

ABSTRACT
Viral infections of the respiratory tract can be complicated by bacterial superinfection, resulting in a significantly longer duration of illness and even a fatal outcome. In this review, we focused on interactions between S. aureus and non-influenza viruses. Clinical data evidenced that rhinovirus infection may increase the S. aureus carriage load in humans and its spread. In children, respiratory syncytial virus infection is associated with S. aureus carriage. The mechanisms by which some non-influenza respiratory viruses predispose host cells to S. aureus superinfection can be summarized in three categories: i) modifying expression levels of cellular patterns involved in S. aureus adhesion and/or internalization, ii) inducing S. aureus invasion of epithelial cells due to the disruption of tight junctions, and iii) decreasing S. aureus clearance by altering the immune response. The comprehension of pathways involved in S. aureus-respiratory virus interactions may help developing new strategies of preventive and curative therapy.
KEYWORDS: Staphylococcus aureus, non-influenza respiratory viruses, human rhinovirus, respiratory syncytial virus, virus-bacterium interaction, nasal carriage
Introduction
The development of upper and lower respiratory tract infections is determined by the interaction between one micro-organism and the host immune response. More recently, the interaction between the resident microbiota and incoming pathogens may also participate in the development of respiratory tract infections.
It has been postulated for a long time that viral infection of the respiratory tract indirectly predispose to bacterial superinfection by disruption of the respiratory mucosal epithelium [1], or passively through anatomical and mechanical changes like Eustachian tube dysfunction [2], ostiomeatal obstruction and reduced mucocilliary clearance [3]. Over the last decades, there was an increasing interest to investigate the contribution of bacterial colonization in the outcome of viral respiratory tract infections. The human respiratory tract is known to be the reservoir of diverse commensals and potential pathogens including mainly Staphylococcus aureus, Streptococcus pneumoniae and Haemophilus influenzae that compose a significant part of the respiratory tract microbiota [4].
The role of interactions between viruses and bacteria in the pathogenesis of respiratory infections have been extensively studied in the literature and notably those between influenza viruses and S. aureus or S. pneumoniae [5–10]. However, interactions between S. aureus and non-influenza respiratory viruses were not reviewed recently.
The aim of this report is to describe the current knowledge on possible interactions between S. aureus and non-influenza viral pathogens in the respiratory tract, with a focus on the mechanisms by which these interactions are potentially mediated.
Impact of viral infections in the respiratory tract on staphylococcus aureus colonization
S. aureus is a commensal bacterium of the skin and mucosa, colonizing 15 to 36% of the whole population [11,12]. The main reservoir of S. aureus carriage in humans remains the nose [12], but other sites of carriage have been reported, such as the skin [11], pharynx [13,14], vagina [15], and rectum [16]. S. aureus is however a Janus-faced bacterium and beyond its commensal status, it is also a life-threatening pathogen. It is in fact considered to be one of the leading causes of nosocomial and community-acquired bacterial infections [17]. In addition to be the most common cause of bacteremia, with a 25% mortality rate despite appropriate treatment [18], it is also known as an etiological agent of other deep-seated infections including osteomyelitis, septic arthritis, endocarditis and device-related infections [17,19].
S. aureus is also an important pathogen in lung infection, mostly implicated in hospital-acquired pneumonia [17]. S. aureus expresses a wide repertoire of surface proteins that recognize cellular adhesive molecules and it is therefore able to adhere to and internalize into lung epithelial cells, which protects the bacteria from the host immune system and facilitate chronic infection [20]. In vitro experiments have demonstrated that the activation of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) signaling pathway in infected pulmonary epithelial cells results in inflammation enhancement via IL-8 expression; furthermore, intracellular S. aureus can lead, after an initial lag period, to the apoptosis of these cells. [21,22] In necrotizing pneumonia, the key virulence factors of S. aureus associated with the apoptosis of lung cells were shown to be pore-forming toxins, namely Panton-Valentine leukocidin (PVL) and alpha-hemolysin [23,24].
Besides, S. aureus is frequently involved in secondary bacterial pneumonia occurring during seasonal influenza outbreaks [8]. During the 2009 A/H1N1 influenza pandemic, bacterial co-infections complicated up to one-third of influenza cases in the United States in which S. aureus was the most common pathogen, accounting for 27% of the cases in both critically ill children and adults [25,26]. S. aureus co-infection was associated with significantly higher morbidity and mortality [26]. To date, the clinical association between the healthy carriage of S. aureus and the secondary staphylococcal pneumonia is still unclear. However, recent evidence from in vitro and in vivo tests showed that host physiologic changes induced by influenza virus can lead to the transition from asymptomatic colonization to invasive disease [27]. In addition to influenza viruses, S. aureus nasopharyngeal carriage has been found to be associated with some other respiratory viruses like rhinovirus and respiratory syncytial virus (RSV).
Staphylococcus aureus and rhinovirus
Rhinovirus is the second respiratory virus that has been most frequently reported to interact with S. aureus. Several studies showed that natural or experimental rhinovirus infection in S. aureus nasal carriers leads to increased S. aureus airborne dispersal, especially when sneezing is a part of the syndrome [28–33]. These studies emphasized that rhinovirus infection may facilitate the spreading of S. aureus from staphylococcal carriers to their environment and the transmission of the bacterium between humans. A recent study showed that rhinovirus infection is associated with changes in the upper respiratory tract microbiota [34]. In this study, healthy adults who were experimentally infected by rhinovirus showed increase in the relative abundance of H. parainfluenzae, Neisseria subflava and S. aureus, and returned to their baseline level after the infection was cleared [34]. It has been also shown that experimental rhinovirus infection significantly increases S. aureus nasal load by 39% compared to baseline bacterial load [30]. These findings suggest that changes in the composition of respiratory microbiota following rhinovirus infection may play a role in the development of bacterial superinfection. However, the role of rhinovirus infection on the onset or increase of staphylococcal carriage in human remains poorly studied.
Staphylococcus aureus and respiratory syncytial virus
A prospective microbiological analysis showed that 40% of children with severe RSV bronchiolitis had a bacterial co-infection in their lower airways and were at increased risk for bacterial pneumonia [35]. Bacterial co-infection in children with RSV bronchiolitis seems to increase inflammatory markers, abnormal radiologic patterns and hospital stay [36,37]. In young infants with RSV bronchiolitis, S. aureus, H. influenzae, M. catarrhalis and S. pneumoniae are the most common pathogens colonizing the nasopharynx and the lower airways [35,36].
A recent prospective study investigated the differences in the nasopharyngeal microbiome during acute respiratory tract infections due to human rhinovirus or RSV in 135 infants aged less than 6 months [38]. By contrast to previous studies, S. aureus was not found among the most abundant bacteria. However, the difference of S. aureus abundance was significantly higher in RSV than in rhinovirus-infected infants [38]. Another prospective study investigated the nasopharyngeal microbiota in young infants with RSV infections by 16S-RNA sequencing; the authors found that RSV infection was positively associated with H. influenzae and S. pneumoniae, but negatively associated with S. aureus nasopharyngeal colonization [39]. In another study concerning nasopharyngeal aspirates from children with RSV infection aged between 6 months and 2 years, S. aureus was shown to colonize 77% of RSV-infected patients with a positive association between RSV and S. aureus nasopharyngeal carriage [40]. The conflicting data from the two latter studies may be explained by differences in target populations, sampling procedures and/or microbiological methods.
To date, most of the studies dedicated to the impact of RSV infection on the bacterial colonization of the upper respiratory airways identified a synergistic interaction between RSV and S. pneumoniae [41,42]. Nevertheless, nasopharyngeal colonization with S. aureus is clearly more than a passive phenomenon during RSV infection and further studies are needed to elucidate the interactions between these 2 pathogens.
There is no doubt that the impact of infection with respiratory non-influenza viruses on S. aureus colonization is important and should not be neglected as this may worsen the disease outcome and even be fatal in some cases [43]. The recent technical breakthrough of molecular diagnostic will be of precious help to investigate the changes in nasopharyngeal microbiota composition and notably in staphylococcal carriage during non-influenza viral infections of the respiratory tract.
Mechanisms involved in interactions between staphylococcus aureus and non-influenza respiratory viruses
The high incidence of staphylococcal superinfection during pandemic and seasonal influenza and the important mortality risk associated to this condition promoted the in-depth investigation of the molecular and immunologic mechanisms that are involved in the bidirectional synergism between both pathogens. Influenza virus is known to promote staphylococcal superinfection by alteration of the host immune system via increased production of pro-inflammatory cytokines and interferons (IFNs), impairment of phagocytic cell functions and suppression of type 17 immunity [9,10]. In addition, influenza virus has been shown to promote S. aureus adhesion and internalization within non-professional phagocytic cells through at least two distinct mechanisms: binding of bacteria to the membrane-associated hemagglutinin of influenza-infected cells, and binding of bacteria to free virions, followed by internalization of virus-coated bacteria into non-infected cells [44]. Besides, S. aureus co-infection promotes influenza virus replication and pathogenicity; indeed, extracellular bacteria secrete staphylokinase that facilitates the binding of influenza virus to the host cells, whereas intracellular S. aureus inhibits influenza virus-induced type I IFN signaling through impaired signal transducers and activators of transcription (STAT1 and STAT2) dimerization [45,46]. While the mechanisms of interaction between S. aureus and influenza virus seem to be deeply understood, the interactions between S. aureus and other respiratory viruses were less investigated. Table 1 summarizes the main mechanisms and pathways potentially involved in the interactions between S. aureus and non-influenza respiratory viruses.
Table 1.
Virulence factors involved in molecular mechanisms of interactions between Staphylococcus aureus and non-influenza respiratory viruses.
| Virulence factors | Cellular target | Effects | Synergism | References |
|---|---|---|---|---|
| Staphylococcus aureus | ||||
| SEA/SEB | Unknown | Increase of IL-1β, IL-6 and IL-8 secretion | Increase of ICAM-1 expression Enhancement of rhinovirus replication |
[53,83–85] |
| LTA | Unknown | Increase of IL-6, IL-12/IL-23 and IFN-γ secretion | Increase of susceptibility to coronavirus | [98] |
| Human rhinovirus | ||||
| Capsid | Membrane TLR2 | NFκB activation Increase of IL-6, IL-8, INFβ and IFN-γ secretion |
Increase of cFn and ICAM-1 expression Increase of S. aureus adhesion and internalization to/in host cells | [48,51–56] |
| ssRNA | Endosomal TLR7/8 | |||
| Cytoplasmic RIG-I | Increase of IFN-β, IFN-γ, RANTES, IL-8, IP-10 and ENA78 secretion | |||
| dsRNA | Cytoplasmic MDA5 | |||
| Endosomal TLR3 | RIG-I and MDA5 upregulation NFκB activation Increase of IL-6, IL-8, INF-β and IFN-γ secretion |
|||
| Unknown | Rac1 activation NOX1 production Stimulation of ROS generation |
Loss of ZO-1 tight junctions Induce S. aureus invasion of the epithelium |
[78,81] | |
| Respiratory Syncytial Virus | ||||
| Viral envelope | Membrane TLR2 | NFκB activation Increase of pro-inflammatory cytokine secretion IRFs activation Increase of type I IFNs secretion |
Alteration of neutrophil recruitment Inhibition of B cells response Decrease of S. aureus clearance |
[87,90,93,94,99] |
| Viral F protein | Membrane TLR4 | |||
| ssRNA | Endosomal TLR3 | |||
| Cytoplasmic RIG-I | ||||
| Cytoplasmic Nod2 | ||||
| dsRNA | Endosomal TLR7 | |||
| Cytoplasmic MDA5 | ||||
| Unknown | NKG2D | Increase of IFN-γ secretion | ||
| Unknown | Unknown | Increase of Lewis blood group antigen expression | Increase of toxin-producing S. aureus binding to epithelial cells | [88,89,91] |
Staphylococcus aureus and rhinovirus
Rhinovirus, the most common cause of upper respiratory tract infections (URTI), primarily targets the nasal and nasopharyngeal epithelial cells [47]. The innate immune system recognizes rhinovirus by different Pattern Recognition Receptors (PRRs) including membrane and endosomal Toll-like Receptors (TLRs) and cytoplasmic inducible RNA helicases like retinoic acid-inducible gene-1 (RIG-I) or melanoma differentiation-associated protein 5 (MDA5) [48]. Rhinovirus infection promotes pro-inflammatory cytokines and IFN production mainly through the activation of NFκB [49,50].
Several potential mechanisms through which rhinovirus increases susceptibility to bacterial infection have been demonstrated in vitro in epithelial cells of the upper and lower airways. In 1981, Selinger, Reed and McLaren developed an in vitro model for studying bacterial adherence to virus-infected epithelial cells [51]. They reported that the adherence of S. aureus was significantly higher in rhinovirus-infected cells compared to uninfected cells. Only recently, various in vitro studies have shown that inflammation due to rhinovirus infection increased cellular patterns that facilitate the adhesion and internalization of S. aureus within host cells [52–56].
Rhinovirus promotes cellular fibronectin expression
In primary human nasal epithelial cells, it has been demonstrated that rhinovirus infection up-regulates the expression of cellular fibronectin [54,56]. The up-regulation of fibronectin was observed at both transcriptional and translational levels and seems to be related to the rhinovirus-induced NFκB activation [56]. Fibronectin is known to mediate the adhesion and the internalization of S. aureus in the presence of epithelial cells [57]. The fibronectin-binding protein A or B (FnBPA/B) are S. aureus cell wall-anchored Microbial Surface Components Recognizing Adhesive Matrix Molecules (MSCRAMMs) that bind type I motif of fibronectin through a tandem beta-zipper interaction [58]. The α5β1 integrin expressed on host cellmembrane binds the RGD motif of fibronectin [59]. Fibronectin is thus acting as a bridge between S. aureus and the host cell, and the complex formed by FnBPA/B, fibronectin and α5β1 integrin leads to S. aureus internalization inside the epithelial cell [57]. Then the up-regulation of fibronectin expression in epithelial cells during rhinovirus infection could explain the increase of S. aureus epithelial cells adhesion [54,56].
Rhinovirus promotes ICAM-1 expression
The intercellular adhesion molecule 1 (ICAM-1), which is the receptor of the major group of rhinoviruses [60], have been found to be overexpressed during rhinovirus infection [52,61]. Adhesion and internalization of S. aureus to epithelial cells is also mediated by ICAM-1 [52,54]. Both immortalized pneumocytes (A549 cell line) and primary human nasal epithelial cells have been found to release higher levels of IL-6 and IL-8 when infected by rhinovirus and subsequently overexpressed ICAM-1. The pro-inflammatory effect of IL-6 and IL-8 also induces an overexpression of ICAM-1 in the surrounding uninfected cells, which increases S. aureus uptake by the epithelial cells [52]. The up-regulation of ICAM-1 and pro-inflammatory cytokines in rhinovirus-infected epithelial cells seems to follow the NFκB pathway [61–63].
Interestingly, the extracellular adherence protein (EAP), an adhesin naturally secreted by S. aureus, was found to bind ICAM-1 [64]. EAP belongs to Secreted Expanded Repertoire Adhesive Molecules (SERAMs) and is involved in bacterial aggregation [65], adhesion and invasion of epithelial and fibroblastic cells [66–68], and in preventing neutrophil recruitment [64,69–72]. In addition to ICAM-1, EAP can also bind a large variety of extracellular matrix and plasma proteins including collagen, fibrinogen, fibronectin, vitronectin, laminin, thrombospondin and prothrombin [64,65,73–75]. Taken together, it can be hypothesized that the ligation of S. aureus to ICAM-1, whose expression is enhanced by rhinovirus infection, is mediated by EAP.
Rhinovirus disrupts tight junctions
The mucosal barrier of the nasal cavity is the first site of exposure to inhaled respiratory pathogens and plays an important role in host defenses in terms of innate immunity. Its integrity is regulated in large part by tight junctions of epithelial cells [76], which is a complex of several internal and membrane proteins including occludin and zona occludens (ZO) proteins [77]. Rhinovirus infection disrupts the barrier function of the airway epithelium through the dissociation of ZO-1 and occludin from the tight junction complex [78]. Rhinovirus infection has been shown to induce oxidative stress in respiratory epithelial cells by generating reactive oxygen species (ROS) [79]. ROS generation is induced by double-strands of viral RNA (dsRNA) that appear transiently during rhinovirus replication, and is produced by NADPH oxidase [78]. In non-phagocytic cells, ROS act as a molecular switch to stimulate pro-inflammatory responses [80]. Nevertheless, ROS generation disrupts the barrier function of rhinovirus-infected cells [78]. Thus, a perturbation of the tight junction barrier function increases paracellular permeability, facilitates translocation of pathogens and their soluble products, and exposes basolateral receptors. It has been found that the infection of primary human airway epithelial cells with both major and minor groups of rhinovirus promoted the paracellular migration of S. aureus and its epithelium invasion [81]. The rhinovirus infection facilitates also the transmigration of other bacterial species like H. influenzae and Pseudomonas aeruginosa [81]. This provides insights into another mechanism by which rhinovirus can predispose the host to secondary bacterial infections.
Staphylococcal enterotoxins a and b promote rhinovirus replication
Interactions between rhinovirus and S. aureus have been shown to be bi-directional: whereas rhinovirus promotes S. aureus adhesion and internalization within host cells, S. aureus enhances the rhinovirus replication [53]. S. aureus secretes several toxins including pore-forming toxins, toxic shock syndrome toxin and enterotoxins [82]. In vitro, staphylococcal enterotoxins A and B (SEA and SEB) were shown to enhance the rhinovirus replication in A549 epithelial cells in a dose-dependent manner; however, they were found able to enhance neither ICAM-1 expression nor Il-1β, IL-6 and IL-8 secretion [53]. In contrast, studies using other cellular models (normal human keratinocytes, coculture of human peripheral blood mononuclear cells and A549 cells, or primary nasal epithelial cell cultures) showed that these enterotoxins induced ICAM-1 expression or the secretion of pro-inflammatory cytokines [83–85]. Therefore, the mechanisms involved in the promotion of rhinovirus replication via staphylococcal enterotoxins need to be studied more in-depth.
All these experimental data provide evidence of bi-directional synergism between rhinovirus and S. aureus. The different mechanisms described above are summarized in Figure 1. Nevertheless, the molecular pathways of these mechanisms remain poorly understood and deserve further investigation.
Figure 1.
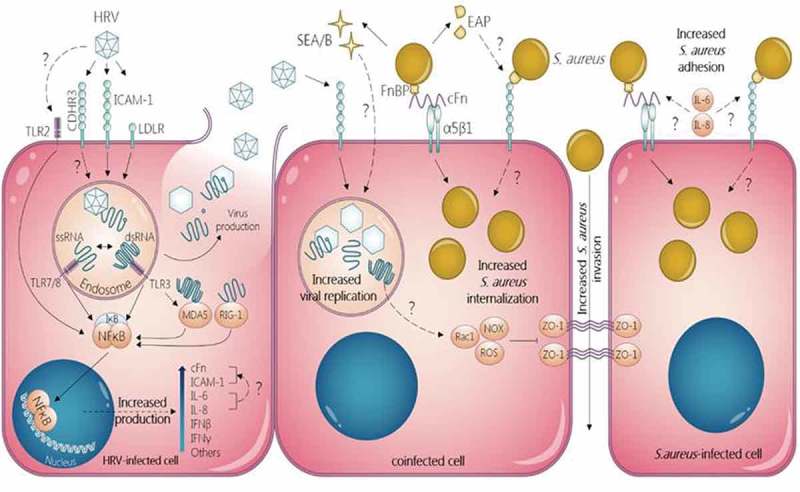
Mechanisms of bi-directional synergism between Staphylococcus aureus and human rhinovirus in non-professional phagocytic epithelial cells. S. aureus increases HRV replication via SEA and SEB. HRV increases adhesion and internalization of S. aureus in both HRV-infected and uninfected cells through increased release of IL-1β, IL-6 and IL-8 and subsequent increase of ICAM-1 and cFn expression via NFkB activation. HRV inhibits ZO-1 tight junctions favoring S. aureus invasion of the epithelium.
Staphylococcus aureus and respiratory syncytial virus
RSV is a worldwide seasonal virus that affects mainly young children. RSV disease manifestations can vary from mild URTI to severe pneumonia or bronchiolitis, which can lead to hospitalization and serious complications like respiratory failure [86]. Despite the high incidence of bacterial and RSV co-infection in both children and adults, only few studies aimed to investigate the possible interactions between S. aureus and this virus [87–90]. RSV is supposed to exacerbate S. aureus co-infection via at least two mechanisms: enhancing the adhesion of S. aureus to the RSV-infected cells [88,89], and decreasing the bacterial clearance by altering the immune response to S. aureus [87,90].
RSV infection enhances staphylococcus aureus adhesion
In vitro studies had shown that RSV-infected epithelial cells bind more S. aureus than uninfected cells [88,89]. Study of nasal washings in patients with respiratory virus diseases has shown that secretion of Lewis blood group antigens is associated with RSV infection [91]. In addition, epithelial cells (HEp2) expressing high concentrations of antigens of the Lewis blood group bound significantly more S. aureus than cells expressing low concentrations of these antigens [88,89]. While RSV infects about 50% of infants by the first year of life, Lewis antigen is expressed in secretions of nearly 90% of 3 month-aged infants [88,89]. This may explain the high susceptibility of RSV-infected infants to secondary staphylococcal infection, sometimes resulting in lethal issue [92]. Nevertheless, the molecular patterns involved in RSV-related increase of S. aureus adhesion to host cells remain unknown.
RSV alters the immune response to staphylococcus aureus
Pro-inflammatory cytokines and type I IFNs are known to be secreted by RSV-infected epithelial cells after viral sensing by different PRRs, mainly through NFκB and Interferon Regulation Factors (IRFs) pathways [93]. In addition, IFN-γ (type II IFN) seems to play an important role in the immune response to RSV as it activates the T cell response [94]. It has been demonstrated that RSV infection of human adult or neonatal mononuclear leukocytes results in significant inhibition of the lymphoproliferative response to heat-killed S. aureus [87]. This inhibitory effect on the development of cell-mediated immune response could be due in part to the increased secretion of IFN-γ[87]. IFN-γ was shown to reduce RSV replication in epithelial cells and also to inhibit B cell responses, which may alter the humoral response to S. aureus infection and decrease the bacterial clearance [87,94].
Murine models had been used to demonstrate that, despite the increased number of inflammatory cells, RSV decreases the clearance of S. aureus and other pathogenic bacteria like S. pneumoniae and P. aeruginosa from the lungs of mice following secondary bacterial infection [90]. It was suggested that RSV alters neutrophil function via changes in inflammatory response and cytokine secretion in the lung. Further studies are needed to delineate the different immunological pathways that sustain the decrease of bacterial clearance and the increase of susceptibility to secondary infections by S. aureus following RSV infection.
Staphylococcus aureus and other respiratory tract viruses
S. aureus interactions with rhinovirus or RSV have been actively, yet insufficiently, investigated. However, bacterial interactions with other respiratory viruses are poorly studied and only partial studies are available. For example, parainfluenza virus has been shown to enhance the ability of non-typeable H. influenzae and S. pneumoniae to adhere to human respiratory epithelial cells [95]. However, there is no information available about the interaction of these agents with S. aureus in humans. Only one in vitro study conducted on bovine embryonic lung cells investigated the effect of bovine parainfluenza virus infection on adherence of several bacterial agents, including S. aureus, and found no virus-specific effect on any of the tested bacteria [96]. Besides, normal human bronchial epithelial cells that were pre-incubated with S. pneumoniae resulted in an increased susceptibility to infection with human metapneumovirus. Nevertheless, this was not the case for cells pre-incubated with H. influenzae, M. catarrhalis or S. aureus [97]. On the contrary, synergistic effect between S. aureus and coronavirus has been demonstrated in vivo in a swine model. Lipoteichoic acid from S. aureus increased the susceptibility to coronavirus infection in pigs via increased secretion of pro-inflammatory cytokines IL-6, IL-12, IL-23 and IFN-γ[98]. To date, the lack of clinical and experimental data about the relationship between S. aureus colonization and other respiratory viruses complicates the understanding of potential interactions between these pathogens.
Concluding remarks
While clinical and mechanistic aspects of the synergism between S. aureus and influenza virus have been deeply studied, the interactions between this bacterium and other common respiratory viruses were only partially investigated [10]. Literature still lacks more data about the relations between S. aureus carriage and non-influenza respiratory virus infections, as well as deeper insights into mechanisms of interactions between these different pathogens. The evaluation of the concrete risk of switch from commensal S. aureus colonization to invasive disease during viral respiratory tract infections may help us to propose efficient strategies of decolonization when needed. Furthermore, the comprehension of the involved molecular pathways may help to develop new strategies of preventive and curative treatments.
CDHR3: Cadherin-Related Family Member 3; cFn: cellular Fibronectin; dsRNA: double-stranded RNA; ENA78: Epithelial Neutrophil Activating peptide-78; FnBP: Fibronectin Binding Protein; HRV: Human Rhinovirus; ICAM-1: InterCellular Adhesion Molecule 1; IFN: Interferon; IL: Interleukin; IRFs: Interferon Regulator Factors; IκB: NFκB Inhibitor; LDLR: Low-Density Lipoprotein Receptor; MDA5: Melanoma Differentiation Associated protein-5; NFκB: Nuclear Factor kappa-light-chain-enhancer of activated B cells; NKG2D: Natural Killer Group 2D receptor; Nod2: Nucleotide-binding oligomerization domain-containing protein 2; NOX: membrane-bound NADPH OXidase; Rac1: Ras-related C3 botulinum toxin substrate 1; RANTES: Regulated on Activation, Normal T cell Expressed and Secreted chemokine; RIG-I: Retinoic acid Inducible Gene-1; ROS: Reactive Oxygen Species; S. aureus: Staphylococcus aureus; SEA: Staphylococcal Enterotoxin A; SEB: Staphylococcal Enterotoxin B; ssRNA: single-stranded RNA; TLR: Toll-Like receptor; ZO-1: Zona Occludens 1.
Funding Statement
M.F.M. was funded by a PhD scholarship from the European Erasmus Mundus–Al Idrisi program supported by the University of Granada, Spain.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Bakaletz LO. Viral potentiation of bacterial superinfection of the respiratory tract. Trends Microbiol. 1995;3:110–114. [DOI] [PubMed] [Google Scholar]
- [2].McBride TP, Doyle WJ, Hayden FG, et al. Alterations of the eustachian tube, middle ear, and nose in rhinovirus infection. Arch Otolaryngol Head Neck Surg. 1989;115:1054–1059. [DOI] [PubMed] [Google Scholar]
- [3].Gwaltney JM. Acute community-acquired sinusitis. Clin Infect Dis. 1996;23:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Robinson J. Colonization and infection of the respiratory tract: what do we know? Paediatr Child Health. 2004;9:21–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].McCullers JA. Insights into the interaction between influenza virus and Pneumococcus. Clin Microbiol Rev. 2006;19:571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].DeLeo FR, Musser JM. Axis of coinfection evil. J Infect Dis. 2010;201:488–490. [DOI] [PubMed] [Google Scholar]
- [7].Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol. 2013;191:2047–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Papanicolaou GA. Severe influenza and S.aureus pneumonia. Virulence. 2013;4:666–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Robinson KM, Kolls JK, Alcorn JF. The immunology of influenza virus-associated bacterial pneumonia. Curr Opin Immunol. 2015;34:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun. 2015;83:3764–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wertheim HF, Melles DC, Vos MC, et al. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis. 2005;5:751–762. [DOI] [PubMed] [Google Scholar]
- [12].Verhoeven PO, Gagnaire J, Botelho-Nevers E, et al. Detection and clinical relevance of Staphylococcus aureus nasal carriage: an update. Expert Rev Anti Infect Ther. 2014;12:75–89. [DOI] [PubMed] [Google Scholar]
- [13].Mertz D, Frei R, Periat N, et al. Exclusive Staphylococcus aureus throat carriage: at-risk populations. Arch Intern Med. 2009;169:172–178. [DOI] [PubMed] [Google Scholar]
- [14].Verhoeven PO, Haddar CH, Grattard F, et al. Does pharyngeal sampling improve the detection of nasopharyngeal persistent carriers of Staphylococcus aureus? J Infect. 2015;70:549–552. [DOI] [PubMed] [Google Scholar]
- [15].Bourgeois-Nicolaos N, Lucet J-C, Daubié C, et al. Maternal vaginal colonisation by Staphylococcus aureus and newborn acquisition at delivery. Paediatr Perinat Epidemiol. 2010;24:488–491. [DOI] [PubMed] [Google Scholar]
- [16].Gagnaire J, Verhoeven PO, Grattard F, et al. Epidemiology and clinical relevance of Staphylococcus aureus intestinal carriage: a systematic review and meta-analysis. Expert Rev Anti Infect Ther. 2017;15:767–785. [DOI] [PubMed] [Google Scholar]
- [17].Tong SYC, Davis JS, Eichenberger E, et al. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28:603–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Melzer M, Welch C. Thirty-day mortality in UK patients with community-onset and hospital-acquired meticillin-susceptible Staphylococcus aureus bacteraemia. J Hosp Infect. 2013;84:143–150. [DOI] [PubMed] [Google Scholar]
- [19].Kern WV. Management of Staphylococcus aureus bacteremia and endocarditis: progresses and challenges. Curr Opin Infect Dis. 2010;23:346–358. [DOI] [PubMed] [Google Scholar]
- [20].Josse J, Laurent F, Diot A. Staphylococcal adhesion and host cell invasion: fibronectin-binding and other mechanisms. Front Microbiol. 2017;8:2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kahl BC, Goulian M, Van Wamel W, et al. Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect Immun. 2000;68:5385–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ratner AJ, Bryan R, Weber A, et al. Cystic fibrosis pathogens activate Ca2+-dependent mitogen-activated protein kinase signaling pathways in airway epithelial cells. J Biol Chem. 2001;276:19267–19275. [DOI] [PubMed] [Google Scholar]
- [23].Labandeira-Rey M, Couzon F, Boisset S, et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science. 2007;315:1130–1133. [DOI] [PubMed] [Google Scholar]
- [24].Bubeck Wardenburg J, Bae T, Otto M, et al. Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med. 2007;13:1405–1406. [DOI] [PubMed] [Google Scholar]
- [25].Randolph AG, Vaughn F, Sullivan R, et al. Critically ill children during the 2009–2010 influenza pandemic in the United States. Pediatrics. 2011;128:e1450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rice TW, Rubinson L, Uyeki TM, et al. Critical illness from 2009 pandemic influenza A (H1N1) virus and bacterial co-infection in the United States. Crit Care Med. 2012;40:1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Reddinger RM, Luke-Marshall NR, Hakansson AP, et al. Host physiologic changes induced by influenza A virus lead to Staphylococcus aureus biofilm dispersion and transition from asymptomatic colonization to invasive disease. mBio. 2016;7:e01235–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sherertz RJ, Reagan DR, Hampton KD, et al. A cloud adult: the Staphylococcus aureus-virus interaction revisited. Ann Intern Med. 1996;124:539–547. [DOI] [PubMed] [Google Scholar]
- [29].Bischoff WE, Bassetti S, Bassetti-Wyss BA, et al. Airborne dispersal as a novel transmission route of coagulase-negative staphylococci: interaction between coagulase-negative staphylococci and rhinovirus infection. Infect Control Hosp Epidemiol. 2004;25:504–511. [DOI] [PubMed] [Google Scholar]
- [30].Bassetti S, Bischoff WE, Walter M, et al. Dispersal of Staphylococcus aureus into the air associated with a rhinovirus infection. Infect Control Hosp Epidemiol. 2005;26:196–203. [DOI] [PubMed] [Google Scholar]
- [31].Bischoff WE, Wallis ML, Tucker BK, et al. “Gesundheit!” sneezing, common colds, allergies, and Staphylococcus aureus dispersion. J Infect Dis. 2006;194:1119–1126. [DOI] [PubMed] [Google Scholar]
- [32].Bischoff WE, Tucker BK, Wallis ML, et al. Preventing the airborne spread of Staphylococcus aureus by persons with the common cold: effect of surgical scrubs, gowns, and masks. Infect Control Hosp Epidemiol. 2007;28:1148–1154. [DOI] [PubMed] [Google Scholar]
- [33].Merler S, Poletti P, Ajelli M, et al. Coinfection can trigger multiple pandemic waves. J Theor Biol. 2008;254:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hofstra JJ, Matamoros S, Van De Pol MA, et al. Changes in microbiota during experimental human rhinovirus infection. BMC Infect Dis. 2015;15:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Thorburn K. High incidence of pulmonary bacterial co-infection in children with severe respiratory syncytial virus (RSV) bronchiolitis. Thorax. 2006;61:611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Suárez-Arrabal MC, Mella C, Lopez SM, et al. Nasopharyngeal bacterial burden and antibiotics: influence on inflammatory markers and disease severity in infants with respiratory syncytial virus bronchiolitis. J Infect. 2015;71:458–469. [DOI] [PubMed] [Google Scholar]
- [37].Jiang W, Wang T, Li L, et al. Impact of bacteria in nasal aspirates on disease severity of bronchiolitis. Infect Dis. 2016;48:82–86. [DOI] [PubMed] [Google Scholar]
- [38].Rosas-Salazar C, Shilts MH, Tovchigrechko A, et al. Differences in the nasopharyngeal microbiome during acute respiratory tract infection with human rhinovirus and respiratory syncytial virus in infancy. J Infect Dis. 2016;214:1924–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].De Steenhuijsen Piters WAA, Heinonen S, Hasrat R, et al. Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am J Respir Crit Care Med. 2016;194:1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fukutani KF, Nascimento-Carvalho CM, Van Der Gucht W, et al. Pathogen transcriptional profile in nasopharyngeal aspirates of children with acute respiratory tract infection. J Clin Virol. 2015;69:190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stensballe LG, Hjuler T, Andersen A, et al. Hospitalization for respiratory syncytial virus infection and invasive pneumococcal disease in Danish children aged <2 years: a population-based cohort study. Clin Infect Dis. 2008;46:1165–1171. [DOI] [PubMed] [Google Scholar]
- [42].Weinberger DM, Klugman KP, Steiner CA, et al. Association between respiratory syncytial virus activity and pneumococcal disease in infants: a time series analysis of US hospitalization data. PLoS Med. 2015;12:e1001776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Metersky ML, Masterton RG, Lode H, et al. Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int J Infect Dis. 2012;16:e321–31. [DOI] [PubMed] [Google Scholar]
- [44].Passariello C, Nencioni L, Sgarbanti R, et al. Viral hemagglutinin is involved in promoting the internalisation of Staphylococcus aureus into human pneumocytes during influenza A H1N1 virus infection. Int J Med Microbiol. 2011;301:97–104. [DOI] [PubMed] [Google Scholar]
- [45].Scheiblauer H, Reinacher M, Tashiro M, et al. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J Infect Dis. 1992;166:783–791. [DOI] [PubMed] [Google Scholar]
- [46].Warnking K, Klemm C, Löffler B, et al. Super-infection with Staphylococcus aureus inhibits influenza virus-induced type I IFN signalling through impaired STAT1-STAT2 dimerization. Cell Microbiol. 2015;17:303–317. [DOI] [PubMed] [Google Scholar]
- [47].Mäkelä MJ, Puhakka T, Ruuskanen O, et al. Viruses and bacteria in the etiology of the common cold. J Clin Microbiol. 1998;36:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gavala ML, Bertics PJ, Gern JE. Rhinoviruses, allergic inflammation, and asthma: rhinovirus infections, allergy, and asthma. Immunol Rev. 2011;242:69–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Triantafilou K, Vakakis E, Richer EAJ, et al. Human rhinovirus recognition in non-immune cells is mediated by Toll -like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence. 2011;2:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Slater L, Bartlett NW, Haas JJ, et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010;6:e1001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Selinger DS, Reed WP, McLaren LC. Model for studying bacterial adherence to epithelial cells infected with viruses. Infect Immun. 1981;32:941–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Passariello C, Schippa S, Conti C, et al. Rhinoviruses promote internalisation of Staphylococcus aureus into non-fully permissive cultured pneumocytes. Microbes Infect Inst Pasteur. 2006;8:758–766. [DOI] [PubMed] [Google Scholar]
- [53].Wang JH, Kwon H-J, Lee B-J, et al. Staphylococcal enterotoxins A and B enhance rhinovirus replication in A549 cells. Am J Rhinol. 2007;21:670–674. [DOI] [PubMed] [Google Scholar]
- [54].Wang JH, Kwon HJ, Jang YJ. Rhinovirus enhances various bacterial adhesions to nasal epithelial cells simultaneously. Laryngoscope. 2009;119:1406–1411. [DOI] [PubMed] [Google Scholar]
- [55].Wang JH, Lee SH, Kwon HJ, et al. Clarithromycin inhibits rhinovirus-induced bacterial adhesions to nasal epithelial cells. Laryngoscope. 2010;120:193–199. [DOI] [PubMed] [Google Scholar]
- [56].Min J-Y, Shin S-H, Kwon HJ, et al. Levocetirizine inhibits rhinovirus-induced bacterial adhesion to nasal epithelial cells through down-regulation of cell adhesion molecules. Ann Allergy Asthma Immunol. 2012;108:44–48. [DOI] [PubMed] [Google Scholar]
- [57].Hauck CR, Ohlsen K. Sticky connections: extracellular matrix protein recognition and integrin-mediated cellular invasion by Staphylococcus aureus. Curr Opin Microbiol. 2006;9:5–11. [DOI] [PubMed] [Google Scholar]
- [58].Schwarz-Linek U, Werner JM, Pickford AR, et al. Pathogenic bacteria attach to human fibronectin through a tandem beta-zipper. Nature. 2003;423:177–181. [DOI] [PubMed] [Google Scholar]
- [59].Sinha B, François PP, Nüs Se O, et al. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin α5β1. Cell Microbiol. 1999;1:101–117. [DOI] [PubMed] [Google Scholar]
- [60].Van Kempen M, Bachert C, Van Cauwenberge P. An update on the pathophysiology of rhinovirus upper respiratory tract infections. Rhinology. 1999;37:97–103. [PubMed] [Google Scholar]
- [61].Jang YJ, Wang JH, Kim JS, et al. Levocetirizine inhibits rhinovirus-induced ICAM-1 and cytokine expression and viral replication in airway epithelial cells. Antiviral Res. 2009;81:226–233. [DOI] [PubMed] [Google Scholar]
- [62].Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-κB-mediated transcription. J Biol Chem. 1999;274:9707–9720. [DOI] [PubMed] [Google Scholar]
- [63].Kim J, Sanders SP, Siekierski ES, et al. Role of NF-kappa B in cytokine production induced from human airway epithelial cells by rhinovirus infection. J Immunol. 2000;165:3384–3392. [DOI] [PubMed] [Google Scholar]
- [64].Chavakis T, Hussain M, Kanse SM, et al. Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med. 2002;8:687–693. [DOI] [PubMed] [Google Scholar]
- [65].Palma M, Haggar A, Flock JI. Adherence of Staphylococcus aureus is enhanced by an endogenous secreted protein with broad binding activity. J Bacteriol. 1999;181:2840–2845.10217776 [Google Scholar]
- [66].Hussain M, Haggar A, Heilmann C, et al. Insertional inactivation of Eap in Staphylococcus aureus strain Newman confers reduced staphylococcal binding to fibroblasts. Infect Immun. 2002;70:2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Haggar A, Hussain M, Lonnies H, et al. Extracellular adherence protein from Staphylococcus aureus enhances internalization into eukaryotic cells. Infect Immun. 2003;71:2310–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bur S, Preissner KT, Herrmann M, et al. The Staphylococcus aureus extracellular adherence protein promotes bacterial internalization by keratinocytes independent of fibronectin-binding proteins. J Invest Dermatol. 2013;133:2004–2012. [DOI] [PubMed] [Google Scholar]
- [69].Lee LY, Miyamoto YJ, McIntyre BW, et al. The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell–mediated responses. J Clin Invest. 2002;110:1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Harraghy N. The adhesive and immunomodulating properties of the multifunctional Staphylococcus aureus protein Eap. Microbiology. 2003;149:2701–2707. [DOI] [PubMed] [Google Scholar]
- [71].Chavakis T, Wiechmann K, Preissner KT, et al. Staphylococcus aureus interactions with the endothelium: the role of bacterial “secretable expanded repertoire adhesive molecules” (SERAM) in disturbing host defense systems. Thromb Haemost. 2005;94:278–285. [DOI] [PubMed] [Google Scholar]
- [72].Athanasopoulos AN, Economopoulou M, Orlova VV, et al. The extracellular adherence protein (Eap) of Staphylococcus aureus inhibits wound healing by interfering with host defense and repair mechanisms. Blood. 2006;107:2720–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Vercellotti GM, McCarthy JB, Lindholm P, et al. Extracellular matrix proteins (fibronectin, laminin, and type IV collagen) bind and aggregate bacteria. Am J Pathol. 1985;120:13–21. [PMC free article] [PubMed] [Google Scholar]
- [74].Bodén MK, Flock JI. Evidence for three different fibrinogen-binding proteins with unique properties from Staphylococcus aureus strain Newman. Microb Pathog. 1992;12:289–298. [DOI] [PubMed] [Google Scholar]
- [75].McGavin MH, Krajewska-Pietrasik D, Rydén C, et al. Identification of a Staphylococcus aureus extracellular matrix-binding protein with broad specificity. Infect Immun. 1993;61:2479–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kojima T, Go M, Takano K, et al. Regulation of tight junctions in upper airway epithelium. Biomed Res Int. 2013;2013:947072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–28. [DOI] [PubMed] [Google Scholar]
- [78].Comstock AT, Ganesan S, Chattoraj A, et al. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J Virol. 2011;85:6795–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Biagioli MC, Kaul P, Singh I, et al. The role of oxidative stress in rhinovirus induced elaboration of IL-8 by respiratory epithelial cells. Free Radic Biol Med. 1999;26:454–462. [DOI] [PubMed] [Google Scholar]
- [80].Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. [DOI] [PubMed] [Google Scholar]
- [81].Sajjan U, Wang Q, Zhao Y, et al. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am J Respir Crit Care Med. 2008;178:1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Otto M. Staphylococcus aureus toxins. Curr Opin Microbiol. 2014;17:32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Morishita Y, Tada J, Sato A, et al. Possible influences of Staphylococcus aureus on atopic dermatitis– the colonizing features and the effects of staphylococcal enterotoxins. Clin Exp Allergy J. 1999;29:1110–1117. [DOI] [PubMed] [Google Scholar]
- [84].Krakauer T. Stimulant-dependent modulation of cytokines and chemokines by airway epithelial cells: cross talk between pulmonary epithelial and peripheral blood mononuclear cells. Clin Diagn Lab Immunol. 2002;9:126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Damm M, Quante G, Rosenbohm J, et al. Proinflammatory effects of Staphylococcus aureus exotoxin B on nasal epithelial cells. Otolaryngol-Head Neck Surg. 2006;134:245–249. [DOI] [PubMed] [Google Scholar]
- [86].Stein RT, Bont LJ, Zar H, et al. Respiratory syncytial virus hospitalization and mortality: systematic review and meta-analysis: incidence of RSV hospitalization and mortality. Pediatr Pulmonol. 2017;52:556–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Paton AW, Goldwater PN. Respiratory syncytial virus modulation of adult and neonatal lymphocyte mitogenic responses and the role of interferon-gamma. Microb Pathog. 1990;9:235–241. [DOI] [PubMed] [Google Scholar]
- [88].Blackwell CC, Saadi AT, Raza MW, et al. Susceptibility to infection in relation to SIDS. J Clin Pathol. 1992;45:20–24. [PubMed] [Google Scholar]
- [89].Saadi AT, Blackwell CC, Raza MW, et al. Factors enhancing adherence of toxigenic Staphylococcus aureus to epithelial cells and their possible role in sudden infant death syndrome. Epidemiol Infect. 1993;110:507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Stark JM, Stark MA, Colasurdo GN, et al. Decreased bacterial clearance from the lungs of mice following primary respiratory syncytial virus infection. J Med Virol. 2006;78:829–838. [DOI] [PubMed] [Google Scholar]
- [91].Raza MW, Blackwell CC, Molyneaux P, et al. Association between secretor status and respiratory viral illness. Br Med J. 1991;303:815–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Highet AR. An infectious aetiology of sudden infant death syndrome. J Appl Microbiol. 2008;105:625–635. [DOI] [PubMed] [Google Scholar]
- [93].Kim TH, Lee HK. Innate immune recognition of respiratory syncytial virus infection. BMB Rep. 2014;47:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lambert L, Sagfors AM, Openshaw PJM, et al. Immunity to RSV in early-life. Front Immunol. 2014;5:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Avadhanula V, Rodriguez CA, DeVincenzo JP, et al. Respiratory viruses augment the adhesion of bacterial pathogens to respiratory epithelium in a viral species- and cell type-dependent manner. J Virol. 2006;80:1629–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Toth TE, Gates C. Lack of virus-specific bacterial adherence to bovine embryonic lung cells infected with bovine parainfluenza virus type 3. J Clin Microbiol. 1983;17:561–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Verkaik NJ, Nguyen DT, De Vogel CP, et al. Streptococcus pneumoniae exposure is associated with human metapneumovirus seroconversion and increased susceptibility to in vitro HMPV infection. Clin Microbiol Infect. 2011;17:1840–1844. [DOI] [PubMed] [Google Scholar]
- [98].Atanasova K, Van Gucht S, Barbé F, et al. Lipoteichoic acid from Staphylococcus aureus exacerbates respiratory disease in porcine respiratory coronavirus-infected pigs. Vet J. 2011;188:210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Li F, Zhu H, Sun R, et al. Natural killer cells are involved in acute lung immune injury caused by respiratory syncytial virus infection. J Virol. 2012;86:2251–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]