

Abstract
The large majority of TACA‐based (TACA=Tumor‐Associated Carbohydrate Antigens) antitumor vaccines target only one carbohydrate antigen, thereby often resulting in the incomplete destruction of cancer cells. However, the morphological heterogeneity of the tumor glycocalix, which is in constant evolution during malignant transformation, is a crucial point to consider in the design of vaccine candidates. In this paper, an efficient synthetic strategy based on orthogonal chemoselective ligations to prepare fully synthetic glycosylated cyclopeptide scaffolds grafted with both Tn and TF antigen analogues is reported. To evaluate their ability to be recognized as tumor antigens, direct interaction ELISA assays have been performed with the anti‐Tn monoclonal antibody 9A7. Although both heterovalent structures showed binding capacities with 9A7, the presence of the second TF epitope did not interfere with the recognition of Tn except in one epitope arrangement. This heterovalent glycosylated structure thus represents an attractive epitope carrier to be further functionalized with T‐cell peptide epitopes.
Keywords: chemoselective ligation, heteroglycodendrimers, synthetic vaccine, TF antigen, Tn antigen
Introduction
Carbohydrate‐based vaccines have recently emerged as a promising approach in cancer immunotherapy. This major achievement is the result of decades of efforts during which immunologists, glycobiologists, and chemists have joined forces to design synthetic or semi‐synthetic structures to selectively eradicate malignant cells by the immune system.1, 2, 3, 4, 5 The identification of aberrant glycosylation patterns at the surface of cancer cells, namely TACAs (Tumor‐Associated Carbohydrate Antigens), which discriminate healthy and malignant tissues is indisputably the source of inspiration in this field.6, 7, 8, 9 The overexpression of these antigens as membrane glycoproteins or glycolipids has consequences in signal transduction and tumor cell metastasis10, 11 and often correlates to poor prognosis.12, 13 Although a small minority of cancer patients has shown a natural production of anti‐TACA antibodies, these glycans are rarely capable of inducing a robust immune response due to their intrinsic nature.14 As a matter of fact, some TACAs are also expressed at low levels in normal tissues, making them to be perceived as “self‐antigens” by the human immune system.15 Moreover, the nature of immune response against TACAs lacks the sustain of activated T‐helper cells, which promotes the proliferation and differentiation of B‐cells, leading to the production of high‐affinity IgG antibodies.16 To induce such a T‐cell‐dependent response, the design of carbohydrate‐based vaccines requires protein or peptide carriers containing known T‐cell epitopes.17, 18, 19, 20 Alternatively, a toll‐like receptor (TLR) ligand can be included in the vaccine formulation to promote antigen‐presenting cells (APC) maturation and subsequent cytokine release to enhance the potency of the antigen‐specific immune response.21
Over the last years, many advances have contributed to the development of TACA‐based conjugate vaccines.22, 23, 24, 25 First, the development of powerful chemical synthesis of oligosaccharides and chemoenzymatic strategies allowed for minimized batch‐to‐batch variations due to the heterogeneity of extracted glycans, to obtain higher quality carbohydrate epitopes.26, 27, 28 Secondly, it was clearly established that TACAs should preferentially be presented in a multivalent display to mimic the native structures expressed on tumors and promote the efficient delivery of the vaccine prototype to B‐cells by virtue of cell receptor cross‐linking and endocytosis.29, 30, 31 In addition, unnatural TACAs can be introduced in conjugated vaccines to increase their resistance towards endogenous glycosylhydrolases and the immunogenicity of the constructs.32, 33 In other studies, protein carriers34, 35 have been replaced with well‐defined, fully synthetic and non‐immunogenic scaffolds featuring clustered carbohydrate epitopes and immuno‐stimulant peptide sequences.36 The development of less toxic adjuvants and self‐adjuvanting multi‐component vaccines has also been investigated, resulting in remarkable immunological effects.37, 38, 39, 40, 41, 42, 43, 44, 45 Finally, when many structures only present the carbohydrate moiety of the TACA instead of the native glycopeptide antigen, more relevant results have been obtained compared to when the TACA are linked to an amino acid of the native peptide fragment.30, 46, 47, 48 However, despite these important improvements, no carbohydrate‐based antitumor synthetic vaccine has succeeded in clinical trials.49
One critical but underestimated aspect in the design of antitumoral vaccines is the morphological heterogeneity of the tumor glycocalix, which is in constant evolution during malignant transformation.50, 51, 52, 53, 54 The large majority of vaccines target only one carbohydrate antigen expressed on tumors, which results in the incomplete destruction of cancer cells and neglect a compelling population of transformed cells. Therefore, it appears essential to consider the heterogeneous expression of TACAs to activate different population of B‐cells and thus stimulate a multi‐faceted response against a large population of tumors present at different stage of the disease.55 Following this idea, the first semi‐synthetic vaccine candidates combining multiple TACAs have been pioneered by the Danishefsky group.56 It was demonstrated that when the highly immunogenic Keyhole Limpeth Hemocyanin (KLH) protein carrier is functionalized with different TACAs (i.e., Globo‐H, Ley, and Tn antigens), a similar antibody response was generated in murine models compared with co‐administration of the individual monomers. More interestingly, the antibodies raised against the multi‐antigenic construct showed equal or higher reactivity towards human cell lines expressing native antigen forms. The same group further extended this approach with an unimolecular pentavalent construct featuring Globo‐H, STn, Tn, GM2, and TF antigens (Figure 1 A).57 This second‐generation vaccine showed promising preclinical results, being capable of producing excellent IgG and IgM antibody titers against all five antigens in mice models, when administered in the presence of QS‐21 adjuvant. This vaccine prototype is currently under phase I clinical trial against ovarian cancer. Although this approach opens promising perspectives in immunotherapy and was recently used only in semi‐synthetic glycoprotein/glycolipid‐based approaches,30, 58 no fully synthetic vaccine prototype displaying different associations of TACAs have been described so far.
Figure 1.
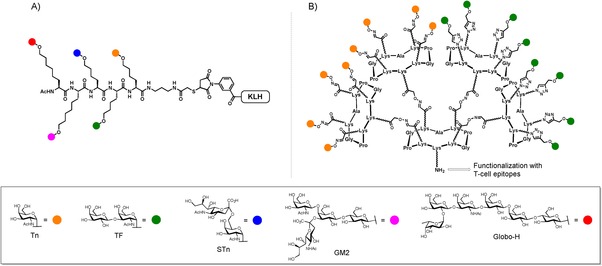
A) Unimolecular pentavalent construct by Danishefsky's group. B) Our multivalent Tn/TF‐based glycocluster.
Herein, we report an efficient synthetic strategy based on orthogonal chemoselective ligations to prepare vaccine candidates combining several carbohydrate antigens. For this purpose, we focused our attention on the preparation of glycosylated cyclopeptide scaffolds grafted with both Tn and TF antigen analogues, and dedicated to be further functionalized with T‐cell peptide epitopes (Figure 1 B).33, 41, 59, 60, 61, 62 We next evaluated through direct interaction ELISA assays the ability of our multivalent glycoclusters to interact with monoclonal anti‐Tn antibody to assess whether the presence of a second epitope (i.e., TF) could interfere with the specific recognition of the Tn moiety.
Results and Discussion
The development of efficient methodologies to prepare heterovalent glycostructures represents not only a synthetic challenge, but also an important progress to deepen the influence of glycoheterogeneity in interaction between carbohydrates and antibodies or lectins.63, 64 In the course of our recent studies in this field, we envisioned to build our multivalent Tn–TF‐based heterovalent glycodendrimers, that is, compounds 1 and 2 (Figure 2), by using two orthogonal reactions: oxime ligation (OL) and copper(I)‐catalyzed azide‐alkyne cycloaddition (CuAAC).65, 66, 67 Considering that our target molecules should contain carbohydrates in a separated (1) or shuffled (2) fashion, we have designed a divergent strategy that requires the synthesis of functionalized building blocks: i) Two “central” cyclopeptidic cores containing two aminooxy groups and two serine residues (3) on the upper domain of the scaffold, or four aldehyde groups (4); ii) Three additional cyclopeptidic “arms” bearing one aminooxy group on the lower domain and four serine residues on the upper domain (5), one aldehyde and four azido groups (6), or one aminooxy, two serines and two azides (7); iii) Aminooxy‐Tn (8) and propargyl‐TF (9) modified antigens (Figure 2).
Figure 2.
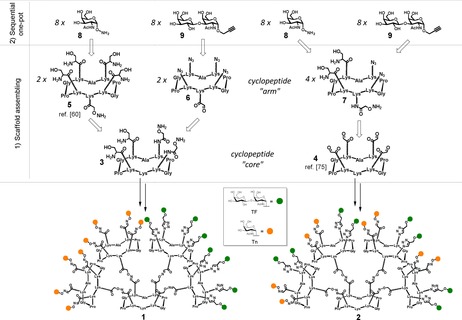
General divergent strategy for the synthesis of heteroglycoclusters 1 and 2.
Synthesis of carbohydrate antigens
The aminooxy‐Tn 8 has been prepared following the strategy reported by our group.68 The propargylated TF antigen 9 was synthesized as shown in Scheme 1. Reaction of 1,3,4,6‐tetra‐O‐acetyl‐2‐azido‐2‐deoxy‐β‐d‐galactopyranose 10 54 with propargyl alcohol yielded compound 11 69 as an inseparable anomeric mixture. Subsequent treatment with thioacetic acid in pyridine led to the desired 2‐acetamido derivative 12.70 Separation of the anomers could be achieved at this stage and compound 12 was then deacetylated and selectively protected on positions 4 and 6 to give the glycosyl acceptor 13. Trimethylsilyl trifluoromethanesulfonate‐promoted glycosylation with trichloroacetimidate 14 yielded disaccharide 15 that was fully deprotected to afford propargyl‐TF 9.
Scheme 1.
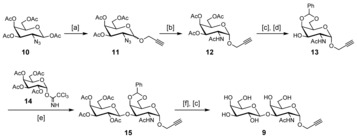
Synthesis of the propargylated TF antigen 9. Reagents and conditions: [a] Propargyl alcohol, BF3⋅Et2O, CH2Cl2, 40 °C, 8 h, 85 %; [b] AcSH, pyridine, r.t., overnight, 70 %; [c] MeONa/MeOH (pH 10), r.t., 2 h, 95 %; [d] PhC(OMe)2, camphorsulfonic acid (CSA), CH3CN, r.t., 72 h, 92 %; [e] Trimethylsilyl trifluoromethanesulfonate (TMSOTf), CH2Cl2, −15 °C, 30 min, 75 %; [f] 70 % AcOH/H2O, 60 °C, 4 h, 85 %.
Synthesis of cyclopeptidic “cores” and “arms”
The synthesis of compounds 1 and 2 is based on a divergent modular approach in which a central scaffold (“core”) has first to be functionalized with four “arms”, and successively with modified TACAs. The first step of the synthetic route involved the solid phase peptide synthesis of the orthogonally protected sequence A on the Fmoc‐Gly‐SASRINTM resin (Fmoc=9‐fluorenylmethoxycarbonyl), in which we introduced the dipeptide building block 16 71 to reduce the number of steps in solution. After cleavage from the resin support in mild acidic conditions and cyclization in diluted DMF/CH2Cl2, compound 17 was obtained with an overall yield of 54 %. Compound 17 underwent palladium‐catalyzed deprotection of allyloxycarbonyl (Alloc) groups; the two resulting free amino groups in the upper domain were reacted with (Boc‐aminooxy)acetic acid N‐hydroxysuccinimide ester72 to afford fully protected intermediate 18. The core scaffold 3 was obtained after treatment of 18 with TFA in the presence of a mixture of triisopropylsilane (TIS), water and hydroxylamine as scavengers (Scheme 2). The key intermediate 3 shows: (i) Two aminooxy groups, prone to react under oxime ligation conditions; (ii) Two serine residues as masked α‐oxo aldehyde residues; and (iii) A free amino group on the lysine side‐chain, on the lower domain.73
Scheme 2.
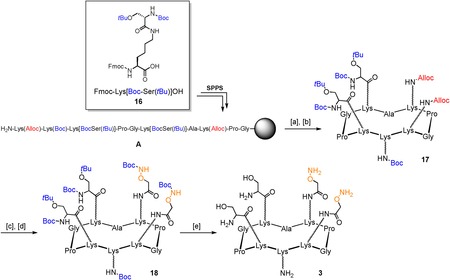
Synthesis of “core” cyclopeptide 3. Reagents and conditions: [a] 1 % TFA/ CH2Cl2, 10×10 min.; [b] PyBOP® (1.2 equiv), DIPEA (2.0 equiv), DMF/CH2Cl2 (1:1), 0.5 mm linear peptide, r.t., 30 min, 54 % overall; [c] Pd(PPh3)4 (0.3 equiv), PhSiH3 (25 equiv), dry DMF/CH2Cl2 (1:1), r.t., 30 min; [d] (Boc‐aminooxy)acetic acid N‐hydroxysuccinimide ester (1.2 equiv), DIPEA (1.5 equiv), dry DMF, r.t., 20 min, 72 % over two steps; [e] 94 % TFA, 2 % TIS, 2 % H2O, 2 % NH2OH, r.t., 3 h, 93 %.
The synthesis of the “right arm” 6 was performed starting from the synthesis of the sequence B, where 6‐azido‐N‐Fmoc‐norleucine 19 74 has been included. Cleavage from the resin and subsequent cyclization gave compound 20 with a 48 % overall yield. Removal of acid‐labile protecting groups of compound 20 afforded intermediate 21, presenting four azido groups on the upper domain and a serine residue in the lower domain. This last residue was treated with sodium periodate to give the α‐oxo aldehyde‐containing intermediate 6. In order to prevent the formation of undesired over‐oxidation side products, the reaction was stopped by RP‐HPLC purification after 40 minutes of reaction (Scheme 3).
Scheme 3.
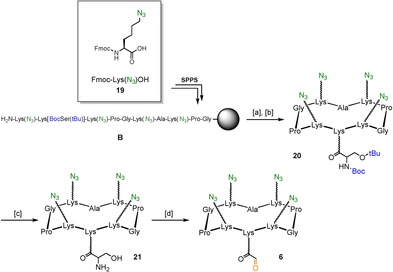
Synthesis of “right arm” cyclopeptide 6. Reagents and conditions: [a] 1 % TFA/ CH2Cl2, 10×10 min; [b] PyBOP® (1.2 equiv), DIPEA (2.0 equiv), DMF/CH2Cl2 (1:1), 0.5 mm linear peptide, r.t., 30 min, 48 % overall; [c] 96 % TFA, 2 % TIS, 2 % H2O, r.t., 3 h, 96 %; [d] NaIO4 (10 equiv), H2O, r.t., 40 min, 71 %.
Synthesis of cyclopeptidic “arms” of scaffold 2 started with compound 22, featuring two protected serine residues, two azido residues, and one amino residue protected with 1‐(4,4‐dimethyl‐2,6‐dioxocyclohex‐1‐ylidene)ethyl (Dde) group in the lower domain, which was obtained from the solid phase peptide synthesis of sequence C. Cleavage from the resin in mild acidic conditions, followed by cyclization afforded the desired compound with an overall yield of 55 %. Deprotection of Dde by means of a 2 % solution of hydrazine monohydrate in N,N‐Dimethylformamide (DMF) afforded compound 23, the free amino group was functionalized with (Boc‐Aminooxy)acetic acid N‐hydroxysuccinimide ester and the resulting crude treated with a strong acidic deprotection cocktail to simultaneously cleave Boc and tert‐butyl groups (Scheme 4). “Cyclopeptide arm” 7 was obtained in good yields (44 % overall) after RP‐HPLC purification.
Scheme 4.
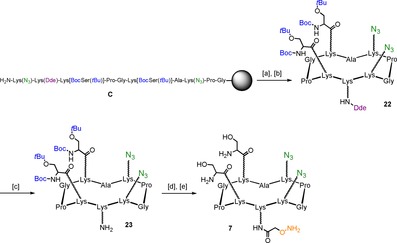
Synthesis of “arm” cyclopeptide 7. Reagents and conditions: [a] 1 % TFA/ CH2Cl2, 10×10 min; [b] PyBOP® (1.2 equiv), DIPEA (2.0 equiv), DMF/CH2Cl2 (1:1), 0.5 mm linear peptide, r.t., 30 min, 55 % overall; [c] 2 % N2H4/DMF, r.t., 20 min., 94 %; [d] (Boc‐aminooxy)acetic acid N‐hydroxysuccinimide ester (1.2 equiv), DIPEA (1.5 equiv), dry DMF, r.t., 20 min; [e] 94 % TFA, 2 % TIS, 2 % H2O, 2 % NH2OH, r.t., 3 h, 86 % over two steps.
Molecular assembly of the dendritic cores by OL
Conjugation between central scaffold 3 and “right arm” 6 was carried out via oxime ligation, in the presence of a 0.1 % TFA solution in H2O/CH3CN (1:1, pH 2.2) at 37 °C for 30 minutes, to afford compound 24 in excellent yield (75 %) and purity. Oxidative cleavage of serine residues of compound 24 by treatment with sodium periodate gave compound 25, which subsequently underwent oxime ligation with the “left arm” 5 60 to afford 26 in good yields (60 % over two steps). Compound 27 was obtained upon treatment of 26 with sodium periodate and subsequent direct purification by HPLC (Scheme 5). The resulting multivalent scaffold 27, featuring eight α‐oxo aldehyde and eight azido functionalities displayed in separated “arms”, was thus ready for the final conjugation step, featuring saccharide epitopes 8 and 9, respectively through both oxime ligation and copper(I)‐catalyzed azide‐alkyne cycloaddition.
Scheme 5.
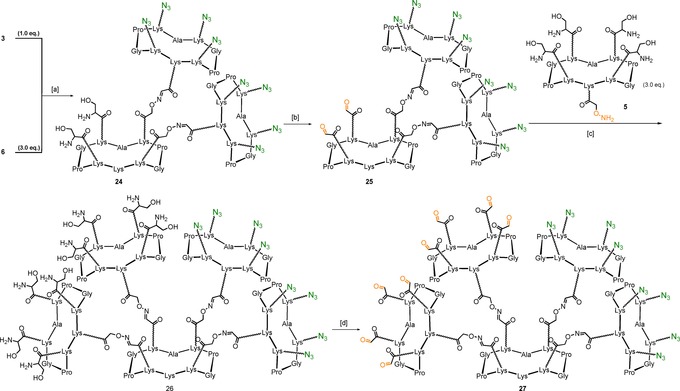
Synthesis of multivalent scaffold 27. Reagents and conditions: [a] 0.1 % TFA in H2O/CH3CN (1:1), 37 °C, 30 min, 75 %; [b] NaIO4 (20 equiv), H2O, r.t., 40 min, 70 %; [c] 0.1 % TFA in H2O/CH3CN (1:1), 37 °C, 30 min, 85 %; [d] NaIO4 (80 equiv), H2O, r.t., 40 min, 78 %.
For the synthesis of the “shuffled” heteroglycocluster 2, we envisaged a divergent strategy employing the “core” scaffold 4 75 displaying four α‐oxo aldehyde groups. This central unit was reacted with 6 equivalents of aminooxy‐containing “cyclopeptide arm” 7 under oxime ligation conditions to give compound 28. The subsequent oxidative cleavage of serine residues afforded the desired compound 29 in good yields (68 % over two steps). (Scheme 6)
Scheme 6.
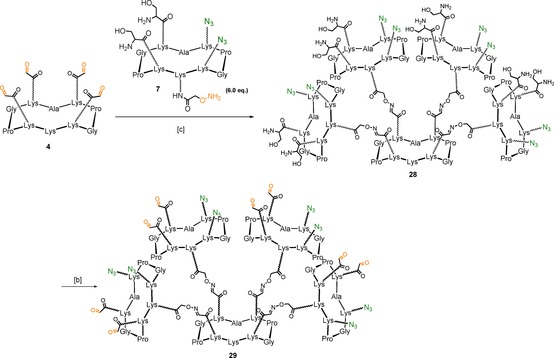
Synthesis of multivalent scaffold 29. Reagents and conditions: [a] 0.1 % TFA/H2O, 37 °C, 30 min, 87 %; [b] NaIO4 (80 equiv), H2O, r.t., 40 min, 78 %.
Functionalization of the dendrimers with TACAs by OL and CuAAC
With multivalent scaffolds 27 and 29 in hand, we proceeded with a sequential one‐pot strategy in which the first coupling involved the oxime ligation between aminooxy‐Tn 8 and the eight aldehyde residues displayed on the multivalent scaffolds. As described above, reaction conditions involved 1.5 equivalents of aminooxy‐saccharide per aldehyde, in a water/acetonitrile mixture containing 0.1 % of TFA. The reaction mixture was incubated at 37 °C and after 30 minutes LC‐MS analysis showed complete conversion in the octa‐Tn intermediate. Phosphate buffer solution (PBS, pH 7.4, 10 mm) was added to the reaction mixture in order to adjust the pH, then propargyl‐TF 9 was added to the crude, along with CuSO4, tris(3‐hydroxypropyltriazolylmethyl)amine) (THPTA) and sodium ascorbate (Scheme 7).76 RP‐UPLC‐MS analysis showed complete conversion after 1.5 hours reaction at room temperature (See the Supporting Information for chromatographic and ESI+ data).
Scheme 7.
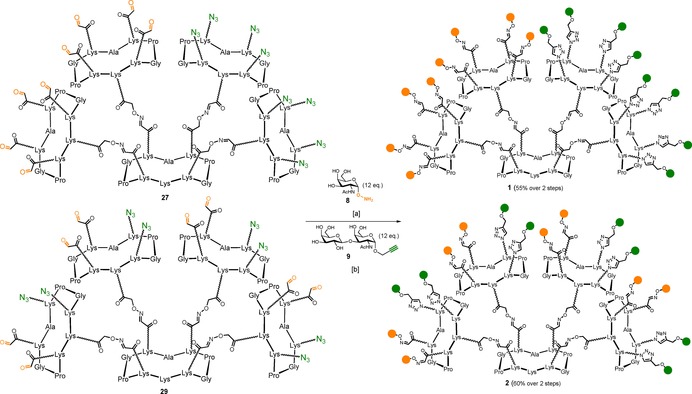
Synthesis of compounds 1 and 2. Reagents and conditions: [a] 0.1 % TFA in H2O/CH3CN, 37 °C, 30 min; [b] CuSO4 (1.5 equiv), Na ascorbate (15 equiv), THPTA (8 equiv), PBS (pH 7.4, 10 mm), r.t., 90 min.
Despite the structural complexity of these compounds, in addition to classic mass spectrometry and analytical chromatography, we have confirmed the correct functionalization of heteroglycoclusters 1 and 2 by 1H‐NMR. Characteristic signals for anomeric protons of Tn (≈5.6 ppm) and TF (≈5.0 ppm), oxime protons (≈7.8 ppm) and 1,4‐triazole protons (≈8.0 ppm) have been detected and integrated to show the expected ratio (Figure 3).
Figure 3.
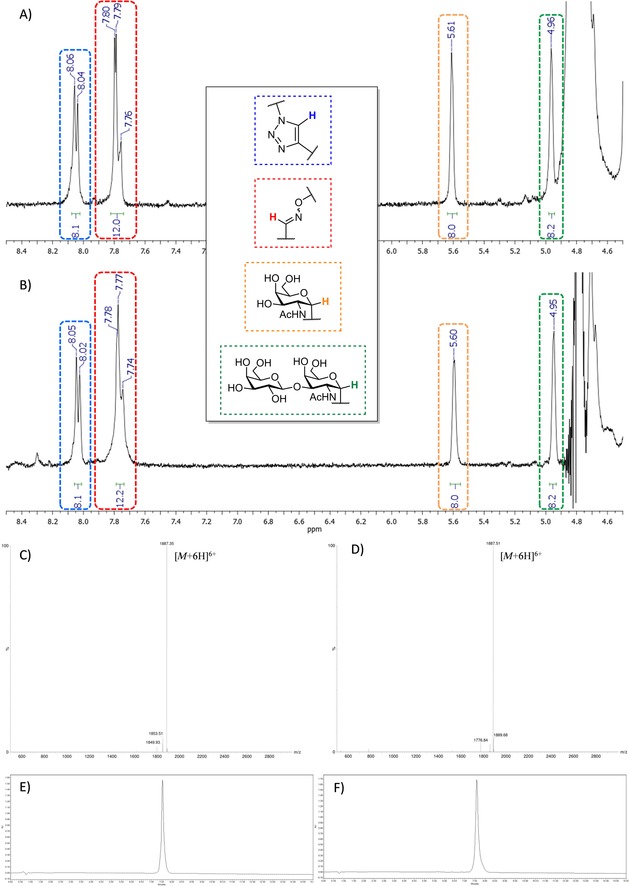
A) 1H NMR (D2O, 400 MHz) zoom of 1; B) 1H NMR (D2O, 400 MHz) zoom of 2; C) ESI+‐MS spectrum of 1; D) ESI+‐MS spectrum of 2; E) Analytical RP‐HPLC profile of 1; F) Analytical RP‐HPLC profile of 2.
ELISA direct interaction assays
Before undertaking immunological assays in mice, we decided to evaluate first the ability of our multivalent glycosylated scaffolds to interact with monoclonal anti‐Tn antibody. This represents an essential step to validate before going further in the synthesis of antitumor vaccines. Firstly because an efficient recognition of membrane‐bound antibodies is a prerequisite for the stimulation of B‐cells,77 and secondly because the presence of the TF epitope could interfere with the specific recognition of the Tn moiety. To perform this binding study with the heteroclusters 1 and 2, we have synthesized the hexadecavalent homocluster 30 (p. S29, Supporting Information), featuring sixteen copies of Tn antigen, the tetravalent homoclusters 32 and 33 (pp. S35 and S37, Supporting Information), grafted with N‐acetyl glucosamine, and the tetravalent heterocluster 31 (p. S32, Supporting Information), that will be used as controls (Figure 4 A).
Figure 4.
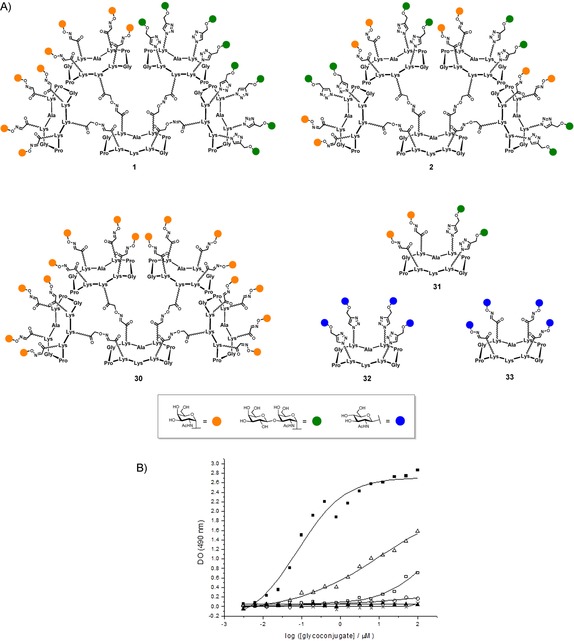
A) Compounds tested in ELISA assay; B) Interaction curves of the monoclonal antibody Anti‐Tn 9A7 with the glycoconjugates 30 (▪), 1 (▵), 2 (□), 31 (○), 32 (×), 33 (▴).
We have selected the monoclonal antibody 9A7, which has been shown to have good to excellent ability to recognize Tn‐positive human cancer cells such as MCF‐7, LS174T, and Jurkat.78 Each molecule have been coated at different concentrations into microtiter wells and their ability to be recognized by the anti‐Tn MAb (9A7) was studied. Compound concentration giving the same absorbance value at 490 nm was used as the indicator of the recognition ability by 9A7 (Figure 4 B). Although the best interaction was observed for the hexadecavalent homoglycocluster 30, hexadecavalent heteroglycoclusters 1 and 2 were both recognized by the anti‐Tn mAb, keeping in mind that 30 displays a 2‐fold increased Tn ratio compared to 1 and 2. More interesting was the high sensitivity of the anti‐Tn mAb to the epitope presentation. A significant difference was observed in the mAb recognition of 1 and 2 varying only by the TF and Tn epitopes distribution on the scaffold. Compound 1 showed a lower efficiency than 30, presumably because the number of Tn antigens (i.e., eight) is half lower than in the most active compound 30 (i.e., sixteen). By contrast, compound 2 which displays a similar number of Tn than 1 was found to be less efficient. These results clearly indicate that the TF epitope interfere with the Tn recognition by 9A7 only in compound 2. These results could be alternatively interpreted as a consequence of the differences in the spatial distribution and interspace distances between the Tn antigens in compounds 1 and 2. Although, the tetravalent heteroglycocluster 31 failed to show a significant interaction with 9A7, suggesting that only two copies of Tn is not sufficient for an efficient recognition. As expected, the tetravalent homoclusters 32 and 33 used as negative control did not show any signal, thus confirming the absence of unspecific interactions between 9A7 and similar carbohydrates, and/or the peptide scaffold itself.
Conclusion
To conclude, we report the first full synthesis of glycosylated scaffolds as carriers of cancer‐related antigens for vaccine design. By using OL and CuAAc ligations, we have prepared two hexadecavalent glycodendrimers with high yield and purity grafted with both Tn and TF antigen analogues in different topological arrangement. To evaluate their ability to be recognized as tumor antigens, direct interaction ELISA assays have been performed with the anti‐Tn monoclonal antibody 9A7. This study has highlighted that either the presence of the second TF epitope or the epitope distribution itself interfere with the recognition of Tn in compound 2 whereas no significant decrease of binding has been observed for compound 1. Considering that an efficient recognition by B‐cell antigen receptor (i.e., membrane‐bound antibody) is crucial for activation of the immune system,76 the identification of this heterovalent glycosylated structure represents a key step toward the design of synthetic vaccines. This epitope carrier will be functionalized with T‐cell peptide epitopes following a procedure described recently in our group.33 Results of immunological evaluation of the derived antitumoral vaccine candidate will be reported in a near future.
Experimental Section
General procedures
All chemical reagents were purchased from Aldrich (Saint Quentin Fallavier, France) or Acros (Noisy‐Le‐Grand, France). All protected amino acids and Fmoc‐Gly‐Sasrin® resin were obtained from Advanced ChemTech Europe (Brussels, Belgium). For peptides and glycopeptides, analytical RP‐HPLC was performed on a Waters system equipped with a Waters 2695 separations module and a Waters 2487 Dual Absorbance UV/Vis Detector. Analytical RP‐HPLC was carried out at 1.23 mL min−1 (Interchim UPTISPHERE X‐SERIE, C18, 5 μm, 125×3.0 mm) with UV monitoring at 214 nm by using a linear A–B gradient (buffer A: 0.09 % CF3CO2H in water; buffer B: 0.09 % CF3CO2H in 90 % acetonitrile). Purifications were carried out at 22.0 mL min−1 (VP 250×21 mm nucleosil 100‐7 C18) with UV monitoring at 214 and 250 nm by using a linear A–B gradient. Analytical RP‐UPLC was carried out at 0.6 mL min−1 (Phenomenex WIDEPORE XB‐ C18, 3.6 μm, 50×2.1 mm) with UV monitoring at 214 nm by using a linear C–D gradient (buffer C: 0.1 % CH2O2 in water; buffer D: 0.1 % CH2O2 in acetonitrile). For carbohydrate derivatives, moisture‐sensitive reactions were performed under an argon atmosphere by using oven‐dried glassware and reactions was monitored by using TLC with silica gel 60 F254 precoated plates (Merck). Spots were inspected by UV light and visualized by charring with 10 % H2SO4 in EtOH for carbohydrates. Silica gel 60 (0.063–0.2 mm or 70–230 mesh, Merck) was used for column chromatography. 1H and 13C NMR spectra were recorded on Bruker Avance 400 MHz or Brucker Avance III 500 MHz spectrometers and chemical shifts (δ) were reported in parts per million (ppm). Spectra were referenced to the residual proton solvent peaks relative to the signal of CDCl3 (δ=7.26 and 77.0 ppm for 1H and 13C) and D2O (4.79 ppm for 1H), assignments were performed by using GCOSY and GHMQC experiments. Standard abbreviations s, d, t, dd, br s, m refer to singlet, doublet, triplet, doublet of doublets, broad singlet, multiplet, respectively. The anomeric configuration was established from J 1,2 coupling constants. HRMS and ESI‐MS spectra of peptides and glycopeptides were measured on an Esquire 3000 spectrometer from Bruker or on an Acquity UPLC/MS system from Waters equipped with an SQ Detector 2. Bovine Serum Albumin (BSA), SIGMA FAST O‐phenylenediamine dihydrochloride (OPD), 96‐well microtiter Nunc‐Immuno plates (Maxi‐Sorp) and goat anti‐Mouse IgG‐HRP‐linked antibody were purchased from Sigma–Aldrich. Monoclonal antibody anti‐Tn (9A7) was kindly given by Prof. R. Lo‐Man (Institut Pasteur, Paris, France). The microtiter plate reader was a POLARstar Omega (BMG LABTECH).
Procedure for solid‐phase peptide synthesis
Assembly of protected linear peptide was performed manually or automatically (Syro II, Biotage) by employing solid‐phase peptide synthesis (SPPS) protocol using the Fmoc/tBu strategy and the Fmoc‐Gly‐Sasrin® resin (loading=0.7 mmol g−1). Coupling reactions were performed using, relative to the resin loading, 1.5–2 equiv of N‐Fmoc‐protected amino acid in situ activated with PyBOP (1.5–2 equiv) and N,N‐diisopropylethylamine (DIPEA, 3–4 equiv) in DMF (10 mL g−1 resin) for 30 min. Coupling reaction was checked by 2,4,6‐Trinitrobenzenesulfonic Acid (TNBS) test using a solution of 1 % trinitrobenzenesulfonic acid in DMF. N‐Fmoc protecting groups were removed by treatment with piperidine/DMF (1:4, 10 mL g−1 resin) for 10 min. The process was repeated three times and the resin was further washed five times with DMF (10 mL g−1 resin) for 1 min. The peptide was released from the resin by treating 10 times with a cleavage solution of TFA:CH2Cl2 (1:99). The combined cleavage fractions were concentrated under reduced pressure, ice‐cold Et2O was added to induce precipitation and the linear peptide was obtained as a white powder after filtration and used without any further purification.
Procedure for peptide cyclization
All linear peptides were dissolved in CH2Cl2 (0.45–0.50 mm) and the pH of the solution was adjusted to 9 by addition of DIPEA. PyBOP (1.2 equiv) was added and the reaction mixture was stirred at room temperature for 30 minutes. The solvent was removed under reduced pressure and precipitation from diethyl ether afforded desired cyclic peptides as white solids.
Compound 1
To a solution of 27 (10.2 mg, 1.6 μmol) in a H2O/CH3CN (1:1, 1.0 mL) mixture containing 0.09 % CF3CO2H, 8 (4.6 mg, 19.5 μmol) was added and the reaction heated at 37 °C without stirring. After 30 minutes, RP‐UPLC showed complete conversion to the octa‐Tn derivative: t R=2.37 min. (5–60 % solv.D in 3.0 min.) (Figure S1). ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C331H530N111O118 [M+5 H]5+: 1590.5, found: 1590.2; calcd for C331H529N111O118 [M+4 H]4+: 1987.9, found: 1987.7.
To this mixture, 9 (8.3 mg, 19.7 μmol) was added, then 1.0 mL of PBS buffer (pH 7.4, 10 mm) was added and the solution degassed by argon bubbling for 15 minutes. A separate solution, containing CuSO4 (0.5 mg, 2.0 μmol), THPTA (5.7 mg, 13.1 μmol) and sodium ascorbate (10.4 mg, 23.9 μmol) in previously degassed PBS buffer (1.0 mL, pH 7.4, 10 mm) was added to the reaction mixture. After 90 minutes stirring at room temperature, RP‐UPLC showed complete conversion to compound 1: t R=1.40 min. (5–60 % solv.D in 3.0 min.) (Figure S3). ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C467H747N119O206 [M+6 H]6+: 1887.4, found: 1887.4.
Chelex® resin was added to the reaction mixture and stirred for 30 minutes at room temperature in order to remove residues of copper. The crude was then purified by preparative RP‐HPLC and lyophilized to afford 10.2 mg of pure 1 (55 % yield). Analytical RP‐HPLC: t R=7.54 min. (0–40 % solv.B in 15 min.).
Compound 2
Compound 2 was obtained in 60 % yield (10.0 mg) by following the same procedure reported above, starting from 9.2 mg (1.5 μmol) of compound 29 (see the Supporting Information). ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C467H747N119O206 [M+6 H]6+: 1887.4, found: 1887.5. Analytical RP‐HPLC: t R=7.60 min. (0–40 % solv. B in 15 min).
Compound 3
A solution containing trifluoroacetic acid (TFA), triisopropylsilane (TIS), water and hydroxylamine (2.0 mL, 94:2:2:2) was added to 18 (22.1 mg, 11.3 μmol) and the mixture stirred at room temperature for 3 hours. After addition of ice‐cold Et2O (10 mL) and filtration of the precipitate, the crude product was purified via preparative RP‐HPLC to give, after lyophilization, compound 3 (14.1 mg) in 93 % yield. ESI+‐MS m/z (Monoisotopic MW) Elemental analysis calcd (%) for C57H102N19O18 [M+H]+: 1340.8, found: 1340.4; Elemental analysis calcd (%) for C57H103N19O18 [M+2 H]2+: 670.9, found: 670.5; C57H107N19O18 Na3 [M+6 H+3 Na]9+: 157.2, found: 157.0. Analytical RP‐HPLC: t R=4.00 min. (0–30 % solv. B in 15 min.).
Compound 6
To a solution of 21 (50.7 mg, 41.9 μmol) in H2O (2.0 mL), sodium periodate (89.6 mg, 419 μmol) was added and the reaction mixture stirred at room temperature for 30 minutes. Direct RP‐HPLC purification, followed by lyophilization afforded compound 6 (35.1 mg) in 71 % yield. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C49H78N23O12 [M+H]+: 1180.6200, found: 1180.6211; Elemental analysis calcd (%) for C49H79N23O13Na [M+H2O+Na]+: 1220.6125, found: 1220.6138. Analytical RP‐HPLC: t R=6.78 min. (5–100 % solv.B in 15 min.).
Compound 7
To a solution of anhydrous DMF (5 mL) containing DIPEA (13 μL, 74.6 μmol), compound 23 (69.4 mg, 44.5 μmol) and (Boc‐Aminooxy)acetic acid N‐hydroxysuccinimide ester56 (14.1 mg, 48.9 μmol) were added. After 20 minutes stirring at room temperature the solvent mixture was concentrated under vacuum and ice‐cold Et2O was added to induce precipitation. The resulting yellowish precipitate was filtered, dried and used for the next step without any further purification. The crude product was added of a cocktail containing TFA, TIS, water and hydroxylamine (5.0 mL, 94:2:2:2) and the reaction stirred at room temperature for 3 hours. After addition of ice‐cold Et2O (30 mL) and filtration of the precipitate, the crude product was purified via preparative RP‐HPLC to give, after lyophilization, compound 7 (50.5 mg) in 86 % yield over two steps. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C55H95N22O16 [M+H]+: 1319.7296, found: 1319.7302; Elemental analysis calcd (%) for C55H94N22O16Na [M+Na]+: 1341.7116, found: 1341.7123. Analytical RP‐HPLC: t R=3.52 min. (5–100 % solv. B in 15 min.).
Propargyl (β‐d‐galactopyranosyl)‐(1–3)‐2‐acetamido‐2‐deoxy‐α‐d‐galactopyranoside (9)
A solution of compound 15 (250 mg, 0.37 mmol) in 70 % aqueous acetic acid (2.5 mL) was stirred at 60 °C for 4 hours. The mixture was concentrated under reduced pressure and co‐evaporated with toluene. The residue was solubilized in dry methanol (5 mL) and a solution of sodium methoxide in methanol was added to reach approximately pH 9. The solution was stirred at room temperature overnight and neutralized with Amberlite IR‐120 H+. The resin was filtered off, washed with methanol and the filtrate was concentrated under reduced pressure to afford the title compound after precipitation (MeOH/CH2Cl2) as a white amorphous solid (147 mg, 0.35 mmol, 95 %); 1H NMR (400 MHz, CD3OD) δ=5.02 (d, 1 H, J=3.7 Hz, H‐1), 4.48 (dd, 1 H, J=3.7, 11.1 Hz, H‐2), 4.40 (d, 1 H, J=7.6 Hz, H‐1′), 4.31 (dd, 1 H, J=2.4, 15.9 Hz, CH2CCH), 4.25 (dd, 1 H, J=2.4, 15.9 Hz, CH2CCH), 4.17 (d, 1 H, J=2.2 Hz, H‐4′), 3.91 (dd, 1 H, 3.0, 11.1 Hz, H‐3), 3.85 (tap, 1 H, J=6.0 Hz, H‐5′), 3.81 (d, 1 H, J=2.9 Hz, H‐4), 3.75 (dd, 2 H, J=6.9, 11.3 Hz, H‐6a, H‐6′a), 3.70 (dd, 2 H, J=5.1, 11.4 Hz, H‐6b, H‐6′b), 3.50–3.56 (m, 2 H, H‐2′, H‐5′), 3.44 (dd, 1 H, J=3.2, 9.7 Hz, H‐3′), 2.85 (t, 1 H, J=2.3 Hz, CH2CCH), 1.97 ppm (s, 3 H, acetyl). Those data are in agreement with the literature.79
Propargyl 2‐acetamido‐4,6‐O‐benzylidene‐2‐deoxy‐α‐d‐galactopyranoside (13)
To a suspension of propargyl 2‐acetamido‐2‐deoxy‐α‐d‐galactopyranoside64 (1.285 g, 4.95 mmol, 1 equiv) and benzaldehyde dimethyl acetal (1.116 mL, 7.43 mmol, 1.5 equiv) in dry acetonitrile was added camphorsulfonic acid (115 mg, 0.49 mmol, 0.1 equiv). The mixture was stirred at room temperature for 48 hours. The reaction was quenched with triethylamine (0.3 mL) and concentrated under reduced pressure. The crude mixture was purified over silica gel chromatography (CH2Cl2/MeOH 98:2) to afford the title compound as a white amorphous solid (1.550 g, 4.46 mmol, 90 %); 1H NMR (400 MHz, [D6]acetone) δ=7.54–7.56 (m, 2 H, H‐Ar), 7.32–7.39 (m, 3 H, H‐Ar), 6.95 (d, 1 H, J=7.8 Hz, NH), 5.65 (s, 1 H, CHPh), 5.07 (d, 1 H, J=3.5 Hz, H‐1), 4.38 (tdap, 1 H, J=8.6, 3.5 Hz, H‐2), 4.30 (dap, 1 H, J=3.4 Hz, H‐4), 4.29 (dd, 1 H, J=16.0, 2.5 Hz, OCH 2CCH), 4.25 (dd, 1 H, J=16.0, 2.5 Hz, OCH 2CCH), 4.17 (dd, 1 H, J=12.4, 1.6 Hz, H‐6a), 4.13 (dd, 1 H, J=12.5, 1.7 Hz, H‐6b), 3.88 (ddd, 1 H, J=11.1, 9.2, 3.4 Hz, H‐3), 3.79 (dap, 1 H, J=1.1 Hz, H‐5), 3.75 (d, 1 H, J=9.2 Hz, OH‐3), 2.97 (t, 1 H, J=2.4 Hz, CH2CCH), 1.91 ppm (s, 3 H, acetyl); 13C NMR (100 MHz, [D6]acetone) δ=170.9 (C=O, acetyl), 139.9 (C‐Ar), 129.4 (CH‐Ar), 128.7 (2×CH‐Ar), 127.3 (2×CH‐Ar), 101.4 (CH‐Ph), 98.3 (C‐1), 80.3 (CH2 CCH), 76.9 (C‐4), 76.1 (CH2CCH), 69.8 (C‐6), 68.2 (C‐3), 64.4 (C‐5), 55.4 (CH2CCH), 51.1 (C‐2), 22.9 ppm (CH3, acetyl); HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C18H22NO6 [M+H] +: 348.1447, found: 348.1445.
Propargyl (2,3,4,6‐tetra‐O‐acetyl‐β‐d‐galactopyranosyl)‐(1–3)‐2‐acetamido‐4,6‐O‐benzilidene‐2‐deoxy‐α‐d‐galactopyranoside (15)
A solution of 13 (430 mg, 1.23 mmol, 1 equiv) and 2,3,4,6‐tetra‐O‐acetyl‐α‐d‐galactopyranosyl trichloroacetimidate (912 mg, 1.85 mmol, 1.5 equiv) in dry dichloromethane (10 mL) was stirred for 1 hour under argon at room temperature in the presence of activated 4 Å molecular sieve. trimethylsilyl trifluoromethanesulfonate (45 μl, 0.25 mmol, 0.2 equiv) was added at −15 °C and the reaction was stirred at −15 °C for 30 minutes, quenched with triethylamine (50 μl), filtered on a celite pad and concentrated under reduced pressure. The crude mixture was purified over silica gel chromatography (CH2Cl2/MeOH 98:2) to afford the title compound as a white amorphous solid (503 mg, 0.74 mmol, 60 %). 1H NMR (400 MHz, CDCl3) δ=7.51–7.54 (m, 2 H, H‐Ar), 7.32–7.38 (m, 3 H, H‐Ar), 5.64 (d, 1 H, J=9.1 Hz, NHAc), 5.54 (s, 1 H, CH‐Ph), 5.37 (d, 1 H, J=3.3 Hz, H‐4′), 5.18 (dd, 1 H, J=10.3, 7.9 Hz, H‐2′), 5.12 (d, 1 H, J=3.5 Hz, H‐1), 4.97 (dd, 1 H, J=10.3, 3.5 Hz, H‐3′), 4.75 (d, 1 H, J=7.9 Hz, H‐1′), 4.70 (ddd, 1 H, J=3.6, 9.1, 11.4 Hz, H‐2), 4.02–4.30 (m, 7 H, H‐4, 2×H‐6, 2×H‐6′, 2×CH2CCH), 3.95 (dd, 1 H, J=3.2, 11.2 Hz, H‐3), 3.89 (tap, 1 H, J=5.9 Hz, H‐5′), 3.69 (tap, 1 H, J=10.3 Hz, H‐5), 2.46 (t, 1 H, J=2.4 Hz, CH2CCH), 2.14, 2.04, 2.03, 1.99, 1.96 ppm (5 s, 15 H, 5×CH3, acetyl); 13C NMR (125 MHz, CDCl3) δ=170.7, 170.6, 170.4, 170.3, 170.2 (5×C, acetyl), 128.5 (C‐Ar), 128.3 (CH‐Ar), 126.7 (2×CH‐Ar), 126.6 (2×CH‐Ar), 100.8 (C‐1′), 97.0 (C‐1), 78.6 (CH2 CCH), 75.4 (CH2CCH), 72.7 (C‐5′), 71.0 (C‐3′), 70.9 (C‐5), 69.0 (C‐3), 68.6 (C‐2′), 68.0 (C‐4), 66.9 (C‐4′), 62.6 (C‐6), 61.3 (C‐6′), 55.3 (CH2CCH), 53.6 (C‐2), 23.6, 20.9, 20.9, 20.8, 20.7 ppm (5×CH3, acetyl); HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C32H40NO15 [M+H]+: 678.6577, found: 678.6570.
Compound 17
Starting from 200 mg of Fmoc‐Gly‐Sasrin® resin (loading=0.7 mmol g−1), linear sequence A was synthesized according to the procedure for solid‐phase peptide synthesis and cleaved to its resin support to give the corresponding linear peptide. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) calcd for C84H146N17O25 [M+H]+: 1794.0706, found: 1794.0641.
Following the procedure for peptide cyclization, cleaved sequence A gave compound 17 (134 mg) in 54 % overall yield after RP‐HPLC purification and lyophilization. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) calcd for C84H144N17O24 [M+H]+: 1776.0600, found: 1776.0605; Elemental analysis calcd (%) for C84H143N17O24Na [M+Na]+: 1797.0390, 1797.0406. Analytical RP‐HPLC: t R=9.62 min. (5–100 % solv. B in 15 min.).
Compound 18
To a solution of 17 (72.3 mg, 40.7 μmol) in a mixture of anhydrous DMF/CH2Cl2 (1:1, 10 mL), phenylsilane (250 μL, 2.0 mmol) and Pd(PPh3)4 (9.4 mg, 8.13 μmol) were added and the reaction stirred at room temperature for 30 minutes. MeOH (5 mL) was added to the mixture and the reaction stirred until production of CO2 ceased. The reaction mixture was concentrated under vacuum and ice‐cold Et2O (20 mL) was added to induce precipitation. The resulting yellowish precipitate was filtrated, dried, and used without any further purification for the subsequent step. To a solution of the crude mixture in anhydrous DMF (5 mL), DIPEA (22 μL, 126 μmol) and (Boc‐Aminooxy)acetic acid N‐hydroxysuccinimide ester71 (25.8 mg, 89.5 μmol) were added. After 20 minutes stirring at room temperature, the reaction mixture was directly purified via preparative RP‐HPLC and lyophilized to give compound 18 (57.2 mg) in 72 % yield over two steps. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C90H157N19O28Na [M+Na]+: 1975.1343, found: 1975.1371. Analytical RP‐HPLC: t R=9.72 min. (5–100 % solv. B in 15 min.).
Compound 20
Starting from 200 mg of Fmoc‐Gly‐Sasrin® resin (loading=0.7 mmol g−1), linear sequence B was synthesized according to the procedure for solid‐phase peptide synthesis and cleaved to its resin support to give the corresponding linear peptide. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C59H101N24O15 [M+H]+: 1385.7878, found: 1385.7856.
Following the procedure for peptide cyclization, cleaved sequence B gave compound 20 (91.9 mg) in 48 % overall yield after RP‐HPLC purification and lyophilization. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C59H99N24O14 [M+H]+: 1367.7773, found: 1367.7745; Elemental analysis calcd (%) for C59H98N24O14Na [M+Na]+: 1389.7592, found: 1389.7561. Analytical RP‐HPLC: t R=8.52 min. (5–100 % solv. B in 15 min.).
Compound 21
A solution containing trifluoroacetic acid (TFA), triisopropylsilane (TIS) and water (2.0 mL, 96:2:2) was added to 20 (78.5 mg, 57.4 μmol). After 3 hours stirring at room temperature the reaction mixture was added to ice‐cold Et2O (10 mL) and the resulting precipitate was filtrated and dried. After RP‐HPLC purification and lyophilization, compound 21 (64.7 mg) was obtained in 96 % yield. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C50H83N24O12 [M+H]+: 1211.6622, found: 1211.6602. Analytical RP‐HPLC: t R=6.59 min. (5–100 % solv. B in 15 min.).
Compound 22
Starting from 200 mg of Fmoc‐Gly‐Sasrin® resin (loading=0.7 mmol g−1), linear sequence C was synthesized according to the procedure for solid‐phase peptide synthesis and cleaved to its resin support to give the corresponding linear peptide. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) calcd for C81H138N21O21 [M+H]+: 1741.0376, found: 1741.0366; calcd for C81H137N21O21Na [M+Na]+: 1763.0196, found: 1763.0176. Following the procedure for peptide cyclization, cleaved sequence C gave compound 22 (132.7 mg) in 55 % overall yield after RP‐HPLC purification and lyophilization. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) calcd for C81H136N21O20 [M+H]+: 1724.0300, found: 1724.0282; Elemental analysis calcd (%) for C81H135N21O20Na [M+Na]+: 1745.0090, found: 1745.0078. Analytical RP‐HPLC: t R=9.60 min. (5–100 % solv. B in 15 min.).
Compound 23
To a 2 % solution of hydrazine monohydrate (5.0 mL), 22 (90.3 mg, 52.4 μmol) was added and the reaction stirred at room temperature for 20 minutes. The solvent mixture was concentrated under vacuum and directly purified via preparative RP‐HPLC to give, after lyophilization, compound 23 (76.8 mg) in 94 % yield. HRMS (ESI+‐TOF) m/z (Monoisotopic MW) Elemental analysis calcd (%) for C71H124N21O18 [M+H]+: 1558.9433, found: 1558.9458. Analytical RP‐HPLC: t R=8.65 min. (5–100 % solv. B in 15 min.).
Compound 24
To a mixture of H2O/CH3CN (1:1, 1.0 mL) containing 0.09 % CF3CO2H, 3 (12.1 mg, 9.0 μmol) and 6 (31.9 mg, 27.0 μmol) were added and the reaction mixture was heated at 37 °C for 30 minutes. RP‐HPLC purification, followed by lyophilization, afforded pure compound 24 (24.7 mg) in 75 % yield. ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C155H253N65O40 [M+2 H]2+: 1833.5, found: 1833.5; calcd for C155H254N65O40 [M+3 H]3+: 1222.7, found: 1222.6; Elemental analysis calcd (%) for C155H254N65O40 [M+3 H+Na]4+: 922.8, found: 922.8. Analytical RP‐HPLC: t R=8.17 min. (5–100 % solv. B in 15 min.).
Compound 25
To a solution of 24 (22.3 mg, 6.1 μmol) in in H2O (2.0 mL), sodium periodate (26.1 mg, 122 μmol) was added and the reaction mixture stirred at room temperature for 30 minutes. Direct RP‐HPLC purification, followed by lyophilization afforded compound 25 (16.5 mg) in 75 % yield. ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C153H246N63O42Na [M+H2O+H+Na]2+: 1831.5, found: 1831.5; Elemental analysis calcd (%) for C153H246N63O42Na2 [M+H2O+H+2 Na]3+: 1228.7, found: 1228.6; Elemental analysis calcd (%) for C153H246N63O42Na3 [M+H2O+H+3 Na]4+: 927.2, found: 927.2. Analytical RP‐HPLC: t R=8.49 min. (5–100 % solv. B in 15 min.).
Compound 26
To a mixture of H2O/CH3CN (1:1, 1.0 mL) containing 0.09 % CF3CO2H, 25 (16.0 mg, 4.4 μmol) and 5 45 (19.0 mg, 13.2 μmol) were added and the reaction mixture was heated at 37 °C for 30 minutes. RP‐HPLC purification, followed by lyophilization, afforded pure compound 26 (24.1 mg) in 85 % yield. ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C275H457N103O78 [M+4 H]4+:1613.6, found: 1613.5; calcd for C275H458N103O78 [M+5 H]5+: 1291.0, found: 1291.0; Elemental analysis calcd (%) for C275H459N103O78 [M+6 H]6+: 1076.0, found: 1076.1; Elemental analysis calcd (%) for C275H460N103O78 [M+7 H]7+: 922.5, found: 922.5. Analytical RP‐HPLC: t R=7.23 min. (5–100 % solv. B in 15 min.).
Compound 27
To a solution of 26 (23.6 mg, 3.7 μmol) in H2O (2.0 mL), sodium periodate (62.6 mg, 293 μmol) was added and the reaction mixture stirred at room temperature for 30 minutes. Direct RP‐HPLC purification, followed by lyophilization afforded pure compound 27 (17.7 mg, 78 % yield). ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C267H416N95O78 [M+3 H]3+: 2068.2, found: 2067.7; Elemental analysis calcd (%) for C267H417N95O78 [M+4 H]4+: 1551.4, found: 1551.1; Elemental analysis calcd (%) for C267H418N95O78 [M+5 H]5+: 1241.4, found: 1241.1. Analytical RP‐HPLC: t R=8.11 min. (5–100 % solv.B in 15 min.).
Compound 28
To a mixture of H2O/CH3CN (1:1, 1.0 mL) containing 0.09 % CF3CO2H, 4 59 (7.6 mg, 6.1 μmol) and 7 (48.1 mg, 36.4 μmol) were added and the reaction heated at 37 °C for 30 minutes. RP‐HPLC purification afforded, after lyophilization, pure compound 28 (34.2 mg) in 87 % yield. ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C275H457N103O78 [M+4 H]4+:1613.6, found: 1613.5; calcd for C275H458N103O78 [M+5 H]5+: 1291.0, found: 1291.1; Elemental analysis calcd (%) for C275H459N103O78 [M+6 H]6+: 1076.0, found: 1076.2. Analytical RP‐HPLC: t R=4.31 min. (5–100 % solv. B in 15 min.).
