

ABSTRACT
Nephronophthisis is an autosomal recessive cystic kidney disease and one of the most common genetic disorders causing end‐stage renal disease in children. Nephronophthisis is a genetically heterogenous disorder with more than 25 identified genes. In 10%–20% of cases, there are additional features of a ciliopathy syndrome, such as retinal defects, liver fibrosis, skeletal abnormalities, and brain developmental disorders. This review provides an update of the recent advances in the clinical features and related gene mutations of nephronophthisis, and novel approaches for therapy in nephronophthisis patients may be needed.
Keywords: cystic kidney disease, nephronophthisis, renal ciliopathy
Summary at a Glance
Nephronophthisis (NPHP) is a renal ciliopathy affecting children and young adults. This review gives an update on the recent advances in the clinical features and related gene mutations of NPHP.
Nephronophthisis (NPHP), the most common monogenic cause of end‐stage renal disease (ESRD) during the first three decades of life, is responsible for 2.4%–15% of ESRD in this population. The estimated incidence varies from 1:50 000 live births in Finland to 1:1 000 000 in the United States.1 It is caused by mutations in many genes that encode nephrocystin protein, which is involved in the function of primary cilia, basal bodies, and centrosomes. These mutations result in renal disease and extra‐renal manifestations.2 This review provides an update about the recent advances in the field of NPHP.
CLINICAL MANIFESTATIONS OF NPHP
Nephronophthisis is characterized by reduced ability of the kidneys to concentrate solutes, chronic tubulointerstitial nephritis, cystic renal disease, and progression to ESRD before age 30. The typical clinical symptoms of NPHP include polyuria, polydipsia with regular fluid intake at night, secondary enuresis, anaemia, and growth retardation. Patients with NPHP typically have a “bland” urinalysis without evidence of proteinuria, hematuria, or cellular elements until the late stage, when proteinuria may develop into secondary glomerulosclerosis.
Clinically, three clinical subtypes of NPHP have been recognized based on the median age of onset of ESRD: infantile, juvenile, and adolescent/adult.3 The main characteristics of these three subtypes of NPHP are summarized in Table 1. However, there have been several case reports of patients with NPHP who progressed to ESRD between the ages of 27 and 56 years.4, 5, 6 These cases of NPHP extend the age of ESRD from birth to the sixth decade of life.
Table 1.
Main features of three clinical subtypes of nephronophthisis (NPHP)
| Item | Infantile NPHP | Juvenile NPHP | Adolescent/adult NPHP |
|---|---|---|---|
| Onset of ESRD (median in years) | 1 year | 13 years | 19 years |
| Clinical manifestations | Oligohydramnios sequence in utero (limb contractures, pulmonary hypoplasia, and facial dysmorphisms), severe renal failure in the first years of life, severe hypertension | Impaired urinary concentrating ability (polyuria and polydipsia), impaired sodium reabsorption (hypovolaemia, hyponatraemia, chronic kidney disease (severe anaemia, growth retardation), proteinuria (late stage), normal blood pressure | Similar to juvenile NPHP |
| Renal ultrasound | Enlarged kidneys, large cortical microcysts, absent medullary cysts | Normal‐sized or smaller hyperechogenic kidneys with corticomedullary cysts and poor corticomedullary differentiation | Similar to juvenile NPHP |
| Renal histology | Tubular atrophy, usually lack tubular basement membrane change, interstitial fibrosis, collecting tubule cystic dilatation, enlarged kidneys | Tubular atrophy, tubular basement membrane disruption, cysts at the corticomedullary border, diffuse interstitial fibrosis with chronic inflammation | Similar to juvenile NPHP |
| Extra‐renal association | Liver fibrosis, severe cardiac valve or septal defects, recurrent bronchial infections | Retinal degeneration, cerebellar vermis aplasia, gaze palsy, liver fibrosis, skeletal defects | Similar to juvenile NPHP |
| Typical gene | NPHP2/INVS, NPHP3, NPHP12/TTC21B/JBTS11, NPHP14 /ZNF423, NPHP18 /CEP83 | All NPHP genes except NPHP2/INVS | NPHP3, NPHP4, NPHP9/NEK8 |
Extra‐renal manifestations occur in approximately 10%–20% of patients, including retinitis pigmentosa,7 skeletal defects,8 hepatic fibrosis,9 neurologic abnormalities,10 and cardiac defects.11 NPHP is also a major clinical finding in several syndromes, including Senior‐Loken, Joubert, Meckel‐Gruber, Cogan, and Sensenbrenner syndromes, and asphyxiating thoracic dystrophy (ATD, also known as Jeune syndrome). A summary of the main extra‐renal manifestations associated with NPHP is described in Table 2.
Table 2.
Extra‐renal manifestations associated with nephronophthisis (NPHP)
| Involved organ | Manifestations | Associated syndrome |
|---|---|---|
| Eye | Retinitis pigmentosa Alstrom |
Senior‐Løken syndrome (retinitis pigmentosa) Arima syndrome (cerebro‐oculo‐hepato‐renal syndrome) Alstrom syndrome (early cone‐rod retinal dystrophy and blindness, hearing loss, childhood obesity, type 2 diabetes mellitus, cardiomyopathy, fibrosis, and multiple organ failure) RHYNS (retinitis pigmentosa, hypopituitarism, skeletal dysplasia) Joubert syndrome (cerebellar vermis hypoplasia) |
| Oculomotor apraxia | Cogan syndrome (congenital oculomotor apraxia (absence or impairment of controlled, voluntary, and retinitis pigmentosa horizontal eye movement) Joubert syndrome |
|
| Nystagmus | Joubert syndrome | |
| Coloboma | Joubert syndrome COACH (cerebellar vermis hypo/aplasia, oligophrenia, ataxia, ocular coloboma and hepatic fibrosis) |
|
| Central nervous system | Encephalocele | Meckel‐Gruber syndrome (central nervous system malformation, bilateral renal cystic dysplasia, cleft palate, polydactyly, ductal proliferation in the portal area of the liver, pulmonary hypoplasia, and situs inversus) |
| Vermis aplasia | Joubert syndrome COACH |
|
| Hypopituitarism | RHYNS | |
| Liver | Liver fibrosis | Boichis syndrome (progressive kidney dysfunction, liver fibrosis, excessive urination, excessive thirst, failure to thrive, retarded growth, progressive kidney insufficiency, anaemia, metabolic acidosis, weakness) Meckel‐Gruber syndrome Arima syndrome Joubert syndrome COACH |
| Bone | Cone‐shaped epiphysis | Mainzer‐Saldino syndrome (phalangeal cone‐shaped epiphyses, chronic renal failure, and early‐onset, severe retinal dystrophy) |
| Polydactyly | Joubert syndrome Meckel‐Gruber syndrome Bardet‐Biedl syndrome (retinitis pigmentosa, obesity, deafness) Ellis van Creveld syndrome (short limbs, short ribs, postaxial polydactyly, dysplastic nails and teeth) Jeune syndrome (asphyxiating thoracic dystrophy) Sensenbrenner syndrome (craniosynostosis, short limbs, brachydactyly, narrow thorax, and facial anomalies) |
|
| Skeletal abnormalities | Sensenbrenner syndrome Ellis van Creveld syndrome |
|
| Heart | Cardiac malformation | |
| Situs inversus | Meckel‐Gruber syndrome | |
| Lung | Bronchiectasis | |
| Other | Ulcerative colitis |
GENOTYPE–PHENOTYPE CORRELATION OF NPHP
To date, more than 25 different genes have been found to be associated with NPHP (Table 3).2, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51 Mutations in the NPHP1 gene are the most common, being reported in approximately 20% of cases. Each of the remaining NPHP genes probably account for 1% or fewer of all cases of NPHP, and around two‐thirds of cases remain genetically unknown.41
Table 3.
Genes mutated in isolated nephronophthisis (NPHP)‐ and NPHP‐associated syndromes
| Gene | Chromosome | Protein | Location | Interaction partners | Functionary mechanism | Disorders associated with mutations | Reference |
|---|---|---|---|---|---|---|---|
| NPHP1 | 2q12.3 | Nephrocystin‐1 | Adherens junction, focal adhesion, transition zone | Inversin, nephrocystin‐3, nephrocystin‐4, filamin A and B, tensin, β‐tubulin, PTK2B, p130 Cas, focal adhesion kinase 2 | Maintains the cellular scaffolding or cytoskeleton, role in cell–cell adhesion and cell signalling | NPHP, SLSN, JBTS | 12 |
| NPHP2/INVS | 9q21‐22 | Inversin | Inversin compartment | Nephrocystin‐1, nephrocystin‐3, calmodulin, catenins, β‐tubulin, APC2 | Acts in Wnt pathway and planar cell polarity | Infantile NPHP, SLSN, Situs inversus, congenital heart defects | 13 |
| NPHP3 | 3q22.1 | Nephrocystin‐3 | Inversin compartment, axoneme | Nephrocystin‐1, inversin, NEK8, ANKS6, PTK2B, BCAR1 | Inhibits Wnt pathway | NPHP, liver fibrosis, RP, Situs inversus, MKS, SLSN, congenital heart defects | 14, 15, 16 |
| NPHP4 | 1p36.31 | Nephrocystin‐4 | Transition zone | Nephrocystin‐1, BCAR1, PTK2B, p130Cas, filamin, tensin | Inhibits Wnt and Hippo pathways | Juvenile NPHP, RP, OMA, SLSN, liver fibrosis | 17 |
| NPHP5/IQCB1 | 3q13.33 | Nephrocystin‐5/IQ motif containing B1 | Transition zone, basal body | Calmodulin, RPGR, nephrocystin‐1, nehrocystin‐4, nephrocystin‐6 | Forms complexes with RPGR | Juvenile NPHP, early‐onset RP, LCA | 18 |
| NPHP6/CEP290 | 12q21.32 | Nephrocystin‐6/centrosomal protein 290 | Transition zone, centrosome | ATF4, nephrocystin‐5, CC2D2A, TMEM67 | Regulates activity of transcription factor ATF4/CREB2, role in cAMP‐dependent renal cyst formation, cell signalling, DNA damage response (DDR), and renal cystogenesis | NPHP, RP, LCA, JBTS, MKS | 19, 20, 21, 22, 23 |
| NPHP7/GLIS2 | 16p13.3 | Nephrocystin‐7/ GLI similar 2 | Nucleus | N/A | Regulates Hedgehog signalling | NPHP | 24, 25 |
| NPHP8/RPGRIP1L/MKS5 | 16q12.2 | Nephrocystin‐8/ RPGRIP1‐like | Transition zone | Nephrocystin‐1, nephrocystin‐4 | Involved in Shh signalling | Juvenile NPHP, JBTS, MKS, RP, LCA, COACH | 26 |
| NPHP9/NEK8 | 17q11.2 | Nephrocystin‐9/ NIMA‐related kinase 8 | Inversin compartment | ANKS6 | Regulates cell cycle, involved in Hippo and DDR signalling | Infantile NPHP | 27, 28 |
| NPHP10/SDCCAG8/SLSN7 | 1q43‐q44 | Nephrocystin‐10/ Serologically defined colon cancer antigen 8 | Basal body | Nephrocystin‐5, OFD1 | Involved in DDR signalling | Juvenile NPHP, RP, SLSN, BBS | 29, 30 |
| NPHP11/ TMEM67/MKS3 | 8q22.1 | Nephrocystin‐11/ Transmembrane protein 67 | Transition zone | Nephrocystin‐1, nephrocystin‐4, nephrocystin‐6, CEP290, MKS1, TMEM216, nesprin‐2 | Maintains cellular structure and mitigates centrosome migration | NPHP, JBTS, MKS, liver fibrosis, COACH | 31, 32 |
| NPHP12/TTC21B/JBTS11 | 2q24.3 | Nephrocystin‐12/ Intraflagellar transport protein 139 | IFT‐A | Ciliopathy modifier | Regulates retrograde trafficking in the primary cilium, regulates Hedgehog signalling | Juvenile NPHP, JS, MKS, JBTS | 33 |
| NPHP13/WDR19 | 4p14 | Nephrocystin‐13/ WD repeat domain 19/IFT protein 144 | IFT‐A | N/A | Participates in retrograde IFT; acts in ciliogenesis | NPHP, JS, RP, Caroli, Sensenbrenner syndrome | 34, 35 |
| NPHP14 /ZNF423 | 16q12.1 | Nephrocystin‐14/ Zinc finger protein 423 | Nucleus | DDR protein PARP1, nephrocystin‐6 | Involved in DDR signalling | Infantile NPHP, JBTS, Situs inversus | 36 |
| NPHP15/ CEP164 | 11q23.3 | Nephrocystin‐15 centrosomal protein 164 | Basal body | Nephrocystin‐3, nephrocystin‐4, TTBK2, ATRIP, CCDC92, CEP83, Dvl3 | Involved in DDR signalling, regulates ciliogenesis | NPHP, liver fibrosis, RP, JBTS | 37 |
| NPHP16/ANKS6 | 9q22.33 | Nephrocystin‐16/ANKS6 | Axoneme, inversin compartment | INVS, nephrocystin‐3, NEK8, ANKS3, NEK7, BICC1, HIF1AN | Connects key components of NEK8, INVS, and NPHP3 | NPHP, liver fibrosis, Situs inversus | 38, 39, 40 |
| NPHP17/IFT172 | 2p23.3 | Nephrocystin‐17/ IFT protein 172 | IFT‐B | IFT80, IFT140 | Involved in intraflagellar transport | NPHP, JS, JBTS, MZSDS | 41 |
| NPHP18 /CEP83 | 12q22 | Nephrocystin‐18/centrosomal protein 83 | Basal body | IFT20, CEP164 | N/A | NPHP, liver fibrosis, mental retardation, hydrocephalus | 42 |
| NPHP19/DCDC2 | 6p22.3 | Doublecortin domain‐containing protein 2 | Axoneme | DVL | Involved in Wnt signalling | NPHP, renal‐hepatic ciliopathy | 43 |
| NPHP20/MAPKBP1 | 15q15.1 | Mitogen‐activated protein kinase binding protein 1 | Cytoplasm | N/A | Involved in DDR signalling and JNK signalling | NPHP | 2 |
| NPHP1L /XPNPEP3 | 22q13 | X‐prolyl aminopeptidase 3 | Mitochondria | Cleaves LRRC50, ALMS1, nephrocystin‐6 | Interferes with cilia function by cleaving certain cilial proteins | NPHP, myocardiosis, epilepsy | 44, 45 |
| NPHP2L /SLC41A1 | 1q32.1 | Solute carrier family 41 member 1 | Tubules at the borders of the cortex and medulla | N/A | Affects Mg2+ transport | NPHP, bronchiectasia | 46 |
| TRAF3IP1 | 2q37.3 | TRAF3 interacting protein 1 | Axonemes, basal bodies | N/A | Affects microtubule stabilization by IFT54 | NPHP, SLSN, RP | 47 |
| AH11/JBTS3 | 6q23.3 | Jouberin | Basal bodies | N/A | Affects cerebellar and cortical development | JBTS, RP | 48, 49 |
| CC2D2A/MKS6 | 4p15.32 | Coiled coil and C2 domain containing 2A | Basal bodies | CEP290 | Acts in ciliogenesis | MKS, COACH, JBST | 50, 51 |
ALMS1, Alstrom Syndrome 1; APC2, anaphase‐promoting complex 2; ATF4, activating transcription factor 4; ATRIP, ATR interacting protein; BBS, Bardet‐Biedl syndrome; BCAR1, breast cancer anti‐estrogen resistance 1; BICC1, Bicaudal‐C1; CAD, cranioectodermal dysplasia; CCDC92, coiled‐coil domain containing 92; CC2D2A, coiled‐coil and C2 domain containing 2A; CEP290, centrosomal protein 290; CHD, congenital heart disease; COACH, cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, ocular coloboma and hepatic fibrosis; DVL3, dishevelled 3; HIF1AN, hypoxia inducible factor 1 alpha subunit inhibitor; IFT, intraflagellar transport; JATD, Jeune asphyxiating thoracic dysplasia; JBTS, Joubert syndrome; JS, Jeune syndrome; LCA, Leber congenital amaurosis; LRRC50, leucine‐rich repeat containing protein 50; MKS, Meckel‐Gruber syndrome; MZSDS, Mainzer‐Saldino syndrome; OFD1, oral‐facial‐digital protein1; OMA, oculomotor apraxia; PTK2B, protein tyrosine kinase 2B; RP, retinitis pigmentosa; RPGR, retinitis pigmentosa GTPase regulator; SBS, Sensenbrenner syndrome; SLSN, Senior‐Loken syndrome; TMEM67,transmembrane protein 67; TTBK2, Tau‐tubulin kinase 2.
Most nephrocystins are located in the transition zone, inversin compartment, or subunits of intraflagellar transport (IFT) complexes.6 However, genome‐wide homozygosity mapping identified pathogenic mutations in NPHP1L and NPHP2L of which the protein product localizes to mitochondria.52 Currently, at least four distinct nephrocystin modules have been found: the NPHP1‐4‐8 module, NPHP2‐3‐9‐ANKS6 module, NPHP5‐6 module, and MKS module (Fig. 1). These nephrocystin modules are related to different signalling pathways, including the Wnt pathway, Hedgehog pathway, DNA damage response (DDR) pathway, Hippo pathway, intracellular calcium signalling pathway, cAMP signalling pathway, and mTOR pathway.
Figure 1.
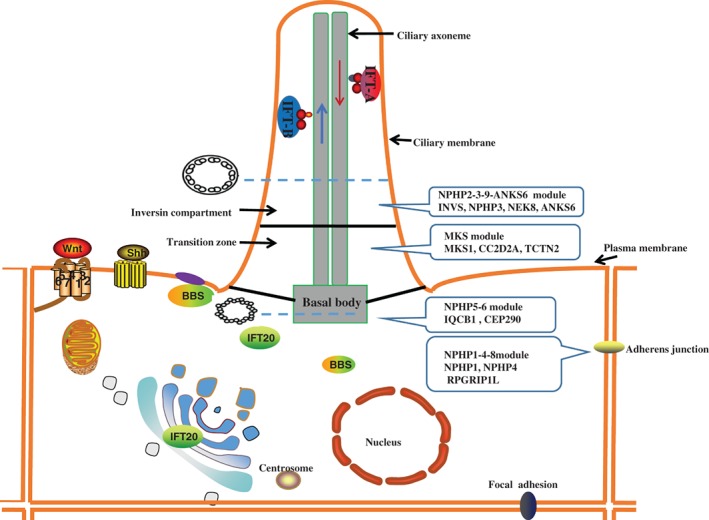
Subcellular localization of different nephrocystin module.
NPHP shows genetic and phenotypic heterogeneity. Mutations in single ciliary genes are often associated with multiple phenotypes (Table 1 and Table 3). Single locus allelism is insufficient to explain the variability in phenotypic heterogeneity in NPHP. Digenic and triallelic inheritance may provide an explanation. Triallelic inheritance was first demonstrated for BBS.53To date, oligogenic inheritance has been noted in some patients with mutations in NPHP1, NPHP5, NPHP6, NPHP8, NPHP9, NPHP11, and TTC21B genes.12, 54, 55, 56
APPROACH TO CLINICAL DIAGNOSIS OF NPHP
The diagnosis of NPHP is suggested by clinical features and confirmed by a positive genetic test (Fig. 2). The role of renal biopsy in diagnosis is controversial. Renal biopsy should be limited to cases in which tissue diagnosis can be used to distinguish it from other differential diagnoses. Molecular genetic analysis is currently the only method available to diagnose NPHP and thus provide patients and families with an unequivocal diagnosis. Due to an increasing number of potentially causative monogenic genes and to advances in next‐generation sequencing, whole‐exome sequencing has mostly replaced targeted‐sequencing panels in the diagnosis of NPHP.57 Using this method, a causative single‐gene mutation can be detected in up to 60% of cases depending on the composition of the cohort. However, the absence of mutation is not sufficient to exclude the diagnosis of NPHP. Most importantly, genetic testing should always be combined with thorough phenotyping and genetic counseling.
Figure 2.
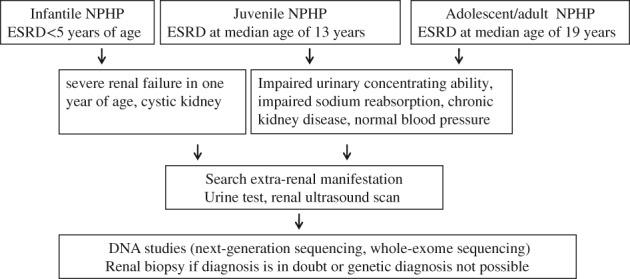
Approach to clinical diagnosis of nephronophthisis (NPHP).
Early onset autosomal dominant polycystic kidney disease and autosomal recessive polycystic kidney disease are often in the main differential diagnosis for patients with NPHP. Renal imaging may be useful in differential diagnosis. But genetic testing is required to make a definite diagnosis.
TREATMENT OF NPHP
There is no specific therapy for NPHP. Management is supportive, focusing on slowing the progression of CKD, controlling complications, and maintaining the promotion of growth. This disease does not recur after transplantation, so renal transplantation is the preferred renal replacement therapy.
Some potential therapeutic interventions have arisen from several lines of investigation into the pathogenesis of NPHP. Various personalized drugs include isosorbide dinitrate and tolvaptan (vasopressin V2 receptor antagonist),58 dimethyl fumarate,59 rapamycin (mTOR inhibitor),60 roscovitine and its analog S‐CR8 (cyclin‐dependent kinases inhibitor),61 purmorphamine (Shh signalling pathway agonist),62 paclitaxel,63 regulation of transcription factor Glis2/NPHP7 by SUMOylation,64 and FR167653 (p38 MAPK pathway inhibitor).65 Despite the many promising interventions that have arisen from preclinical studies, no clinical trials have yet been conducted in NPHP patients. Furthermore, large numbers of compounds which may be potential therapies are being screened in the zebrafish models of NPHP.66
The lack of a clear‐cut genotype–phenotype correlation remains a major challenge for physicians treating children with NPHP, even though the development of a single comprehensive histopathology and the discovery of specific disease genes and molecular mechanisms have significantly improved our understanding of NPHP. Only about 30% of NPHP patients have clear genetic mutations, suggesting that more NPHP genes have yet to be discovered. Novel genes will enable us to better understand the pathogenesis and relationship between cilia and cystic diseases. It is necessary to find new therapeutic strategies and develop alternative treatments other than conservative approaches and renal replacement therapy.
CONFLICTS OF INTEREST
There authors declare that they have no potential or actual competing interests.
Acknowledgement
No.
REFERENCES
- 1. Ala‐Mello S, Koskimies O, Rapola J, Kääriäinen H. Nephronophthisis in Finland: Epidemiology and comparison of genetically classified subgroups. Eur. J. Hum. Genet. 1999; 7(2): 205–11. [DOI] [PubMed] [Google Scholar]
- 2. Macia MS, Halbritter J, Delous M et al Mutations in MAPKBP1 cause juvenile or late‐onset cilia‐independent Nephronophthisis. Am. J. Hum. Genet. 2017; 100(2): 323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: Disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol. 2009; 20(1): 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Georges B, Cosyns JP, Dahan K et al Late‐onset renal failure in Senior‐Loken syndrome. Am. J. Kidney Dis. 2000; 36(6): 1271–5. [DOI] [PubMed] [Google Scholar]
- 5. Hoefele J, Nayir A, Chaki M et al Pseudodominant inheritance of nephronophthisis caused by a homozygous NPHP1 deletion. Pediatr. Nephrol. 2011; 26(6): 967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Srivastava S, Sayer JA. Nephronophthisis. J. Pediatr Genet. 2014; 3(2): 103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kang HG, Ahn YH, Kim JH et al Atypical retinopathy in patients with nephronophthisis type 1: An uncommon ophthalmological finding. Clin. Experiment. Ophthalmol. 2015; 43(5): 437–42. [DOI] [PubMed] [Google Scholar]
- 8. Baujat G, Huber C, El HJ et al Asphyxiating thoracic dysplasia: Clinical and molecular review of 39 families. J. Med. Genet. 2013; 50(2): 91–8. [DOI] [PubMed] [Google Scholar]
- 9. Gunay‐Aygun M. Liver and kidney disease in ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2009; 151C(4): 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barker AR, Thomas R, Dawe HR. Meckel‐Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis 2014; 10(1): 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Otto EA, Schermer B, Obara T et al Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left‐right axis determination. Nat. Genet. 2003; 34(4): 413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tory K, Lacoste T, Burglen L et al High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: Potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J. Am. Soc. Nephrol. 2007; 18(5): 1566–75. [DOI] [PubMed] [Google Scholar]
- 13. O'Toole JF, Otto EA, Frishberg Y, Hildebrandt F. Retinitis pigmentosa and renal failure in a patient with mutations in INVS. Nephrol. Dial. Transplant. 2006; 21(7): 1989–91. [DOI] [PubMed] [Google Scholar]
- 14. Olbrich H, Fliegauf M, Hoefele J et al Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto‐retinal degeneration and hepatic fibrosis. Nat. Genet. 2003; 34(4): 455–9. [DOI] [PubMed] [Google Scholar]
- 15. Sun L, Tong H, Wang H et al High mutation rate of NPHP3 in 18 Chinese infantile nephronophthisis patients. Nephrology (Carlton) 2016; 21(3): 209–16. [DOI] [PubMed] [Google Scholar]
- 16. Tory K, Rousset‐Rouvière C, Gubler MC et al Mutations of NPHP2 and NPHP3 in infantile nephronophthisis. Kidney Int. 2009; 75(8): 839–47. [DOI] [PubMed] [Google Scholar]
- 17. Bakkaloğlu SA, Kandur Y, Bedir‐Demirdağ T, Işık‐Gönül İ, Hildebrandt F. Diverse phenotypic expression of NPHP4 mutations in four siblings. Turk. J. Pediatr. 2014; 56(4): 423–6. [PMC free article] [PubMed] [Google Scholar]
- 18. Otto EA, Loeys B, Khanna H et al Nephrocystin‐5, a ciliary IQ domain protein, is mutated in Senior‐Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005; 37(3): 282–8. [DOI] [PubMed] [Google Scholar]
- 19. Frank V, den Hollander AI, Brüchle NO et al Mutations of the CEP290 gene encoding a centrosomal protein cause Meckel‐Gruber syndrome. Hum. Mutat. 2008; 29(1): 45–52. [DOI] [PubMed] [Google Scholar]
- 20. Helou J, Otto EA, Attanasio M et al Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior‐Løken syndrome. J. Med. Genet. 2007; 44(10): 657–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leitch CC, Zaghloul NA, Davis EE et al Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet‐Biedl syndrome. Nat. Genet. 2008; 40(4): 443–8. [DOI] [PubMed] [Google Scholar]
- 22. Valente EM, Silhavy JL, Brancati F et al Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006; 38(6): 623–5. [DOI] [PubMed] [Google Scholar]
