

Summary:
The structural, dynamical and functional characterization of the small GTPase K-Ras has become a research area of intense focus due to its high occurrence in human cancers. Ras proteins are only fully functional when they interact with the plasma membrane. Here we present all atom molecular dynamics simulations (totaling 5.8 μs) in order to investigate the K-Ras4A protein at membranes that contain anionic lipids (POPS or PIP2). We find that similarly to the homologous and highly studied K-Ras4B, K-Ras4A prefers a few distinct orientations at the membrane. Remarkably, the protein surface charge and certain lipids can strongly modulate the orientation preference. In a novel analysis, we reveal that the electrostatic interaction (attraction but also repulsion) between the protein’s charged residues and anionic lipids determines the K-Ras4A orientation, but that this is also influenced by the topology of the protein, reflecting the geometry of its surfaces.
Keywords: Ras, K-Ras4A, signaling lipids, molecular dynamics simulation, protein topology, protein-membrane interaction, protein orientation
Introduction
Ras is a small GTPase which transmits cellular signals in order to regulate key events in cells (Cox and Der, 2010; Vetter and Wittinghofer, 2001). As a molecular switch it participates in cascades of molecular interactions which normally convey information from the cell’s exterior to the interior. Three main isoforms, N-Ras, H-Ras and K-Ras, are expressed in human cells (Cox and Der, 2010). Except for the C-terminal -so called hypervariable region (HVR, Fig. 1a)-, the proteins have a high degree of sequence identify in the region encompassing a globular α/β-fold, here referred to as the catalytic domain, or CD. Activation or inactivation of Ras is to a large part determined by the nucleotide (GTP or GDP) that is bound to the conformation sensitive switch regions of the catalytic domain. In pathology, mutations typically disable GTP hydrolysis. Such constitutively activated Ras (especially K-Ras mutations at residue G12) are often over-expressed in many different types of cancer cells (accounting for 30 % human cancers) (Bryant et al., 2014; Prior et al., 2012; Schubbert et al., 2007). There have been intense efforts to find potent and specific Ras inhibitors to treat tumors. These efforts were made difficult because the molecular surface of isolated Ras is relatively smooth for lack of evident drug target pockets, and Ras was, therefore, regarded as undruggable (Nussinov et al. 2013). However, promising inhibitors which typically aim at prevention of Ras-GTP association or blocking of Ras-effector interactions are emerging (Athuluri-Divakar et al., 2016; Bollag and Zhang, 2013; Lito et al., 2016; Milroy and Ottmann, 2014; Ostrem et al., 2013; Shima et al., 2013). Another strategy follows the realization that conformational states of Ras, which present binding pockets, may be sampled even in absence of effectors and regulatory proteins. While their population may be low, the equilibrium would be shifted by inhibitor binding.
Figure 1:
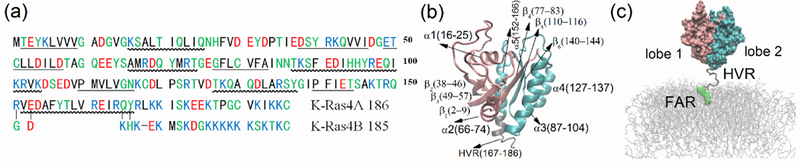
(a) Sequence of human K-Ras4A and K-Ras4B. Blue for basic residues, red for acid residues, green for polar residues and black for non-polar residues. Note that only four residues differ in the CD while the HVR is considerably different. (b) Structural model of K-Ras4A. Residues 1 to 86 (lobe 1) are colored in orange and residues 87 to 166 (lobe 2) in cyan. HVR (residue 167 to 186) in grey. (c) Initial simulation system. CD (residues 1 to 166) in surface representation. HVR in trace representation except the FAR group is highlighted by green beads. The lipid membrane is denoted by gray lines. See also Table. S1.
Ras-membrane interactions are of great significance since Ras is only functional when it is localized to the inside leaflet of the plasma membrane. The membrane and its composition not only is critical for Ras localization and diffusion, clustering/oligomer formation, but also is a platform for the association with effector and regulatory proteins (Chung et al., 2016; Li et al., 2012; Weise et al., 2011). The localization of Ras is mainly achieved though lipidation and processing of the C-terminus of the HVR by palmitoylation or farnesylation, and by the direct insertion of this attached hydrophobic group into the membrane (Hancock et al., 1990; Williams, 2003; Zhou et al., 2017) (Fig. 1b). K-Ras4A, which is a close homologue of the widely studied K-Ras4B, was only recently found to be widely expressed in cancer cells (Tsai et al., 2015). It has different sequences, mainly in the HVR, compared with K-Ras4B (Fig. 1a) and may use a different mechanism to localize to the plasma membrane. Apart from the anchoring of the HVR to the membrane, interestingly, there is recent evidence that the K- and H-Ras catalytic domain can also bind to the membrane (Abankwa et al., 2010; Li et al., 2016). Even more intriguingly, the binding of the Ras catalytic domain to the membrane is neither fully random nor, typically, single state. Instead, Ras exhibits several preferred orientations with respect to the membrane (Gorfe et al., 2007; Jang et al., 2016; Kapoor et al., 2012a, 2012b; Mazhab-Jafari et al., 2015; Prakash et al., 2016a). Importantly, the orientation of the Ras catalytic domain relative to the membrane was shown to affect the function of Ras. For example, in both the experiments of Mazhab-Jafari et al. (2015) and the simulations of Prakash et al. (2016a), it was observed that the very region of K-Ras4B that is involved in effector protein binding also interacts with the membrane. Thus, although the Ras protein was in the GTP bound/active state, this orientation prevented the interaction with an effector protein, phenocopying the inactive state of Ras (Prior and Hancock, 2001).
The emerging role of lipids in regulating Ras-membrane interaction is also receiving intense attention, most dramatically, by the recent demonstration of a link between the membrane’s phosphatidylserine content, an electrophysiological gradient and K-Ras activation level in cancer cells (Zhou et al., 2015). Considering the asymmetry of the membrane, the cytoplasmic side of the plasma membrane is often enriched with anionic phospholipid such as phosphatidylserine (PS), and also with certain cell signaling lipid molecules including phosphatidylinositols (PI). The anionic PS and also PI lipids are expected to affect the association of the Ras catalytic domain with the membrane. However, it is still unclear how such membrane components likely influence the orientation of the Ras catalytic domain relative to the lipid bilayer. Characterization of the Ras-membrane interactions and their effect on orientation preferences will lead to further understanding of the structure-function relationship and possibly inform the design of inhibitors that interrupt Ras function.
Here we present a multi-microsecond all-atom molecular dynamics (MD) simulation study of K-Ras4A bound to different model membranes. It is found that the K-Ras4A catalytic domain populates several orientations with respect to the membrane, and remarkably, that the orientation preference is modulated by the specific lipid molecules---POPS or PIP2. Based on the extent and shape of the Ras surface, we propose that at most five major orientations are possible for the K-Ras catalytic domain at the membrane, and that the interactions between the catalytic domain and the membrane will determine the balance among these different orientation states.
Results
Binding of K-Ras4A catalytic domain to membranes
The goal of the project was to characterize K-Ras4A catalytic domain (CD)-membrane interactions and how they are affected by different membrane compositions. Thus, we started several lengthy all atom MD simulations of the GTPase with the lipid anchor membrane embedded, but with the CD placed around 6.0 nm away from the different bilayers (Fig. 1c). The simulations are denoted PC1–4, PS1–4 and PI1–4 for the four independent runs at the POPC, POPC/POPS and POPC/PIP2 membranes, respectively. PI5–8, denote additional simulations at the latter membrane. In all of the simulations, the K-Ras4A CD binds to the membrane (see Movie S1 for a typical binding process). Fig. 2 shows the time evolution of the distance between the center of mass of the K-Ras4A CD and that of the membrane and the simulation averaged contact map of residue-membrane interactions. The contacts of the CD with the POPC membrane were typically established within 100–150 ns, while it took less time (50–100 ns) for the CD to bind to mixed membranes. For all the simulation systems, there are residues from the CD contacting with the membrane as shown in the contact maps. However, in the simulations of the POPC system (PC1-PC3, but not in PC4), after association with the membrane, the CD temporarily detaches from the membrane (distance larger than 5.5 nm in Fig. 2a) and does not contact with the membrane for a while before it rebinds to the membrane (see Movie S2). This observation suggests that the CD only has a weak attraction to a pure POPC membrane and that it interacts with a charge neutral membrane in a dynamic and less persistent way.
Figure 2:
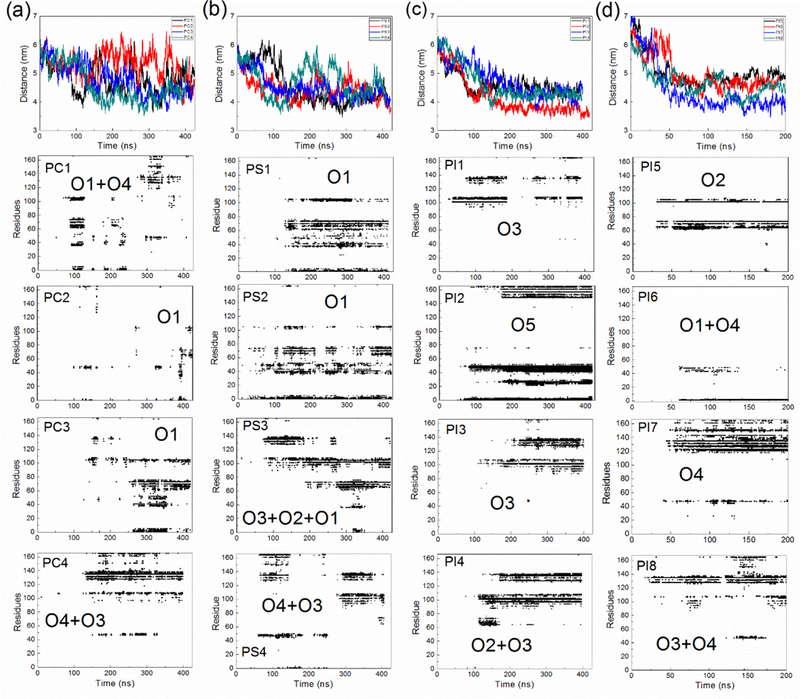
Top row 1: Time evolution of the distance in the Z direction between center of mass of the K-Ras4A CD and the center of mass of the membrane. See also Movie S1 and Movie S2. Rows 2–5: Contact map of residues of the CD with the membrane. Residues within 0.3 nm of membrane lipids are treated as contact residues. (a) Results for the POPC, (b) for the POPC/POPS, and (c-d) for the POPC/PIP2 systems, respectively.
For a mixed membrane system, once established, the association of the CD with the membrane is relatively stable, as no dissociation events are evident (except PS4). Thus, anionic lipids appear to enhance the binding of K-Ras4A CD to the membrane. However, the contacts of individual residues with the membrane are considerably dynamic and transient---residues at a time involved in contact with the membrane may be moved away from the contact area at a later time. Comparing the contact maps from different simulations in Fig. 2, it is clear that these maps differ from one another and the CD utilizes different surface residues to contact the membrane. For example, in cases of PS1 and PS2, mainly the lobe 1 domain (residues from 1 to 86) is contacting the membrane, while in cases of PI1 and PI3, the lobe 2 domain (residues from 87 to 166) is the region involved. Thus, the K-Ras4A CD can present different orientations with respect to the membrane.
Compared to the CD, the HVR is highly dynamic as seen in the RMSF plotted in Fig. S1, and while at the same time, being proximate, it is able to form much more stable contacts with the membrane. On the side of the HVR-membrane interactions this is accomplished through the anchoring of its farnesyl group into the lipid acyl region of the bilayer and the contact of several hydrophobic and positively charged HVR residues with the membrane. Fig. 3a shows the average distance between the center of mass of the membrane and that of residues from the HVR. Residues following T177 (PGCVKIKK) are about 1.4 to 2.5 nm away from the membrane (itself with an average thickness of 1.95 nm from the center), corresponding mostly to an insertion depth at the level of polar/charged region of the membrane. Apart from the FAR group, the side chain of residues I183 and C180 also have a deeper insertion into the membrane, meeting the non-polar acyl-chains. In the study of Chavan et al. (2015) and Chakrabarti et al. (2016), the HVR of K-Ras was found to be able to interact with residues of the CD. Similarly, in our simulations, the HVR is able to form transient contacts with the CD when K-Ras4A is simulated in solvent (Fig. S2), but when it is placed near the membrane, no substantial contacts between the HVR and CD were observed. An exception to this is residue R167 which is found to interact with E3. In addition, positively charged residues K169, K170 and K176 are able to form transient contacts with residues D108 and less so with S106 mainly due to an electrostatic/hydrogen bonding attraction (Fig. 3b).
Figure 3:
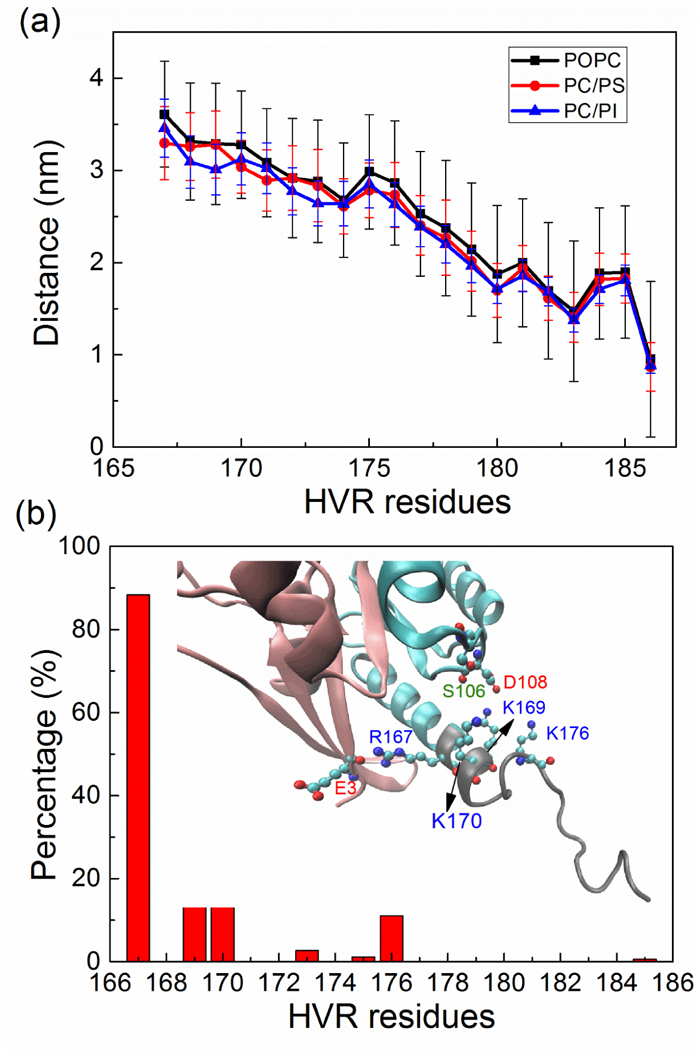
(a) Average distance and standard deviations in the Z direction between the center of mass of residues in the HVR and that of membrane. (b) The contact probability (% time of contact at < 0.3 nm) of HVR with the CD averaged over all the simulations. The inserted snapshot (final structure from the PI1 system) highlights the interaction residues. See also Figure S1 and Figure S2.
Different orientations of K-Ras4A relative to the membrane
In this work we introduce several new analyses tools to examine the orientation of a protein relative to the membrane. As the HVR of Ras is anchored to the membrane, the orientational freedom of the Ras CD is reduced. The different orientations of the CD mainly originate from its rotation around a principle axis. We devised two variables, i. e., a distance between lobe 1 Ras subdomain and the membrane (D1), and the angle between a directional vector related to both Ras subdomains and the normal direction to the membrane plane (θ). By analyzing the trajectories in terms of these two parameters, the definition of orientation states is considerably enhanced. The contour maps show the population of the different orientation states of Ras in Fig 4. For each kind of model membrane we find that several “islands” clearly appear on the contour maps. These island regions denote dominant orientations of the K-Ras4A CD relative to the membrane. Totally five major orientations (denoted as O1, O2, O3, O4 and O5 in Fig. 5) were identified. The residues involved in binding to the membrane are evident from Fig. 5 and are discussed below.
Figure 4:
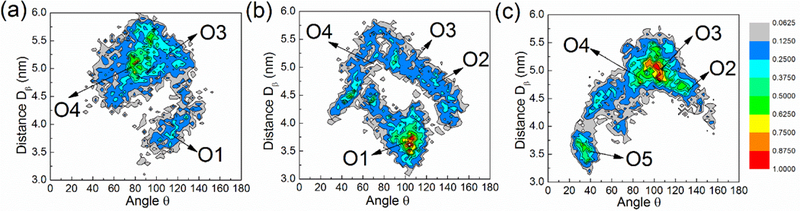
Contour maps of the relative orientation of K-Ras4A with respect to the membrane for (a) the POPC, (b) the POPC/POPS and (c) the POPC/PIP2 system. D1 is the distance between the vertical center of membrane and that of lobe 1. θ is the cross angle between the normal direction of the membrane and a directional vector of K-Ras4A CD as explained in Fig. S3. The color scale from gray to red denotes the relative population of K-Ras4A in different orientation states (D1, θ). The probability is calculated by dividing the number of each state by the total number of sampling frames and then scaled to 1 for the maximally populated state at each membrane. See also Figure S3.
Figure 5:
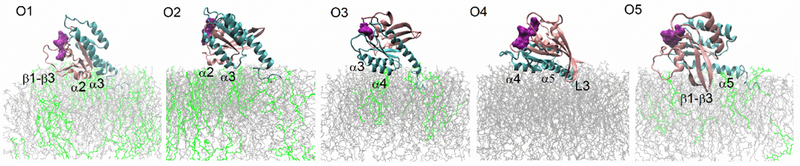
Typical orientations of K-Ras4A relative to membranes. The lobe 1 domain is colored in pink, lobe 2 and HVR in cyan, POPC in grey, POPS and PIP2 in green. The main secondary structure elements in contact with the membrane are marked. GTP nucleotide (shown as surface representation in purple) and the two switch regions are typically located at least 1.0 nm away from the membrane surface.
One major finding based on Fig.4 is that the relative populations of different membrane orientations are significantly different for different model membranes. Specifically, O1, O3 and O4 appear in the POPC systems, but these three orientations have a low population when compared to those seen at the anionic membranes. As discussed above, this is because there are only weak attractions with an overall uncharged membrane and, therefore, fewer contacts and a wider distribution of orientations. For the POPC/POPS systems, O1, and to a lesser extent O3, O2 and O4 are the dominant orientations. However, O1 is absent in the simulations with the POPC/PIP2 membrane. We have performed four additional simulations where the protein is started from different orientations and distances, but still O1 is not populated. Thus, in the POPC/PIP2 system, the orientation preference shows a strong bias to O3 and its two adjacent orientations (O2 and O4).
To further unveil the underlying mechanism how lipids modulate the orientation of K-Ras4A, the properties of both the protein CD and of different model membranes were analyzed in further detail.
Charge of lipid membranes and K-Ras4A
First, we compare the charge distributions that arise from the location of negatively charged phosphate in the lipid head groups for the different model membranes (Fig. 6a). For a PIP2 lipid, apart from the normal phosphate group which connects the lipid acyl chain, the two inositol phosphate groups carry another three negative charges and the position of inositol in a membrane differs significantly from the position of normal phosphate groups (Li et al, 2009). Thus, as shown in Fig. 6a, the charge distribution of phosphate groups for the POPC/PIP2 system is broader than for the POPC or POPC/POPS system. According to Fig. 6b, which shows the difference in net charge to the POPC membrane, there are approx. 12.8 more negative charges for the POPC/PIP2 system than for the POPC/POPS system beyond a distance of 2.3 nm from the membrane center. Following this observation, it is clear that charge of the anionic lipids is partly shielded by other groups and also by ions. Fig. 6c shows the radial distribution of Na+ and Cl- ions around a POPC, POPS or PIP2 lipid. Due to the net negative charge of the anionic lipid head-group, sodium ions will be attracted to POPS or PIP2 lipids, significantly shielding the negative charge potential. When a POPS or PIP2 molecule interacts with a nearby positively charged residue of the protein, however, sodium atoms bound to anionic lipids are usually released to solvent, gaining considerable entropy. We suggest that the difference in charge distribution between 2.3–3.0 nm is vital in regulating protein-membrane interactions.
Figure 6:
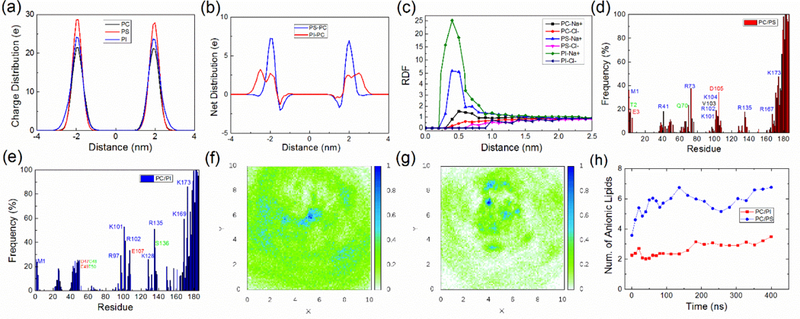
(a) Distribution of charges due to placement of phosphate in the lipid headgroups at the surface of the three model membranes in a 100 ns simulation (without protein). (b) Net distribution of charge of lipid head-phosphate groups in the POPC/POPS system or POPC/PIP2 system by subtracting the distribution of charges in the POPC system. (c) Radial distribution function of ions (sodium and chlorine ion) relative to the center of mass of the headgroup of a POPC, POPS or PIP2 lipid in the membrane only simulations. (d-e) Protein residues that are in frequent contact with the membrane for the POPC/POPS and POPC/PIP2 system respectively. The frequency is calculated by taking the time average of all the coordinate frames for each kind of membrane system. (f-g) show, respectively, the density of POPS or of PIP2 from the center of the K-Ras4A CD (density scaled to 1 for maximum). The CD is centered at (5.0 nm, 5.0 nm), and the direction vector connecting the center of CD and HVR is aligned in the direction of the X axis. (h) Charged lipid molecules gather within 2.5 nm of the center of the Ras CD (which itself has radius of gyration about 1.54 nm) in the POPC/POPS or POPC/PIP2 systems. See also Figure S4.
Since the major difference of the different model membranes comes from the presence or absence of anionic lipids, it is reasonable to speculate that charge plays a major role in modulating the K-Ras4A membrane interaction. In Fig. 6d-e, we identify CD residues which make frequent contacts with the membrane. Most of them are charged residues and the remaining residues are usually adjacent to the charged residues. Simply, the proximity of positively charged residues to the bilayer also brings their adjacent residues to the membrane, even when they are negatively charged. Only very few polar and essentially no non-polar residues are involved in membrane contact in O1 to O4. However, O5, which is seen in only 1 of 16 simulations, inserts several residues into the membrane. The residues belong to a segment of residues 24–28 (IQNHF) and a loop region around β2 and β3 (residues 43–50, QVVIDGET). Compared to K-Ras4B at a POPC/POPS membrane (Prakash et al., 2016a), similar residues are used in the K-Ras4A CD in forming contacts with the membrane. The conservative mutation D153E (K-Ras4B to 4A) is expected to have a modest influence, but mutation G151R may have a role in stabilizing the O4 orientation, as R151 was found to be able to contact the membrane in O4.
The contacts between charged groups have lengthy persistence times, being present 20% to 60% of simulation time after the first 100 ns. The stability of the contacts results in a clustering of anionic lipids. Fig. 6f-g shows the distribution of POPS and PIP2 around K-Ras4A respectively, which clearly present an enrichment of POPS and especially PIP2 molecules under or nearby the K-Ras4A-membrane contact region. Also clustering of anionic lipids around K-Ras4A is observed as a function of simulation time (see Fig. 6h), however, the clustering of anionic lipids is rather modest, and may have reached a plateau by 200–300 ns. This behavior is in accord with the limited number of protein binding sites presented to the membrane for each orientation of K-Ras4A (Table S2). Meanwhile, membrane thickness is slightly compressed under the GTPase, especially for the POPC/PIP2 system (Fig. S4)
Typical contacts of anionic lipids and nearby charged protein residues from several simulations and orientation states are detailed in Fig. 7a-b, where M1 (NH3+ of the N-terminus), R41 and R73 interact with the -COO- moiety of the POPS lipid. Interestingly, D105 was also found to interact with a POPS lipid when this interaction is bridged by a sodium atom, Na+. Further, Fig. 7c-e shows several PIP2 interacting with positively charged residues such as R97, K101, R102, and R135. Similar to the POPS-Na+-negative residue pairing, PIP2 was also found to interact with a negative residue E107 mediated by sodium ions. Interestingly, since the inositol phosphate group has several substituents, a PIP2 molecule is able to interact with several different residues simultaneously (see Fig. 7c-d). Table S2 lists the residues forming interactions with POPS or PIP2 lipids for each simulation. Typically, there are 2–9 residue-anionic lipid contacts, and while negatively charged protein residues can also participate, most of the contacts are formed between positive residues of the CD and anionic lipids.
Figure 7:
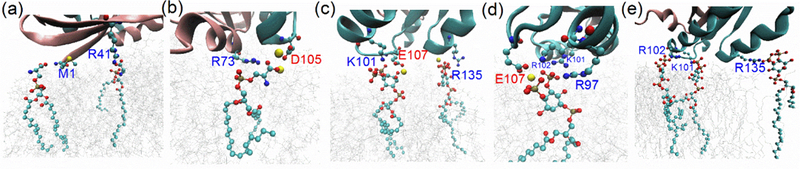
Snapshot of anionic lipid interacting with nearby charged protein residues. Coordinate frames for PS1 (a) and PS2 (b) in, O1 like orientation; for PI1 (c), PI3 (d), and PI4 (e) in, O3 like orientation. Na+ ions that were found to mediate the interaction of negative residues with anionic lipids in (b-d) are shown as yellow beads. See also Table S2.
Electrostatic Interaction in Regulating Membrane Orientation
The electrostatic attraction between charged residues and the lipids is a main driving force in determining orientation preferences. Charged residues close to the membrane surface when Ras adopts different orientation states are shown in Fig. 8a. These residues that are close to the membrane are considered as “membrane exposed residues” and the pair electrostatic interaction energies between these residues and the membrane (also including membrane proximate surface ions) are calculated and shown in Fig. S5 and Table S3. The interaction energy of the different orientation states (O1, O2, O3, O4) are comparable in the POPC/POPS systems (−902, −930, −966 and −878 kcal/mol respectively). Although O3 has the lowest energy, Fig. 4b shows that O1 is the most populated state. Nevertheless, O2, O3 and O4 blend into a wider “island” as seen in Fig. 4c. This implies that the energy barrier between these orientation states is low. Indeed, O3 is able to interconvert with its two adjacent orientation states O2 and O4, as revealed in simulation systems PS3, PI4, and PS4, PI8 respectively (Fig. 2). Taking O3, O2 and O4 together, the relative population of O1 and O3/O2/O4 in a POPC/POPS system is comparable.
The dominant orientation shifts to O3 in the POPC/PIP2 system, while O1 is absent there and is, essentially, replaced by O5. We have shown that the POPC/PIP2 membrane has a broader distribution of the negative charges carried by the PIP2 head-group, and there are also more negative charges at the membrane-water interface (2.3 nm-3.0 nm, see Fig. 6b). Apart from the electrostatic attraction with positively charged protein sidechains, we suggest that there also exist repulsive interactions between the negatively charged membrane and negatively charged residues when they are brought close to the membrane, noting the exception when interactions are bridged by sodium ions. Lobe 1 domain has 15 negative charges versus 6 positive residues (net −9), while lobe 2 domain has 13 negative residues versus 15 positive residues (net +2). On average, lobe 1 domain is highly negatively charged. Therefore, this region is able to establish contacts with positive residues of several regulatory proteins, such as c-Raf (Fetics et al., 2015). When Ras uses this region of the protein to contact the membrane (corresponding to orientation state O1), many of the negatively charged residues will also be brought to the membrane interface.
As shown in Fig. 8a, the orientation state O1 in particular has many of the negatively charged residues exposed to the membrane compared to the other orientations (8, 3, 5, 5, 4 for O1, O2, O3, O4, O5 respectively), therefore, there is an enhanced repulsive interaction between POPC/PIP2 membrane and these negative residues. Since the O1 orientation is not populated in the simulations starting from an initial displacement of K-Ras4A detached from the membrane, we performed an extra simulation by directly starting a simulation with K-Ras4A in the O1 orientation at a POPC/PIP2 membrane. In a simulation of 400 ns, the K-Ras4A CD maintained the O1 orientation state and this did not convert to the other orientation states, suggesting that the energy barrier for such transition is high. However, the calculated electrostatic interaction of K-Ras4A in O1 state with the membrane is less favorable than the orientation state O3 (−800 and −1132 kcal/mol respectively, see Fig. S5 and Table S3.) As illustrated in Fig. 8b, both O1 and O3 can use multiple positively charged residues to interact with anionic lipids, however, when POPS lipids are replaced by PIP2 lipids, an enhanced repulsion interaction of K-Ras4A in the O1 state will make this orientation less favorable. Therefore, O1 is absent while O3 becomes dominant in a POPC/PIP2 system.
Figure 8:
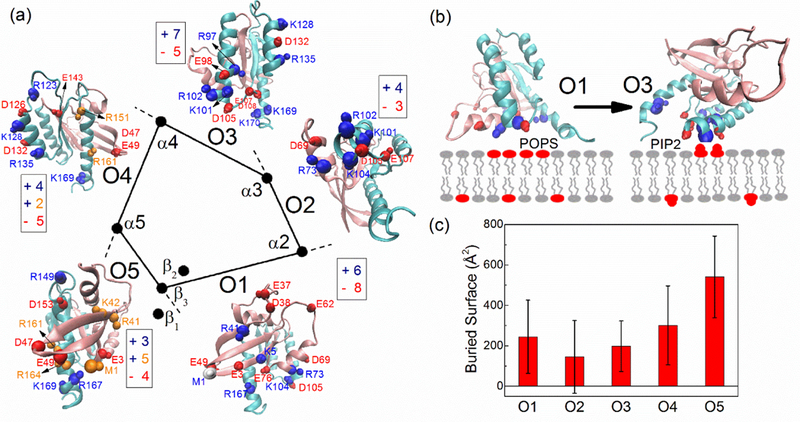
(a) Arrangement of surface secondary structure elements of K-Ras4A. The center of mass of β1-β3, α2-α5 are projected on the membrane surface respectively with direction vector Vy (see Fig. S3) perpendicular to the membrane surface. Each two adjacent elements constitute a surface, with a certain orientation. Charged residues exposed to the membrane in each kind of orientation (contact probability larger than 10 %) are denoted. Residues are shown as balls for Cα and Cβ atoms, indicating the mainchain and the sidechain. Negatively charged residues are denoted in red. Positively charged residues are denoted in blue or orange. The blue residues but not the orange residues are found to directly interact with anionic lipids. (b) Competitive interaction of the anionic membranes interacting with positive and negative residues. O1 has more negatively charged residues exposed to the membrane, while PIP2 preferred O3 orientation has more negative charges above the membrane surface. Rather than a rotation, the protein is flipped, using the opposite surface to contact the membrane. (c) Buried solvent accessible surface between protein and lipid bilayers (on site of the protein) for different orientations, averaged over all simulation systems. See also Figure S5 and Table S3.
Topology of Ras Surface in Modulating K-Ras4A-Membrane Interaction
As a second novel analysis method, we considered the shape of the protein and extent of the interacting surfaces. We found that the surface mainly comprised of secondary structural elements, including β1-β3 and α2-α5 of K-Ras4A, are involved in contacts with the membrane. For the GTPase fold, the mutual orientation of secondary structure, i.e. topology, appears to determine the five major orientations seen in the simulations. In Fig. 8a, the center of mass of these secondary structure elements are projected onto the membrane surface. Typically K-Ras4A uses two adjacent secondary structural elements to contact the membrane. Therefore, considering all such possible pairs, i. e., β(1–3)/α2, α2/α3, α3/α4, α4/α5, α5/β(1–3), at most five orientations of K-Ras4A can become major orientations (Fig. 8a). This, indeed, corresponds to the five major orientations seen in the simulations. Transitions between the different orientations mainly originate from a rotation of Ras and the level of rotation required between different orientation states should be related to the angle between the surfaces. An orientation state is typically able to convert to its two adjacent orientation states, e. g. O3 to O2 and O4 (Fig. 4).
The extent of the surface area presented by such elements of secondary structure is likely to increase the interaction, depending on the contact groups involved and prevents the detachment of the CD from the membrane due to thermal fluctuations. In order to quantify the extent of the surfaces, we calculated the average solvent accessible surface (SASA) that is buried at the membrane (Fig. 8c). O2, associated with the surface made by α2 and α3 is very small. α2 and α3 are almost perpendicular to each other (Fig. 8a), therefore when α2 is parallel to the membrane surface, α3 is significantly inclined to the membrane and can only use a part of its surface to interact with the membrane, resulting in a small surface area. O2 is found to be able to interconvert with its adjacent O3, and therefore it can also be treated as an extended orientation of O3. The surface area of O5 made by β(1–3) and α5 that are perpendicular to each other, is also very small. However, as O5 has many residues inserted into the membrane, it gives the largest value of the surface that is buried. Insertion into the membrane likely needs to overcome a significant energy barrier (e. g. creating a defect in the membrane/moving lipid head-groups apart). Therefore, we suggest that only when there is a strong attraction between Ras and membrane (i. e., in POPC/PIP2 system), O5 could appear. Both O2 and O5 don’t show up at the POPC membrane due to their unfavorable surface. Both of them appear under the influence of anionic lipid molecules, but at lesser extent.
Based on the geometric grounds, the K-Ras surfaces of O4, O1 and O3 facing the membrane are smooth and extended, well matching the flat membrane. Of the three orientations, O4 has the largest buried surface, then O1 and O3. At a pure POPC membrane where the electrostatic interaction with K-Ras is not very strong, the structural features of the protein become the primary determinant in modulating membrane orientations; therefore O3, O1 and especially O4 become the major orientations. In the presence of anionic lipids, these three orientations (especially O1 and O3) are still the dominant orientations as they have many charged residues exposed to the membrane. However, the presence of certain anionic lipids can greatly tilt the orientation balance due to the increased electrostatic interactions between protein and membrane. The POPS lipid molecules lead to an orientation preference towards to O3-centered and O1 states, while a clear shift in the population of orientation states towards the O3-centered orientation is seen in the presence of PIP2 lipid molecules.
DISCUSSION
Our main findings are as follows: We propose that the topology of the small GTPase protein, K-Ras4A, catalytic domain provides a limited number of five orientations/surfaces that can interact with lipid membrane. We particularly emphasize that three of the orientations (O1, O3 and O4) are the most dominant states when Ras interacts with membranes. As mentioned by others, it is remarkable that the catalytic domain of small GTPases can interact with uncharged membranes (Karpoor et al. 2012a, 2012b). Although these interactions are weak, they might help GTPase-membrane association prior to C-terminal processing/lipidation or in cases where the lipid group is occluded by lipid binding proteins, such as PDE (Suladze et al., 2014). However, we also find that anionic lipid molecules can greatly alter the preference for certain orientations due to electrostatic interactions with the membrane. For example, a clear shift in the population of orientation states towards the O3-centered orientation is seen in the presence of PIP2 lipid molecules. The additional repulsion between PIP2 and the negatively charged residues likely lead to the absence of O1 at the POPC/PIP2 membrane. Altogether, our analysis provides a semi-quantitative explanation for the major orientation preferences of a K-Ras GTPase catalytic domain at different model membranes.
The membranes of our study are essentially flat and only experience modest perturbations due to interactions with the GTPase and concomitant anionic lipid clustering. This contrasts with the behavior of several scaffold proteins which have an intrinsically curved shape (such as BAR domains) and which are able to remodel the shape of the membrane (Zimmerberg and Kozlov, 2006). Here, for a Ras GTPase, K-Ras4A, we propose that the topology of the membrane anchored soluble protein provides flat surfaces that match, in charge localization, the character of the different membranes. From the nature of the contacts we conclude that electrostatic interactions dominate, especially at anionic membranes. This is consistent with the absence of persistent hydrophobic and non-polar interactions and suggests that waters and, as we have seen, ions play a significant role as they remain lodged between significant parts of the protein–membrane interacting surfaces. This finding has implications for the design of possible therapeutic agents which may be sought to disrupt/or to stabilize these interactions. No dramatic conformational or allosteric changes are indicated for the Ras catalytic domain. As such the membrane binding surfaces of K-Ras may be difficult to target by small molecules, except perhaps in the case of molecules that could also pre-organize PIP2 groups at the membrane and bridge the repulsive interaction between Asp/Glu and PIP2 groups in manner that is similar to sodium ions.
Despite having a very different hypervariable region, our results for K-Ras4A at the POPC/POPS membrane are in good agreement with recent simulations of Gorfe and co-workers on K-Ras4B in a similar setting. Of all the five orientations predicted here, O1 and O3 are similar to the two principal orientations reported by Prakash et al 2016a. Besides, O5 is similar to one of the simulation conformations in the work of Jang et al., 2016. Because of the similarity between K-Ras4A and −4B we anticipate that cancer associated mutants, will have also similar effects on the orientation preferences of the catalytic domain. More significantly different behavior is likely when considering other family members (H-Ras, N-Ras, etc.), as the residue changes are greater in number in the catalytic domain and include residues in the membrane interacting regions; this topic of GTPase isoform specificity has recently been reviewed by Prakash and Gorfe (2016b).
The proposal that K-Ras samples multiple orientations relative to the membrane is strengthened by several experimental studies that suggest the protein’s membrane interactions, including its orientation preference, is a critical regulator for Ras function. K-Ras only has a single globular catalytic domain but it needs to interact with many regulatory and effector proteins. The binding of the protein to the membrane will hinder the interaction of Ras with its regulatory and effector proteins at certain surfaces, especially near/at the GTP binding site and switch regions, hence have an effect on Ras function. For example, it was recently proposed from an NMR study that the orientation preference of K-Ras at anionic membranes is nucleotide dependent (Mazhab-Jafari et al., 2015), with GTP stabilizing an occluded state, whereas GDP bound Ras (the inactive state) has the switch regions exposed to solvent. This may be beneficial for interactions with exchange factor proteins, GEFs, which turn Ras into the active form. It may also promote a prolonged K-Ras activity as the approach of GTPase Activating proteins, GAPs, is hindered together with the hydrolysis which returns the protein to the in active, GDP state. However, as far as interactions with effector proteins are concerned this behavior at the membrane, observed by Ikura and colleagues, appeared to be counterintuitive. We make similar observations in the simulations presented here, although we only studied the GTP bound state of a constitutively active cancer driving mutant, G12D. For example, O1 which is prominent at POPC/POPS, is expected to be geometrically unable to bind an effector protein. By contrast, the switch regions in O3 and O4, which are dominant in the POPC/PIP2 system, are more available for effector protein association (Fig. 5). Consequently, higher levels of PIP2 would be expected to enhance Ras signaling.
Specifically, it was recently shown that membrane depolarization, which is associated with some cancer cells such as neuroblastoma cells, results in an increase in PS and PIP2 nanoclustering (Zhou et al., 2015). Before, PS had been shown by the same group (and others) to stimulate K-Ras4B clustering and signaling (Zhou et al., 2014). The recent MD study of the Gorfe group with K-Ras4B.G12V at a POPC/POPS membrane suggested an almost equal population of an O1- and O3-like state, similar to our work. By contrast, the experimental work by Ikura and colleagues indicate an O1-like/switch region occluded state, whose population shifts to an accessible state upon certain cancer associated mutations. In our laboratory we have carried out similar NMR experiments with PIP2 doped nanodiscs in K-Ras4B (in preparation) and overall find PIP2 interaction sites that are consistent with our simulations for K-Ras4A, presented here. However, presenting this data and comparison here, is beyond the scope of the paper.
The situation in cancer cells could be even more complicated, likely depending also on cancer cell type. For instance, Fleischmann et al., (1986) have shown that, in Ras transformed fibroblast cells, the DAG to PIP2 ratio was increased 2.5 to 3 fold compared to normal cells. Increased activation of PI3K and/or loss of PTEN will result in increased PIP3 levels at the expense of PIP2. As noted above, the response by both K-Ras, in terms of its orientation/accessibility to effector proteins, seems against expectation. However, as pointed out in recent reviews, the correlation between GTPase-membrane orientation and level of activity is not straightforward (Li and Buck, 2016; Lu et al., 2016). Effector proteins, such as c-Raf, may, for example, bind to K-Ras dimers or small clusters and/or effector protein binding may itself alter the orientation preference of the GTPase at the membrane, especially if the effector protein interacts with the membrane as well. Further experiments as well as computational studies with Ras dimers and oligomers, cancer mutants as well as wild type proteins forming complexes with effector proteins will be needed to resolve the exact mechanism of K-Ras hyperactivation in cancer at the local level. However, the analyses presented here is a useful tool to light the way. Beyond Ras dimerization at the membrane, it is known that K-Ras can form small clusters. This formation as well as the microlocalization of Ras into particular membranes is now becoming understood, with the HVR playing a key role (Winter et al., 2009; Zhou et al., 2017). The apparently dominant role of the HVR is likely integrated, at least to some extent, with the local orientation preferences of the Ras catalytic domains at membranes.
In summary, using all atom simulations, we have characterized the interaction of K-Ras4A with different membranes and analyzed the role of lipids in modulating GTPase catalytic domain-membrane interactions and in determining orientation preferences. We introduced a novel method to analyze Ras-membrane interaction that is based on subdomain distance as well as the angle of protein surfaces (underlying secondary structural elements) relative to the membrane. The catalytic domain of K-Ras4A was found to bind to the membrane and populate only a few orientation states with respect to the lipid bilayer. The presence of anionic lipids (POPS or PIP2) enhanced the binding to, and population of certain orientation states of K-Ras4A at the membrane. Importantly, POPS and PIP2 have different effects on the orientation preferences, due to the difference in distribution of charge density of diffusing POPS or PIP2 in the membrane (and arrangement/orientation of these lipid head-groups). We find that the competition between attraction of positively charged protein residues with the anionic lipid groups and repulsion of negatively charged protein residues with the membrane is a major determinant of K-Ras4A orientation, but this is also influenced by the extent of the surfaces of K-Ras (in relation to the protein’s topology). The study greatly enhances our understanding of specific molecular interactions of K-Ras with physiologically relevant lipid molecules in model membranes.
Experimental Procedures
Three different model membranes were built for this study. First, a bilayer with 300 POPC (Palmitoyloleoyl-phospatidyl-choline) lipids; second, a bilayer consisting of 284 POPC and 64 POPS (1-Palmitoyl-2-oleoylphosphatidylserine) lipids (18.4 % POPS); and third, a bilayer composed of 284 POPC and 16 PIP2 (Phosphatidylinositol 4,5-bisphosphate) lipids (5.3 % PIP2). Anionic lipids are equally distributed in each monolayer of the membrane. Since a PIP2 lipid possesses four negative charges while a POPS has one negative charge, here the POPC/POPS and POPC/PIP2 ratios were chosen to give these membranes an equal amount of negative charges. All model membranes were created by the CHARMM-GUI (Wu et al., 2014) and equilibrated for 100 ns using NAMD.
K-Ras4A shares a high sequence similarity with K-Ras4B in the catalytic domain, but it is quite different from K-Ras4B in the HVR, where it has less positively charged residues and is slightly more hydrophobic than K-Ras4B (Fig. 1a). Since no crystal structure is available, the structure of GTP bound K-Ras4A was based on the crystal structure of G12D K-Ras4B (PDB ID: 4DSO) by mutating the residues that differ, i. e., four residues in the catalytic domain (G151R, D153E, K165Q, H166Y) and most residues in the flexible HVR (Fig. 1a). The modeled structure of K-Ras4A was solvated in a box of TIP3P water and briefly subjected to 1 ns simulation with a harmonic constraint on protein atoms (force constant of 1 kcal/mol*Å2), followed by an unrestrained simulation about 450 ns in length. The final K-Ras4A structure from this simulation of the protein in solvent was then chosen to be the initial conformation for the simulations of Ras-membrane interactions. The HVR for the chosen conformation was extended for anchoring the protein to the membrane and the C-terminal cysteine of the HVR was modified by a farnesyl group and carboxymethylated (named as residue FAR). The parameter for this group was obtained by the CHARMM generalized force field (CGenFF) (Vannommeslaeghe and Mackerell, 2012). It should be noted that K-Ras4A has a second site for lipidation, however it is not yet clear whether both sites are utilized for membrane association (Nussinov, et al, 2016). A simulation with K-Ras4B, which investigated the flexibility of the HVR with one or two lipidations suggested that a second lipidation has no or only little effect on the flexibility of the HVR or the orientation states of the catalytic domain (Prakash et al., 2016a). The farnesyl group was pre-inserted into the membrane, leaving the rest of the HVR almost perpendicular to the membrane. The center of mass of the catalytic domain was placed about 6 nm away from the center of membrane in the Z direction, in order not to interact with the membrane at the beginning of simulations (Fig. 1c).
In total, 16 simulation systems of K-Ras4A and model membranes were prepared, with 4 for the POPC system, 4 for the PC/PS system, and 8 for the PC/PI systems (denoted by PC1-PC4, PS1-PS4, and PI1-PI8). Most of the simulations used the same initial structure by assigning different initial velocities. In order to examine whether the initial placement of K-Ras4A has an influence on the final orientation at the membrane, 4 additional simulations (PI5-PI8) were carried out for the POPC/PIP2 system, where the K-Ras4A starting structure was chosen from alternate coordinate frames towards the end of the simulation of the protein in solution, effectively resulting in slightly different starting orientations towards the membrane. The CHARMM36 force field including the CMAP correction was used to describe all the protein, lipids and GTP nucleotide (Buck et al., 2006; Huang and Mackerell, 2013; Klauda et al., 2010). Sodium and chloride ions were added to neutralize the system and maintain a near-physiological ion concentration of 150 mM. The long distance electrostatic interaction was treated by the Particle-Mesh Ewald (PME) method. A 1.2 nm cut-off was used for van der Waals and for local electrostatic interactions. Lengths of all bonds involving hydrogens were controlled via the SHAKE algorithm. A time step of 2 fs was employed and neighbor lists updated every 10 steps. The temperature was coupled by using the Langevin thermostat at 310 K, whereas the pressure control was achieved by a semi-isotropic Langevin scheme at 1 bar. All these systems were simulated using the NAMD/2.10 package (Phillips et al., 2005). The initial 100 ns of all simulations (in case of the additional simulations, the entire 200 ns) were run at the Ohio Supercomputer Center while trajectories from 100 to 400 ns were run on Anton, a supercomputer that is optimized for MD simulation (Shaw et al., 2009).
For ease of analysis, in Fig. 1c the catalytic domain is further divided into two regions, as done in a previous study (Prakash et al., 2016a). The lobe 1 subdomain involves residues 1 to 86, while the lobe 2 subdomain encompasses residues 87 to 166. In analyzing the simulations, in most cases, the data after the first 100 ns were used unless stated otherwise. The analyses were carried out using VMD (Humphrey et al., 1996) and scripts written in the lab.
Supplementary Material
Acknowledgement
We thank Dr. S. Cao and other members of the Buck lab. for discussion. This work is supported by NIGMS grant R01GM112491 to the Buck lab. We thank to the Ohio Super Computer center located in Columbus for providing the computational resource on this work. Anton Computer time was provided by the Pittsburgh Supercomputing Center (PSC) through Grant R01GM116961 from the National Institutes of Health.
References
- Abankwa D, Gorfe AA, Inder K, and Hancock JF (2010). Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. U. S. A. 107,1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athuluri-Divakar SK, Vasquez-Del CR, Dutta K, Baker SJ, Cosenza SC, Basu I, Gupta YK, Reddy MVR, Ueno L, Hart JR, et al. (2016). A small molecule Ras-mimetic disrupts Ras association with effector proteins to block signaling. Cell 165, 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, and Zhang C (2013). Drug discovery: pocket of opportunity. Nature 503, 475–476. [DOI] [PubMed] [Google Scholar]
- Bryant KL, Mancias JD, Kimmelman AC, and Der CJ (2014). KRAS: feeding pancreatic cancer proliferation. Trends Biochem. Sci. 39, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M, Bouguet-Bonnet S, Pastor RW, and MacKerell AD (2006). Importance of the CMAP correction to the CHARMM22 protein force field: dynamics of Hen lysozyme. Biophys. J. 90, L36–L39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti M, Jang H, and Nussinov R (2016). Comparison of the conformations of KRAS isoforms, K-Ras4A and K-Ras4B, points to similarities and significant differences. J. Phys. Chem. B 120, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan TS, Jang H, Khavrutskii L, Abraham SJ, Banerjee A, Freed BC, Johannessen L, Tarasov SG, Gaponenko V, Nussinov R, et al. (2015). High-affinity interaction of the K-Ras4B hypervariable region with the Ras active site. Biophys. J. 119, 2602–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JK, Lee YK, Lam HYM, and Groves JT (2016). Covalent Ras dimerization on membrane surfaces through photosensitized oxidation. J. Am. Chem. Soc. 138, 1800–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, and Der CJ (2010). Ras history: the saga continues. Small GTPase 1, 2–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, and Matthos C (2015). Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 23, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischman LF, Chahwala SB, and Cantley L (1986). Ras-transformed cells: altered levels of phosphatidylinositol-4,5-bisphosphate and catabolites. Science 231, 407–410. [DOI] [PubMed] [Google Scholar]
- Gorfe AA, Hanzal-Bayer M, Abankwa D, Hancock JF, and McCammon JF (2007). Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-Dimyristoy-3 phosphocholine bilayer. J. Med. Chem. 50, 674–684. [DOI] [PubMed] [Google Scholar]
- Hancock J, Paterson H, and Marshall CJ (1990). A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize ~p21RAS to the plasma membrane. Cell 63, 133–139. [DOI] [PubMed] [Google Scholar]
- Huang J, and MacKerell AD (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD-visual molecular dynamics. J. Molec. Graphics. 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Jang H, Banerjee A, Chavan TS, Lu S, Zhang J, Gaponenko V, and Nussinov R (2016). The higher level of complexity of K-Ras4B activation at the membrane. FASEB J. 30, 1643–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor S, Triola G, Vetter IR, Erlkamp M, Waldmann H, and Winter R (2012a). Revealing conformational substates of lipidated N-Ras protein by pressure modulation. Proc. Natl. Acad. Sci. U. S. A. 109, 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor S, Weise K, Erlkamp M, Triola G, Waldmann H, and Winter R (2012b) The Role of G-Domain Orientation and Nucleotide State on the Ras Isoform-Specific Membrane Interaction. Eur. Biophys. J. 41, 801–813. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O Connor JW, Mondragon-Ramirez C, Vorobyov I, Tobias DJ, MacKerell AD, and Pastor RW (2010). Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Janosi L, and Gorfe AA (2012). Formation and domain-partitioning of H-ras peptide nanoclusters: effects of peptide concentration and lipid composition. J. Am. Chem. Soc. 134, 17278–17285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Venable RM, Rogers LA, Murray D, and Pastor RW (2009). Molecular dynamics simulations of PIP2 and PIP3 in lipid bilayers: determination of ring orientation, and the effects of surface roughness on a Poisson-Boltzmann description. Biophys. J. 97, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZL, Cao S, and Buck M (2016). K-RAS at anionic membrane: orientation, orientation-orientation. Recent simulations and experiments. Biophys. J. 5, 1033–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Solomon M, Li LS, Hansen R, and Rosen N (2016). Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Jang H, Muratcioglu S, Gursoy A, Keskin O, Nussinov R, and Zhang J (2016). Ras conformational ensembles, allostery, and signaling. Chem. Rev. 116, 6607–6665. [DOI] [PubMed] [Google Scholar]
- Mazhab-Jafari MT, Marshall CB, Smith MJ, Gasmi-Seabrook GM, Stathopulos PB, Inagaki F, Kay LE, Neel BG, and Ikura M (2015). Oncogenic and Rasopathy-associated K-Ras mutations relieve membrane-dependent occlusion of the effector binding site. Proc. Natl. Acad. Sci. U. S. A. 112, 6625–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Milroy LG, and Ottmann C (2014). The renaissance of Ras. ACS Chem. Biol. 9, 2447–2458. [DOI] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, Chakrabarti M, and Jang H (2016). A new view of Ras isoforms in cancers. Cancer Res. 76, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ, and Mattos C (2013). Pathway drug cocktail: targeting Ras signaling based on structural pathways. Trends. Mol. Med. 19, 695–704. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, Zhou Y, Liang H, Hancock JF, and Gorfe AA (2016. a). Oncogenic K-RAS binds to an anionic membrane in two distinct orientations: a molecular dynamics simulations. Biophys. J. 110, 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P, and Gorfe AA (2016b). Membrane orientation dynamics of lipid-modified small GTPases. Small GTPase DOI: 10.1080/21541248.2016.1211067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulter K (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, and Hancock J,F. (2001). Compartmentalization of Ras protein. J. Cell. Sci. 107, 1603–1608. [DOI] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, and Mattos CA (2012). Comprehensive surgey of Ras mutation in cancer. Cancer. Res. 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S, Shannon K, and Bollag G (2007). Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, Bank JA, Young C, Deneroff MM, Batson B, Bowers KJ, et al. (2009). Anton, a special-purpose machine for molecular dyanmics simulaiton. Proceeding of the ACM/IEEE Conference on Supercomputing (SC09), Portland, Oregon. [Google Scholar]
- Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, et al. (2013). In silico discovery of small molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. U. S. A. 110, 8182–8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suladze S, Ismail S, and Winter R (2014). Thermodynamic, dynamic and solvational properties of PDEδ binding to farnesylated cystein: a model study for uncovering the molecular mechanism of PDEδ interaction with prenylated proteins. J Phys Chem B 118, 966–975. [DOI] [PubMed] [Google Scholar]
- Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM, and Philips MR (2015). K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. U. S. A. 112, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, and Mackerell AD (2012). Automation of the CHARMM general force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter I, and Wittinghofer A 2001. The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- Weise K, Kapoor S, Denter M, Nikolaus J, Opitz N, Koch S, Triola G, Herrmann A, Waldmann H, and Winter R (2011). Membrane-mediated induction and sorting of K-Ras microdomain signaling platforms. J. Am. Chem. Soc. 133, 880–887. [DOI] [PubMed] [Google Scholar]
- Weise K, Triola G, Brunsveld L, Waldmann H, and Winter R (2009). Influence of the lipidation motif on the partitioning and asociation of N-Ras in model membrane subdomains. J. Am. Chem. Soc. 131, 1557–1564. [DOI] [PubMed] [Google Scholar]
- Williams CL (2003). The polybasic region of Ras and Rho family small GTPases: a regulator of protein interactions and membrane association and a site of nuclear loalizaiotn signal sequences. Cell Signal 15, 1071–1080. [DOI] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, et al. (2014). CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liang H, Rodkey T, Ariotti N, Parton RG, and Hancock JF (2014). Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Mol. Cell Biol. 34, 862–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Prakash P, Liang H, Cho KJ, Gorfe AA, and Hancock JF (2017). Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 168, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wong C, Cho K, vad der Hoeven D, Liang H, Thakur DP, Luo J, Babie M, Zinsmaier KE, Zhu MX, et al. (2015). Membrane potential modulates plasma membrane phosphplipid dynamics and K-Ras signaling. Science 349, 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J and Kozlov MM (2006). How proteins produce celluar membrane curvature. Nat. Rev. Mol. Cell Biol. 7, 9–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.