

Abstract
Essentials.
Fibrinogen Disorders are characterized by variable expressivity.
Patients with fibrinogen disorders can present with bleeding, thrombosis, or both.
As previously reported, genotype‐phenotype correlations are difficult to establish.
Molecular modeling may help to further understand the effects of mutations on the mature fibrinogen protein.
Introduction
Fibrinogen is a complex molecule comprised of two sets of Aα, Bβ, and γ chains. Fibrinogen deficiencies can lead to the development of bleeding or thromboembolic events. The objective of this study was to perform DNA sequence analysis of patients with clinical fibrinogen abnormalities, and to perform genotype‐phenotype correlations.
Materials and Methods
DNA from 31 patients was sequenced to evaluate disease‐causing mutations in the three fibrinogen genes: FGA,FGB, and FGG. Clinical data were extracted from medical records or from consultation with referring hematologists. Fibrinogen antigen and functional (Clauss method) assays, as well as reptilase time (RT) and thrombin time (TT) were obtained for each patient. Molecular modeling was used to simulate the functional impact of specific missense variants on the overall protein structure.
Results
Seventeen mutations, including six novel mutations, were identified in the three fibrinogen genes. There was little correlation between genotype and phenotype. Molecular modeling predicted a substantial conformational change for a novel variant, FGG p.Ala289Asp, leading to a more rigid molecule in a region critical for polymerization and alignment of the fibrin monomers. This mutation is associated with both bleeding and clotting in the two affected individuals.
Conclusions
Robust genotype‐phenotype correlations are difficult to establish for fibrinogen disorders. Molecular modeling might represent a valuable tool for understanding the function of certain missense fibrinogen mutations but those should be followed by functional studies. It is likely that genetic and environmental modifiers account for the incomplete penetrance and variable expressivity that characterize fibrinogen disorders.
Keywords: afibrinogenemia, dysfibrinogenemia, fibrinogen disorders, fibrinogen mutations, molecular modeling
1. INTRODUCTION
Fibrinogen is a 340‐kDa glycoprotein with numerous and diverse functions in hemostasis. The mature fibrinogen molecule is composed of two sets of three polypeptide chains, Aα, Bβ, and γ, which are joined by disulfide bridges. Mature fibrin is formed by thrombin‐mediated cleavage of specific fibrinopeptides from the Aα and Bβ chains to expose polymerization sites (EA), which interact with complementary binding pockets on the γ and β chains (the terminal D regions). Interaction of thrombin‐cleaved EA regions with the D regions, as well as interactions between D regions (D:D) allow for end‐to‐middle and end‐to‐end assembly of the fibrin molecule, respectively.1, 2 Mature fibrin supports platelet aggregation, provides structural stability to the clot, promotes interactions with platelets and endothelial cells, and serves as a binding site for circulating plasma factors such as factor XIII, thrombin, and fibrinolytic proteins (eg, tissue‐type plasminogen activator [tPA] and plasminogen).1, 3, 4
The mature fibrinogen molecule is encoded by three genes, fibrinogen alpha chain (FGA), fibrinogen beta chain (FGB), and fibrinogen gamma chain (FGG), located contiguously on chromosome 4q23.5 Mutations in these genes can lead to quantitative (afibrinogenemia and hypofibrinogenemia) or qualitative (dysfibrinogenemia and hypodysfibrinogenemia) fibrinogen abnormalities which are diagnosed by plasma fibrinogen antigen and activity assays.6 Congenital afibrinogenemia (CAF) is characterized by undetectable plasma fibrinogen antigen and bleeding. It is inherited in an autosomal recessive manner with mutations or deletions commonly located in FGA.7, 8 Hypofibrinogenemia is characterized by a decreased plasma fibrinogen antigen level that is proportionally related to decreased fibrinogen function, with phenotypes ranging from asymptomatic to mild or moderate bleeding. Patients with hypofibrinogenemia are often heterozygous carriers of afibrinogenemia mutations.6 Approximately 30% of patients with CAF or severe hypofibrinogenemia develop thrombosis, which may be due to embolism of an unstable thrombus.9, 10 Congenital dysfibrinogenemia (CD) is a qualitative defect classified by decreased functional activity despite normal antigen levels.11 CD mutations are often inherited in an autosomal dominant manner and are commonly found in exon 2 of FGA, which encodes the thrombin cleavage site for EA exposure enabling EA‐D interactions, or in exon 8 of FGG, which is involved in D:D end‐to‐end assembly of fibrin.12 Clinical presentation is highly variable13 and approximately 50% of CD patients are diagnosed incidentally.14, 15 Hypodysfibrinogenemia is defined by disproportionately decreased fibrinogen antigen and decreased function.5 Patients with hypodysfibrinogenemia patients can be asymptomatic or have bleeding and/or clotting events.
Clinically, the diagnosis of fibrinogen disorders is based on antigen and functional assays, often in addition to thrombin time (TT) and reptilase time (RT). However, phenotypic variability and lack of correlation between laboratory values, symptoms manifestation and disease severity complicate diagnosis and consequently, clinical management.
In this study, we evaluated 31 patients with fibrinogen disorders for disease‐causing mutations, performed molecular modeling to elucidate the function of amino acid residues from novel variants, and attempted to correlate these genetic variants with fibrinogen levels and clinical presentation. These data contribute to understanding the function of fibrinogen in thrombosis and hemostasis.
2. METHODS
2.1. Subject identification
Subjects with fibrinogen disorders were identified for this study by the presence of: (i) a previously diagnosed congenital fibrinogen deficiency; (ii) abnormal laboratory results (either symptomatic or asymptomatic and diagnosed during routine or presurgical screenings); and/or (iii) a positive family history of a fibrinogen disorder. Thirty‐one subjects (20 subjects from 7 families, in addition to 11 other unrelated individuals), were identified at the Hemostasis and Thrombosis Centers in Denver, Colorado; Houston, Texas; and North Carolina in the United States; and in Cordoba, Argentina.
2.2. Fibrinogen measurements
Whole blood samples were drawn from all consenting patients in accordance with the Declaration of Helsinki and University of Colorado‐Denver Institutional Review Boards. Whole blood was processed to platelet‐poor plasma via centrifugation (15 min, 2500 ×g), re‐centrifugation of the plasma supernatant at the same settings, and then flash frozen at −80°C. Buffy coat samples were extracted after the initial centrifugation following plasma removal. Research tests were performed on plasmas thawed at 37°C. Fibrinogen antigen was assayed by ELISA, ZYMUTEST (Aniara Diagnostica, Inc, West Chester, OH, USA). Fibrinogen activity was determined by the Clauss method.16 TT and RT were performed on the Diagnostica Stago Compact Max Analyzer, according to the manufacturer's instructions (Diagnostica Stago, Inc., Parsippany, NJ, USA).
2.3. Clinical diagnoses
Clinical information regarding the type and severity of both bleeding and thromboembolic events were collected from electronic medical records and from communication with treating physicians. Initially, bleeding status was determined by the treating physician and was classified as mild, moderate, or severe and bleeding sites were defined as either mucosal, joint, muscle, intracranial, or other internal bleeding. In order to make the diagnosis of bleeding more consistent and since patients were from different hemophilia centers, clinical bleeding severity grades were assigned based on categories defined by the European network of Rare Bleeding Disorders (EN‐RBD).17 Thrombosis was defined as any venous or arterial thromboembolic event. A diagnosis of thromboembolism was determined using radiological evidence: ultrasonography with Doppler imaging for objective confirmation of extremity DVT, spiral computed tomography (CT), or high‐resolution computed tomography (HRCT) for PE and CT or magnetic resonance imaging (MRI) for intracranial hemorrhage (ICH). Disease classification (CAF, CD, hypofibrinogenemia, hypodysfibrinogenemia, and normal fibrinogen levels) was based on clinical laboratory values consistent with the definitions previously described.18
2.4. DNA sequencing
Sanger sequencing was performed for FGA, FGB, and FGG on genomic DNA to evaluate for disease‐causing mutations. DNA from buffy coat or whole blood samples was isolated using a commercially available FlexiGene DNA kit (Qiagen Sciences Inc., Germantown, MD, USA) following instructions from the published protocol. The exons, intron‐exon boundaries, and core promoter regions of the fibrinogen genes were amplified by Polymerase Chain Reaction (PCR) using the G‐storm thermal cycler (G‐storm, Somerset, UK). Primers were designed using the published FGA, FGB, and FGG sequences and are available upon request. PCR products were purified using ExoSap‐IT (Affymetrix, Santa Clara, CA, USA) according to the manufacturer's instructions. Nucleotide sequences were determined using the Sanger sequencing method as previously described.19 Mutations were compared against previously described mutations as reported by Ensembl (http://www.ensembl.org) and Groupe d'Etude sur l'Hemostase et la Thrombose (GEHT) (http://www.geht.org) databases, as well as against known non disease‐causing single nucleotide polymorphisms (SNPs) described in the dbSNP database (http://www.ncbi.nlm.nih.gov). Variants were reported according to guidelines determined by the International Society of Thrombosis and Hemostasis (ISTH).20 For the novel splice site variant, Human Splicing Finder (http://www.umd.be/HSF/) was used to evaluate the potential effect of the variant.
2.5. In silico molecular modeling
Molecular modeling simulations were conducted using the Accelrys Discovery Studio 4.0 (Accelrys Software, Inc., San Diego, CA; http://accelrys.com). The crystal structure coordinates of the human fibrinogen molecule were obtained from the protein data bank (http://www.pdb.org; PDB ID: 3GHG and 1FZB). Structures of the γ and Bβ chain mutants were constructed by altering the relevant wild‐type residues to the mutant form within Discovery Studio. Predicted mutation‐induced changes in energy and/or protein conformation of the fibrinogen complex and the fragment D dimer were determined by subjecting the wild‐type and mutant structures to solvent‐based energy minimization using the smart minimization protocol with a CHARMM forcefield21 and the Generalized Born with Simple Switching (GBSW) implicit solvent model.22 Predicted protein–protein interactions between the minimized wild‐type and/or mutant fibrinogen fragment D structures were calculated using the ZDock algorithm.23 The ZRank scoring function was used to calculate the interaction energies of the resulting predicted protein complexes and represents a combination of van der Waals attractive and repulsive energies, short and long range repulsive and attractive energies, and desolvation.24 Similarity of the predicted protein complexes to the crystal structure (1FZB) was determined by calculating the root mean square deviation (RMSD) using the crystal structure as the reference.25 The three poses with the lowest RMSD values (ie, the greatest similarity to the crystal structure) were selected from each group for the energy comparison. Protein structures were rendered in Lightwave 3D 2015.3 (NewTek, Inc., San Antonio, TX; http://www.lightwave3d.com).
3. RESULTS
3.1. Clinical data (Table 1)
Table 1.
Genetic mutations, fibrinogen laboratory data and clinical categorization for each individual subject
| Disease category | Patient ID | Gene(s) | Native protein mutation | Clauss (1.50‐4.00 g/L) | Fgn Ag (1.50‐4.00 g/L) | Ratio Clauss:Antigen | TT (15‐19s) | RT (15‐19s) | Age (years) at Diaqnosis | Sex | Phenotype | Bleeding grade |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Afibrinogenemia | 1 | FGA/ FGA | p. Met(‐19)Val/ IVS4+1G>T | <0.10 | <0.10 | N/A | >120 | >120 | 1 | M | B | III |
| 2 | FGA/ FGA | p.Leu169PhefsX4/ p.Leu169PhefsX4 | <0.10 | <0.10 | N/A | >120 | >120 | 2 | F | B/T | III | |
| 3 | FGG/ FGG | p. Lys85X/ IVS6‐12A>G | <0.10 | <0.10 | N/A | 83.1 | >120 | 1 | F | B/T | III | |
| Dysfibrinogenemia | 4 | FGG | p. Ala82Gly | 1.42 | 3.01 | 0.47 | 16.5 | 31.0 | 18 | h | A | N/A |
| 5 | FGG | p. Arg275His | 0.67 | 1.88 | 0.36 | 24.0 | 26.0 | 58 | M | T | N/A | |
| 6 | FGG | p. Arg275His | 0.88 | 2.01 | 0.44 | 22.0 | 28.0 | 23 | M | A | N/A | |
| 7 | FGG | p. Arg275His | 0.52 | 2.10 | 0.25 | 34.0 | 75.0 | 62 | F | T | N/A | |
| 8 | FGG | p.Arg275His | 0.89 | 1.67 | 0.53 | 79.9 | 37.4 | 71 | M | A | N/A | |
| 9 | FGG | p. Arg275His | 0.55 | 1.58 | 0.35 | 33.0 | 28.0 | 33 | M | B | I | |
| 10* | FGG | p. Ala289Asp | 0.78 | 1.01 | 0.77 | 25.5 | 34.1 | 45 | M | B/T | I | |
| 11* | FGG | p. Ala289Asp | 1.46 | 1.69 | 0.86 | 19.6 | 24.6 | 0 | F | B/T | II | |
| 12 | FGG | p. Cys326Ser | 0.82 | 1.48 | 0.55 | 34.0 | 27.0 | 40 | M | T | N/A | |
| 13 | FGA | p.Arg16His | 0.98 | 3.22 | 0.30 | ND | ND | 22 | F | B | II | |
| 14 | FGA | p.Arg16His | 0.71 | 2.75 | 0.26 | 36.7 | >120 | 5 | F | B | II | |
| 15 | FGA | p. Pro192Thr | 1.02 | 1.46 | 0.70 | ND | ND | 23 | F | B | I | |
| 16 | N/A | N/A | 0.49 | 1.46 | 0.34 | 35.5 | 23.6 | 33 | F | B | II | |
| Hypodysfibrinogenemia | 17 | FGA | p. Arg16Cys | 0.31 | 0.56 | 0.55 | 51.0 | 45.0 | 40 | F | A | N/A |
| 18 | FGA/ FGA | p. Arg16Cys/ p.Trp33X | <0.10 | 0.31 | N/A | >120 | >120 | 4 | F | B | II | |
| 19 | FGB | p. Cys326Ser | 0.22 | 0.43 | 0.51 | 49.0 | 36.0 | 40 | F | A | N/A | |
| 20 | N/A | N/A | 0.22 | 0.33 | 0.67 | 29.5 | 53.0 | 8 | F | B | II | |
| Hypofibrinogenemia | 21 | FGB | p. Gln189Arg | 1.47 | 1.56 | 0.94 | 17.2 | 19.4 | 27 | F | A | N/A |
| 22 | FGB | p. Gly272Arg | 1.19 | 1.49 | 0.80 | 17.0 | 25.0 | 42 | F | A | N/A | |
| 23 | FGB | p. Arg455Lys | 1.27 | 1.50 | 0.85 | 17.5 | 19.0 | 2 | M | B | II | |
| 24 | FGB | p.Tyr345Cys | 1.16 | 1.22 | 0.95 | ND | ND | 28 | M | A | N/A | |
| Normal fibrinogen levels | 25 | FGG | p. Lys85X | 1.54 | 1.54 | 0.99 | 19.6 | 20.0 | 52 | M | T | N/A |
| 26 | FGG | IVS6‐12A>G | 1.75 | 1.87 | 0.93 | 18.7 | 19.6 | 55 | F | T | N/A | |
| 27 | FGA | p. Met(‐19)Val | 2.59 | 2.95 | 0.88 | 16.6 | 16.7 | 45 | M | A | N/A | |
| 28 | FGA | IVS4+1G>T | 2.40 | 2.47 | 0.97 | 17.5 | 17.8 | 47 | F | B | II | |
| 29 | FGA | p. Trp33X | 1.70 | 1.85 | 0.92 | 12.9 | 17.9 | 41 | M | A | N/A | |
| 30 | FGA | p.Leu169PhefsX4 | 3.50 | 3.73 | 0.94 | ND | ND | 18 | M | A | N/A | |
| 31 | FGB | p. Arg455Lys | 2.17 | 2.44 | 0.89 | 16.8 | 19.4 | 36 | M | A | N/A |
Disease categories are classified based on fibrinogen antigen and activity. Phenotype is simplified to A for asymptomatic, B for bleeding symptoms, T for thrombosis, and B/T for both bleeding and thrombosis. ND is indicated when no data for the subject could be obtained.
3.1.1. Patients with congenital afibrinogenemia
All of the subjects with CAF demonstrated bleeding early in life. Subject 1 manifested with moderate bleeding since early childhood with soft tissue and muscle hemorrhages. Subject 2 developed six recurrent intracranial hemorrhages over 20 years. She has remained on prophylactic replacement of fibrinogen twice weekly. Subject 3 has a moderately severe bleeding tendency with recurrent hemarthrosis, muscle and soft tissue hemorrhages and menorrhagia. She developed a spontaneous pulmonary embolism (PE) at age 21, five weeks after receiving fibrinogen replacement. Both parents developed DVT in their 50s. Interestingly, both parents have normal fibrinogen levels.
3.1.2. Patients with congenital dysfibrinogenemia
Subject 5 suffered from an acute myocardial infarction at age 59, while on oral anticoagulation. Subject 7 suffered from a lower extremity DVT after knee surgery at age 62. Subject 9 had excessive perioperative bleeding. Subject 11, suffered a left cerebral arterial stroke (likely in utero) and was diagnosed with fibrinogen deficiency shortly after birth. She also showed spontaneous and perioperative bleeding requiring fibrinogen replacement. The father, subject 10, suffered from mild mucosal bleeding and developed an unprovoked lower extremity DVT at age 49. They are both negative for other known thrombophilia. Subject 12 had a spontaneous PE at age 43. He remains on lifelong anticoagulation. Subject 13 had bleeding with surgeries and menorrhagia. Subject 14 had oral bleeding and mild epistaxis. Subject 15 suffered a knee hemarthrosis injury necessitating fibrinogen replacement after minor trauma. Subject 16 has a long history of menorrhagia and suffered excessive bleeding post‐partum. Subjects 4 and 8 were asymptomatic and were diagnosed with fibrinogen abnormalities incidentally or during pre‐surgical screenings.
3.1.3. Patients with hypodysfibrinogenemia
Subject 18 experienced excessive bleeding necessitating a transfusion following a frenulum tear at age 5. Both the mother, and father, subjects 17 and 29, respectively, are asymptomatic. Subject 19 is asymptomatic. Subject 20 has excessive oral bleeding and menorrhagia.
3.1.4. Patients with hypofibrinogenemia
Subjects 21, 22, and 24 are asymptomatic. Subject 23 had profuse bleeding during circumcision.
3.2. Sequencing results
Mutations were identified in 29 out of 31 patients (93.5%) (Table 2). Seventeen different mutations in 29 patients were found: 6 novel and 11 previously described variants, in all 3 of the fibrinogen genes (Figure 1). There were 12 missense mutations, 2 nonsense mutations, 2 splice site mutations, and 1 insertion/frameshift mutation. Novel mutations identified were: FGA Met(‐19)Val, FGA p.Trp33Stop, FGA p. Pro192Thr, FGB p.Arg455Lys, FGG p.Ala289Asp, and FGG IVS6‐12A>G. Consistent with the literature, most of the predicted deleterious mutations (insertions, deletions, nonsense variants) were located in FGA.26 In contrast, most of the missense mutations were predominately found in FGG and FGB were clustered in conserved protein domains that are critical for dimerization.
Table 2.
Genetic sequencing results for each of the 31 subjects
| Disease Category | Patient ID | Family ID | Gene | Intron/ Exon | Native protein mutation | Mature protein mutation | Type of mutation | Genotype status | Novel |
|---|---|---|---|---|---|---|---|---|---|
| Afibrinogenemia | 1 | 1 | FGA/ FGA | Exon 1/ Intron 4 | p.MetlVal/ IVS4+1G>T | p. Met(‐19)Val/ IVS4+1G>T | Missense/ Splice Site | Compound Heterozygous | Yes/ No |
| 2 | 2 | FGA/ FGA | Exon 5/ Exon 5 | p.Leul88PhefsX4/ p.Leul88PhefsX4 | p.Leul69PhefsX4/ p.Leul69PhefsX4 | Frameshift | Homozygous | No | |
| 3 | 3 | FGG/FGG | Exon 4/ Intron 6 | p.LyslllX/ IVS6‐12A>G | p. Lys85X/ IVS6‐12A>G | Nonsense/ Splice Site | Compound Heterozygous | No/Yes | |
| Dysfibrinogenemia | 4 | N/A | FGG | Exon 4 | p. Alal08Gly | p. Ala82Gly | Missense | Heterozygous | No |
| 5 | 4 | FGG | Exon 8 | p. Arg301His | p. Arg275His | Missense | Heterozygous | No | |
| 6 | 4 | FGG | Exon 8 | p. Arg301His | p. Arg275His | Missense | Heterozygous | No | |
| 7 | N/A | FGG | Exon 8 | p. Arg301His | p. Arg275His | Missense | Heterozygous | No | |
| 8 | N/A | FGG | Exon 8 | p.Arg301His | p.Arg275His | Missense | Heterozygous | No | |
| 9 | N/A | FGG | Exon 8 | p. Arg301His | p. Arg275His | Missense | Heterozygous | No | |
| 10 | 5 | FGG | Exon 8 | p. Ala315Asp | p. Ala289Asp | Missense | Heterozygous | Yes | |
| 11 | 5 | FGG | Exon 8 | p. Ala315Asp | p. Ala289Asp | Missense | Heterozygous | Yes | |
| 12 | N/A | FGG | Exon 8 | p. Cys352Ser | p. Cys326Ser | Missense | Heterozygous | No | |
| 13 | N/A | FGA | Exon 2 | p.Arg35His | p.Argl6His | Missense | Heterozygous | No | |
| 14 | N/A | FGA | Exon 2 | p.Arg35His | p.Argl6His | Missense | Heterozygous | No | |
| 15 | N/A | FGA | Exon 5 | p. Pro222Thr | p. Prol92Thr | Missense | Heterozygous | Yes | |
| Hypodysfibrinogenemia | 17 | 6 | FGA | Exon 2 | p. Arg35Cys | p. Argl6Cys | Missense | Heterozygous | No |
| 18 | 6 | FGA/ FGA | Exon 2/ Exon 2 | p. Arg35Cys/ p.Trp52X | p. Argl6Cys/ p.Trp33X | Missense/ Nonsense | Compound Heterozygous | No/ Yes | |
| 19 | N/A | FGB | Exon 8 | p. Cys352Ser | p. Cys326Ser | Missense | Heterozygous | No | |
| Hypofibrinogenemia | 21 | N/A | FGB | Exon 4 | p. Gln219Arg | p. Glnl89Arg | Missense | Heterozygous | Yes |
| 22 | N/A | FGB | Exon 6 | p. Gly302Arg | p. Gly272Arg | Missense | Heterozygous | No | |
| 23 | 7 | FGB | Exon 8 | p. Arg485Lys | p. Arg455Lys | Missense | Heterozygous | Yes | |
| 24 | N/A | FGB | Exon 7 | p. Tyr375Cys | p.Tyr345Cys | Missense | Heterozygous | No | |
| Normal fibrinogen levels | 25 | 3 | FGG | Exon 4 | p.LyslllX | p. Lys85X | Nonsense | Heterozygous | No |
| 26 | 3 | FGG | Intron 6 | IVS6‐12A>G | IVS6‐12A>G | Splice Site | Heterozygous | Yes | |
| 27 | 1 | FGA | Exon 1 | p.MetlVal | p. Met(‐19)Val | Missense | Heterozygous | Yes | |
| 28 | 1 | FGA | Intron 4 | IVS4+1G>T | IVS4+1G>T | Splice Site | Heterozygous | No | |
| 29 | 6 | FGA | Exon 2 | p. Trp52X | p. Trp33X | Nonsense | Heterozygous | Yes | |
| 30 | 2 | FGA | Exon 5 | p.Leul88PhefsX4 | p.Leul69PhefsX4 | Frameshift | Heterozygous | No | |
| 31 | 7 | FGB | Exon 8 | p. Arg485Lys | p. Arg455Lys | Missense | Heterozygous | Yes |
Subjects with normal fibrinogen levels and fibrinogen mutations were all family members of individuals with clinical fibrinogen abnormalities.
Figure 1.
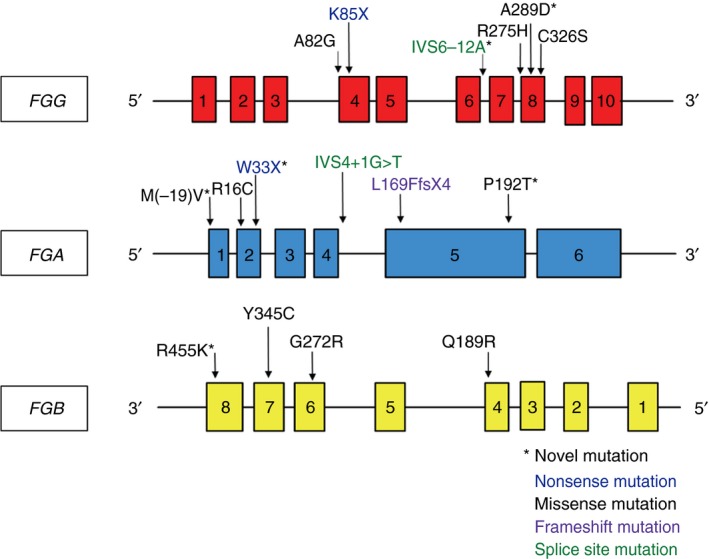
Location of mutations in relation to the exons of the three fibrinogen genes. FGG,FGA, and FGB are located contiguously on chromosome 4q23. FGG and FGA are transcribed in the reverse direction, opposite of FGB. FGG,FGA, and FGB consist of 10, 6, and 8 exons, respectively. Novel mutations are indicated by *, nonsense mutations are colored blue, missense mutations are black, frameshift mutations are purple, and splice site mutations are green. There are missense mutations located throughout the three genes, and the more deleterious mutations (nonsense, frameshift and splice site) are located only in FGG and FGA
Table 3.
Clinical and laboratory aggregate data classified by disease category
| Afibrinogenemia | Dysfibrinogenemia | Hypofibrinogenemia | Hypodysfibrinogenemia | Normal fibrinogen levels | |
|---|---|---|---|---|---|
| Patients (N=31) | 3 | 13 | 3 | 4 | 8 |
| Age | 27 (21‐37) | 39.5 (25‐71) | 27.5 (2‐45) | 25.5 (5‐66) | 45 (27‐59) |
| Fibrinogen Antigen (150‐400 mg/dL) | <10 | 169 (101‐322) | 148 (116‐220) | 38 (31‐56) | 244 (154‐373) |
| Clauss Functional (150‐400 mg/dL) | <10 | 82 (49‐146) | 124.5 (119‐156) | 22 (9‐31) | 217 (154‐350) |
| Thrombin Time (15‐19 s) | >120 s | 33 (19.6‐79.9) | 17.2 (17‐17.5) | 50 (49‐>120) | 17.2 (12.9‐19.6) |
| Bleeding phenotype only | 1 | 5 | 0 | 2 | 2 |
| Thrombotic phenotype only | 0 | 2 | 0 | 0 | 2 |
| Bleeding and thrombosis | 2 | 2 | 0 | 0 | 0 |
| Asymptomatic | 0 | 4 | 3 | 2 | 4 |
Medians and (ranges) are listed for each variable, except for afibrinogenemia subjects when all results fell below the detectable limit. Phenotypes vary widely. Subjects with afibrinogenemia, dysfibrinogenemia and normal fibrinogen levels experienced thrombotic events. Thirteen subjects (42%) overall were asymptomatic, across disease categories.
3.3. FGA
There were two novel mutations in FGA (p. Met[‐19]Val and p. Trp33X). Subjects 1 and 18 had compound heterozygosity involving these variants, p.Met(‐19)Val/IVS4 + 1G>T and p.Trp33X/p.Arg16Cys, and had CAF and hypodysfibrinogenemia, respectively.27 Heterozygous carriers of these novel mutations were asymptomatic and had no fibrinogen laboratory deficiencies (subjects 27 and 29). We found that patients heterozygous for the Arg16Cys mutation presented with dysfibrinogenemia (patients 13 and 14), but interestingly we also found a patient heterozygous for the Arg16Cys (patient 17) with hypodyisfibrinogenemia that did not have additional mutations raising the possibility of either a deletion, a non‐coding variant in the other allele, or other genetic defects outside of the fibrinogen genes. The FGA IVS4 + 1G>T variant, found in subject 1, is the most common afibrinogenemic mutation, activating a cryptic splice site leading to truncation of the protein.28 FGA p.Met(‐19)Val is a substitution of the start codon, methionine to a valine, (ATG>GAG), which could potentially activate an alternative translation initiation site three amino acids downstream. Tirefort et al. published a similar afibrinogenemic mutation, FGA Met(‐19)Leu, reporting that the sequence surrounding the alternative initiation codon three amino acids downstream is not consistent with the Kozak consensus sequence that is necessary for initiation of translation.29, 30
The FGA Arg16 mutation has been previously described as a mutation hotspot in dysfibrinogenemia as it lies in the thrombin cleavage site of fibrinopeptide A, delaying EA exposure and fibrin formation. Interestingly, patients who were heterozygous for this mutation have a similar phenotype to the one patient who was compound heterozygous for this mutation and a null mutation on the other FGA allele, indicating that a heterozygous FGA Arg16 mutation might be sufficient to induce a phenotype, as has been previously reported.31
3.4. FGB
A novel mutation found in FGB was a heterozygous p.Arg455Lys mutation. The majority of subjects with heterozygous FGB mutations had hypofibrinogenemia (67%), one had hypodysfibrinogenemia, and one had normal fibrinogen levels. Two out of six patients with FGB mutations were symptomatic with bleeding, the remainder were asymptomatic.
3.5. FGG
Novel mutations found in FGG were: IVS6‐12A>G and p.Ala289Asp. Although two out of the three patients with CAF had mutations in the FGA gene,32 one subject with CAF had a compound heterozygous mutations in FGG. One missense (p.Lys85X), previously reported and one novel splice‐site mutation (IVS6‐12A>G), both predicted to result in non‐functional gene products of FGG.9 Seven patients had mutations in the C‐terminus region of FGG, a mutation hotspot for variants causing dysfibrinogenemia and a region that functions in D:D interactions, which occur in amino acids 275‐300 of the FGG.6, 33 Two family members and three other unrelated patients had the previously described Bergamo II mutation (FGG p.Arg275His) and two had a novel mutation (FGG p.Ala289Asp) in this region.34 The p.Arg275His mutation disrupts D:D interactions and thus interrupts the fibrin alignment in the conversion from fibrinogen to fibrin.8 Patients with the Bergamo II FGG p.Arg275His mutation (subjects 5‐9) all had normal measurable fibrinogen levels and decreased function (0.52‐0.89 g/L and 1.58‐2.10 g/L, respectively) indicating a qualitative deficiency. Interestingly, these five patients had similar laboratory values and an identical mutation, but exhibited different clinical phenotypes: bleeding, (n = 1), clotting (n = 2), or no symptoms (n = 2), suggesting variable expressivity or other genetic or environmental modifiers of the phenotype. The two related patients, 10 and 11, had a heterozygous FGG p.Ala289Asp mutation located in the same C‐terminus region as the Bergamo II mutation and had hypofibrinogenemia (defined by laboratory standards prior to genetic studies) with mild bleeding and unprovoked thrombotic events.
3.6. Molecular modeling
To gain mechanistic insight into the functional relevance of the four missense mutations FGG p.Ala289Asp, FGB p.Gln189Arg, FGB p.Gly272Arg, FGB p.Arg455Lys, computational molecular modeling was performed to examine the potential structural and functional effects of each mutant. Subjects with the FGG p.Ala289Asp mutation had both mild bleeding and unprovoked clotting events. The mutation substitutes a negatively charged aspartate residue for a neutral, nonpolar alanine residue, introducing a new, much stronger hydrogen bond and pi‐anion interactions with the surrounding residues that were predicted to induce a substantial conformational change in the D:D polymerization interface (Figure 2A). The predicted interaction energy was significantly lower for FGG p.Ala289Asp mutant protein compared with either the WT protein or WT‐mutant protein interaction (average Zrank scores of ‐83.0, ‐65.9, and ‐69.8 kcal/mol, respectively), suggesting more favorable interaction for FGG p.Ala289Asp mutant dimers (Figure 2B). Decreased potential energy for p.Ala289Asp mutant homodimers suggests a more stable structure for complexes containing one or two mutants (data not shown). Overall, the in silico modeling suggests that p.Ala289Asp mutation might lead to a more rapid or stronger interaction between fibrinogen molecules, and could account for the thrombotic propensity in the patients with this mutation. Increased rigidity of the fibrin clot has been found to impair the access of fibrinolytic enzymes to fibrin, resulting in a higher incidence of thrombosis.35 In contrast, more porous, flexible fibrin networks have been associated with increased bleeding tendencies.36
Figure 2.
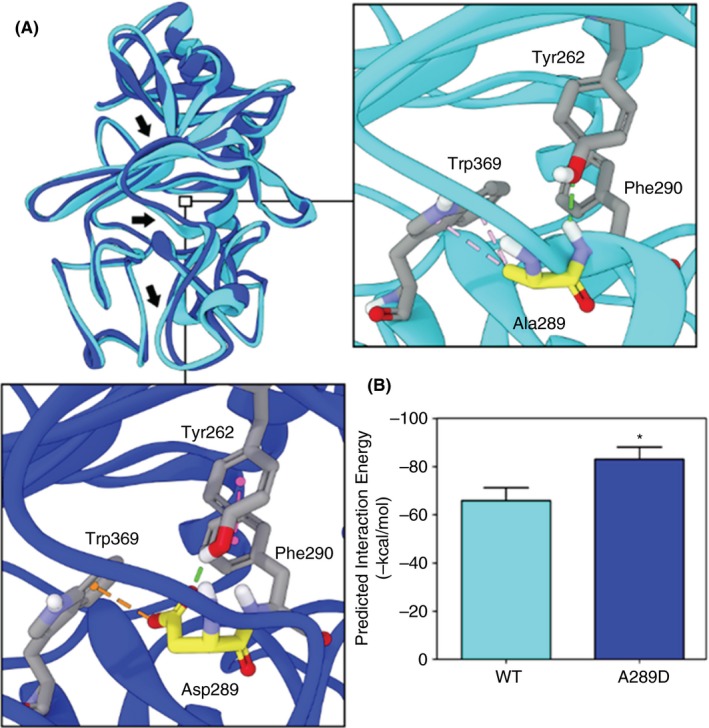
Predicted structural and functional changes of the FGG mutant. (A) Forward facing view of the overlay of the energy minimized WT (cyan) and FGG A289D mutant (blue) human fibrinogen polymerization interface. Arrows highlight major predicted structural alterations in response to the mutation. Insets highlight the predicted local differences between the WT (right) and mutant (bottom). Dashed lines indicate predicted non‐bond interactions (green = hydrogen bonds, orange = pi‐anion, magenta = pi‐pi) (B) Predicted protein–protein interaction energy between two WT or two A289D human fibrinogen molecules. *P = .016
The two Bβ chain mutants FGB p.Gly272Arg, FGB p.Arg455Lys were predicted by in silico modeling to induce conformational changes to the fibrinogen structure by altering electrostatic and hydrogen bond interactions. However, the observed changes do not appear to impact the protein–protein interactions between fibrin(ogen) molecules at the D‐D interaction interface (Figure 3 and data not shown). This is consistent with the clinical phenotype of a mild bleeding tendency and no thrombotic events observed in the subjects with these specific FGB mutations. Interestingly, the FGB p.Gly272Arg mutant, despite representing a drastic change in residue identity, occurs in a region of the protein that can accommodate the much bulkier side chain.
Figure 3.
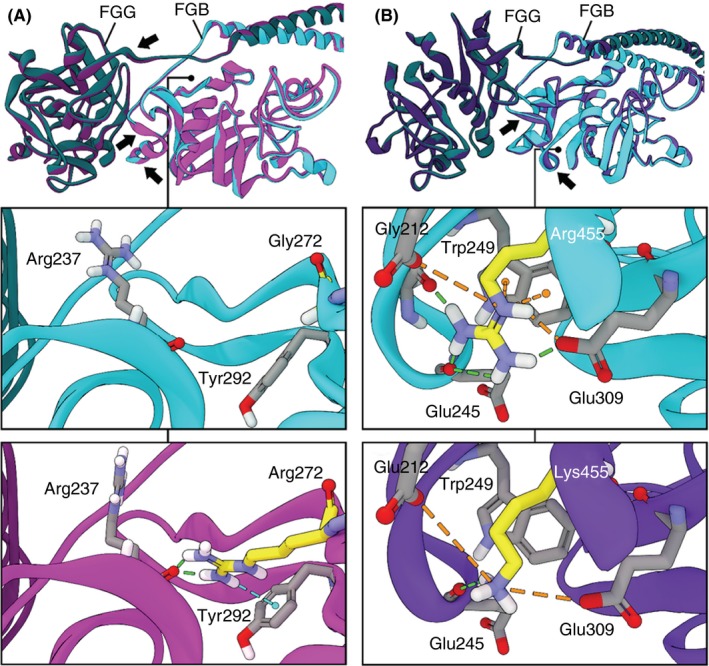
Molecular modeling simulation of WT and FGB mutants. Structural overlays of energy minimized WT (cyan) and (A) Gly272Arg (magenta) or (B) Arg455Lys human fibrinogen. Substantial conformational changes observed between the mutant and WT structures are indicated with arrows. Insets depict the regions surrounding the mutations of each structure separately. Mutated residues are highlighted in yellow and dashed lines indicate predicted non‐bond interactions (green = hydrogen bond, cyan = hydrogen‐pi, orange = electrostatic and pi‐cation)
Conversely, the FGB p.Arg455Lys mutant represents a relatively conservative change, yet it decreases the coordination of the surrounding charged Glu residues via the loss of electrostatic, pi‐cation, and hydrogen bond interactions and would likely result in a decrease in the cohesiveness (increase in flexibility) of this region and could potentially account for the mild bleeding tendency seen in the subject with this mutation.
Finally, the arginine in the FGB p.Gln189Arg mutant induces substantial changes to the protein structure. The bulkiness of the arginine distorts the region and induces additional non‐bond interactions that could result in reduced flexibility or stiffening of that region. The new H‐bonds predicted from the mutation interacts with the backbone carbonyl of Arg167 of the Aα chain.
3.7. Splice site prediction
According to Human Splicing Finder (http://umd.be/HSF3/), the FGG IVS6‐12A>G variant effects the acceptor sites at the 3’ end of intron 6. It alters the wildtype acceptor site and is predicted to activate an intronic cryptic acceptor site 12 nucleotides upstream of exon 7, which could potentially alter splicing.
4. DISCUSSION
The clinical presentation of fibrinogen disorders does not show a consistent correlation with fibrinogen levels, functional assays, or genotype. Results in our cohort were not much different. We did not find clear correlation between the mutations described and any of the functional laboratory analyses or clinical presentation. In the work presented here, subjects with CAF had excessive bleeding or both bleeding and clotting events, while subjects with hypodysfibrinogenemia experienced bleeding or no symptoms. There were asymptomatic subjects in every disease category, except for CAF.
Interestingly, there were seven subjects who had normal laboratory values but harbored heterozygous fibrinogen mutations (individuals 25 to 31, Table 1); four of these individuals were asymptomatic, one experienced excessive bleeding and two had venous thromboembolic events. These individuals were all family members of patients with clinical fibrinogen disorders. The normal fibrinogen levels are somewhat surprising since the mutations detected in these patients have been previously associated with disease in the heterozygous state (FGB p.Arg455Lys), or found in subjects with afibrinogenemia in a homozygous or compound heterozygous state (FGG p. Lys85X, FGG IVS6‐12A>G, FGA p. Met[‐19]Val, FGA IVS4 + 1G>T, FGA p.Trp33X, FGA p.Leu169PhefsX4). Normal fibrinogen levels in these individuals could be attributed to the low sensitivity of the antigen and function assays as these subjects had normal or only slightly prolonged times for the more sensitive, TT and RT assays. This might suggest that these assays are not always efficient measures of laboratory deficiencies.18 Interestingly, of the clinically asymptomatic subjects with fibrinogen mutations, 50% and 70% had prolonged TT and RT, respectively with an average TT of 29.89 seconds and RT of 27.58 seconds (normal range 15‐19 seconds). Fibrinogen antigen and function levels also showed little correlation to phenotype. Excluding CAF subjects, lower function or antigen levels did not correlate with bleeding or thrombotic tendency.
There can be considerable phenotypic variability even when comparing subjects with an identical mutation. For example, all the patients with the FGG p. Arg275His mutation had similar laboratory values, with fibrinogen levels indicative of dysfibrinogenemia, but they all displayed a range of clinical phenotypes from asymptomatic to bleeding and/or thrombosis. FGG p. Arg275His, located in the C‐terminus, or the aC, region of FGG, disrupts gamma dimerization interactions and thus interrupts the fibrin alignment in the conversion from fibrinogen to fibrin, and has been previously associated with both a bleeding and clotting tendency (http://site.geht.org/base-de-donnees-fibrinogene/).34 Hence for the individual patient with a FGG p.Arg275His variant, it is difficult to make any clinical decisions based solely on genotype.
The characterization of four missense mutations by molecular modeling, allowed us to infer the potential functional consequences of these genetic changes. Further functional studies will be needed to characterize these changes. The two individuals with the FGG p.Ala289Asp mutation had fibrinogen levels indicative of hypofibrinogenemia (patient 10) and dysfibrinogenemia, respectively (patient 11), but both patients had unprovoked thrombotic events. Based on molecular modeling, the missense mutation is predicted to induce substantial conformational changes that potentially enhance the protein–protein interactions between fibrinogen molecules, making the ends “stickier.” A more rigid mutant molecule is predicted, which may be the potential mechanism for propensity for clot formation in these individuals. Therefore, our molecular modeling results suggest a dysfunctional protein with thrombotic potential. However, individuals with this mutation in our cohort presented with both bleeding and thrombosis, indicating the limitations of this approach. Other fibrinogen mutations have been reported in this part of the D region and have been associated with bleeding and thrombosis.37, 38
There are several limitations to this study. For example, the rigidity and flexibility assessments derived from molecular modeling experiments in this study reflect the properties of fibrinogen in the static state and there could be differences when extrapolating the static function and applying it to the dynamic environment. DNA microarrays were not used to look for larger deletions and insertions that have been reported in some patients with fibrinogen deficiencies (http://site.geht.org/base-fibrinogene/) so there is the possibility that these types of mutations were undetected in some of these individuals. Finally, there is a possibility that the two patients for which a mutation could not be found or the patients with mutations without predicted damaging effects have an acquired fibrinogen deficiency, although this is less likely when taking age of presentation and clinical history into account.
5. CONCLUSION
Fibrinogen disorders are difficult to characterize due to their phenotypic variability within clinical disease categories. Determination of DNA variants and their molecular function does yield valuable information for understanding the biogenesis and diverse functions of fibrinogen. Functional studies are needed to understand the behavior of these mutations. Meanwhile, molecular modeling may represent a valuable tool for predicting the function of fibrinogen variants in vivo.
RELATIONSHIP DISCLOSURE
The authors declare no conflict of interest relevant to this paper.
AUTHOR CONTRIBUTIONS
NS, LB, LN, ASW, MM, and JDP designed the study, performed experiments and wrote manuscript. HG, SM, CDT, and ME contributed with patients’ samples and clinical information and with the writing of the manuscript. DB performed all molecular modeling simulations. TCW‐A performed genetic sequencing and LJ performed biochemical assays.
Smith N, Bornikova L, Noetzli L, et al. Identification and characterization of novel mutations implicated in congenital fibrinogen disorders. Res Pract Thromb Haemost. 2018;2:800–811. 10.1002/rth2.12127
Funding information
This work was supported by supported by the National Institutes of Health (R01HL120728 to JDP) and the Mountain States Hemophilia Network from the Maternal and Child Health Bureau/HRSA/DHHS H30MC24049 to MMK and JDP. Computational modeling was conducted at the University of Colorado Computational Chemistry and Biology Core Facility which is funded in part by NIH/NCATS Colorado CTSA Grant Number UL1 TR0018.
Natalie Smith and Larissa Bornikova contributed equally
REFERENCES
- 1. Mosesson MW, Siebenlist KR, Hainfeld JF, Wall JS. The covalent structure of factor XIIIa crosslinked fibrinogen fibrils. J Struct Biol. 1995;115:88–101. [DOI] [PubMed] [Google Scholar]
- 2. Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. 2013;121:1712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meh DA, Siebenlist KR, Mosesson MW. Identification and characterization of the thrombin binding sites on fibrin. J Biol Chem. 1996;271:23121–5. [DOI] [PubMed] [Google Scholar]
- 4. Wolberg AS, Campbell RA. Thrombin generation, fibrin clot formation and hemostasis. Transfus Apher Sci. 2008;38:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kant JA, Fornace AJ, Saxe D, Simon MI, McBride OW, Crabtree GR. Evolution and organization of the fibrinogen locus on chromosome 4: gene duplication accompanied by transposition and inversion. Proc Natl Acad Sci U S A. 1985;82:2344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. de Moerloose P, Neerman‐Arbez M. Congenital fibrinogen disorders. Semin Thromb Hemost. 2009;35:356–66. [DOI] [PubMed] [Google Scholar]
- 7. Lak M, Keihani M, Elahi F, Peyvandi F, Mannucci PM. Bleeding and thrombosis in 55 patients with inherited afibrinogenaemia. Br J Haematol. 1999;107:204–6. [DOI] [PubMed] [Google Scholar]
- 8. Peyvandi F, Kaufman RJ, Seligsohn U, et al. Rare bleeding disorders. Haemophilia. 2006;12(Suppl 3):137–42. [DOI] [PubMed] [Google Scholar]
- 9. Zawilska K, Undas A, Fish RJ, Molendowicz‐Portala L, De Moerloose P, Neerman‐Arbez M. Characterisation of a novel nonsense mutation in FGG (Fibrinogen Poznan) causing hypofibrinogenaemia with a mild bleeding tendency. Thromb Haemost. 2010;103:677–9. [DOI] [PubMed] [Google Scholar]
- 10. Ni H, Denis CV, Subbarao S, et al. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000;106:385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Casini A, Neerman‐Arbez M, Ariëns RA, de Moerloose P. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. J Thromb Haemost. 2015;13:909–19. [DOI] [PubMed] [Google Scholar]
- 12. Neerman‐Arbez M, de Moerloose P. Williams Hematology. New York: McGraw‐Hill; 2010. [Google Scholar]
- 13. Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost. 1995;73:151–61. [PubMed] [Google Scholar]
- 14. Casini A, Blondon M, Lebreton A, et al. Natural history of patients with congenital dysfibrinogenemia. Blood. 2015;125:553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shapiro SE, Phillips E, Manning RA, et al. Clinical phenotype, laboratory features and genotype of 35 patients with heritable dysfibrinogenaemia. Br J Haematol. 2013;160:220–7. [DOI] [PubMed] [Google Scholar]
- 16. Clauss A. Rapid physiological coagulation method in determination of fibrinogen. Acta Haematol. 1957;17:237–46. [DOI] [PubMed] [Google Scholar]
- 17. Peyvandi F, Di Michele D, Bolton‐Maggs PH, Lee CA, Tripodi A, Srivastava A; Project on Consensus Definitions in Rare Bleeeding Disorders of the Factor VIII/Factor IX Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis . Classification of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity. J Thromb Haemost. 2012;10:1938–43. [DOI] [PubMed] [Google Scholar]
- 18. Verhovsek M, Moffat KA, Hayward CP. Laboratory testing for fibrinogen abnormalities. Am J Hematol. 2008;83:928–31. [DOI] [PubMed] [Google Scholar]
- 19. Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94:441–8. [DOI] [PubMed] [Google Scholar]
- 20. Medved L, Weisel JW. Haemostasis FaFXSoSSCoISoTa. Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost. 2009;7:355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brooks BR, Brooks CL 3rd, Mackerell AD Jr, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL 3rd. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem. 2004;25:265–84. [DOI] [PubMed] [Google Scholar]
- 23. Chen R, Weng Z. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins. 2002;47:281–94. [DOI] [PubMed] [Google Scholar]
- 24. Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–86. [DOI] [PubMed] [Google Scholar]
- 25. Ryan K, Backos DS, Reigan P, Patel M. Post‐translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J Neurosci. 2012;32:11250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neerman‐Arbez M, de Moerloose P, Bridel C, et al. Mutations in the fibrinogen aalpha gene account for the majority of cases of congenital afibrinogenemia. Blood. 2000;96:149–52. [PubMed] [Google Scholar]
- 27. Henschen A, Kehl M, Southan C. Genetically abnormal fibrinogens—strategies for structure elucidation, including fibrinopeptide analysis. Curr Probl Clin Biochem. 1984;14:273–320. [PubMed] [Google Scholar]
- 28. Attanasio C, de Moerloose P, Antonarakis SE, Morris MA, Neerman‐Arbez M. Activation of multiple cryptic donor splice sites by the common congenital afibrinogenemia mutation, FGA IVS4 + 1 G–>T. Blood. 2001;97:1879–81. [DOI] [PubMed] [Google Scholar]
- 29. Tirefort Y, Alson OR, de Moerloose P, Neerman‐Arbez M. Mutation of the translation initiation codon in FGA causes congenital afibrinogenemia. Blood Coagul Fibrinolysis. 2012;23:556–8. [DOI] [PubMed] [Google Scholar]
- 30. Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 2005;361:13–37. [DOI] [PubMed] [Google Scholar]
- 31. Galanakis DK, Henschen A, Peerschke EI, Kehl M. Fibrinogen Stony Brook, a heterozygous A alpha 16Arg—Cys dysfibrinogenemia. Evaluation of diminished platelet aggregation support and of enhanced inhibition of fibrin assembly. J Clin Invest. 1989;84:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neerman‐Arbez M, de Moerloose P. Mutations in the fibrinogen gene cluster accounting for congenital afibrinogenemia: an update and report of 10 novel mutations. Hum Mutat. 2007;28:540–53. [DOI] [PubMed] [Google Scholar]
- 33. Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature. 1997;389:455–62. [DOI] [PubMed] [Google Scholar]
- 34. Reber P, Furlan M, Henschen A, et al. Three abnormal fibrinogen variants with the same amino acid substitution (gamma 275 Arg—His): fibrinogens Bergamo II. Essen and Perugia. Thromb Haemost. 1986;56:401–6. [PubMed] [Google Scholar]
- 35. Weisel JW. Structure of fibrin: impact on clot stability. J Thromb Haemost. 2007;5(Suppl 1):116–24. [DOI] [PubMed] [Google Scholar]
- 36. Sugo T, Endo H, Matsuda M, et al. A classification of the fibrin network structures formed from the hereditary dysfibrinogens. J Thromb Haemost. 2006;4:1738–46. [DOI] [PubMed] [Google Scholar]
- 37. Dear A, Dempfle CE, Brennan SO, Kirschstein W, George PM. Fibrinogen Mannheim II: a novel gamma307 His–>Tyr substitution in the gammaD domain causes hypofibrinogenemia. J Thromb Haemost. 2004;2:2194–9. [DOI] [PubMed] [Google Scholar]
- 38. Bantia S, Mane SM, Bell WR, Dang CV. Fibrinogen Baltimore I: polymerization defect associated with a gamma 292Gly—Val (GGC—GTC) mutation. Blood. 1990;76:2279–83. [PubMed] [Google Scholar]