- 23. den Hollander AI, Koenekoop RK, Yzer S et al Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006; 79(3): 556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Attanasio M, Uhlenhaut NH, Sousa VH et al Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007; 39(8): 1018–24. [DOI] [PubMed] [Google Scholar]
- 25. Ramachandran H, Yakulov TA, Engel C, Müller B, Walz G. The C175R mutation alters nuclear localization and transcriptional activity of the nephronophthisis NPHP7 gene product. Eur. J. Hum. Genet. 2016; 24(5): 774–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wolf MT, Saunier S, O'Toole JF et al Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int. 2007; 72(12): 1520–6. [DOI] [PubMed] [Google Scholar]
- 27. Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F. NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J. Am. Soc. Nephrol. 2008; 19(3): 587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rajagopalan R, Grochowski CM, Gilbert MA et al Compound heterozygous mutations in NEK8 in siblings with end‐stage renal disease with hepatic and cardiac anomalies. Am. J. Med. Genet. A 2016; 170(3): 750–3. [DOI] [PubMed] [Google Scholar]
- 29. Airik R, Slaats GG, Guo Z et al Renal‐retinal ciliopathy gene Sdccag8 regulates DNA damage response signaling. J. Am. Soc. Nephrol. 2014; 25(11): 2573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Otto EA, Hurd TW, Airik R et al Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal‐renal ciliopathy. Nat. Genet. 2010; 42(10): 840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doherty D, Parisi MA, Finn LS et al Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J. Med. Genet. 2010; 47(1): 8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Otto EA, Tory K, Attanasio M et al Hypomorphic mutations in meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11). J. Med. Genet. 2009; 46(10): 663–70. [DOI] [PubMed] [Google Scholar]
- 33. Davis EE, Zhang Q, Liu Q et al TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 2011; 43(3): 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fehrenbach H, Decker C, Eisenberger T et al Mutations in WDR19 encoding the intraflagellar transport component IFT144 cause a broad spectrum of ciliopathies. Pediatr. Nephrol. 2014; 29(8): 1451–6. [DOI] [PubMed] [Google Scholar]
- 35. Park E, Lee JM, Ahn YH, Kang HG, Ha IIS, Lee JH, Park YS, Kim NKD, Park WY, Cheong HII Hepatorenal fibrocystic diseases in children. Pediatr. Nephrol. 2016; 31(1): 113–19. [DOI] [PubMed] [Google Scholar]
- 36. Chaki M, Airik R, Ghosh AK et al Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012; 150(3): 533–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Slaats GG, Ghosh AK, Falke LL et al Nephronophthisis‐associated CEP164 regulates cell cycle progression, apoptosis and epithelial‐to‐mesenchymal transition. PLoS Genet. 2014; 10(10): e1004594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoff S, Halbritter J, Epting D et al ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3. Nat. Genet. 2013; 45(8): 951–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramachandran H, Engel C, Müller B, Dengjel J, Walz G, Yakulov TA. Anks3 alters the sub‐cellular localization of the Nek7 kinase. Biochem. Biophys. Res. Commun. 2015; 464(3): 901–7. [DOI] [PubMed] [Google Scholar]
- 40. Taskiran EZ, Korkmaz E, Gucer S et al Mutations in ANKS6 cause a nephronophthisis‐like phenotype with ESRD. J. Am. Soc. Nephrol. 2014; 25(8): 1653–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Halbritter J, Bizet AA, Schmidts M et al Defects in the IFT‐B component IFT172 cause Jeune and Mainzer‐Saldino syndromes in humans. Am. J. Hum. Genet. 2013; 93(5): 915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Failler M, Gee HY, Krug P et al Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am. J. Hum. Genet. 2014; 94(6): 905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schueler M, Braun DA, Chandrasekar G et al DCDC2 mutations cause a renal‐hepatic ciliopathy by disrupting WNT signaling. Am. J. Hum. Genet. 2015; 96(1): 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Böttinger EP. Lights on for aminopeptidases in cystic kidney disease. J. Clin. Invest. 2010; 120(3): 660–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Toole JF, Liu Y, Davis EE et al Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis‐like nephropathy. J. Clin. Invest. 2010; 120(3): 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hurd TW, Otto EA, Mishima E et al Mutation of the Mg2+ transporter SLC41A1 results in a nephronophthisis‐like phenotype. J. Am. Soc. Nephrol. 2013; 24(6): 967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bizet AA, Becker‐Heck A, Ryan R et al Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat. Commun. 2015; 6: 8666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ferland RJ, Eyaid W, Collura RV et al Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat. Genet. 2004; 36(9): 1008–13. [DOI] [PubMed] [Google Scholar]
- 49. Fleming LR, Doherty DA, Parisi MA et al Prospective evaluation of kidney disease in Joubert Syndrome. Clin. J. Am. Soc. Nephrol. 2017; 12(12): 1962–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Al‐Hamed MH, Kurdi W, Alsahan N et al Genetic spectrum of Saudi Arabian patients with antenatal cystic kidney disease and ciliopathy phenotypes using a targeted renal gene panel. J. Med. Genet. 2016; 53(5): 338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gorden NT, Arts HH, Parisi MA et al CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy‐associated basal body protein CEP290. Am. J. Hum. Genet. 2008; 83(5): 559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Simms RJ, Eley L, Sayer JA. Nephronophthisis. Eur. J. Hum. Genet. 2009; 17(4): 406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Katsanis N, Ansley SJ, Badano JL et al Triallelic inheritance in Bardet‐Biedl syndrome, a Mendelian recessive disorder. Science 2001; 293(5538): 2256–9. [DOI] [PubMed] [Google Scholar]
- 54. Hoefele J, Wolf MT, O'Toole JF et al Evidence of oligogenic inheritance in nephronophthisis. J. Am. Soc. Nephrol. 2007; 18(10): 2789–95. [DOI] [PubMed] [Google Scholar]
- 55. Penchev V, Boueva A, Kamenarova K et al A familial case of severe infantile nephronophthisis explained by oligogenic inheritance. Eur. J. Med. Genet. 2017; 60(6): 321–5. [DOI] [PubMed] [Google Scholar]
- 56. Louie CM, Caridi G, Lopes VS et al AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 2010; 42(2): 175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gee HY, Otto EA, Hurd TW et al Whole‐exome resequencing distinguishes cystic kidney diseases from phenocopies in renal ciliopathies. Kidney Int. 2014; 85(4): 880–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Strong A, Muneeruddin S, Parrish R, Lui D, Conley SB. Isosorbide dinitrate in nephronophthisis treatment. Am. J. Med. Genet. A 2018; 176(4): 1023–6. [DOI] [PubMed] [Google Scholar]
- 59. Oey O, Rao P, Luciuk M et al Effect of dimethyl fumarate on renal disease progression in a genetic ortholog of nephronophthisis. Exp. Biol. Med. (Maywood) 2018; 243(5): 428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tobin JL, Beales PL. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 2008; 23(11): 2095–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov‐Beskrovnaya O. Long‐lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature 2006; 444(7121): 949–52. [DOI] [PubMed] [Google Scholar]
- 62. Srivastava S, Ramsbottom SA, Molinari E et al A human patient‐derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum. Mol. Genet. 2017; 26(23): 4657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Slaats GG, Lilien MR, Giles RH. Nephronophthisis: Should we target cysts or fibrosis. Pediatr. Nephrol. 2016; 31(4): 545–54. [DOI] [PubMed] [Google Scholar]
- 64. Ramachandran H, Herfurth K, Grosschedl R, Schäfer T, Walz G. SUMOylation blocks the ubiquitin‐mediated degradation of the Nephronophthisis gene product Glis2/NPHP7. PLoS One 2015; 10(6): e0130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sugiyama N, Kohno M, Yokoyama T. Inhibition of the p38 MAPK pathway ameliorates renal fibrosis in an NPHP2 mouse model. Nephrol. Dial. Transplant. 2012; 27(4): 1351–8. [DOI] [PubMed] [Google Scholar]
- 66. Westhoff JH, Giselbrecht S, Schmidts M et al Development of an automated imaging pipeline for the analysis of the zebrafish larval kidney. PLoS One 2013; 8(12): e82137. [DOI] [PMC free article] [PubMed] [Google Scholar]