET (JMJD1C‐RET) were found. Furthermore, we observed a high prevalence (63.9%, 23/36) of potentially targetable genomic alterations in our cohort. Finally, we identified germline mutations in BPIFB1 (rs6141383, p.V284M), CHD4 (rs74790047, p.D140E), PARP1 (rs3219145, p.K940R), NUDT1 (rs4866, p.V83M), RAD52 (rs4987207, p.S346*), and MFI2 (rs17129219, p.A559T) were significantly enriched in the young never‐smoker patients with LUAD when compared with the in‐house noncancer database (p < 0.05). Our study provides a detailed mutational portrait of LUAD occurring in young never‐smokers and gives insights into the molecular pathogenesis of this distinct subgroup of NSCLC.
Keywords: lung adenocarcinoma, young age, genomics, genetic predisposition, next generation sequencing
Short abstract
What's new?
Young patients with non‐small‐cell lung cancer (NSCLC) represent a distinct disease entity: they are often female, never smoked and usually present with lung adenoma carcinomas. Here the authors performed whole‐genome sequencing in patients with early‐onset NSCLC who never smoked and find an overall lower mutation burden and fewer classic driver substitutions. However, oncogenic fusions were found more frequently, underscoring that a unique molecular make‐up defines this specific subgroup of cancer patients.
Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- BMR
background mutation rate
- BWA
Burrows‐Wheeler Aligner
- CNVs
copy number variations
- ExAC
Exome Aggregation Consortium
- FDR
false discovery rate
- IGV
Integrative Genomics Viewer
- InDels
insertions and deletions
- kb
kilobase
- lncRNA
long noncoding RNA
- LUAD
lung adenocarcinoma
- Mb
megabase
- miRNA
microRNA
- NGS
next generation sequencing
- NSCLC
non‐small‐cell lung cancer
- snoRNA
small nucleolar RNA
- snRNA
small nuclear RNA
- SNVs
single nucleotide variations
- SVs
structural variations
- WGS
whole genome sequencing
Lung cancer is the most common cancer and the leading cause of cancer‐related death worldwide, accounting for over 1 million deaths per year. Among all lung cancer cases, 85% are non‐small‐cell lung cancer (NSCLC) with two main histological types of adenocarcinoma and squamous cell carcinoma.1, 2 With the development of precision medicine, NSCLC has been increasingly recognized as a highly heterogeneous disease with phenotypic and genotypic diversity in each subgroup.3, 4, 5, 6
While tobacco smoking is the most important risk factor for lung cancer, there is a distinct subset of patients (∼10–40%) who develop the disease with no history of smoking.6 Previous studies have characterized the genomic alterations of NSCLC in never‐smoker patients using next generation sequencing (NGS). Never‐smoker patients with lung adenocarcinoma (LUAD) harbor significantly lower somatic mutation burden than smoker patients with the same disease.7 Besides, C > T transitions are more common in never‐smoker patients, while C > A transversions occur more often in smoker patients.8 Moreover, EGFR activating mutations and EML4‐ALK fusions have been identified to be more frequent in never‐smoker patients than smoker patients. Thanks to targeted tyrosine kinase inhibitors, patients with the two genetic alterations have experienced a better survival.9
Aging is another fundamental factor for the development of lung cancer. Recently, it has been demonstrated that young patients have unique disease biology among a number of cancers. For instance, colon cancer arising at young age has been identified to be characterized with high frequency of microsatellite instability.10 Breast cancer diagnosed at a young age has a higher proportion of BRCA1/BRCA2 mutations and ERBB2 overexpression than the older ones.11 Although only 1.3–5.3% of patients with lung cancers are 45 years or younger at diagnosis, there is a trend of increasing incidence of lung cancer among young adults.12, 13, 14, 15 Many recent studies have suggested that NSCLC occurring in young patients constitutes a disease entity with distinct clinicopathologic characteristics.4, 5, 16, 17 Early‐onset NSCLC occurs more often in women and never‐smokers, presents a predominance of LUAD. However, only a few studies have investigated the genomic alterations of NSCLC occurring in young patients, and all of them focused on the mutational frequency of several certain driver events involved in lung cancer. Compared with older patients with NSCLC, higher incidence of ALK, ROS1 and RET fusions exist among the younger patients.4, 5, 16, 17
Despite these progresses, the landscape of genomic alterations of LUAD in young never‐smoker patients remains to be characterized. In this study, we elucidated the both somatic and germline alterations of 36 never‐smoker patients with LUAD aged 45 years or younger through whole genome sequencing (WGS). Our aim was to identify the molecular features of LUAD in young never‐smoker patients and to explore their clinical implications.
Material and Methods
Study population and sample collection
Thirty‐six never‐smoker (defined as <100 cigarettes in a life time) patients, who were diagnosed with LUAD at 45 years or younger were included for this study from West China Hospital from 2011 to 2016. None of them underwent neoadjuvant therapy before surgery. Tumors and matched distal normal lung tissues were obtained during surgery, snap‐frozen in liquid nitrogen and stored at −80°C until sequencing. All samples were reviewed by two pathologists to determine the histological subtype and tumor cellularity. The tumor tissues containing at least 60% of tumor cells were included. All patients provided informed consent, and this study was approved by the Institutional Review Board of West China Hospital, Sichuan University, Chengdu, China. The retrospective study of 1,296 patients with LUAD that received ALK‐Ventana immunohistochemistry testing was also approved by the Institutional Review Board of West China Hospital, Sichuan University, Chengdu, China.
Genomic DNA preparation and whole genome sequencing
The genomic DNA from frozen tissues was extracted using the DNeasy blood and tissue kit (Qiagen, USA) following the manufacturer's protocol. Degradation and contamination were monitored on 1% agarose gel, while the concentration was measured by Qubit DNA Assay Kit in Qubit 2.0 Flurometer (Life Technologies, USA).
For WGS, a total amount of 0.5 μg genomic DNA per sample with high‐molecular weight (>20 kb single band) was used for the DNA library preparation. Sequencing library was generated using Truseq Nano DNA HT Sample Prep Kit (Illumina, USA) following the manufacturer's recommendations, and index codes were added to each sample. Briefly, genomic DNA sample was fragmented by a Covarias sonication system to a size of ∼350 bp. Then DNA fragments were endpolished, A‐tailed and ligated with the full‐length adapter for Illumina sequencing, followed by further PCR amplification. After PCR products were purified (AMPure XP system), libraries were analyzed for size distribution by Agilent 2100 Bioanalyzer and quantified by real‐time PCR (3 nmol/L). The clustering of the index‐coded samples was performed on a cBot Cluster Generation System using HiSeq X PE Cluster Kit V2.5 (Illumina, USA) according to the manufacturer's instructions. After cluster generation, the DNA libraries were sequenced on Illumina HiSeq X platform and 150 bp paired‐end reads were generated.
The original fluorescence image files obtained from HiSeq platform were transformed to short reads (raw data) by base calling and recorded in FASTQ format, which contained sequence information and corresponding sequencing quality information. After excluding reads containing adapter contamination and low‐quality/unrecognizable nucleotides, clean data were applied for downstream bioinformatical analyses. Meanwhile, the total reads number, sequencing error rate, percentage of reads with average quality >20 and with average quality >30 and GC content distribution were calculated (Supporting Information, Table 1).
Reads mapping and somatic genetic alteration detection
Valid sequencing data were mapped to the reference human genome (UCSC hg19) by Burrows‐Wheeler Aligner (BWA) software to get the original mapping results stored in BAM format.18 Then, SAMtools,19 Picard (http://broadinstitute.github.io/picard/) and GATK20 were used to sort BAM files and do duplicate marking, local realignment and base quality recalibration to generate final BAM file for computing the sequence coverage and depth.
To call somatic single nucleotide variations (SNVs) and small insertions and deletions (InDels) from paired tumor‐normal samples, MuTect and Strelka were used respectively.21, 22 In addition to default filters, polymorphisms of somatic SNVs and InDels referenced in the 1000 Genomes Project23 or Exome Aggregation Consortium (ExAC)24 with a minor allele frequency over 1% were removed. Subsequently, VCF (Variant Call Format) was annotated by ANNOVAR.25
Somatic copy number variations (CNVs) were identified by Control‐FREEC,26 while GISTIC27 algorithm was used to infer recurrently amplified or deleted genomic regions. G‐scores were calculated for genomic and gene‐coding regions on the basis of the frequency and amplitude of amplification or deletion affecting each gene. A significant CNV region was defined as having amplification or deletion with a G‐score >0.1, corresponding to a p value threshold of 0.05 from the permutation‐derived null distribution.
For somatic structural variations (SVs) detection based on the soft‐clipped reads, CREST28 was implemented to directly map SVs at the nucleotide level of resolution. Only breakpoint pairs with at least three supporting clipped reads spanning the breakpoint were selected for further analysis.
PCR and Sanger sequencing
To validate somatic SNVs, InDels and SVs identified from the WGS data, we used PCR to amplify genomic DNA spanning mutation sites with specific primers. PCR products were electrophoresed through 1.0% agarose gel and sequenced by Sanger method. For ALK and RET fusions detected by WGS, Chimeric reads covering breakpoints were visualized and carefully evaluated using Integrative Genomics Viewer (IGV).26 A total of 29 identified somatic nonsynonymous SNVs/InDels were successfully verified (93.5%, 29/31) (Supporting Information, Table 2) and 9 SVs were verified (Supporting Information, Table 3).
Identification of significantly mutated genes and pathways
Significantly mutated genes were identified using MuSiC and MutSigCV,29, 30 which estimate the background mutation rate (BMR) for each gene‐patient‐category combination based on the observed silent mutations in the gene and noncoding mutations in the surrounding regions. Significant levels (p values) were determined by testing whether the observed mutations in a gene occurred more frequently than expected by random chance based on the background model. False discovery rates (FDR, q value) were then calculated, and candidate driver genes with q value <0.1 were exhibited after the elimination of apparent false‐positive findings and genes encoding proteins with >4,000 amino acids.30
Pathway enrichment analysis was carried out using PathScan algorithm to identify known cellular pathways with significant accretions of somatic mutations in lung tumors.31 Regardless of the frequency of mutation in specific genes, the entire nonsynonymous mutation was investigated to figure out the distribution of genes within the KEGG database.
Interpretation of germline variants
Genetic predisposition was estimated by variants considered as “Pathogenic” or “Likely Pathogenic” using the American College of Medical Genetics and Genomics (ACMG) guidelines.32 Germline SNVs/InDels were detected by SAMtools,19 followed by classification assignment of the input variant. The mutation frequency of positive genes assigning “pathogenic” in our cohort were compared with those disclosed in the in‐house noncancer WGS database (Novo‐Zhonghua Non‐Cancer Genomes Database from Novogene Co., Ltd, Beijing, China), which recorded germline variants found in 568 Chinese noncancer individuals. The mutation frequency in the controls included not only candidate variants identified in cases but also the other pathogenic loci located in the gene of attention.
Statistical analysis
The difference of somatic mutation rate between two groups was tested by Wilcoxon test. The other statistical comparisons between two groups were determined by Fisher's exact test. The difference of log10 of number of SNVs per kilobase (kb) among different gene types was tested by Fisher's least significant difference (LSD) test. Overall survival was estimated using Kaplan–Meier method with log rank test. All statistical analyses were performed using R software.
Results
Clinical and whole‐genome sequencing data
Among the 36 young never‐smoker patients with LUAD, 30 patients (83.3%) were female; the median age was 40 years (range, 28–45). Detailed clinicopathologic data were in Supporting Information, Table 4. The mean sequencing coverage was 53× (range, 50.47–57.04×) for the tumors and 34× (range, 30.28–45.88×) for the matched normal tissues. Overall, we detected 4,344–60,259 somatic mutations per tumor (Supporting Information, Table 5). Among them, 2,739 nonsynonymous mutations were identified, including 2,412 missense mutations, 138 nonsense mutation, 1 nonstop mutation, 8 in‐frame InDels, 27 frame‐shift InDels and 153 splicing mutations (Fig. 1 a Supporting Information, Table 2).
Figure 1.
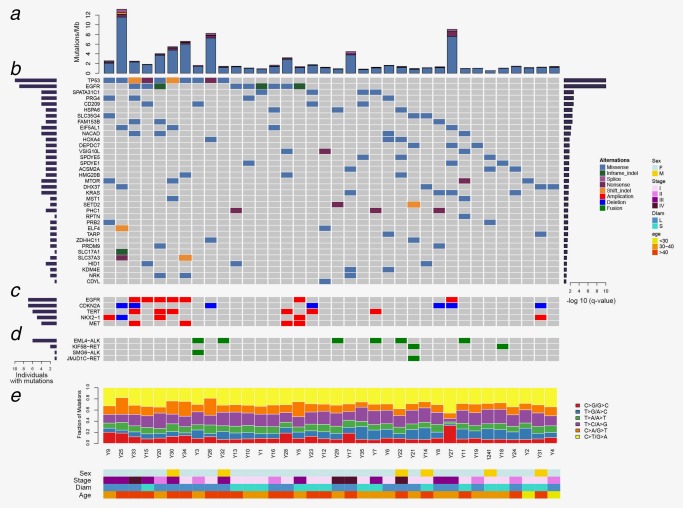
Mutation landscape of lung adenocarcinoma in young never‐smoker patients. (a) Nonsynonynous mutation rates (number of mutations per Mb) in 36 tumor samples. (b) Genes predicted to be significantly mutated by MuSiC. Asterisk indicated that the genes were noted by both MuSiC and MutSigCV. Genes were sorted by significant FDR value (right panel); the frequency was indicated by the number of mutated samples (left panel). (c) Focal CNVs in known lung cancer genes in 36 tumor samples. (d) SVs previously implicated in lung cancer in 36 tumor samples. (e) Percentage of six types of single nucleotide substitutions in each tumor sample. The samples were ordered on the horizontal axis based on the clustering of their mutated genes. The colors denoted different types of somatic events. [Color figure can be viewed at http://wileyonlinelibrary.com]
The mean somatic mutation rate in our cohort was 4.7 per megabase (Mb), lower than that of LUAD from the TCGA cohort (8.87 per Mb).8 Patients with somatic TP53 mutation harbored higher somatic mutation rate than patients with TP53 wild type (p < 0.001), patients aged older than 40 carried higher somatic mutation rate than patients aged at 40 years or younger (p = 0.035), and patients with larger tumor size (maximum diameter of tumor >3 cm) bore heavier mutational burdens with statistically significance, when compared with that of patients with smaller tumor size (p < 0.001). Mutational spectrum analysis revealed that except for five patients (Y5, Y34, Y30, Y26 and Y25) carrying dominant C > A transversions, the most common somatic nucleotide substitution in our cohort was C > T transitions, which had been implicated in LUAD in never‐smokers (Fig. 1 e).8
Recurrent somatic mutations in protein‐coding genes
To identify potential driver mutations, we characterized somatic mutations and identified 35 significantly mutated genes (q < 0.1) (Fig. 1 b). Apart from well‐known gene mutations (e.g., TP53, EGFR, MTOR, KRAS and SETD2),33 our analysis also identified several potential lung cancer associated gene mutations that were rarely reported (e.g., HOXA4, MST1 and CD209).34, 35, 36, 37 When comparing with the mutation frequency of top 50 most frequently mutated genes reported in 412 LUAD cases from the TCGA cohort,8 our cohort exhibited lower prevalence of recurrent mutations, especially in TP53 (27.78% vs. 47.57%, p = 0.024), KRAS (11.11% vs. 27.91%, p = 0.030), STK11 (0.00% vs. 15.53%, p = 0.005) and KEAP1 (0.00% vs. 14.81%, p = 0.009) (Supporting Information, Table 6). Conversely, EGFR and RB1 were relatively enriched in our study, although the difference did not reach the statistical significance (p = 0.16 and p = 0.19, respectively). Given the genetic heterogeneity between different ethnic populations, the distinct mutation patterns of our cohort were further investigated through comparing with previous study on Asians.38 Fewer EGFR mutations were observed in our cohort than previous data from Asians under the similar genetic background (25.00% vs. 39.40%, p = 0.1). Consistent with the previous studies,8, 38 EGFR activating mutations in our cohort occurred recurrently at the most common sites, including exon 19 deletions (3/9, 33.3%) and exon 21 L858R mutations (6/9, 66.7%).
As to CNVs detected in our cohort, only deletion in CDKN2A located on 9p21.3 was found statistically significant (p < 0.05). Additionally, we figured out 4 substantially amplified or deleted genes (EGFR, TERT, NKX2‐1 and MET) by examining the copy number of reported CNV hotspots in our sequencing data.33 A total of 15 patients (41.7%) carried lung cancer‐related CNVs (Fig. 1 c Supporting Information, Table 7).
Among the SVs detected in our cohort, the lung cancer related structural variations (e.g., EML4‐ALK and KIF5B‐RET) were commonly observed (22.2%, 8/36). Apart from two well‐known gene fusions, EML4‐ALK and KIF5B‐RET, we also identified two novel gene fusions, which were SMG6‐ALK and JMJD1C‐RET (Fig. 1 d Supporting Information, Table 3). Interestingly, the different fusion partners of ALK co‐occurred in a single patient, the same with RET. In addition, the frequency of ALK fusions in our cohort were higher than previous studies on never‐smoker patients with LUAD from China (16.7% vs. 7.0%, p = 0.039).39 In the six EML4‐ALK fusions, all breakpoints of ALK were generated in intron 19 and three of EML4 in intron 12, intimating that these two segments might be hotspots. To validate the high frequency of ALK fusions in young never‐smokers with LUAD, we reviewed the records of the 1296 patients with LUAD that received ALK‐Ventana immunohistochemistry testing between January 1, 2016 and January 1, 2017 in West China Hospital. Among the 839 patients with no history of smoking, the positive rate of ALK fusions in patients aged 45 years or younger was significantly higher than that in older patients (17.1% vs. 5.8%, p < 0.001). Meanwhile, among the 457 patients having smoking history, the positive rate of ALK fusions in patients aged 45 years or younger was also significantly higher than that in older patients (15.9% vs. 3.4%, p < 0.001).
Recurrent somatic mutations in noncoding regions
To investigate the role of noncoding RNA in LUAD occurring in young never‐smokers, we compared the frequency of mutations of different gene types in our samples. First, we calculated the number of SNVs in per kilobase (kb). Our results indicated that the number of SNVs in long noncoding RNAs (lncRNAs) and other types of noncoding genomic regions were higher than that in protein coding genes (Fig. 2 a). Among the gene types with >100 mutational genes, lncRNA had the highest average frequency of mutations (32.65 SNVs/kb), while the average frequency of protein‐coding genes was 6.16 SNVs/kb.
Figure 2.
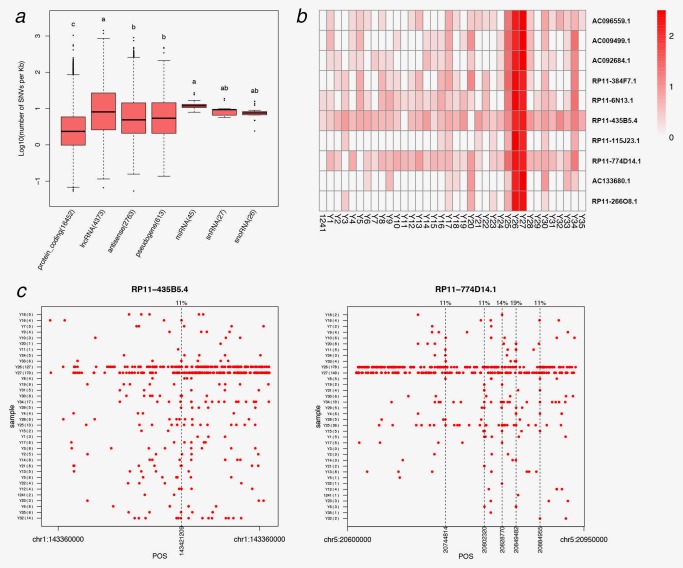
Mutations of noncoding regions in lung adenocarcinoma occurring in young never‐smoker patients. (a) The log10 of number of SNVs per Kb in the six noncoding regions and protein‐coding genes, and the number of mutational genes displayed in the parentheses behind every gene types. Items with different letters were significantly different (LSD test, p < 0.01). (b) The mutation frequency heatmap of the top 10 genes with highest mutation frequency in 36 tumor samples. The legend showed the meaning of the heatmap color; the depth of the color represented the size of the log10 (number of SNVs per kb). (c) RP11–774D14.1 and RP11–435B5.4 mutations in each tumor. The x‐axis indicated the absolute position of the gene and the dotted line showed the recurrent mutations, which had a high mutation percentage (>10% in all samples). And the mutation percentage of the recurrent mutations was showed in the upper of the figure. [Color figure can be viewed at http://wileyonlinelibrary.com]
We next accessed whether the SNVs in these lncRNA genes were recurrent in our patients. Patient Y25, Y26 and Y27, who carried a large amount of somatic mutations across the whole genome level (Fig. 1 a), also harbored high level of variants within the lncRNA genes. When plotting the top 10 lncRNA genes with most mutations in our study, we found that RP11–435B5.4 and RP11–774D14.1 showed mutation in majority of the patients (Fig. 2 b). Among the loci in RP11–774D14.1 gene, five SNVs were found in >10% of the patients (Fig. 2 c), and one SNV of RP11–435B5.4 were found in 11% of the patients, suggesting these recurrent SNVs in the two lncRNAs may be associated with biological function or can be used as potential biomarkers.
Somatically altered pathways and clinical implications
Integrative analysis of altered key pathways affected by SNVs/InDels, CNVs and SVs was performed to construct a comprehensive view of genomic characteristics of LUAD in young never‐smokers (Fig. 3). The most frequently aberrant pathways were RTK/RAS/PI3K pathway, affecting 32 patients (88.9%), which was in accordance with the TCGA study on LUAD.8 The p53 pathway was also frequently aberrant, with 21 patients (58.3%) harboring genomic alterations involved in this pathway.
Figure 3.
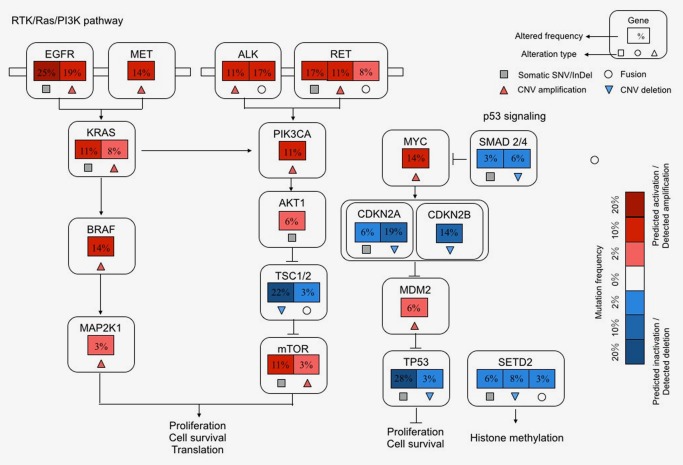
Somatically altered pathways in lung adenocarcinoma in young never‐smoker patients. Components and inferred functions of p53 signaling/cell cycle process, RTK/Ras/PI3K pathway and histone/chromatin modification were summarized in the main text. Percentage presented alteration frequencies in 36 tumor samples. Pathway alterations including somatic SNVs, CNVs and SVs were shown. Activated and inactivated pathways/genes and activating or inhibitory symbols were based on predicted effects of genome alterations and/or pathway function. [Color figure can be viewed at http://wileyonlinelibrary.com]
To comprehensively identify potentially targetable genomic alterations in our cohort, we matched SNVs/InDels, CNVs and SVs with previous data from published clinical trials.40, 41 As a result, we identified 5 genes (EGFR, ALK, RET, MET and MTOR) with potentially targetable alterations that were responsive to specific kinase inhibitors or antibodies in 23 patients (63.9%) (Fig. 4). The most frequent potentially targetable genomic alterations were EGFR activating mutations and ALK fusions.
Figure 4.
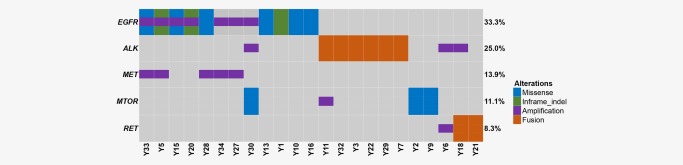
Therapeutic targets in young never‐smoker patients with lung adenocarcinoma. Missense mutations, in‐frame indel, copy number amplifications and gene fusions that were regarded as potential targets of specific kinase inhibitors or antibodies were investigated thoroughly. Tumors with at least one alteration were shown. [Color figure can be viewed at http://wileyonlinelibrary.com]
Genetic predisposition
To explore the genetic factor for early‐onset of LUAD, germline variants from the current 36 cases and 28 additional unpublished LUAD cases occurring in never‐smokers were evaluated according to ACMG guideline (Fig. 5 and Supporting Information, Table 8). These cases were classified into two groups: the young group (patients aged at 45 or younger, n = 46) and the old group (patients aged older than 55 years, n = 18). Pathogenic or likely pathogenic germline mutations in 35 cancer genes were identified in 36 patients of the young Group (36/46, 78.3%) (Fig. 5 and Supporting Information, Table 9). Notably, germline mutations in BPIFB1 (rs6141383, p.V284M), CHD4 (rs74790047, p.D140E), PARP1 (rs3219145, p.K940R), NUDT1 (rs4866, p.V83M), RAD52 (rs4987207, p.S346*) and MFI2(rs17129219, p.A559T) were significantly enriched in the young group when compared with the in‐house noncancer database (p < 0.05). Among them, BPIFB1, CHD4 and RAD52 susceptibility loci were also not detected in the old group.
Figure 5.
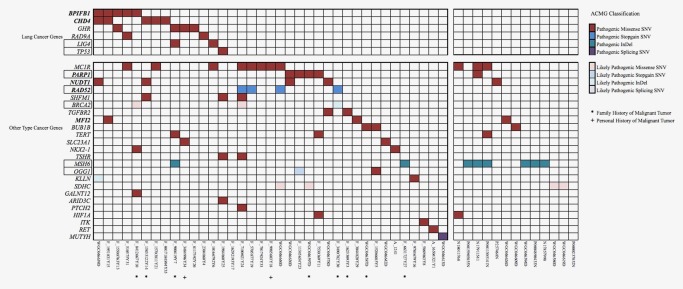
Pathogenic and likely pathogenic germline alterations classified by ACMG. Genes were categorized into “lung cancer genes” (top) and “other type cancer genes” (below). The square frame indicated genes related to DNA reparation. The color denoted different tiers of germline events. Patients with assumable predisposition of cancer were indicated by * (multiple primary cancers) or # (immediate family member diagnosed with cancer). [Color figure can be viewed at http://wileyonlinelibrary.com]
The patient (Y25) that had a germline TP53 missense mutation (rs121912664, p. R205H) was found to be hypermutated when compared with the others. However, it seemed that germline defections may not be served as a predictor for prognosis, as neither identified lung susceptibility genes nor genes significantly enriched in cases showed obvious effects on clinical outcome according to Kaplan–Meier curves for overall survival (p = 0.318 and p = 0.827).
Discussion
WGS provides a unique opportunity to perform an integrated analysis concerning not only point mutation but also structural alteration. That, combined with the restriction of sample included, enabled us to identify recurrent somatic mutations with tumorigenic ability in the special group of lung cancer (young never‐smoker patients with LUAD). In contrast to common sense that LUAD was labeled with high mutation burden,8, 42 young never‐smoker patients possessed the characteristics of lower mutation load and fewer classic driver substitutions. Nevertheless, oncogenetic fusions occurred more frequently, emphasizing the importance of more study and special consideration of non‐SNV aberrations in the carcinogenic processes of this distinct subgroup of lung cancer.
It was well known that the prevalence of EGFR mutations varied hugely in different settings, according to age, smoking status and ethics. Previous researches agreed that EGFR activating mutations was most commonly found in the Asian decent and never‐smokers, and the mutation rate climbed extremely high (60.7%, 462/761) when focusing on Asian never‐smokers.43 In striking contrast to the driver landscape of Asian never‐smokers dominated by EGFR mutations, only 25.0% patients in our cohort were featured with activating EGFR. The discrepancy might be ascribed to younger age; however, previous studies concerning the difference of proportion of EGFR mutations between young and old patient groups were inconclusive.4, 5, 13, 17 In addition, other fairly frequent events (e.g., TP53, KRAS and KEAP1) in the TCGA cohort did not appear frequent in our cohort, which also demonstrated heterogeneity exist in the specific subgroup of NSCLC. Furthermore, among the recurrent somatic mutations in protein‐coding genes in the study, HOXA4 and MST1 are labeled as lung cancer‐associated genes. HOXA4 belongs to the HOX family of transcription factors that has been implicated in regulating gene expression. Previous studies have found that HOX overexpression exist in LUAD and result in enhanced motile and invasive properties.36 MST1 encodes serine threonine kinase, which has been identified to perform tumor‐suppressor function involving in cell growth, proliferation and apoptosis. A recent study has shown that MST1 overexpression inhibit the growth of NSCLC A549 cells both in vitro and in vivo.37
Apart from identifying novel fusions of ALK and RET (i.e., SMG6‐ALK and JMJD1C‐RET), we also found that the prevalence of EML4‐ALK in young LUAD patients was significantly higher than that in older ones regardless of the smoking status. Intriguingly, other malignancies that harbor ALK fusions, including anaplastic large cell lymphomas, neuroblastoma and inflammatory myofibroblastic tumor, mainly occur in young adults and children.44, 45, 46 In addition, other gene fusions in LUAD, such as ROS1 and RET fusions, are also reported to be associated with earlier onset.4, 5 These findings suggest that SVs result in more aggressive tumors that require less time to become overt phenotypes, and highlight the need for performing genetic SVs testing in young patients with LUAD.
Another major finding of this study was that young never‐smoker patients with LUAD harbored a high frequency (63.9%) of potentially targetable genomic alterations in EGFR, ALK, RET, MET and MTOR. Consistent with our finding, Sacher et al. evaluated molecular features of 2,237 patients with NSCLC and found that patients aged 50 years or younger were significantly more likely to carry a targetable genomic alteration than the older ones (78% vs. 49%, p < 0.001).5 These data suggest that young never‐smoker patients with LUAD represent a distinct subgroup of NSCLC that was enriched with targetable genotypes, and deserve extensive screening of targetable genomic alterations and subsequently benefit from personalized medicine strategies with specific targeted therapy.
Moreover, we had the opportunity to better understand the genetic predisposition to LUAD with stringent requirement for enrollment of the study. We found that young never‐smoker patients with LUAD harbored a unique pattern of germline mutations in cancer predisposition genes, including BPIFB1 (rs6141383, p.V284M), CHD4 (rs74790047, p.D140E), PARP1 (rs3219145, p.K940R), NUDT1 (rs4866, p.V83M), RAD52 (rs4987207, p.S346*) and MFI2 (rs17129219, p.A559T). Among them, BPIFB1 and CHD4 susceptibility loci have been identified to be associated with lung cancer risk (p = 1.79 × 10−7 and p < 0.001, respectively) and the former was also linked to age of onset of lung cancer (p = 0.006).47, 48 Meanwhile, studies have been shown that PARP1, NUDT1, RAD52 and MFI2 susceptibility loci would increase the risk of other cancers, for example, PARP1 in gastric and breast cancer and MFI2 in colorectal cancer.49, 50, 51 These findings suggest that the existence of mutations in cancer predisposition genes may be a possible reason for their early onset of LUAD without smoking history and that a better understanding of lung cancer risk will depend on evaluation of cancer predisposition genes. Although we failed to find considerable differences in clinical presentation and overall survival across the patients with or without genetic defects, it seemed that predisposition genes had an impact on the initiation rather than the development of tumor. It was arbitrary to negate the bridge from germline mutations to clinical features of disease. More patients and longer follow‐up period would help to explore the relative contributions of inherited genetic factors to prognosis.
This study is the first to characterize the genomic alterations of LUAD in young never‐smokers through WGS. Our study provides insights into understanding the genomic landscape and molecular basis for this specific subgroup of NSCLC. A limitation of this study is its small sample size. Future studies should include validation of these findings in a larger size of samples.
Supporting information
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5
Supplementary Table 6
Supplementary Table 7
Supplementary Table 8
Supplementary Table 9
Conflict of Interest: None of the authors have conflict of interest to disclose.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008;359:1367–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y, Wang DC, Shi L, et al. Genome analyses identify the genetic modification of lung cancer subtypes. Semin Cancer Biol 2017;42:20–30. [DOI] [PubMed] [Google Scholar]
- 4. Tanaka K, Hida T, Oya Y, et al. Unique prevalence of oncogenic genetic alterations in young patients with lung adenocarcinoma. Cancer 2017;123:1731–40. [DOI] [PubMed] [Google Scholar]
- 5. Sacher AG, Dahlberg SE, Heng J, et al. Association between younger age and targetable genomic alterations and prognosis in non‐small‐cell lung cancer. JAMA Oncol 2016;2:313–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee YJ, Kim JH, Kim SK, et al. Lung cancer in never smokers: change of a mindset in the molecular era. Lung Cancer (Amsterdam, Netherlands). 2011;72:9–15. [DOI] [PubMed] [Google Scholar]
- 7. Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150:1107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Collisson EA, Campbell JD, Brooks AN, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Swanton C, Govindan R. Clinical implications of genomic discoveries in lung cancer. N Engl J Med 2016;374:1864–73. [DOI] [PubMed] [Google Scholar]
- 10. Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med 2000;342:69–77. [DOI] [PubMed] [Google Scholar]
- 11. Azim HA, Jr , Partridge AH. Biology of breast cancer in young women. Breast Cancer Res. 2014;16:427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The National Institutes of Health National Cancer Institute. SEER Cancer Statistics Review, 1975–2014. 2017. https://seer.cancer.gov/csr/1975_2014/results_merged/sect_01_overview.pdf.
- 13. Hsu CL, Chen KY, Shih JY, et al. Advanced non‐small cell lung cancer in patients aged 45 years or younger: outcomes and prognostic factors. BMC Cancer 2012;12:241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Chen SF, Zhen Y, et al. Multicenter analysis of lung cancer patients younger than 45 years in Shanghai. Cancer 2010;116:3656–62. [DOI] [PubMed] [Google Scholar]
- 15. Fidler MM, Gupta S, Soerjomataram I, et al. Cancer incidence and mortality among young adults aged 20–39 years worldwide in 2012: a population‐based study. Lancet Oncol 2017;18:1579–89. [DOI] [PubMed] [Google Scholar]
- 16. Ye T, Pan Y, Wang R, et al. Analysis of the molecular and clinicopathologic features of surgically resected lung adenocarcinoma in patients under 40 years old. J Thorac Dis 2014;6:1396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. VandenBussche CJ, Illei PB, Lin MT, et al. Molecular alterations in non‐small cell lung carcinomas of the young. Hum Pathol 2014;45:2379–87. [DOI] [PubMed] [Google Scholar]
- 18. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics (Oxford, England). 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAM tools. Bioinformatics (Oxford, England). 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saunders CT, Wong WS, Swamy S, et al. Strelka: accurate somatic small‐variant calling from sequenced tumor‐normal sample pairs. Bioinformatics (Oxford, England). 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 22. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boeva V, Popova T, Bleakley K, et al. Control‐FREEC: a tool for assessing copy number and allelic content using next‐generation sequencing data. Bioinformatics (Oxford, England). 2012;28:423–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy‐number alteration in human cancers. Genome Biol 2011;12:R41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang J, Mullighan CG, Easton J, et al. CREST maps somatic structural variation in cancer genomes with base‐pair resolution. Nat Methods 2011;8:652–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dees ND, Zhang Q, Kandoth C, et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res 2012;22:1589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature 2013;499:214–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wendl MC, Wallis JW, Lin L, et al. PathScan: a tool for discerning mutational significance in groups of putative cancer genes. Bioinformatics (Oxford, England). 2011;27:1595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Devarakonda S, Morgensztern D, Govindan R. Genomic alterations in lung adenocarcinoma. Lancet Oncol 2015;16:e342–51. [DOI] [PubMed] [Google Scholar]
- 34. van Gisbergen KP, Aarnoudse CA, Meijer GA, et al. Dendritic cells recognize tumor‐specific glycosylation of carcinoembryonic antigen on colorectal cancer cells through dendritic cell‐specific intercellular adhesion molecule‐3‐grabbing nonintegrin. Cancer Res 2005;65:5935–44. [DOI] [PubMed] [Google Scholar]
- 35. Ling P, Lu TJ, Yuan CJ, et al. Biosignaling of mammalian Ste20‐related kinases. Cell Signal 2008;20:1237–47. [DOI] [PubMed] [Google Scholar]
- 36. Omatu T. Overexpression of human homeobox gene in lung cancer A549 cells results in enhanced motile and invasive properties. Hokkaido J Med Sci 1999;74:367–76. [PubMed] [Google Scholar]
- 37. Xu CM, Liu WW, Liu CJ, et al. Mst1 overexpression inhibited the growth of human non‐small cell lung cancer in vitro and in vivo. Cancer Gene Ther 2013;20:453–60. [DOI] [PubMed] [Google Scholar]
- 38. Wu K, Zhang X, Li F, et al. Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas. Nat Commun 2015;6:10131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gou LY, Niu FY, Wu YL, et al. Differences in driver genes between smoking‐related and non‐smoking‐related lung cancer in the Chinese population. Cancer 2015;121: 3069–79. [DOI] [PubMed] [Google Scholar]
- 40. Hirsch FR, Suda K, Wiens J, et al. New and emerging targeted treatments in advanced non‐small‐cell lung cancer. Lancet (London, England). 2016;388:1012–24., Jr., [DOI] [PubMed] [Google Scholar]
- 41. Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annu Rev Med 2016;67:11–28. [DOI] [PubMed] [Google Scholar]
- 42. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shi Y, Au JS, Thongprasert S, et al. prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non‐small‐cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol 2014;9:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carpenter EL, Mosse YP. Targeting ALK in neuroblastoma–preclinical and clinical advancements. Nat Rev Clin Oncol 2012;9:391–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Minard‐Colin V, Brugieres L, Reiter A, et al. Non‐Hodgkin lymphoma in children and adolescents: progress through effective collaboration, current knowledge, and challenges ahead. JCO. 2015;33:2963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pomari E, Basso G, Bresolin S, et al. NPM‐ALK expression levels identify two distinct subtypes of paediatric anaplastic large cell lymphoma. Leukemia 2017;31:498–501. [DOI] [PubMed] [Google Scholar]
- 47. Jin G, Zhu M, Yin R, et al. Low‐frequency coding variants at 6p21.33 and 20q11.21 are associated with lung cancer risk in Chinese populations. Am J Hum Genet 2015;96:832–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamada M, Sato N, Ikeda S, et al. Association of the chromodomain helicase DNA‐binding protein 4 (CHD4) missense variation p.D140E with cancer: potential interaction with smoking. Genes Chromosomes Cancer 2015;54:122–8. [DOI] [PubMed] [Google Scholar]
- 49. Webb EL, Rudd MF, Sellick GS, et al. Search for low penetrance alleles for colorectal cancer through a scan of 1467 non‐synonymous SNPs in 2575 cases and 2707 controls with validation by kin‐cohort analysis of 14 704 first‐degree relatives. Hum Mol Genet 2006;15:3263–71. [DOI] [PubMed] [Google Scholar]
- 50. Ma X‐B, Wang X‐J, Wang M, et al. Impact of the PARP1 rs1136410 and rs3219145 polymorphisms on susceptibility and clinicopathologic features of breast cancer in a Chinese population. Transl Cancer Res 2016;5:520–8. [Google Scholar]
- 51. He W, Liu T, Shan Y, et al. PARP1 polymorphisms increase the risk of gastric cancer in a Chinese population. Mol Diagn Ther 2012;16:35–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Table 4
Supplementary Table 5
Supplementary Table 6
Supplementary Table 7
Supplementary Table 8
Supplementary Table 9