Compound 29
Compound 29 was obtained in 73 % yield (15.3 mg) by following the same procedure reported above, starting from 21.8 mg (3.4 μmol) of compound 28. ESI+‐MS m/z (Average MW) Elemental analysis calcd (%) for C267H416N95O78 [M+3 H]3+: 2068.3, found: 2068.5; calcd for C267H417N95O78 [M+4 H]4+: 1551.4, found: 1551.7; Elemental analysis calcd (%) for C267H418N95O78 [M+5 H]5+: 1241.4, found: 1241.6; C267H423N95O78 [M+10 H]10+: 621.2, found: 620.9. Analytical RP‐HPLC: t R=8.08 min. (5–100 % solv. B in 15 min.).
Enzyme‐linked immunosorbent assay
96‐well microtiter Nunc‐Immuno plates (Maxi‐Sorp) were coated with serial two‐fold dilutions of each glycoclusters in PBS buffer pH 7.4 (from 100 μm to 3 nm, 100 μL per well,) for 1 h at 37 °C. The wells were then washed with T‐PBS (3×100 μL per well, PBS pH 7.4 containing 0.05 % (v/v) Tween 20). This washing procedure was repeated after each incubation step. The coated microtiter plates were then blocked with BSA in PBS (3 % w/v, 1 h at 37 °C, 100 μL per well). Primary mouse anti‐Tn monoclonal antibody (9A7) was then added (100 μL per well) and plates were incubated for 1 h at 37 °C. The Anti‐Tn antibody interaction with the coated glycoclusters was revealed by using goat anti‐mouse IgG peroxidase conjugate 1:1000 (100 μL per well, incubation 1 h at 37 °C) and o‐phenyldiamine/H2O2 substrate (OPD 100 μL per well). The reaction was stopped after 10 min by adding H2SO4 (30 % v/v, 50 μL per well) and the absorbance was measured at 490 nm. Glycoclusters presenting four GlcNAc residues (32, 33) were used as control for specificity. The optical density (OD at 490 nm) was plotted against the logarithm of the concentration for each glycocluster. The sigmoidal curves were fitted using Origin v6.1 software.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was partly supported by CNRS, Université Grenoble Alpes, the “Communauté d'agglomération Grenoble‐Alpes Métropole” (Nanobio program), the French National Research Agency in the framework of the “Investissements d'avenir” program Glyco@Alps (ANR‐15‐IDEX‐02) and Labex ARCANE (ANR‐11‐LABX‐0003‐01). O.R. acknowledges the French National Research Agency (ANR‐12‐JS07‐0001‐01 “VacSyn”) and the European Research Council Consolidator Grant “LEGO” (647938). We thank Dr R. Lo‐Man for providing the anti‐Tn monoclonal antibody 9A7.
C. Pifferi, B. Thomas, D. Goyard, N. Berthet, O. Renaudet, Chem. Eur. J. 2017, 23, 16283.
References
- 1. Cipolla L., Peri F., Airoldi C., Anticancer Agents Med. Chem. 2008, 8, 92–121. [DOI] [PubMed] [Google Scholar]
- 2. Guo Z., Boons G.-J., in Carbohydrate-Based Vaccines and Immunotherapeutics (Ed.: B. Wang), 2009, pp. 1–416. [Google Scholar]
- 3. Astronomo R. D., Burton D. R., Nat. Rev. Drug Discovery 2010, 9, 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng D., Shaikh A. S., Wang F., ACS Chem. Biol. 2016, 11, 850–863. [DOI] [PubMed] [Google Scholar]
- 5. Gaidzik N., Westerlind U., Kunz H., Chem. Soc. Rev. 2013, 42, 4421–4442. [DOI] [PubMed] [Google Scholar]
- 6. Meezan E., Wu H. C., Black P. H., Robbins P. W., Biochemistry 1969, 8, 2518–2524. [DOI] [PubMed] [Google Scholar]
- 7. Hakomori S., Cancer Res. 1985, 45, 2405–2414. [PubMed] [Google Scholar]
- 8. Lloyd K. O., Semin. Cancer Biol. 1991, 2, 421–431. [PubMed] [Google Scholar]
- 9. Livingston P. O., Immunol. Rev. 1995, 145, 147–166. [DOI] [PubMed] [Google Scholar]
- 10. Fukuda M., Cancer Res. 1996, 56, 2237–2244. [PubMed] [Google Scholar]
- 11. Hakomori S., Zhang Y., Chem. Biol. 1997, 4, 97–104. [DOI] [PubMed] [Google Scholar]
- 12. Bresalier R. S., Niv Y., Byrd J. C., Duh Q. Y., Toribara N. W., Rockwell R. W., Dahiya R., Kim Y. S., J. Clin. Invest. 1991, 87, 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Werther J. L., Tatematsu M., Klein R., Kurihara M., Kumagai K., Llorens P., Guidugli N. J., Bodian C., Pertsemlidis D., Yamachika T., et al., Int. J. Cancer 1996, 69, 193–199. [DOI] [PubMed] [Google Scholar]
- 14. Livingston P. O., Wong G. Y., Adluri S., Tao Y., Padavan M., Parente R., Hanlon C., Calves M. J., Helling F., Ritter G., J. Clin. Oncol. 1994, 12, 1036–1044. [DOI] [PubMed] [Google Scholar]
- 15. Speiser D. E., Miranda R., Zakarian A., Bachmann M. F., Mckall-Faienza K., Odermatt B., Hanahan D., Zinkernagel R. M., Ohashi P. S., J. Exp. Med. 1997, 186, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kennedy R., Celis E., Immunol. Rev. 2008, 222, 129–144. [DOI] [PubMed] [Google Scholar]
- 17. Germain R. N., Margulies D. H., Annu. Rev. Immunol. 1993, 11, 403–450. [DOI] [PubMed] [Google Scholar]
- 18. Haurum J. S., Arsequell G., Lellouch A. C., Wong S. Y. C., Dwek R. A., McMichael A. J., Elliott T., J. Exp. Med. 1994, 180, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dudler T., Altmann F., Carballido J. M., Blaser K., Eur. J. Immunol. 1995, 25, 538–542. [DOI] [PubMed] [Google Scholar]
- 20. Danishefsky S. J., Allen J. R., Angew. Chem. Int. Ed. 2000, 39, 836–863; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 882–912. [Google Scholar]
- 21. Temizoz B., Kuroda E., Ishii K. J., Int. Immunol. 2016, 28, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fernandez E. M. S., Navo C. D., Martinez-Saez N., Goncalves-Pereira R., Somovilla V. J., Avenoza A., Busto J. H., Bernardes G. J. L., Jimenez-Oses G., Corzana F., et al., Org. Lett. 2016, 18, 3890–3893. [DOI] [PubMed] [Google Scholar]
- 23. Rojas-Ocáriz V., Companon I., Aydillo C., Castro-Lopez J., Jimenez-Barbero J., Hurtado-Guerrero R., Avenoza A., Zurbano M. M., Peregrina J. M., Busto J. H., et al., J. Org. Chem. 2016, 81, 5929–5941. [DOI] [PubMed] [Google Scholar]
- 24. Martínez-Sáez N., Castro-Lopez J., Valero-Gonzalez J., Madariaga D., Companon I., Somovilla V. J., Salvado M., Asensio J. L., Jimenez-Barbero J., Avenoza A., et al., Angew. Chem. Int. Ed. 2015, 54, 9830–9834; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9968–9972. [Google Scholar]
- 25. Madariaga D., Martinez-Saez N., Somovilla V. J., Coelho H., Valero-Gonzalez J., Castro-Lopez J., Asensio J. L., Jimenez-Barbero J., Busto J. H., Avenoza A., et al., ACS Chem. Biol. 2015, 10, 747–756. [DOI] [PubMed] [Google Scholar]
- 26. Seeberger P. H., Werz D. B., Nature 2007, 446, 1046–1051. [DOI] [PubMed] [Google Scholar]
- 27. Wilson R. M., Danishefsky S. J., J. Am. Chem. Soc. 2013, 135, 14462–14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Townsend Y. E. C. A., in Comprehensive Natural Products II, Chemistry and Biology, Vol. 1 (Eds.: L. Mander, H.-W. Liu), 2010, pp. 95–96. [Google Scholar]
- 29. Gaidzik N., Kaiser A., Kowalczyk D., Westerlind U., Gerlitzki B., Sinn H. P., Schmitt E., Kunz H., Angew. Chem. Int. Ed. 2011, 50, 9977–9981; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10153–10157. [Google Scholar]
- 30. Cai H., Sun Z., Chen M., Zhao Y., Kunz H., Li Y., Angew. Chem. Int. Ed. 2014, 53, 1699–1703; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1725–1729. [Google Scholar]
- 31. Kiessling L. L., Grim J. C., Chem. Soc. Rev. 2013, 42, 4476–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fiore M., Thomas B., Daskhan G. C., Renaudet O., in Carbohydrates Chemistry: State-of-the-Art and Challenges for Drug Development, (Ed.: L. Cipolla), 2015, chapter 14, pp. 357–378. [Google Scholar]
- 33. Richichi B., Thomas B., Fiore M., Bosco R., Qureshi H., Nativi C., Renaudet O., Ben Mohamed L., Angew. Chem. Int. Ed. 2014, 53, 11917–11920; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 12111–12114. [Google Scholar]
- 34. Palitzsch B., Gaidzik N., Stergiou N., Stahn S., Hartmann S., Gerlitzki B., Teusch N., Flemming P., Schmitt E., Kunz H., Angew. Chem. Int. Ed. 2016, 55, 2894–2898; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2944–2949. [Google Scholar]
- 35. Jeon I., Lee D., Krauss I. J., Danishefsky S. J., J. Am. Chem. Soc. 2009, 131, 14337–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lo-Man R., Bay S., Vichier-Guerre S., Deriaud E., Cantacuzene D., Leclerc C., Cancer Res. 1999, 59, 1520–1524. [PubMed] [Google Scholar]
- 37. Gungor B., Yagci F. C., Tincer G., Bayyurt B., Alpdundar E., Yildiz S., Ozcan M., Gursel I., Gursel M., Sci. Transl. Med. 2014, 6, 235ra61/1–235ra61/12, 12. [DOI] [PubMed] [Google Scholar]
- 38. Fernández-Tejada A., Chea E. K., George C., Pillarsetty N. V. K., Gardner J. R., Livingston P. O., Ragupathi G., Lewis J. S., Tan D. S., Gin D. Y., Nat. Chem. 2014, 6, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Di Pasquale A., Preiss S., Tavares Da Silva F., Garcon N., Vaccine 2015, 3, 320–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buskas T., Ingale S., Boons G.-J., Angew. Chem. Int. Ed. 2005, 44, 5985–5988; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 6139–6142. [Google Scholar]
- 41. Bettahi I., Dasgupta G., Renaudet O., Chentoufi A. A., Zhang X., Carpenter D., Yoon S., Dumy P., BenMohamed L., Cancer Immunol. Immunother. 2009, 58, 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sarkar S., Lombardo S. A., Herner D. N., Talan R. S., Wall K. A., Sucheck S. J., J. Am. Chem. Soc. 2010, 132, 17236–17246. [DOI] [PubMed] [Google Scholar]
- 43. Wilkinson B. L., Day S., Malins L. R., Apostolopoulos V., Payne R. J., Angew. Chem. Int. Ed. 2011, 50, 1635–1639; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1673–1677. [Google Scholar]
- 44. Abdel-Aal A.-B. M., El-Naggar D., Zaman M., Batzloff M., Toth I., J. Med. Chem. 2012, 55, 6968–6974. [DOI] [PubMed] [Google Scholar]
- 45. Yin X.-G., Chen X.-Z., Sun W.-M., Geng X.-S., Zhang X.-K., Wang J., Ji P.-P., Zhou Z.-Y., Baek D. J., Yang G.-F., et al., Org. Lett. 2017, 19, 456–459. [DOI] [PubMed] [Google Scholar]
- 46. Ingale S., Wolfert M. A., Gaekwad J., Buskas T., Boons G.-J., Nat. Chem. Biol. 2007, 3, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cai H., Degliangeli F., Palitzsch B., Gerlitzki B., Kunz H., Schmitt E., Fiammengo R., Westerlind U., Bioorg. Med. Chem. 2016, 24, 1132–1135. [DOI] [PubMed] [Google Scholar]
- 48. Glaffig M., Palitzsch B., Hartmann S., Schüll C., Nuhn L., Gerlitzki B., Schmitt E., Frey H., Kunz H., Chem. Eur. J. 2014, 20, 4232–4236. [DOI] [PubMed] [Google Scholar]
- 49. Miles D., Roche H., Martin M., Perren T. J., Cameron D. A., Glaspy J., Dodwell D., Parker J., Mayordomo J., Tres A., et al., Oncologist 2011, 16, 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang S., Cordon-Cardo C., Zhang H. S., Reuter V. E., Adluri S., Hamilton W. B., Lloyd K. O., Livingston P. O., Int. J. Cancer 1997, 73, 42–49. [DOI] [PubMed] [Google Scholar]
- 51. Zhang S., Zhang H. S., Cordon-Cardo C., Reuter V. E., Singhal A. K., Lloyd K. O., Livingston P. O., Int. J. Cancer 1997, 73, 50–56. [DOI] [PubMed] [Google Scholar]
- 52. Zhang S., Zhang H. S., Reuter V. E., Slovin S. F., Scher H. I., Livingston P. O., Clin. Cancer Res. 1998, 4, 295–302. [PubMed] [Google Scholar]
- 53. Livingston P., Clin. Cancer Res. 2001, 7, 1837–1838. [PubMed] [Google Scholar]
- 54. Dube D. H., Bertozzi C. R., Nat. Rev. Drug Discovery 2005, 4, 477–488. [DOI] [PubMed] [Google Scholar]
- 55. Ortiz Mellet C., Nierengarten J.-F., Garcia Fernandez J. M., J. Mater. Chem. B 2017, 5, 6428–6436. [DOI] [PubMed] [Google Scholar]
- 56. Ragupathi G., Coltart D. M., Williams L. J., Koide F., Kagan E., Allen J., Harris C., Glunz P. W., Livingston P. O., Danishefsky S. J., Proc. Natl. Acad. Sci. USA 2002, 99, 13699–13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhu J., Wan Q., Lee D., Yang G., Spassova M. K., Ouerfelli O., Ragupathi G., Damani P., Livingston P. O., Danishefsky S. J., J. Am. Chem. Soc. 2009, 131, 9298–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pett C., Cai H., Liu J., Palitzsch B., Schorlemer M., Hartmann S., Stergiou N., Lu M., Kunz H., Schmitt E., Westerlind U., Chem. Eur. J. 2017, 23, 4018. [DOI] [PubMed] [Google Scholar]
- 59. Pifferi C., Berthet N., Renaudet O., Biomater. Sci. 2017, 5, 953–965. [DOI] [PubMed] [Google Scholar]
- 60. Grigalevicius S., Chierici S., Renaudet O., Lo-Man R., Deriaud E., Leclerc C., Dumy P., Bioconjugate Chem. 2005, 16, 1149–1159. [DOI] [PubMed] [Google Scholar]
- 61. Renaudet O., BenMohamed L., Dasgupta G., Bettahi I., Dumy P., ChemMedChem 2008, 3, 737–741. [DOI] [PubMed] [Google Scholar]
- 62. Renaudet O., Dasgupta G., Bettahi I., Shi A., Nesburn A. B., Dumy P., BenMohamed L., PLoS One 2010, 5, e11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jiménez Blanco J. L., Ortiz Mellet C., García Fernández J. M., Chem. Soc. Rev. 2013, 42, 4518–4531. [DOI] [PubMed] [Google Scholar]
- 64. García-Moreno M. I., Ortega-Caballero F., Risquez-Cuadro R., Ortiz Mellet C., Garcia Fernandez J. M., Chem. Eur. J. 2017, 23, 6295–6304. [DOI] [PubMed] [Google Scholar]
- 65. Galibert M., Renaudet O., Dumy P., Boturyn D., Angew. Chem. Int. Ed. 2011, 50, 1901–1904; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1941–1944. [Google Scholar]
- 66. Berthet N., Thomas B., Bossu I., Dufour E., Gillon E., Garcia J., Spinelli N., Imberty A., Dumy P., Renaudet O., Bioconjugate Chem. 2013, 24, 1598–1611. [DOI] [PubMed] [Google Scholar]
- 67. Thomas B., Fiore M., Daskhan G. C., Spinelli N., Renaudet O., Chem. Commun. 2015, 51, 5436–5439. [DOI] [PubMed] [Google Scholar]
- 68. Renaudet O., Dumy P., Org. Biomol. Chem. 2006, 4, 2628–2636. [DOI] [PubMed] [Google Scholar]
- 69. Goddard-Borger E. D., Stick R. V., Org. Lett. 2007, 9, 3797–3800. [DOI] [PubMed] [Google Scholar]
- 70. Thomas B., Pifferi C., Daskhan G. C., Fiore M., Berthet N., Renaudet O., Org. Biomol. Chem. 2015, 13, 11529–11538. [DOI] [PubMed] [Google Scholar]
- 71. Foillard S., Ohsten Rasmussen M., Razkin J., Boturyn D., Dumy P., J. Org. Chem. 2008, 73, 983–991. [DOI] [PubMed] [Google Scholar]
- 72. Lelle M., Peneva K., Amino Acids 2014, 46, 1243–1251. [DOI] [PubMed] [Google Scholar]
- 73. El-Mahdi O., Melnyk O., Bioconjugate Chem. 2013, 24, 735–765. [DOI] [PubMed] [Google Scholar]
- 74. Byrne C., McEwan P. A., Emsley J., Fischer P. M., Chan W.-C., Chem. Commun. 2011, 47, 2589–2591. [DOI] [PubMed] [Google Scholar]
- 75. Singh Y., Renaudet O., Defrancq E., Dumy P., Org. Lett. 2005, 7, 1359–1362. [DOI] [PubMed] [Google Scholar]
- 76. Hong V., Presolski S. I., Ma C., Finn M. G., Angew. Chem. Int. Ed. 2009, 48, 9879–9883; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 10063–10067. [Google Scholar]
- 77. Kiessling L. L., Gestwicki J. E., Strong L. E., Angew. Chem. Int. Ed. 2006, 45, 2348–2368; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2408–2429. [Google Scholar]
- 78. Mazal D., Lo-Man R., Bay S., Pritsch O., Dériaud E., Ganneau C., Medeiros A., Ubillos L., Obal G., Berois N., et al., Cancer Immunol. Immunother. 2013, 62, 1107–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nagel L., Budke C., Erdmann R. S., Dreyer A., Wennemers H., Koop T., Sewald N., Chem. Eur. J. 2012, 18, 12783–12793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary