

Abstract
The mechanism and selectivity of terminal alkyne coupling reactions promoted by rhodium(I) complexes of NHC‐based CNC pincer ligands have been investigated. Synthetic and kinetic experiments support E‐ and gem‐enyne formation through a common reaction sequence involving hydrometallation and rate‐determining C−C bond reductive elimination. The latter is significantly affected by the ligand topology: Employment of a macrocyclic variant enforced exclusive head‐to‐head coupling, contrasting the high selectivity for head‐to‐tail coupling observed for the corresponding acyclic pincer ligand.
Keywords: C−C coupling, enynes, macrocyclic ligands, pincer ligands, rhodium
The transition‐metal‐catalysed dimerisation of terminal alkynes into conjugated enynes is an attractive, atom‐economic method for the preparation of versatile organic building blocks from readily accessible starting materials.1, 2 These reactions involve the formal addition of the C(sp)−H bond of one alkyne across the C≡C bond of the other, a process that can in principle result in three different regio‐ or stereochemical isomers: gem‐, E‐, and Z‐enynes. With the additional possibility for the substrates to be consumed through competing metal‐catalysed reaction pathways—for instance leading to butatrienes, arenes, or other polyenes—the widespread application of terminal alkyne coupling reactions in organic synthesis rests on the development of catalysts that can enforce high reaction control.1, 3 Despite the evaluation of a variety of transition‐metal complexes, this remains a largely unfulfilled aspiration, with few catalysts capable of producing single enyne isomers with sufficiently high selectivity.4
Building on early landmarks,5 rhodium‐based catalysts are of notable contemporary interest.6, 7, 8, 9 Recent examples bearing anionic pincer ligands, in particular, show promising activity and product fidelity. For instance, Rh(CNC) complex A has been shown to be a highly selective precatalyst for the production of a range of gem‐enynes (1 mol %, 80 °C),6 while Rh(PNP) complex B principally affords E‐enynes under similar conditions (1 mol %, 100 °C).7 Although closely related Rh(PNP) and Rh(PCP) systems demonstrate reduced selectivity, they predominantly give mixtures of only gem‐ and E‐enynes.7, 8 As previously asserted by Ozerov,8 these observations implicate a common hydrometallation–reductive elimination mechanism for the aforementioned rhodium pincer complexes, a scheme that bifurcates on coordination of the second alkyne to afford either “head‐to‐tail” (gem) or “head‐to‐head” (E) coupled products (i.e., II→III vs. II→V in Scheme 1). The formation of Z‐enynes instead typically evokes vinylidene intermediates.1
Scheme 1.
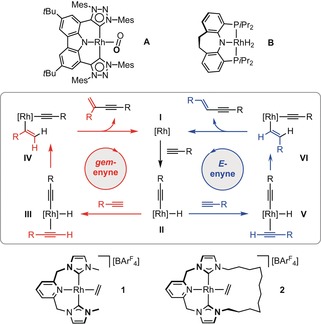
Selected complexes and proposed mechanistic pathways for the rhodium pincer catalysed formation of gem‐ and E‐enynes from terminal alkynes. ArF=3,5‐(CF3)2C6H3.
Motivated by these precedents and as part of our research exploring the organometallic chemistry of NHC‐based pincer ligands,10, 11 we set about evaluating the use of rhodium complexes containing neutral lutidine‐based CNC ligands in terminal alkyne coupling reactions. In particular, we were interested in ascertaining the capacity of macrocyclic variants for imparting additional reaction control. To this end, we herein describe our work contrasting the reactions of RhI ethylene complexes [Rh(CNC‐Me)(C2H4)][BArF 4] (1) and [Rh(CNC‐12)(C2H4)][BArF 4] (2) with a bulky terminal alkyne (Scheme 1; R=3,5‐tBu2C6H3=Ar′). These novel and appreciably air‐sensitive complexes were synthesised by employing reactions of the corresponding CuI transfer agents [Cu(CNC)][BArF 4] with [Rh(C2H4)2Cl]2,11 and fully characterised in solution and the solid state (see the Supporting Information and Figure 1). Reinforcing the electronic similarities of the CNC‐Me and CNC‐12 ligands evident from these data, the corresponding RhI carbonyl derivatives [Rh(CNC‐Me)(CO)][BArF 4] (3) and [Rh(CNC‐12)(CO)][BArF 4] (4) have directly comparable carbonyl stretching frequencies (ν(CO)=1980 cm−1, 3; 1978 cm−1, 4).12
Figure 1.
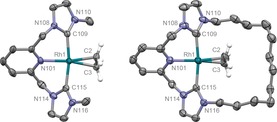
Solid‐state structures of 1 and 2. Thermal ellipsoids drawn at 50 % probability; anions and most hydrogen atoms omitted for clarity. Selected data for 1: Rh1–Cnt(C2,C3) 2.033(3) Å, C2–C3 1.373(4) Å, Rh1–N101 2.116(2) Å, Rh1–C109 2.037(2) Å, Rh1–C115 2.042(2) Å; Py‐Rh‐C=C twist 16.0(2)°; for 2: Rh1–Cnt(C2,C3) 1.996(3) Å, C2–C3 1.363(5) Å, Rh1–N101 2.116(2) Å, Rh1–C109 2.042(3) Å, Rh1–C115 2.061(3) Å; Py‐Rh‐C=C twist 31.7(2)°.13
Under comparably milder conditions to those employed for A and B, complex 1 was found to be an effective and selective precatalyst for the homocoupling of HC≡CAr′ in CD2Cl2, affording the gem‐product Ar′C≡CC(CH2)Ar′ exclusively until alkyne conversion reached >90 % (5 mol %, t 1/2=4.2 h, 25 °C; Scheme 2). At this point, nearing complete consumption of the alkyne, there was evidence for a subsequent metal‐catalysed reaction of the enyne.14 Ar′C≡CC(CH2)Ar′ was, however, readily isolated in practically useful yield (73 %) upon quenching the reaction with carbon monoxide at high alkyne conversion, sequestering the catalyst as 3. Alternative use of [Rh(CNC‐Me)(SOMe2)][BArF 4] (5) as the precatalyst maintained the exclusive gem‐selectivity observed for 1, but the homocoupling was an order of magnitude slower under equivalent conditions (5 mol %, 25 °C), presumably owing to reversible binding of dimethyl sulfoxide. Whilst use of 5 is detrimental to catalytic activity, it advantageously facilitated in situ investigation of the organometallic intermediates involved by NMR spectroscopy and ESI‐MS. In this way, a single and persistent species was identified and, following additional interrogation under conditions more amenable to characterisation (33 mol %, 5 °C), assigned as RhIII gem‐alkenyl complex 6 (Scheme 2; cf. IV in Scheme 1). Notable spectroscopic features of 6 include C 1 symmetry, two 18H tBu resonances, geminal alkene 1H resonances at δ 3.21 and 4.79, and 13C resonances at δ 101.9 (RhC≡C, 1 J RhC=55 Hz) and 153.8 (RhC(CH2)Ar′, 1 J RhC=38 Hz) that display large 103Rh coupling.15 Isolation of 6 from solution was encumbered by facile reductive elimination of Ar′C≡CC(CH2)Ar′.
Scheme 2.
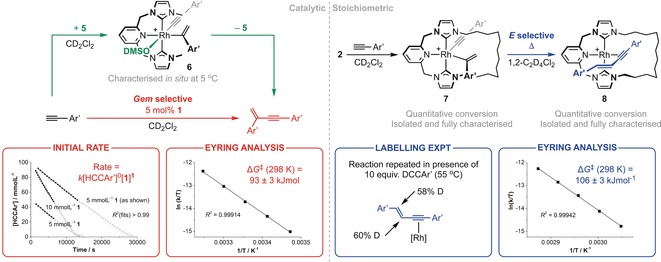
Reactions of 1 and 2 with HC≡CAr′. [BArF 4]− counteranions omitted for clarity.
These observations can be reconciled by a hydrometallation mechanism for the HC≡CAr′ homocoupling with the resting state IV (cf. 6) and turnover‐limiting C−C bond reductive elimination (i.e., IV→I in Scheme 1). Consistent with this suggestion, a kinetic analysis using the more active precatalyst 1 indicated that the homocoupling reaction is zero order in the alkyne substrate and first order in 1 (Scheme 2). Working within our conjecture, the activation barrier for the reductive elimination step was also determined through measurement of the temperature dependence of the reaction, affording ΔG ≠ (298 K)=93±3 kJ mol−1 (ΔH ≠=97±1 kJ mol−1, ΔS ≠=15±5 J K−1 mol−1; Scheme 2).
Paralleling the formation of 6, reaction of 2 with 2.1 equiv of HC≡CAr′ in CD2Cl2 led to rapid and quantitative formation of RhIII gem‐alkenyl complex 7, which was subsequently isolated and fully characterised (Scheme 2 and Figure 2). Presumably as a consequence of the confined metal coordination sphere evident in the solid state, 7 is appreciably dynamic in solution on the NMR timescale (500 MHz; see the Supporting Information). The spectroscopic characteristics nevertheless corroborate the structure of 7 in the solution phase, for instance, the presence of germinal 1H resonances at δ 5.57 and 5.76 alongside doublets in the 13C NMR spectrum at δ 85.0 (RhC≡C, 1 J RhC=72 Hz) and 157.7 (RhC(CH2)Ar′, 1 J RhC=27 Hz). While the intricacies of the spectroscopic data diverge, the most meaningful difference between 6 and 7 is the significantly enhanced stability of the latter, macrocyclic variant. Reminiscent of active‐metal‐template methods used in the construction of interlocked molecules,16 7 does however undergo C−C bond reductive elimination through the annulus of the bound CNC‐12 ligand when heated in the higher‐boiling‐point solvent 1,2‐C2D4Cl2. Curiously, the resulting mechanically entrapped hydrocarbon product is not the gem‐enyne Ar′C≡CC(CH2)Ar′, expected for a single reaction step and through extrapolation of the reactivity established for 1 and 5, but instead the alternative E‐regioisomer E‐Ar′C≡CCH=CHAr′ (Scheme 2).17 The associated RhI adduct 8 is formed quantitatively and was isolated from solution and comprehensively characterised, including in the solid state by single‐crystal X‐ray diffraction (see the Supporting Information and Figure 2).
Figure 2.
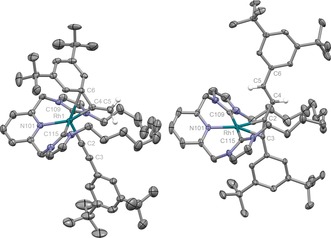
Solid‐state structures of 7 and 8. Thermal ellipsoids drawn at 50 % and 30 % probability, respectively; anions, minor disordered components (1×tBu group in 7; 1×Ar′ and 1×CH2CH2 group in 8), solvent molecules, and most hydrogen atoms omitted for clarity. Selected data for 7: Rh1–C2 1.940(5) Å, C2–C3 1.204(8) Å; Rh1‐C2‐C3 176.6(5)°; Rh1–C4 1.983(5) Å, C4–C5 1.310(8) Å; Rh1‐C4‐C5 140.1(4)°, C2‐Rh1‐C4 96.6(2)°; Rh1–C6 2.435(5) Å, Rh1–N101 2.228(5) Å, Rh1–C109 2.066(5) Å, Rh1–C115 2.075(6) Å; for 8: Rh1–Cnt(C2,C3) 1.981(3) Å, C2–C3 1.255(4) Å, C2–C4 1.425(4) Å, C4–C5 1.338(4) Å; C2‐C4‐C5 124.7(3)°; Rh1–N101 2.105(2) Å, Rh1–C109 2.052(3) Å, Rh1–C115 2.042(3) Å; Py‐Rh‐C≡C twist 38.7(2)°.13
The formation of 8 from 7 necessitates a multistep mechanism starting with β‐H abstraction and terminating with reductive elimination from a RhIII E‐alkenyl alkynyl species. Moreover, on the basis of a labelling experiment, which involved heating 7 in the presence of excess DC≡CAr′ and resulted in significant D incorporation into both positions of the enyne core, exchange and reversible C(sp)−H activation of both alkyne components must occur: IV⇌III⇌II(⇌I)⇌V⇌VI→8 (Scheme 1 and Scheme 2). No intermediates were observed when the conversion of 7 into 8 was followed in situ by 1H NMR spectroscopy, with the reaction following ideal first‐order kinetics across a wide temperature range (328–348 K). On the basis of these data, we assign reductive elimination as the rate‐determining step, with an associated barrier of ΔG ≠ (298 K)=106±3 kJ mol−1 (ΔH ≠=119±1 kJ mol−1, ΔS ≠=44±4 J K−1 mol−1).
Reflecting on these results from a fundamental perspective, the observation, structural elucidation, and onward (orthogonal) reactivity of 6 and 7 provide convincing evidence for a common hydrometallation–reductive elimination mechanism for the formation of gem‐ and E‐enynes by rhodium pincer promoted terminal alkyne coupling. In the case of the lutidine‐based CNC ligands employed in this study, product‐forming reductive elimination appears to be rate‐determining. The energetics of this step are, however, significantly perturbed when performed through the aperture of the macrocyclic CNC‐12 ligand. By comparison to the acyclic congener CNC‐Me, the steric constraints imposed by the flexible ring raise the barrier for C−C bond reductive elimination from a RhIII gem‐alkenyl alkynyl species relative to the alternative RhIII E‐alkenyl alkynyl intermediate by at least ΔG ≠ (298 K)=13±6 kJ mol−1, triggering a switch in product selectivity. These observations provide new insight into how terminal alkyne coupling reactions can be controlled and, more generally, showcase an unconventional approach for tuning the reactivity of pincer ligands.18 As a proof‐of‐concept demonstration of how this knowledge could be applied, the orthogonal selectivity of acyclic 1 and macrocyclic 2 can be exploited to prepare both PhC≡CC(CH2)Ph (catalytically) and E‐PhC≡CCH=CHPh (stoichiometrically) from HC≡CPh in high yield (Scheme 3, see the Supporting Information for full details). Our future work is focused on exploring the application of these lutidine‐based CNC ligands in catalysis, supramolecular, and organometallic chemistry.
Scheme 3.
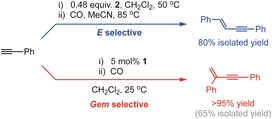
Preparation of PhC≡CC(CH2)Ph and E‐PhC≡CCH=CHPh.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313; C.M.S., M.R.G., A.B.C.), the University of Warwick (R.E.A.), and the Royal Society (UF100592, UF150675; A.B.C.) for financial support. Crystallographic (1, 3, 8) and high‐resolution mass‐spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data for 2, 5, 7, and [Cu(CNC‐12)][BArF 4] were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (Grant Agreement No. 637313).
C. M. Storey, M. R. Gyton, R. E. Andrew, A. B. Chaplin, Angew. Chem. Int. Ed. 2018, 57, 12003.
In memory of Andrew Brodie
Contributor Information
Caroline M. Storey, http://go.warwick.ac.uk/abchaplin
Dr. Adrian B. Chaplin, Email: a.b.chaplin@warwick.ac.uk.
References
- 1. Trost B. M., Masters J. T., Chem. Soc. Rev. 2016, 45, 2212–2238; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhou Y., Zhang Y., Wang J., Org. Biomol. Chem. 2016, 14, 6638–6650; [DOI] [PubMed] [Google Scholar]; Nishiura M., J. Mol. Catal. A 2004, 213, 101–106; [Google Scholar]; Trost B. M., Toste F. D., Pinkerton A. B., Chem. Rev. 2001, 101, 2067–2096; [DOI] [PubMed] [Google Scholar]; Ritleng V., Sirlin C., Pfeffer M., Chem. Rev. 2002, 102, 1731–1770. [DOI] [PubMed] [Google Scholar]
- 2. Trost B. M., Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- 3. Leroyer L., Maraval V., Chauvin R., Chem. Rev. 2012, 112, 1310–1343; [DOI] [PubMed] [Google Scholar]; Liu J., Lam J. W. Y., Tang B. Z., Chem. Rev. 2009, 109, 5799–5867; [DOI] [PubMed] [Google Scholar]; Batrice R. J., McKinven J., Arnold P. L., Eisen M. S., Organometallics 2015, 34, 4039–4050; [Google Scholar]; Esteruelas M. A., Herrero J., López A. M., Oliván M., Organometallics 2001, 20, 3202–3205; [Google Scholar]; Saito S., Yamamoto Y., Chem. Rev. 2000, 100, 2901–2916. [DOI] [PubMed] [Google Scholar]
- 4.Notable examples not otherwise cited herein: Rivada-Wheelaghan O., Chakraborty S., Shimon L. J. W., Ben-David Y., Milstein D., Angew. Chem. Int. Ed. 2016, 55, 6942–6945; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7056–7059; [Google Scholar]; Alós J., Bolaño T., Esteruelas M. A., Oliván M., Oñate E., Valencia M., Inorg. Chem. 2014, 53, 1195–1209; [DOI] [PubMed] [Google Scholar]; Jahier C., Zatolochnaya O. V., Zvyagintsev N. V., Ananikov V. P., Gevorgyan V., Org. Lett. 2012, 14, 2846–2849; [DOI] [PubMed] [Google Scholar]; Trost B. M., Chan C., Ruhter G., J. Am. Chem. Soc. 1987, 109, 3486–3487. [Google Scholar]
- 5. Singer H., Wilkinson G., J. Chem. Soc. A 1968, 849–853. [Google Scholar]
- 6. Kleinhans G., Guisado-Barrios G., Liles D. C., Bertrand G., Bezuidenhout D. I., Chem. Commun. 2016, 52, 3504–3507. [DOI] [PubMed] [Google Scholar]
- 7. Weng W., Guo C., Çelenligil-Çetin R., Foxman B. M., Ozerov O. V., Chem. Commun. 2006, 197. [DOI] [PubMed] [Google Scholar]
- 8. Pell C. J., Ozerov O. V., ACS Catal. 2014, 4, 3470–3480. [Google Scholar]
- 9. Rubio-Pérez L., Azpíroz R., Di Giuseppe A., Polo V., Castarlenas R., Pérez-Torrente J. J., Oro L. A., Chem. Eur. J. 2013, 19, 15304–15314; [DOI] [PubMed] [Google Scholar]; Xu H.-D., Zhang R.-W., Li X., Huang S., Tang W., Hu W.-H., Org. Lett. 2013, 15, 840–843; [DOI] [PMC free article] [PubMed] [Google Scholar]; Peng H. M., Zhao J., Li X., Adv. Synth. Catal. 2009, 351, 1371–1377; [Google Scholar]; Katagiri T., Tsurugi H., Satoh T., Miura M., Chem. Commun. 2008, 3405–3407. [DOI] [PubMed] [Google Scholar]
- 10. Apps S. L., Alflatt R. E., Leforestier B., Storey C. M., Chaplin A. B., Polyhedron 2018, 143, 57–61; [Google Scholar]; Andrew R. E., Ferdani D. W., Ohlin C. A., Chaplin A. B., Organometallics 2015, 34, 913–917; [Google Scholar]; Andrew R. E., Chaplin A. B., Inorg. Chem. 2015, 54, 312–322; [DOI] [PubMed] [Google Scholar]; Andrew R. E., Chaplin A. B., Dalton Trans. 2014, 43, 1413–1423. [DOI] [PubMed] [Google Scholar]
- 11. Andrew R. E., Storey C. M., Chaplin A. B., Dalton Trans. 2016, 45, 8937–8944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carbonyl complexes 3 and 4 are readily prepared by reaction of 1 and 2, respectively, with carbon monoxide (see the Supporting Information for details and the solid-state structure of 3).
- 13.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201807028 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
- 14.Balance tetramer formed. Full details will be published in due course.
- 15.For a structurally related rhodium complex, see: Schäfer M., Wolf J., Werner H., Organometallics 2004, 23, 5713–5728. [Google Scholar]
- 16. Denis M., Goldup S. M., Nat. Rev. Chem. 2017, 1, 0061; [Google Scholar]; Lewis J. E. M., Beer P. D., Loeb S. J., Goldup S. M., Chem. Soc. Rev. 2017, 46, 2577–2591; [DOI] [PubMed] [Google Scholar]; Crowley J. D., Goldup S. M., Lee A.-L., Leigh D. A., McBurney R. T., Chem. Soc. Rev. 2009, 38, 1530–1541. [DOI] [PubMed] [Google Scholar]
- 17. E-Ar′C≡CCH=CHAr′ was quantitatively retained within the macrocyclic complex after heating at 85 °C in MeCN under CO (1 atm) for 16 h. Under equivalent conditions, E-PhC≡CCH=CHPh was liberated alongside concomitant formation of 4 (Scheme 3, Supporting Information).
- 18. Peris E., Crabtree R. H., Chem. Soc. Rev. 2018, 47, 1959–1968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary