

Summary
CRISPR-Cas endonucleases directed against foreign nucleic acids mediate prokaryotic adaptive immunity and have been tailored for broad genetic engineering applications. Type VI-D CRISPR systems contain the smallest known family of single effector Cas enzymes, and their signature Cas13d ribonuclease employs guide RNAs to cleave matching target RNAs. To understand the molecular basis for Cas13d function and explain its compact molecular architecture, we resolved cryo-electron microscopy structures of Cas13d-guide RNA binary complex and Cas13d-guide-target RNA ternary complex to 3.4 and 3.3 Å resolution, respectively. Furthermore, a 6.5 Å reconstruction of apo Cas13d combined with hydrogen-deuterium exchange revealed conformational dynamics that have implications for RNA scanning. These structures, together with biochemical and cellular characterization, provide insights into its RNA-guided, RNA-targeting mechanism and delineate a blueprint for the rational design of improved transcriptome engineering technologies.
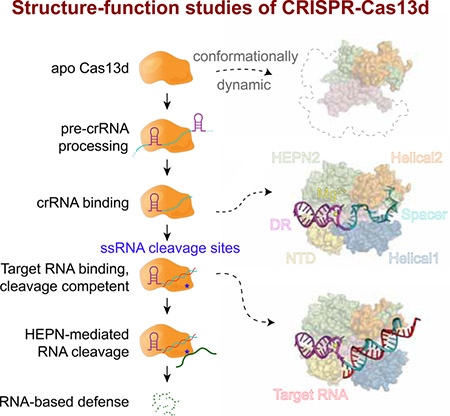
Introduction
Bacterial life employs diverse CRISPR systems to protect themselves against predatory phage, engaging Cas nucleases with programmable guide RNAs to target invading nucleic acids and endow the host cell with adaptive immunity (Barrangou et al., 2007; Brouns et al., 2008). CRISPR systems are broadly divided into two classes, each with multiple types and subtypes, wherein Class 1 systems (types I and III) coordinate multiple proteins that cooperate for target surveillance and defense, while Class 2 systems integrate both functions into a single effector enzyme (Koonin et al., 2017).
Class 2 CRISPR-Cas systems include types II, V, and VI, with types II and V shown to target DNA. Adapted over the last half-decade into a remarkably flexible genetic engineering toolbox, Class 2 DNA-targeting enzymes such as CRISPR-Cas9 (type II) and CRISPR-Cas12a/Cpf1 (type V) have facilitated many applications, from gene editing to lineage tracing, multi-color chromosomal imaging, and gene drives. Although some Class 1 CRISPR systems can target RNA (Hale et al., 2009; Jiang et al., 2016; Kazlauskiene et al., 2017; Niewoehner et al., 2017; Samai et al., 2015), Type VI systems have been recently described as the only known single effector CRISPR nucleases that exclusively target RNA (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Konermann et al., 2018; Shmakov et al., 2015; Smargon et al., 2017; Yan et al., 2018). Cas13, the signature single-effector enzyme family, comprises guide RNA-directed ribonucleases with 4 subtypes (Cas13a-d) that each exhibit significant sequence divergence apart from two consensus HEPN (Higher eukaryotes and prokaryotes nucleotide-binding domain) RNase motifs, R-X4–6-H. Domains belonging to the HEPN superfamily are frequently found in ribonucleases involved in immune defense (Anantharaman et al., 2013), including in Class 1 CRISPR RNases such as Csm6 or the homologous Csx1 (Jiang et al., 2016; Kazlauskiene et al., 2017; Niewoehner and Jinek, 2016), as well as prokaryotic Abi and T-AT defense systems or the anti-viral mammalian RNase L (Han et al., 2014). To defend against viral infection, Cas13 enzymes process pre-crRNA into mature crRNA guides in a HEPN-independent manner, followed by HEPN-dependent cleavage of a complementary “activator” target RNA in cis. Upon target-dependent activation, Cas13 is also able to cleave bystander RNAs in trans, reflecting a general RNase activity capable of both cis- and trans-cleavage.
Despite functional similarities of crRNA-dependent activation of HEPN-mediated RNA cleavage, Cas13 subtypes characterized to date exhibit key differences beyond their significant divergence at the primary sequence level. Using a computational pipeline for identifying novel Class 2 CRISPR-Cas loci from genome and metagenome sequences sourced from large-scale microbiome sequencing efforts, we recently described a Cas13 subtype designated as Cas13d (Konermann et al., 2018). Cas13d enzymes are 20–30% smaller than other Cas13 subtypes, facilitating flexible packaging into size-constrained therapeutic viral vectors such as adenoassociated virus (AAV) (Konermann et al., 2018; Yan et al., 2018).
Cas13 enzymes provide a rich resource for new RNA targeting technologies, and have been recently developed for RNA knockdown (Abudayyeh et al., 2017; Cox et al., 2017; Konermann et al., 2018), editing (Cox et al., 2017), splicing (Konermann et al., 2018), and viral delivery (Konermann et al., 2018). Remarkably, Cas13 subtypes and individual orthologs exhibit highly variable activity in human cells, with Cas13d displaying robust activity for both target cleavage and binding (Cox et al., 2017; Konermann et al., 2018). Here, we sought to understand the molecular and structural basis for Cas13d function, including both guide and target RNA recognition.
Results
Determination of a high-resolution cryo-EM structure of Cas13d in complex with crRNA
To gain structural insight into Cas 13d function, we purified the catalytically active EsCas13d (Konermann et al., 2018) and formed a binary complex containing Cas13d bound to CRISPR RNA (crRNA) followed by cryo-EM imaging (Figure S1A-B). A large data collection, followed by a computational analysis and refinement of 43,786 particles led to the derivation of a coulombic potential map of the binary complex bound to crRNA, resolved to a mostly homogeneous resolution of ~3.4 Å (Figure S1C, and Table S1). Using recently described procedures for characterizing anisotropy in cryo-EM density maps (Tan et al., 2017), we found that the structure maintained approximately even distribution of directional resolution (Figure S1D).
At 954 amino acids (molecular weight ~105 kDa), EsCas13d is considerably smaller than average members of the Type VI-A, -B, and -C subtypes. Most residues could be built into the density, with the exception of several flexible loops. All 52 nucleotides spanning the crRNA were observed in the EM density and 51 of them could be confidently modeled (Figure S1E). The final model is consistent with the cryo-EM map, with good geometry and model statistics (Table S1).
The structure of crRNA-bound Cas13d reveals a compact protein architecture surrounding solvent-exposed RNA
The Cas 13d binary complex (Figure 1A-D) maintains a bilobed architecture with five distinct domains organized around the central crRNA guide (Figure 1A). The domains include an N-terminal domain (NTD), a HEPN1 catalytic domain that is split into two distinct regions in sequence space, a first linker domain termed Helical-1, a second HEPN2 catalytic domain, and a second linker domain termed Helical-2. With the exception of the NTD, which is composed of two short α-helices flanking a β-sandwich region formed by two antiparallel 3-stranded β-sheets, the protein is predominantly alpha-helical.
Figure 1. Structures of Cas13d-crRNA and Cas13d-crRNA-target RNA.

(A) Domain organization of EsCas13d, with boundaries and catalytic sites of the HEPN domains (R295, H300, R849, H854) indicated.
(B) Schematic representation of the crRNA used for binary complex assembly. The DR (5’ handle) and spacer of crRNA are colored magenta and cyan, respectively.
(C) Opposing views of the cryo-EM binary reconstruction, resolved to ~3.4 Å resolution.
(D) Opposing views of the binary atomic model derived from the cryo-EM density.
(E) Schematic representation of the crRNA used for ternary complex assembly. The DR and spacer of crRNA are colored as in (B), target RNA is red.
(F) Opposing views of the cryo-EM ternary reconstruction, resolved to ~3.3 Å resolution.
(G) Opposing views of the ternary atomic model derived from the cryo-EM density. See also Figure S1, S2
The overall binary ribonucleoprotein architecture is reminiscent of a half-open clam shape surrounding the solvent exposed crRNA channel. The mature crRNA is divided into a constant direct repeat (DR) region (nt 1–30), derived from the characteristic repeat of CRISPR arrays, and a spacer region (nt 31–52) complementary to the target protospacers (Figure 1B). In the binary complex, the 5’ crRNA handle (also referred to as the DR) is clamped by NTD and HEPN2, with the first two base pairs and its 5-nt loop protruding away from protein density (Figure 1C,D, Figure S1E). Immediately downstream of the DR, the spacer region resides within a cleft and is sandwiched between Helical-1 and Helical-2. HEPN1 provides a structural scaffold connecting the two lobes of Cas13d, reminiscent of a hinge around the largely solventexposed RNA density. In this compact configuration, Cas13d forms a “surveillance complex”, poised for searching and identifying complementary target sites (Figure 1D).
Determination of a high-resolution cryo-EM structure of Cas13d in complex with crRNA and target RNA
Type VI CRISPR-Cas RNases catalyze degradation of ssRNA through a process that is mediated by the formation of an activated ternary complex containing both spacer and complementary protospacer (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Konermann et al., 2018; Smargon et al., 2017; Yan et al., 2018). To understand the molecular basis for nuclease activation, we sought to determine the ternary structure composed of Cas 13d bound to both crRNA and its complementary target RNA. To stabilize the ternary complex in a pre-cleavage state, we mutated all four predicted catalytic HEPN domain residues to alanine (R295A/H300A/R849A/H854A). We previously reported that this “catalytically dead” Cas13d (dCas13d) retains the ability to bind both crRNA and target RNA, but cannot cleave ssRNA (Konermann et al., 2018).
Using similar procedures, we refined 51,885 particles to determine the structure of the Cas13d ternary complex to an average resolution of ~3.3 Å (Figure 1E-G and Figure S1F-H), with a more anisotropic distribution of directional resolution (Figure S1I). The quality and overall features of the map were sufficient to derive an atomic model of the ternary complex consistent with the cryo-EM density (Table S1) and resolve most of the polypeptide chain, the entire 52 nt crRNA, and all complementary nucleotides of the target RNA (nt 5–26) (Figure 1G, Figure S1J).
Cas13d binds target RNA within a large central cleft opposite to the catalytic site
The structure of the Cas13d-crRNA-target RNA ternary complex shows a similar compact architecture as the binary complex, with the protein subunits of both lobes wrapped around a spacer:protospacer duplex (Figure 1G). All 22 complementary nucleotides of the target RNA (nt 5–26) base-pair with the spacer within crRNA (Figure 1E, Figure S1J), and only the terminal two bases of the RNA duplex extend outside of the central cleft (Figure 1G). The 5’ handle maintains a solvent-exposed organization as in the binary state, while the guide-target duplex assembles into an A-form RNA helix within the cleft bound by HEPN1, Helical-1, and Helical-2 domains.
On the outside face of the protein, opposite of the central cleft, HEPN1 and HEPN2 form an endoRNase heterodimer. HEPN1 is subdivided in sequence space (residues ~150–344 termed HEPN1-I and ~495–577 termed HEPN1-II), but forms a contiguous tertiary fold. The α1 of HEPN1-I and the C-terminal portion of HEPN2 form the structural backbone of the bipartite active site and position the four catalytic residues (R295A/H300A/R849A/H854A) of the R-X4- H motif outward on the external face of Cas13d. This orientation of the HEPN active site primes the Cas13d ternary complex for cleavage of both target and collateral RNAs.
Bilobed organization is conserved across Class 2 CRISPR effectors
Class 2 CRISPR-Cas effectors are characterized by their bilobed architectures containing a nucleic acid recognition (REC) lobe that binds crRNA, as well as a nuclease (NUC) lobe that is responsible for cleavage of target nucleic acids (Garcia-Doval and Jinek, 2017) (Figure S2). In contrast to Cas13a, which buries the DR within multiple REC lobe domains (Liu et al., 2017a), Cas13d features a more compact REC lobe, and the DR prominently protrudes from the effector. A similar compaction, coupled to solvent-exposure of crRNA and spacer:protospacer duplex, has been observed among other small Cas nucleases such as SaCas9 (Nishimasu et al., 2015).
Cas13d maintains an extensive nucleoprotein interface throughout the length of bound RNA
In both its binary and ternary forms, Cas13d makes extensive nucleoprotein interactions with spacer and complementary target protospacer, with all five protein domains contributing to RNA stabilization. Both complexes maintain a similar configuration of the DR (Figure 1D, G). In the binary complex, most of the 22-nt single-stranded spacer interfaces with key residues of Helical-1, Helical-2, HEPN1, and HEPN2 (Figure 2). The majority of interactions are comprised of backbone contacts, typically with phosphates but also ribose hydroxyl groups. Individual base interactions occur at lower frequency and are typically constrained to conserved bases within the DR (Figure 2, 3A). These extensive contacts stabilize the spacer region in a primed pseudohelical conformation within the solvent-exposed central channel (Figure 3B-D).
Figure 2. Cas13d nucleoprotein interactions reconfigure between its binary and ternary forms.

Schematic of protein:DNA interactions within binary and ternary forms of Cas13d. Arrows point to the specific interactions. Standard amino acid and nucleotide abbreviations apply to all residues and bases. Conservation of interacting residues and types of interactions are indicated in the legend.
Figure 3. Recognition of crRNA.

(A) Close-up view of the nucleoprotein interactions at the DR-spacer interface in the binary complex.
(B-D) Close-up view of the nucleoprotein interactions at the (B) 5’ end, (C) middle region, and
(D) 3’ end of spacer RNA in the binary complex.
(E) Sequence logo of processed DR from seven Cas13d orthologs (top) with sequences of four selected orthologs shown (bottom). Star, protein:base interactions; dot, protein:backbone interactions. Red bases, mutations used in (F). Blue bases, critical residue differences in (G) between orthologs.
(F) Denaturing gel indicating EsCas13d RNase activity with crRNAs carrying mutant DR.
(G) Denaturing gel indicating EsCas13d RNase activity with non-cognate crRNAs. See also Figure S3
Upon target binding, most of the spacer interactions are shifted towards the 3’ end, following formation of the A-form dsRNA helix. Only four residues (K443, Y447, K376, Y680) interface with RNA in both enzymatic forms, forming backbone contacts with the 5’ end of the spacer in binary form. Overall, interactions along the RNA duplex are sparser in ternary and exclusively confined to the RNA backbone (Figure 2).
Cas13d recognizes the 5’ handle of crRNA
Specific recognition and binding of the constant 5’ handle within their cognate crRNAs is a key requirement for all class 2 CRISPR effectors. In the Cas13d binary structure, we identified multiple residues that interact in either a base- or backbone-specific manner with the DR of crRNA. (Figure 2, Figure 3A-B). The base-specific contacts are concentrated within the unpaired, conserved terminal nucleotides of the DR (nt 22–30) and include G22, U23 from the 2- nt bulge region, as well as A26, A27, A28, and A29 within the 5 terminal nt. The 2-nt bulge (nt 22–23) appears to be an invariant feature among Type VI RNA-guided RNases. Mutagenesis of each of the 6 crRNA nucleotides forming base-specific contacts abolished Cas13d-mediated ssRNA cleavage, confirming the importance of these interactions for proper crRNA binding and positioning (Figure 3E, F).
Given the absence of base-specific contacts along the 5’ region of the DR (nt 1–21), we reasoned that EsCas13d can utilize distinct crRNA of other Cas13d orthologs containing conserved 3’ terminal nucleotides. As predicted, EsCas13d maintained full target cleavage activity with the UrCas13d cognate crRNA, which contains numerous DR mutations relative to the EsCas13d crRNA but maintains the necessary base-specific contacts. In contrast, crRNAs from RffCas13d and RfxCas13d were predicted to disrupt the critical base-specific G22 and G26 contacts; accordingly, target cleavage activity was abolished (Figure 3G). These data provide a structural basis for defining key base requirements and likely crRNA exchangeability across the Cas13d family, facilitating multiplexed effector applications.
A prominent feature characterizing the nucleoprotein interface in both the binary and ternary Cas13d forms is the highly ordered, albeit irregularly shaped single-stranded RNA at the 3’ end of the DR (Figure 2, 3A, 4A). This region (nt 23–29) forms a hairpin loop surrounding a density visible in the cryo-EM map that is likely a centrally located Mg2+ ion in both binary (Figure 3A) and ternary complexes (Figure 4A). While Mg2+ is indispensable for target cleavage for all Cas13 subtypes, it is generally not required for pre-crRNA processing by Cas13a and Cas13b. In contrast to a previous report of Mg2+ -dependent array processing by Cas13d (Yan et al., 2018), we find that Mg2+ is not essential for pre-crRNA processing (Figure S3A-C). Rather, Mg2+ can increase processing efficiency at lower protein: pre-crRNA ratios likely by improving binding affinity of crRNA and Cas13d (Figure S3D), similar to recent observations for Type V Cas12a/Cpf1 (Swarts et al., 2017), highlighting its role across diverse Class 2 effectors.
Figure 4. Complementarity requirements for Cas13d ribonuclease activation.

(A) Close-up view of the nucleoprotein interactions at the DR-spacer interface in the ternary complex.
(B) Close-up view of the nucleoprotein interactions surrounding the spacer:protospacer duplex.
(C) Schematic of competitor cleavage assay describing three possible scenarios and expected results. MM, mismatched competitor target RNAs. NT, non-targeting competitors.
(D) RNA cleavage assay containing Cas13d, crRNA and competitor RNAs only (no matching target). Signal is normalized to a negative control containing Cas13d and crRNA only. M, fully matched competitor. Each condition represents activity across four distinct target sequences (n = 3, mean ± SD).
(E) RNA cleavage assay containing Cas13d, crRNA, target RNA, and competitor RNA at 25X molar excess to target. Signal is normalized to the condition with Cas13d, crRNA and target without competitor. Each condition represents activity across four distinct target sequences (n=3, mean ± SD).
(F) Illustration of parallel readout for Cas13d cleavage and binding in human cells using a fluorescent splicing reporter with the.RfxCas13d-NLS (CasRx) ortholog.
(G) CasRx cleavage and dCasRx splicing (binding) activity as a function of crRNA spacer length. Cleavage and splicing were each normalized to a non-targeting crRNA and represented relative to the activity of the full-length (22 nt) crRNA (n=3, mean ± SD).
See also Figure S3.
Target duplex formation and lack of PFS requirement
In the binary Cas13d surveillance complex, most of the single-stranded spacer is solvent-exposed and structurally poised for base-pairing with potential targets to enter a ternary state (Figure 4B). For DNA-targeting class 2 CRISPR effectors, target interaction is initiated by protein-PAM (protospacer adjacent motif) interactions. Cas13d lacks the analogous protospacer flanking sequence (PFS) requirements (Konermann et al., 2018; Yan et al., 2018). In contrast, some Cas13a orthologs (including LshCas13a) have been reported to display a single base 3’ H (non-G) PFS. This was previously proposed to be caused by base-pairing of the terminal conserved C(30) of crRNA DR with a complementary target G, which would then destabilize critical contacts with the HEPN-1 domain following rotation of the C(30) base away from the protein density (Figure S3E) (Liu et al., 2017b). In EsCas13d, the C(30) base is already rotated toward the target in its ternary form despite a mismatch with the target base (U) (Figure S3F). A complementary G nucleotide would therefore not be expected to cause any additional rearrangement, suggesting a structural rationale for the absence of a PFS requirement in Cas13d.
Cas13d binding and cleavage are interlinked
Given the lack of an overt PFS requirement, we sought to understand Cas13d target binding and cleavage in the context of target complementarity. Class 2 CRISPR-Cas nucleases exhibit distinct binding and cleavage mechanisms. Cas9 from Streptococcus pyogenes exploits a sequential target binding mechanism with stable binding after ~12 nt of PAM-proximal spacer complementarity (Dahlman et al., 2015; Kiani et al., 2015; Sternberg et al., 2014), while mismatches in this “seed” region are poorly tolerated (Hsu et al., 2013). Cas9 target cleavage, however, requires ~4 nt of additional matches to activate the HNH catalytic domain (Sternberg et al., 2015). In contrast to SpCas9, Cas12a/Cpf1 requires extended complementarity of at least 17 nt for stable binding (Singh et al., 2018). Cas13 binding and cleavage requirements are largely unclear, although a central seed region has been proposed for both Cas13a and Cas13b based on the observation that mismatches are least tolerated in the center of the spacer (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Knott et al., 2017; Liu et al., 2017a; Smargon et al., 2017) and can affect HEPN domain activation (Tambe et al., 2018).
To investigate Cas13d target binding and cleavage complementarity, we conducted cleavage assays with a panel of target RNA competitors carrying 4-nt mismatches at different positions along the target sequence (Figure 4C, Table S2). We sought to distinguish between three possible scenarios when a competitor RNA is introduced at high molar excess relative to the target RNA: Cas13d 1) directly cleaves competitor, increasing fluorescent signal relative to target alone, 2) binds competitor without forming a catalytically active ternary complex, thereby sequestering Cas13d from target RNA and decreasing fluorescent signal, or 3) is unable to bind or cut competitor, with no resulting change in fluorescence.
In the absence of target, perfectly matched competitors triggered a robust increase in fluorescence as expected (Figure 4D). Competitors carrying 5’-proximal mismatches activated bystander cleavage in 2/4 guides, indicating that complementarity in the DR-distal region of the crRNA spacer is not strictly required for ternary activation (scenario 1). Bystander activity was not activated by other competitors (scenario 3), suggesting a lack of a consistent seed region within the crRNA spacer for target cleavage. In the presence of target, we observed a similar pattern of Cas13d activity across all competitor mismatch positions to the target-free condition (Figure 4E). In particular, none of the mismatch competitors mediated a decrease in fluorescence (in contrast to scenario 2), suggesting that stable target binding requires at least 18 nt of complementarity and that binding and cleavage are closely coupled in Cas13d.
To further explore the interdependence between nucleotide complementarity and cleavage efficiency, we tested a closely related Cas 13d ortholog (RfxCas13d) in a cell-based reporter to assay for Cas13 knockdown and splicing as a proxy for RNA binding and cleavage activity (Konermann et al., 2018) (Figure 4F). Using a series of crRNA spacer truncations progressing from 22-nt to 10-nt, we observed a simultaneous decrease in knockdown and splicing as a function of decreasing spacer length (Figure 4G). Half-maximal activity occurred between 18 and 20-nt spacer length.
Taken together, the lack of a clear seed is consistent with the observation of a solvent exposed spacer stabilized in an accessible configuration along the open channel in the binary complex, positioning crRNA to initiate base-pairing with protospacer at multiple possible locations. Beginning with ~18 nt of complementarity, Cas13d likely undergoes a partial reconfiguration of its relevant nucleoprotein interface. Upon reaching complete 22 nt guide-target duplex complementarity, the enzyme transitions to a maximally active and cleavage-competent state.
Target RNA binding reconfigures Cas13d into an ssRNA cleavage complex and allosterically activates the HEPN domains
The transition from the binary surveillance complex to the ternary complex activates Cas 13d for RNA cleavage. Numerous conformational rearrangements occur during this transition, stabilizing the activated cleavage-competent state within the catalytic HEPN domain dimer. HEPN domains function as obligate dimers, with 2 R-X4–6-H motifs forming a bipartite active site to mediate RNA hydrolysis (Anantharaman et al., 2013). Target RNA binding to Cas13d triggers enzyme activation. The most dramatic polypeptide rearrangements occur within the Helical-1 domain, which shifts outward by an average Cα-Cα RMSD of ~12 Å to accommodate target RNA binding within an expanded cleft (Figure 5A,B). To a lesser extent, portions of the HEPN1, Helical-2, and HEPN2 domains, particularly regions proximal to the 3’ end of spacer (5’ end of protospacer), undergo subtler structural changes. Whereas most of the DR remains unperturbed, the majority of the spacer reorganizes from a single-stranded pseudohelical conformation into a double-stranded A-form RNA helix (Figure 5C-D). Collectively, such rearrangements widen the RNA-binding cleft, ranging from ~13Å at the narrowest point between NTD and Helical-1 (Figure 5E) and from 12Å-44Å between Helical-1 and HEPN2 at the 3’ end of crRNA (Figure 5F, Movie S1). Mutagenesis of key residues forming critical crRNA and target RNA backbone contacts abolished target RNA cleavage but did not affect pre-crRNA processing (Figure S3G,H), confirming their role in stabilizing the spacer:target duplex.
Figure 5. Conformational rearrangements of Cas13d upon target substrate recognition.

(A) Schematic representation of Cas13d domain movement. Each vector represents a Cα atom translation from its binary to ternary forms. Vector length corresponds to the translational distance. The directions of movement are indicated by arrows.
(B) Quantitative measurement of Cas13d domain movement (mean ± SD). The residues that undergo largest translations in HEPN2 are highlighted with a dashed box.
(C) Superposition of the crRNA in the ternary complex (DR region in magenta and spacer in cyan) with that in the binary complex (in gray).
(D) Schematic representation of Cas13d crRNA movement. Vector representations as in (A). The position of the phosphate atom in the ternary structure are highlighted in red.
(E-F) Target RNA binding widens the central channel and (E) opens the cleft between NTD and Helical-1 and (F) increases distance between Helical-1 and HEPN2.
(G-I) Stabilization of the HEPN catalytic domain dimer. (G) Surface representation of Cas13d ternary complex, with HEPN catalytic residues highlighted in white. (H) Superposition of HEPN1 and HEPN2, and (I) a close-up view of the catalytic center. The four catalytic residues advance inward and a nearby loop is stabilized upon target binding.
See also Figure S4
We also observe some intra-domain reorganization within Cas13d (Figure S4A). Most prominently, a kink is introduced into the C-terminal region of HEPN1–1, while the N-terminal loop of HEPN1-II (and, correspondingly, the C-terminal loop residues of Helical-1) reposition by an average Cα-Cα RMSD of ~4 Å. Both of these correspond to the two flexible linkers connecting Helical-1 to HEPN1-I and HEPN1-II, which reposition Helical-1 as it cradles incoming target RNA (Figure S4B-C).
The HEPN2 domain, which resides on the “back side” of Cas13d (Figure 5G), undergoes several rearrangements to facilitate target cleavage. A structural alignment of HEPN1-I between binary and ternary forms indicates the catalytic residues of HEPN2 (R849 and H854) reposition by ~4 Å, shifting closer to the corresponding catalytic residues of HEPN1-I (R295 and H300) (Figure 5H-I). Given that target RNA resides on the opposite side of the HEPN catalytic site, these rearrangements suggest allosteric HEPN activation modulated by target RNA binding. Together, the structural changes serve to (1) accommodate target RNA binding and (2) reconfigure the catalytic site into its cleavage-competent form.
Apo Cas13d utilizes multiple dynamic domains to form an RNA-binding cleft that is stabilized upon guide binding
To better understand the mechanism of crRNA binding, we examined the apo form of Cas13d. Using similar experimental strategies as before, we obtained 154,889 particles for 3D classification and refinement. We observed variable density within the 2D class averages (Figure S5A), in contrast with the sharp signal present for both the binary and ternary data sets (Figure S1). Despite the apparent heterogeneity, an ab initio 3D reconstruction led to a map that was refined to ~6.5 Å resolution (Figure 6A-B, Figure S5C-D, Table S1). Strikingly, the apo Cas13d reconstruction accounts for only part of the mass in comparison to both binary and ternary forms, with the majority of homogeneous density corresponding to a stable α-helical core (Figure 6C). To verify that the remaining density was present in the data, we derived 2D class averages ab initio and found that many of these captured protein density that was otherwise unresolved in the 3D reconstruction (Figure S5E).
Figure 6. Apo Cas13d contains mobile domains that are stabilized upon RNA binding.

(A) Domain organization of full-length &Cas13d and the visible portion of apo EsCas13d by cryo-EM (see below). Domain boundaries and catalytic sites of the HEPN domains (R295, H300, R849, H854) are indicated. Undetectable regions in the apo Cas13d reconstruction (see B-D) are represented with dashed lines
(B) Cryo-EM reconstruction of apo EsCas13d, resolved to 6.5 Å resolution.
(C) Rigid-body docked model of EsCas13d based on the cryo-EM density.
(D) Superposition of the structured portion of apo EsCas13d evident within the cryo-EM reconstruction (surface representation) with that of EsCas13d in the binary form.
(E) Differential HDX colored onto the binary protein form of Cas13d. Regions that exhibit statistically significant changes in amide exchange are colored onto the structure, and onto a domain organization (top). Insets show four distinct close-up views. White, no statistically significant difference; gray, no coverage in the differential HDX map.
We could readily assign the known domain boundaries by docking the binary model into the apo reconstruction. Portions of HEPN1, Helical-2, and HEPN2 (NUC lobe) account for the mass derived by cryo-EM, whereas the entire REC lobe (NTD and Helical-1) was invisible (Figure 6D). The domain organization of NUC lobe in the apo enzyme remains largely unchanged relative to the binary complex at this resolution. These data suggest that the REC lobe (NTD, Helical-1) and portions of HEPN2 may be dynamically arranged in the absence of RNA. Further, because cryo-EM is performed under solution conditions, the observed dynamics are likely an inherent property of the enzyme.
To understand Cas13d conformational dynamics between its apo and binary forms on a residue-by-residue level, we conducted hydrogen-deuterium exchange with mass spectrometry (HDX-MS) to probe for unstructured and flexible regions of Cas13d that undergo deuterium exchange more rapidly than those that are stable and hydrogen-bonded (Englander, 2006). Individual exchange profiles of apo and binary Cas13d overall showed similar patterns at the sequence level (Figure S6A,B), indicating that the constituent domains, including the REC lobe, exhibit similar overall folds. However, several regions of Cas13d were clearly stabilized upon RNA binding, as indicated by reduced exchange in the differential profiles (Figure 6E). Three of these regions bind distinct segments within crRNA. Residues ~81–131 form part of the NTD, where residues K68, G85 and R139 make critical base-specific interactions with the 3’ end of the DR (Figure 2, Figure 3F-G), while residues ~371–390 and ~594–613 form an RNA-binding interface with the phosphate backbones of the 5’ and 3’ ends of spacer RNA, respectively. Finally, residues ~325–360 represent the interface between HEPN1-I and Helical-1, with residues 335–340 forming a hinge-like structure connecting Helical-1 to the NUC lobe (Figure 6E).
These data, under solution conditions, indicate that both Cas13d lobes are appropriately folded in the apo configuration and the REC lobe is likely to be mobile relative to the NUC lobe. 2D class averages indicate the presence of unresolved density in the 3D reconstruction, and HDX analysis indicates increased mobility of the NTD and the linker region (residues ~325–360) connecting Helical-1 to the NUC lobe. Upon RNA binding, the Cas13d binary complex is stabilized in multiple regions, and a central, positively charged RNA binding cleft is formed between the REC and NUC lobes (Figure S6C). Analogous reconfigurations of NUC and REC lobes upon crRNA binding and formation of the central positively charged cleft in the binary complex have been reported for other Class 2 CRISPR enzymes, including Cas9 (Jinek et al., 2014; Nishimasu et al., 2015), and may have implications for sampling distinct RNA features for facilitating efficient crRNA recognition and binary transition.
Structure-guided Cas13d truncations for minimal coding sequence
The compact size of Cas 13d is accompanied by an integration of multiple distinct functions into each individual constituent domain. Each of the protein domains within Cas13d contribute key protein:RNA contacts in addition to their structural and catalytic functions (Figure 2), with many conserved residues among Cas13d orthologs (Figure S7A). As a consequence, regions of high conservation between Cas13d orthologs are dispersed throughout the linear protein sequence and separated by only short stretches of low conservation.
We predicted that all five domains of Cas13d would be essential for its RNase activity, in contrast to our previous demonstration that the REC2 domain of SpCas9 is largely dispensable for target DNA cleavage (Nishimasu et al., 2014). We designed 6 deletions in the closely related RfxCas13d ortholog (CasRx) and evaluated the ability of these truncation mutants to knockdown a fluorescent reporter in human cells (Figure 7A, B). One deletion in Helical-2 (Δ3), which is located on the external surface of Cas13d and avoids the removal of highly conserved residues, exhibited full activity relative to the wild-type enzyme.
Figure 7. Cas13d truncations and protein conservation.

(A) Schematic of reporter assay for CasRx-mediated RNA knockdown in human cells.
(B) Knockdown activity of CasRx deletions. Left panel, consensus sequence of the multiple sequence alignment of Cas13d orthologs (Figure S8) with domain boundaries and sequence identity as indicated. Pink bars denote conserved protein:crRNA interactions. Deletion junctions are denoted as a linked interval. Knockdown activity is represented relative to non-targeting guide (mean ± SD, n = 3). Deletions exhibiting > 95% reduction of RFP+ cells are bolded and the corresponding deletion junction is marked in blue.
(C) Surface view of the EsCas13d ternary complex colored by conservation percentage. All truncated regions retaining high knockdown activity (blue) are located on external surface of Cas13d with low (<40%) conservation. Regions with high degree of conservation are concentrated in the RNA-binding cleft (left panel) as well as the HEPN active site (right panel).
See also Figure S7
A second round of 7 small, surface-localized deletions on top of Δ3 successfully generated three additional variants with >95% knock-down activity (Δ3.1, Δ3.3 and Δ3.7, Figure 7B,C). The most active resulting variant (Δ3.3) removes 50 aa, facilitating AAV-mediated CasRx delivery with increased flexibility (Figure S7B,C).
Discussion
The structural, biochemical, and functional analysis of Type VI EsCas13d presented here reveals 3 distinct states of the Cas13d enzyme as it transitions from its inactive apo (Cas13d) to a surveillance (Cas13d-crRNA) and cleavage-competent (Cas13d-crRNA-target RNA) form. These transitions are accompanied by numerous structural rearrangements that accommodate and stabilize these enzymatic forms. Our data suggests a model whereby REC lobe dynamics within Cas13d may facilitate scanning for the crRNA for its proper recognition within a CRISPR array.
Cas13d is among the smallest CRISPR-Cas single effectors, with 20–30% less mass than other Type VI Cas 13 endoRNases (Konermann et al., 2018; Yan et al., 2018). Compared to Cas13a (Knott et al., 2017; Liu et al., 2017a; Liu et al., 2017b), Cas13d compacts the analogous NTD and Helical-1 domains of Cas13a into a single, 150 aa NTD (Figure S2) within the REC lobe of Cas13d.
Upon satisfying multiple base-specific contacts within the 3’ end of the DR, crRNA binding triggers stabilization of the binary complex and formation of a central positively-charged cleft between the REC and NUC lobes of Cas13d. The solvent-exposed, single-stranded spacer region takes on a stabilized pseudo-helical conformation which appears to be poised for target binding at multiple positions. Stable RNA duplex formation requires ~18 nt of complementarity and triggers large conformational rearrangements primarily in Helical-1 and HEPN2 to activate the bipartite HEPN domain. Unlike some other Type VI RNases, Cas13d does not require any PFS for target recognition, possibly mediated by the flipped C(30) base within crRNA that avoids base-specific pairing with target RNA. Upon Cas13d ternary formation, the HEPN catalytic residues within the HEPN1 and HEPN2 domains migrate toward one another to generate an external-facing active site. This facilitates direct RNA hydrolysis of both guide-complementary activator RNA and non-complementary bystander RNA for antiviral defense.
Although class 2 CRISPR-Cas nucleases share many common traits for nucleic acid sensing, they also vary substantially in size, shape, domain architecture, and organization. Within individual Cas types (e.g. Type VI), there is often minimal sequence conservation across subtypes. Cas13d enzymes, for example, do not share sequence homology with Cas13a apart from the minimal 6 aa HEPN catalytic motifs, despite overall similarity of their RNase activities. Independent origins of type V subtypes from mobile genetic elements, as previously suggested for DNA-targeting type II and V effectors and subtypes via distinct TnpB transposase subfamilies (Koonin et al., 2017), may also explain the convergence and divergence of Cas13 superfamily function and structural organization.
Overall, our data elucidates the structural basis of Cas13d RNA-guided RNase activity and its compaction of these properties into a minimal effector size, providing a blueprint for improving Cas13d-based RNA targeting tools. Further engineering of smaller Cas13d variants, as shown here, will enable flexible packaging into size-limited viral vectors with large regulatory elements for optimal transgene expression and activity. Furthermore, base-specific contacts of Cas13d with the 5’ handle of crRNA were sufficient to delineate crRNA exchangeability across distinct Cas13d orthologs, defining functionally orthogonal subfamilies that could be exploited to facilitate Cas 13-based multiplexing applications in both cellular (Abudayyeh et al., 2017; Konermann et al., 2018) and cell-free systems (Gootenberg et al., 2017). Some Cas13d orthologs have accessory proteins (Yan et al., 2018) that enhance activity and could provide clues for improving Cas13d binding or cleavage. In analogy to engineered variants of Cas9 and related nucleases, structure-guided engineering of diverse CRISPR-Cas13d enzymes can be expected to enable improved properties for diverse biomolecular applications of RNA targeting.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dmitry Lyumkis (dlyumkis@salk.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture of Human Embryonic Kidney (HEK) cell line 293FT
The human embryonic kidney (HEK) cell line 293FT (female, Thermo Fisher) was cultured in DMEM (4.5 g/L glucose), supplemented with 10% FBS (GE Life Sciences) and 10 mM HEPES at 37°C with 5% CO2. Cells were passaged before reaching 90% confluency using TrypLE Express (Life Technologies) at a ratio of 1:2. The cell line was purchased directly from Thermo Fisher and maintained within the lab for less than 20 passages total following purchase. It was not otherwise authenticated.
METHOD DETAILS
Protein expression and purification
Recombinant £sCas13d proteins were cloned into a pET-based vector with an N-terminal His-MBP fusion and TEV protease cleavage site. The resulting plasmids were transformed into Rosetta2(DE3) cells (Novagen), induced with 200 qM IPTG at OD600 0.5, and grown for 20 hours at 18°C. Cells were then pelleted, freeze-thawed, and resuspended in Lysis Buffer (50 mM HEPES, 500 mM NaCl, 2 mM MgCl2, 20 mM Imidazole, 1% v/v Triton X-100, 1 mM DTT) supplemented with 1X protease inhibitor tablets, 1 mg/mL lysozyme, 2.5U/mL Turbo DNase (Life Technologies), and 2.5U/mL salt active nuclease (Sigma Aldrich). Lysed samples were then sonicated and clarified via centrifugation (18,000 × g for 1 hour at 4°C), filtered with 0.45 qM PVDF filter and incubated with 50 mL of Ni-NTA Superflow resin (Qiagen) per 10 L of original bacterial culture for 1 hour. The bead-lysate mixture was applied to a chromatography column, washed with 5 column volumes of Lysis Buffer, and 3 column volumes of Elution Buffer (50 mM HEPES, 500 mM NaCl, 300 mM Imidazole, 0.01% v/v Triton X-100, 10% glycerol, 1 mM DTT). The samples were then dialyzed overnight into TEV Cleavage Buffer (50 mM Tris-HCl, 250 mM KCl, 7.5% v/v glycerol, 0.2 mM TCEP, 0.8 mM DTT, TEV protease) before cation exchange (HiTrap SP, GE Life Sciences) and gel filtration (Superdex 200 16/600, GE Life Sciences). Purified, eluted protein fractions were pooled and frozen at 4 mg/mL in Protein Storage Buffer (50 mM Tris-HCl, 1M NaCl, 10% glycerol, 2 mM DTT).
Preparation of guide and target RNAs
In vitro transcription template oligos carrying the T7 promoter were synthesized (IDT) and either annealed with an antisense T7 oligo for crRNAs or PCR amplified for targets and arrays. In vitro transcription was performed using the Hiscribe T7 High yield RNA synthesis kit (New England Biolabs) at 31°C for 12 hours. For in vitro cleavage reactions, targets were body-labeled through incorporation of Aminoallyl-UTP-ATTO-680 (Jena Biosciences) during the in vitro transcription. crRNAs and short competitors were purified using RNAclean Agencourt AMPure XP beads (Beckman Coulter) with addition of 50% volume of isopropanol for retention of small RNAs. Longer targets and arrays were purified with the MEGAclear Transcription Clean-Up Kit (Thermo Fisher) and frozen at −80°C. The short 30nt target for ternary complex formation for cryo-EM imaging was synthesized by Synthego.
Binary and ternary complex formation.
For cryo-EM binary complex formation, 200 μg &Cas13d was incubated with a 3x molar excess of crRNA in complex formation buffer at 37°C for 1 hr (25mM Tris-HCl, 50mM NaCl, 1mM DTT, 1mM MgCl2, pH 7.5). The resulting binary complex was purified by size exclusion chromatography on a Superdex 200 16/600 column (GE Life Sciences) in S200 complex buffer (25mM Tris HCl, 100mM NaCl, 1mM DTT, 1mM MgCl2, 5% glycerol, pH 7.5). Binary peak fractions were pooled and concentrated to 1.5 mg/mL and processed for cryo-EM sample preparation. Ternary formation was performed sequentially, with 15 minutes of binary complex formation at 1:2 ratio of dEsCas13d:crRNA followed by 45 minutes of incubation with target at 1:3 ratio (protein: target). Ternary size exclusion purification and concentration was performed analogous to the binary complex. For HDX binary sample preparation, the purification was scaled up 5X relative to the cryo-EM samples but was otherwise identical. Apo-EsCas13d protein was buffer exchanged into S200 complex buffer and normalized to 1.5 mg/mL prior to Cryo-EM sample preparation.
Electron microscopy sample preparation and data acquisition
All samples, including binary, ternary, and apo, were concentrated to ~1.5 mg/ml prior to vitrification. In all 3 cases, Amphipol A8–35 was added to the sample to a final concentration of 0.1% (w/v) immediately before vitrification on cryo-EM grids, in order to ameliorate preferential specimen orientation, which was established in earlier attempts to collect the data (Lu et al., 2014). Cryo-EM grids were prepared under >80% humidity at 4°C inside a cold room, and a multi-blotting approach was used to increase particle density (Snijder et al., 2017). Initially, 2ul of sample was applied to an UltrAuFoil R1.2/1.3 300-mesh grid (Quantifoil) after plasma-cleaning (75% argon/25% oxygen atmosphere, 15 W for 7s using a Gatan Solarus). Next, the grid was side-blotted manually with a filter paper (Whatman No.1) followed by a second round of sample loading and side-blot. Finally, another 2ul sample was added to the grid and blotted immediately before plunging into liquid ethane using a manual plunger. Leginon was used for automated EM image acquisition (Suloway et al., 2005). Micrographs of Cas13d-apo and Cas13d binary complex were collected on a Talos Arctica microscope (FEI) operating at 200kV and equipped with a K2 Summit direct electron detector (Gatan). A nominal magnification of 57,000x was used for data collection, providing a pixel size of 0.73 Å at the specimen level, with a defocus range of −0.5 μm to −2.0 μm. Micrographs of Cas13d ternary complex were acquired on a Titan Krios microscope (FEI) operating at 300kV and equipped with a K2 Summit direct electron detector. A nominal magnification of 37,000x was used for data collection, corresponding to a pixel size of 0.79 Å at the specimen level, with the defocus ranging from −1.0 μm to −3.0 μm Movies were recorded in counting mode, with a total dose of ~57e- per Å2 for all three samples and under a dose rate of ~2.5 – 3 electrons per pixel per second. All details corresponding to individual datasets are summarized in Supplementary Table 1.
Image processing of Cas13d binary and Cas13d ternary complex
All pre-processing was performed within the Appion suite (Lander et al., 2009). Motion correction was performed using the program MotionCor2 (Zheng et al., 2017) and exposure-filtered in accordance with the relevant radiation damage curves (Grant and Grigorieff, 2015). For processing of Cas13d binary complex and Cas13d ternary complex datasets, structures of LbuCas13a-crRNA complex (PDB:5XWY) and Cas13d-crRNA complex were used as the templates for automatic particle picking in Appion, respectively, using FindEM (Roseman, 2004). The Contrast transfer function (CTF) was estimated using CTFFind4 during data collection on whole micrographs (Rohou and Grigorieff, 2015). After selecting particle coordinates, per-particle CTF estimation was refined using the program GCTF (Zhang, 2016). Stacks containing 400K (Cas13d binary) and 680K (Cas13d ternary) particles were subjected to two rounds of 2D classification, followed by one round of 3D classification in GPU-enabled Relion (Kimanius et al., 2016). The best classes containing 49K (Cas 13d binary) and 52K (Cas 13d ternary) particles were selected for Relion refinement. Lastly, the parameters were imported into cisTEM (Grant et al., 2018), and the last several rounds of orientation and per- particle CTF refinement were performed to improve the resolution by ~0.2 Å for the binary dataset and ~0.3 Å for the ternary data dataset. The spectral amplitudes for each reconstruction were flattened inside cisTEM between 8 Å and 3.4 Å or 3.3 Å for the binary and ternary complexes, respectively. The resolutions for both maps were evaluated using conventional Fourier Shell Correlation analysis to evaluate global resolution and directional Fourier Shell Correlation analysis to obtain 3D FSCs and evaluate directional resolution anisotropy (Tan et al., 2017). Due to the manner in which the particles adhered to the air-water interface, the ternary map is characterized by more anisotropic directional resolution.
Image processing of apo Cas13d
Cryo-EM data was processed in a conceptually similar manner as in binary/ternary. The same templates used for particle picking of Cas13d binary complex were also used to select 330,986 particles from the apo Cas13d dataset. After CTF estimation in GCTF (Zhang, 2016)) and 2 rounds of Relion 2D classification to remove bad particles, the extracted stack containing 154,889 particles was imported into cryoSPARC for ab initio reconstruction. We used the following parameters in the reconstruction: Number of Ab-initio classes=1, Initial resolution=20 and Maximum resolution=5, which resulted in a map with clearly distinguishable secondary structure elements from 16K particles. Numerous other attempts were performed to obtain an ab initio reconstruction, but the particle heterogeneity from the large amount of conformational flexibility precluded our ability to improve the map or use a greater subset of particles at this stage. After a map was generated, the orientations were imported into cisTEM (Grant et al., 2018), and the orientations, as well as per-particle CTF parameters were refined for several rounds, resulting in a ~0.2 Å increase in resolution. The final global resolution was estimated at 6.5 Å. Further attempts to classify the data, either through cisTEM, or through other processing packages, did not result in visual improvements to the map. We believe that the reasons for the challenges within this dataset have to do with the heterogeneity associated with the apo form of the protein, coupled to its small size. The homogeneous part of the protein only accounts for ~60 kDa of total mass.
Model building and refinement
The model of Cas13d binary complex was built de novo in Coot (Emsley et al., 2010). A poly-ala model with gaps in looped region was first built based on the EM density, and then residues having bulky side chains (Phe, Trp, and Tyr) were registered to facilitate sequence assignment of the remaining protein. The register of crRNA was conducted based on prior knowledge: that the DR region will form a base-paired stem-loop structure and that the spacer is single stranded. This allowed for unambiguous registration of the N-terminal residues 1–57, as well as certain loops scattered throughout the structure and the C-terminal residues 950–954 were poorly ordered, and were thus omitted from the final model. Most of these regions are not conserved among Cas13d orthologs, with ~50% of Cas13d orthologs missing the N-terminal residues (Konermann et al., 2018). For building the model of the Cas13d ternary complex, the binary model was first docked into the ternary cryo-EM map and individual domains were repositioned according to the relevant conformational rearrangements. The NTD, HEPN1, HEPN2 domains and DR of crRNA remain constant, whereas the Helical-1 and Helical-2 domains, as well as the crRNA spacer required repositioning. All connecting loops and any atoms outside of density were rebuilt accordingly. Watson-Crick base pairing between the spacer and target protospacer allowed unambiguous RNA registration. Each model was independently refined in PHENIX (Adams et al., 2010) using phenix.real_space_refine against separate EM half-maps with geometrical, secondary structure, and hydrogen bond restraints. The maps were refined into a working half-map, and improvement of the model was monitored using the free half map. The geometry parameters of the final models were validated in Coot and using MolProbity (Chen et al.. 2010). These refinements were performed iteratively until no further improvements were observed. The Cas13d-apo model was generated by rigid-body docking of the Cas13d binary complex structure into the Cas13d-apo cryo-EM map without further refinement or modification, and the parts of the model having poor densities were removed. All the structure figures were prepared in Pymol and UCSF Chimera.
Biochemical cleavage assays
For DR mutant analysis, purified EsCas13d protein and guide RNA were mixed at 2:1 molar ratio in RNA Cleavage Buffer on ice (25mM Tris pH 7.5, 15mM Tris pH 7.0, 1mM DTT, 6mM MgCl2). Protein and and guide RNA were incubated at 37°C for 15 minutes for binary complex formation prior to the addition of target at 1:2 molar ratio relative to EsCas13d. For pre-crRNA cleavage reactions, purified EsCas13d and EsdCas13d proteins were mixed with purified pre- crRNA at 0.5:1, 1:1 and 5:1 molar ratios in RNA cleavage buffer containing 6mM MgCl2 or EDTA. Reactions were prepared on ice and incubated 37°C for 1 hour. Both cleavage reactions were quenched with 1 μL of enzyme stop solution (10 mg/mL Proteinase K, 4M Urea, 80mM EDTA, 20mM Tris pH 8.0) at 37°C for 15 minutes. The in vitro cleavage reaction was finally denatured in 2X RNA loading buffer (2X: 13mM Ficoll, 8M Urea, 25 mM EDTA), at 85°C for 10 minutes. Cleavage products were separated on a 10% TBE-Urea gel (Life Technologies). Reactions containing and unlabeled guide RNA and a fluorescently labeled target (Atto-680, Jena Biosciences) were visualized on the Odyssey Clx Imaging System (Li-Cor); pre-crRNA cleavage gels were stained with SYBR Gold prior to imaging via Gel Doc EZ system (Bio-Rad).
For collateral fluorescent ssRNA reporter assays (Figure 4C-E), EsCas13d protein was mixed with guide RNA at 1:1 ratio in RNA cleavage buffer on ice and then assembled into protein:guide complexes at 37°C for 15 minutes. Reactions were put on ice and competitor RNAs containing 4nt mismatches at different positions were mixed in at 25X molar ratio to target RNA. Target RNAs were added at a 1:5 ratio to EsCas13d, and 150nM RNAse-Alert substrate (Thermo-Fisher) was added and mixed. Reactions were then incubated in a real-time PCR machine (Bio-Rad, CFX384 Real-Time System) for 180 minutes at 32 °C and measurements were taken every 5 minutes. Fluorescence values are an average of the last 5 measurements for each condition.
Cell-based reporter assays
Engineered .RfxCas13d mutants were cloned into pXR002: EF1a-dCasRx-2A-EGFP (Addgene #109050) and prepared using the Nucleobond Xtra Midi EF Kit (Machery Nagel) according to the manufacturer’s protocol. RfxCas13d gRNAs with variable spacer lengths were cloned into pXR003: CasRx gRNA cloning backbone (Addgene #109053) by golden gate assembly.
HEK 293FT cells were transfected in 96-well format with 200 ng of Cas13d expression plasmid, 200 ng of guide expression plasmid, and 20 ng of the bichromatic reporter plasmid with Lipofectamine 2000 (Life Technologies). Cells were harvested in FACS Buffer (1X DPBS−/−, 0.2% BSA, 2 mM EDTA) after 72 hours, then analyzed in 96-well plate format using a MACSQuant VYB (Miltenyi Biotec) followed by analysis using FlowJo 10. RG6 was a gift from Thomas Cooper (Addgene plasmid # 80167) and modified to replace EGFP with mTagBFP2. All represented samples were assayed with three biological replicates.
Hydrogen-deuterium exchange detected by mass spectrometry
Differential Hydrogen-deuterium exchange mass sepctrometry (HDX-MS) experiments were conducted as previously described with a few modifications (Chalmers et al., 2006).
Peptide Identification: Peptides were identified using tandem MS (MS/MS) with an Orbitrap mass spectrometer (Fusion Lumos, ThermoFisher). Product ion spectra were acquired in data-dependent mode with the top five most abundant ions selected for the product ion analysis per scan event. The MS/MS data files were submitted to Mascot (Matrix Science) for peptide identification. Peptides included in the HDX analysis peptide set had a MASCOT score greater than 20 and the MS/MS spectra were verified by manual inspection. The MASCOT search was repeated against a decoy (reverse) sequence and ambiguous identifications were ruled out and not included in the HDX peptide set.
HDX-MS analysis: Cas13d (10 μM) was incubated with or without guide RNA at a 1:5 protein- to-RNA molar ratio for 1 h at room temperature. Next, 5 μ! of sample was diluted into 20 μl D2O buffer (50 mM Phosphate, pH 7.5; 150 mM NaCl; 2 mM TCEP) and incubated for various time points (0, 10, 30, 90, 300, 900, and 3600 s) at 4°C. The deuterium exchange was then slowed by mixing with 25 μl of cold (4°C) 6 M urea, 200 mM TCEP and 1% trifluoroacetic acid. Quenched samples were immediately injected into the HDX platform. Upon injection, samples were passed through an immobilized pepsin column (1mm × 2cm) at 50 μl min−1 and the digested peptides were captured on a 1mm × 1cm C8 trap column (Agilent) and desalted. Peptides were separated across a 1mm × 5cm C18 column (1.9 μl Hypersil Gold, ThermoFisher) with a linear gradient of 4% - 40% CH3CN and 0.3% formic acid, over 5 min. Sample handling, protein digestion and peptide separation were conducted at 4°C. Mass spectrometric data were acquired using an Orbitrap mass spectrometer (Fusion Lumos, Thermo Fisher). HDX analyses were performed in triplicate, with single preparations of Cas13d and the Cas13d-gRNA complex. The intensity weighted mean m/z centroid value of each peptide envelope was calculated and subsequently converted into a percentage of deuterium incorporation. This is accomplished determining the observed averages of the undeuterated and fully deuterated spectra and using the conventional formula described elsewhere (Zhang and Smith, 1993). Statistical significance for the differential HDX data is determined by an unpaired t-test for each time point, a procedure that is integrated into the HDX Workbench software (Pascal et al., 2012). Corrections for back-exchange were made on the basis of an estimated 70% deuterium recovery, and accounting for the known 80% deuterium content of the deuterium exchange buffer.
Data Rendering: The HDX data from all overlapping peptides were consolidated to individual amino acid values using a residue averaging approach. Briefly, for each residue, the deuterium incorporation values and peptide lengths from all overlapping peptides were assembled. A weighting function was applied in which shorter peptides were weighted more heavily and longer peptides were weighted less. Each of the weighted deuterium incorporation values were then averaged to produce a single value for each amino acid. The initial two residues of each peptide, as well as prolines, were omitted from the calculations. This approach is similar to that previously described (Keppel and Weis, 2015).
crRNA filter binding assays
crRNA oligos were synthesized by Synthego, resuspended in RNase-free water at 100 μM and 3’-end labeled with radioactive cordycepin using yeast poly(A) polymerase in a 10 μL reaction (1 pmol crRNA oligo, 2 pmol [α−32P] cordycepin 5’-triphosphate, 2 μL 5X yeast poly(A) polymerase buffer, 1 μL yeast poly(A) polymerase, 5 μL RNase-free water). The reaction was incubated at 37°C for 30 minutes, then 40 μL RNase-free water was added to increase volume and the 50 μL sample was passed through a P-30 column to remove free cordycepin. The RNA was extracted with 300 μL acid-phenol:chloroform and precipitated in 700uL 100% ethanol with 30 μL 3M Sodium Acetate, pH 5.5, and 1 μL GlycoBlue (15 mg/mL). The pellet was resuspended in 100 μL RNase-free water, containing ~10 nM 3’-end labeled RNA.
Each binding reaction was performed in 10 μL containing 1 μL 3’-end labeled crRNA (final concentration ~1 nM), 1 μL Cas13d at various concentrations, and 1x cleavage buffer (25mM Tris pH 7.5, 15mM Tris pH 7.0, 1mM DTT, 6mM MgCl2 or 6 mM EDTA). Reactions were incubated for 2 hours at 37°C and then filtered through stacked nitrocellulose and nylon membranes. Circular membranes (0.5-inch diameter) were punched from stock, pre-equilibrated with 1x cleavage buffer, and stacked with the nitroceullulose membrane atop the nylon membrane onto the internal pedestal of a Whatman filter holder (Sigma Aldrich #WHA420100) that was inserted into a closed valve of a Visiprep™ vacuum manifold (Sigma Aldrich #57250- U). For filter binding, 100 μL of 1x cleavage buffer was applied to the top filter, the valve was opened, the binding reaction was applied, and the membrane stack was immediately washed with 100 μL ice-cold 1x cleavage buffer. The two membranes were then allowed to air dry and later separated and exposed to a phosphorimaging screen overnight. The detection and quantification was done using Typhoon and ImageQuant, respectively. Curve fitting and KD calculation was done using Prism 7.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values reported are the average at least three independent replicates from separate cleavage or reporter experiments with the exact number of replicates indicated in the individual figure legends. HDX analyses were performed in triplicate, with single preparations of each purified protein/complex. Error bars represent SEM or SD, also as indicated in the individual legends.
For determination of significant differences, one-way ANOVA was used. Statistical significance for the differential HDX data is determined by t test for each time point, and is integrated into the HDX Workbench software. No a priori sample size estimation for statistical power was performed.
Supplementary Material
Movie S1. Conformational rearrangements between Cas13d binary complex and Cas13d ternary complex, including the binding of target RNA. Related to Figure 5
Supplementary Table 1. Cryo-EM structures of the Cas13d-guide RNA binary complex and Cas13d-guide-target RNA ternary complex offer insight for engineering this editing system.
Supplementary Table 2. Oligonucleotides used in this study. Related to Figures 1-4, and S3
Figure S1. Cryo-EM data collection, analysis, and modeling of Cas13d-crRNA binary complex and Cas13d-crRNA-target RNA ternary complex. Related to Figure 1
(A) Representative 2D class averages of Cas13d-crRNA complex calculated using Relion.
(B) Classification strategy and refined maps of Cas13d-crRNA complex, colored by local resolution, calculated using “sxlocres.py” implemented within Sparx. The central RNA-binding groove is characterized by a higher resolution (closer to ~3 Å) than the peripheral regions of the protein (closer to ~4 Å).
(C) Fourier Shell Correlation (FSC) curves for cross-validation between the maps and models of Cas13d-crRNA complex. Curves calculated between two half maps (red), between the model and the working half map used for model refinement (blue), between the model and the free half map used for validation (magenta), and between the model and the full map (green).
(D) Euler angle distribution plot (left) showing the relative orientation of the particles used in the final 3D reconstruction of Cas13d-crRNA complex.3D FSC isosurfaces (right), thresholded at a cutoff of 0.75 and displayed in three axial orientations, describe the directional resolution (isotropy) of the refined map.
(E) EM map carved around crRNA and Mg2+ of Cas13d-crRNA complex
(F) Representative 2D class averages of Cas13d-crRNA-target RNA complex calculated using Relion.
(G) Classification strategy and refined maps of Cas13d-crRNA-target RNA complex, colored by local resolution, calculated using “sxlocres.py” implemented within Sparx. The central RNA-binding groove is characterized by a higher resolution (closer to ~3 Å) than the peripheral regions of the protein (closer to ~4 Å).
(H) Fourier Shell Correlation (FSC) curves for cross-validation between the maps and models of Cas13d-crRNA-target RNA complex. Curves calculated between two half maps (red), between the model and the working half map used for model refinement (blue), between the model and the free half map used for validation (magenta), and between the model and the full map (green).
(I) Euler angle distribution plot (left) showing the relative orientation of the particles used in the final 3D reconstruction of Cas13d-crRNA-target RNA complex.3D FSC isosurfaces (right), thresholded at a cutoff of 0.75 and displayed in three axial orientations, describe the directional resolution (isotropy) of the refined map.
(J) EM map carved around crRNA, target RNA and Mg2+ of Cas13d-crRNA-target RNA complex
Figure S2. Bilobed structures of representative Class 2 CRISPR effectors. Related to Figure 1
(A) Domain organizations of Class 2 CRISPR effectors: SpCas9, SaCas9, AsCas12a, AacCas12b, LbuCas13a and EsCas13d. The Helical-1 domain of Cas13a has been redefined here to include Cas13 regions The REC and NUC domains are colored wheat and light blue, respectively.
(B-I) Surface representations of crystal structures of (B) SpCas9 (PDB: 4OO8), (C) SaCas9 (PDB: 5CZZ), (D) AsCas12a (PDB: 5B43), (E) AacCas12b (PDB: 5U31) (F) cryo-EM structure of LbuCas13a in the binary form (PDB: 5XWY), (G) crystal structure of LbuCas13a in the ternary form (PDB:5XWP), (H) EsCas13d in binary form, (I) EsCas13d in ternary forms. Nucleic acid components and orthogonal views of each structure are shown. The REC lobe of LbuCas13a was proposed to include NTD, Helical-1, and Helical-2, and the NUC lobe to include HEPN1, Helical3/Linker and HEPN2 domains. In our representation, to be consistent with grouping on each side of crRNA, we defined the REC lobe of LbuCas13a as including NTD and Helical-2, whereas the NUC lobe would contain Helical-1, HEPN1, HEPN2, and Helical3/Linker. In EsCas13d, The NTD and Helical1 domains are grouped into the REC lobe, and its NUC lobe contains HEPN1, Helical-2 and HEPN2 domains.
Figure S3. Pre-crRNA and target RNA cleavage properties of Cas13d. Related to Figures 3 and 4
(A) Cas13d pre-crRNA cleavage is enhanced in the presence of Mg2+ . Denaturing gel of EsCas13d pre-crRNA array cleavage in the presence (M, Mg2+ ) or absence (E, EDTA) of 6 mM Mg2+ given an increasing ratio of protein:pre-crRNA (0.5:1, 1:1 and 5:1 molar ratios of EsCas13d to pre-crRNA) at a 1 hour timepoint. Red arrowheads indicate mature crRNA 1, which requires two cleavage events to be generated and is abolished by the addition of EDTA at lower protein ratios. Catalytic HEPN residues are not required for pre-crRNA processing as dEsCas13d exhibits equivalent activity as active EsCas13d.
(B) Possible cleavage products are indicated as i-v.
(C) Denaturing gel depicting timecourse of pre-crRNA processing and sensitivity of mature crRNA generation to EDTA. Pre-crRNA processing is conducted at a limiting protein:effector ratio (1:1). In the absence of Mg2+ , processing of mature crRNA 1 is largely eliminated, while processing of crRNA 2 is only minimally delayed.
(D) Filter binding assays were conducted to determine the binding affinity of Cas13d to mature crRNA 1 in the presence or absence of magnesium. Measured KD (mean ± SEM, n = 3) were 25.6 ± 5.3 (crRNA-Mg2+) and 262.1 ± 76.3 (crRNA-EDTA).
(E-F) Structural rationale for the 3’ PFS requirement in LbuCas13a and EsCas13d. Close-up views of 3’ PFS region of LbuCas13a (E) and EsCas13d (F). Key interactions are shown as dashed lines. Unlike LbuCas13a, whose nt-30 of crRNA DR (magenta) faces toward the HEPN-1 domain , nt-30 of crRNA DR in EsCas13d faces toward the solvent and is free to make base-pairing interactions with incoming target RNA. These would be treated as normal spacer:protospacer interactions or, as in this structure, non-Watson-Crick base-pairs.
(G-H) Point mutations of conserved spacer:target RNA interacting residues in EsCas13d impair ssRNA target cleavage but not pre-crRNA processing. Denaturing gel (G) of ssRNA target (arrowhead) cleavage by EsCas13d wild type (+) and mutant (1–3) proteins, guided by crRNA or pre-cRNA. “1” = N86A/T524A/N641A, “2” = R386A/R679A/Y680A, “3” = K376A/K443A/Y447A. (H) Denaturing gel of pre-crRNA array cleavage by EsCas13d wild type and triple mutant proteins. Full length pre-crRNA (i) is cleaved into smaller products (ii-v) as in B.
Figure S4. Domain movement of EsCas13d from binary to ternary transition. Related to Figure 5
(A) Superposition of individual EsCas13d domains between binary and ternary forms.
(B) Local rearrangement of the hinged region that connects the HEPN1 and Helical-1 domains.
(C) Superposition of the Helical-1 and HEPN1 domains between binary and ternary forms. The structures are aligned to HEPN1.
Figure S5. Cryo-EM data collection, analysis, and modeling of Apo EsCas13d. Related to Figure 6
(A) Representative 2D class averages calculated using Relion. Scale bar, 100 nm.
(B) Ab initio cryo-EM reconstruction colored by local resolution, calculated using “sxlocres.py” implemented within Sparx.
(C) Euler angle distribution plot (left) showing the relative orientation of the particles used in the final 3D reconstruction. 3D FSC isosurfaces (right), thresholded at a cutoff of 0.75 and displayed in three axial orientations, describe the directional resolution (isotropy) of the refined map.
(D) Fourier Shell Correlation (FSC) curves calculated between two half maps.
(E) Comparison of ab initio 2D class averages calculated using cATEM from the raw data with 2D projections from the apo-EsCas13d model, calculated along optimally assigned Euler angles. Densities that are missing in the 2D projections are often seen within ab initio 2D classes averages and are indicated by red arrows.
Figure S6. HDX and electrostatic potential maps for EsCas13d. Related to Figure 6
(A) Percentage of deuterium incorporation was consolidated from peptides and plotted as a function of residue number. The amount of deuterium incorporation for each residue, for apo and binary samples, is colored red and black, respectively. Regions with high Deuterium incorporation are indicated.
(B) The deuterium uptake for individual apo or binary samples is mapped onto the respective structural models. Blue and red colors represent low and high deuterium uptake.
(C) Electrostatic surface maps of &Cas13d in its apo, binary and ternary states. The RNA components are removed for clarity. Red and Blue represent negatively and positively charged regions, respectively.
Figure S7. Multiple sequence alignment and viral packaging schematic of Cas13d. Related to Figure 7
(A) Sequence alignment was conducted for seven Cas13d sequences: Es, Rfx, Adm, P1E0, Ur, Rff and Ra. Residue conservation is indicated by grey-scale shading according to Blosum62. Secondary structural elements observed in EsCas13d binary structure are shown above the sequence. Residues that interact with nucleic acids in the binary and ternary states are labeled with blue and red bars, respectively. The same color scheme as Figure 6 was used to highlight the differential HDX onto EsCas13d protein residues. Magenta triangles, catalytic residues of HEPN domains.
(B) Five CasRx deletion variants from Figure 7 exhibiting minimal cleavage activity were tested on the splicing reporter illustrated in Figure 4F to assay for binding activity. All variants displayed reduced or minimal splicing activity compared to full-length dCasRx. This finding is in agreement with close association of binding and cleavage activities in Cas13d (Figure 4) and the participation of HEPN1 and HEPN2 in target binding.
(C) AAV construct designs with Δ3.3 CasRx and a full-length WPRE post-transcriptional element for enhanced transgene expression with payload size <4.7 kb, the AAV packaging limit.
Structures of the smallest Type VI CRISPR effector in guide and target-bound states
Mechanistic insights into guide RNA and target RNA recognition
Insights into apo Cas13d structural dynamics through cryo-EM and HDX/MS
Rational engineering of Cas13d for minimal coding sequence
Acknowledgements
We thank Bill Anderson at TSRI and Youngmin Jeon at Salk for help with EM data collection, and Bruce D. Pascal for assistance in analysis and rendering of HDX data. We also thank Tony Hunter, as well as members of the Hsu and Lyumkis labs for critical reading of the manuscript. This work was supported by the Howard Hughes Medical Institute Hannah H. Gray Fellowship (to S.K.), Helen Hay Whitney Foundation Fellowship (to X.W.), NIH DP5 OD021369 (to P.D.H.), NIH DP5 OD021396 and U54GM103368 (to D.L.), and the Helmsley Charitable Trust (to P.D.H. and D.L.).
Footnotes
Declaration of Interests
S.K. and P.D.H. are inventors on patent applications relating to CRISPR-Cas13, as well as other patents on CRISPR technology.
DATA AND SOFTWARE AVAILABILITY
The cryo-EM structures for corresponding to the binary, and ternary, and apo forms of Cas13d have been deposited into Electron Microscopy Databank under accession codes EMD-9013, EMD-9014, and EMD-9015, respectively. The models for the binary and ternary forms have been deposited into the Protein Databank under accession codes PDB-6E9E and PDB-6E9F, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al. (2017). RNA targeting with CRISPR-Cas13. Nature 550, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, and Aravind L (2013). Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol Direct 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, He Y, Hendrickson CL, Marshall AG, and Griffin PR (2006). Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal Chem 78, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, and Zhang F (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman JE, Abudayyeh OO, Joung J, Gootenberg JS, Zhang F, and Konermann S (2015). Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol 33, 1159–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, and Doudna JA (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander SW (2006). Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom 17, 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Doval C, and Jinek M (2017). Molecular architectures and mechanisms of Class 2 CRISPR-associated nucleases. Curr Opin Struct Biol 47, 157–166. [DOI] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, and Grigorieff N (2015). Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. Elife 4, e06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, and Terns MP (2009). RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, and Korennykh A (2014). Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343, 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine J, Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Samai P, and Marraffini LA (2016). Degradation of Phage Transcripts by CRISPR-Associated RNases Enables Type III CRISPR-Cas Immunity. Cell 164, 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, et al. (2014). Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskiene M, Kostiuk G, Venclovas C, Tamulaitis G, and Siksnys V (2017). A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357, 605–609. [DOI] [PubMed] [Google Scholar]
- Keppel TR, and Weis DD (2015). Mapping residual structure in intrinsically disordered proteins at residue resolution using millisecond hydrogen/deuterium exchange and residue averaging. J Am Soc Mass Spectrom 26, 547–554. [DOI] [PubMed] [Google Scholar]
- Kiani S, Chavez A, Tuttle M, Hall RN, Chari R, Ter-Ovanesyan D, Qian J, Pruitt BW, Beal J, Vora S, et al. (2015). Cas9 gRNA engineering for genome editing, activation and repression. Nat Methods 12, 1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, and Lindahl E (2016). Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, East-Seletsky A, Cofsky JC, Holton JM, Charles E, O’Connell MR, and Doudna JA (2017). Guide-bound structures of an RNA-targeting A-cleaving CRISPR-Cas13a enzyme. Nat Struct Mol Biol 24, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, and Hsu PD (2018). Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, et al. (2009). Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 166, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li X, Ma J, Li Z, You L, Wang J, Wang M, Zhang X, and Wang Y (2017a). The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 170, 714–726 e710. [DOI] [PubMed] [Google Scholar]
- Liu L, Li X, Wang J, Wang M, Chen P, Yin M, Li J, Sheng G, and Wang Y (2017b). Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell 168, 121–134 e112. [DOI] [PubMed] [Google Scholar]
- Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SHW, et al. (2014). Three-dimensional structure of human gamma-secretase. Nature 512, 166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewoehner O, Garcia-Doval C, Rostol JT, Berk C, Schwede F, Bigler L, Hall J, Marraffini LA, and Jinek M (2017). Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548. [DOI] [PubMed] [Google Scholar]
- Niewoehner O, and Jinek M (2016). Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA 22, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Cong L, Yan WX, Ran FA, Zetsche B, Li Y, Kurabayashi A, Ishitani R, Zhang F, and Nureki O (2015). Crystal Structure of Staphylococcus aureus Cas9. Cell 162, 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, and Nureki O (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, and Griffin PR (2012). HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseman AM (2004). FindEM--a fast, efficient program for automatic selection of particles from electron micrographs. J Struct Biol 145, 91–99. [DOI] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, and Marraffini LA (2015). Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 161, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, et al. (2015). Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol Cell 60, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Mallon J, Poddar A, Wang Y, Tippana R, Yang O, Bailey S, and Ha T (2018). Real-time observation of DNA target interrogation and product release by the RNA-guided endonuclease CRISPR Cpf1 (Cas12a). Proc Natl Acad Sci U S A 115, 5444–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargon AA, Cox DB, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, Abudayyeh OA, Essletzbichler P, Shmakov S, Makarova KS, et al. (2017). Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol Cell 65, 618–630 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder J, Borst AJ, Dosey A, Walls AC, Burrell A, Reddy VS, Kollman JM, and Veesler D (2017). Vitrification after multiple rounds of sample application and blotting improves particle density on cryo-electron microscopy grids. J Struct Biol 198, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, LaFrance B, Kaplan M, and Doudna JA (2015). Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, and Doudna JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Swarts DC, van der Oost J, and Jinek M (2017). Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol Cell 66, 221–233 e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambe A, East-Seletsky A, Knott GJ, Doudna JA, and O’Connell MR (2018). RNA Binding and HEPN-Nuclease Activation Are Decoupled in CRISPR-Cas13a. Cell Rep 24, 1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YZ, Baldwin PR, Davis JH, Williamson JR, Potter CS, Carragher B, and Lyumkis D (2017). Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat Methods 14, 793–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, and Scott DA (2018). Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Smith DL (1993). Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein science : a publication of the Protein Society 2, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Conformational rearrangements between Cas13d binary complex and Cas13d ternary complex, including the binding of target RNA. Related to Figure 5
Supplementary Table 1. Cryo-EM structures of the Cas13d-guide RNA binary complex and Cas13d-guide-target RNA ternary complex offer insight for engineering this editing system.
Supplementary Table 2. Oligonucleotides used in this study. Related to Figures 1-4, and S3
Figure S1. Cryo-EM data collection, analysis, and modeling of Cas13d-crRNA binary complex and Cas13d-crRNA-target RNA ternary complex. Related to Figure 1
(A) Representative 2D class averages of Cas13d-crRNA complex calculated using Relion.
(B) Classification strategy and refined maps of Cas13d-crRNA complex, colored by local resolution, calculated using “sxlocres.py” implemented within Sparx. The central RNA-binding groove is characterized by a higher resolution (closer to ~3 Å) than the peripheral regions of the protein (closer to ~4 Å).
(C) Fourier Shell Correlation (FSC) curves for cross-validation between the maps and models of Cas13d-crRNA complex. Curves calculated between two half maps (red), between the model and the working half map used for model refinement (blue), between the model and the free half map used for validation (magenta), and between the model and the full map (green).
(D) Euler angle distribution plot (left) showing the relative orientation of the particles used in the final 3D reconstruction of Cas13d-crRNA complex.3D FSC isosurfaces (right), thresholded at a cutoff of 0.75 and displayed in three axial orientations, describe the directional resolution (isotropy) of the refined map.
(E) EM map carved around crRNA and Mg2+ of Cas13d-crRNA complex
(F) Representative 2D class averages of Cas13d-crRNA-target RNA complex calculated using Relion.
(G) Classification strategy and refined maps of Cas13d-crRNA-target RNA complex, colored by local resolution, calculated using “sxlocres.py” implemented within Sparx. The central RNA-binding groove is characterized by a higher resolution (closer to ~3 Å) than the peripheral regions of the protein (closer to ~4 Å).
(H) Fourier Shell Correlation (FSC) curves for cross-validation between the maps and models of Cas13d-crRNA-target RNA complex. Curves calculated between two half maps (red), between the model and the working half map used for model refinement (blue), between the model and the free half map used for validation (magenta), and between the model and the full map (green).
(I) Euler angle distribution plot (left) showing the relative orientation of the particles used in the final 3D reconstruction of Cas13d-crRNA-target RNA complex.3D FSC isosurfaces (right), thresholded at a cutoff of 0.75 and displayed in three axial orientations, describe the directional resolution (isotropy) of the refined map.
(J) EM map carved around crRNA, target RNA and Mg2+ of Cas13d-crRNA-target RNA complex
Figure S2. Bilobed structures of representative Class 2 CRISPR effectors. Related to Figure 1
(A) Domain organizations of Class 2 CRISPR effectors: SpCas9, SaCas9, AsCas12a, AacCas12b, LbuCas13a and EsCas13d. The Helical-1 domain of Cas13a has been redefined here to include Cas13 regions The REC and NUC domains are colored wheat and light blue, respectively.
(B-I) Surface representations of crystal structures of (B) SpCas9 (PDB: 4OO8), (C) SaCas9 (PDB: 5CZZ), (D) AsCas12a (PDB: 5B43), (E) AacCas12b (PDB: 5U31) (F) cryo-EM structure of LbuCas13a in the binary form (PDB: 5XWY), (G) crystal structure of LbuCas13a in the ternary form (PDB:5XWP), (H) EsCas13d in binary form, (I) EsCas13d in ternary forms. Nucleic acid components and orthogonal views of each structure are shown. The REC lobe of LbuCas13a was proposed to include NTD, Helical-1, and Helical-2, and the NUC lobe to include HEPN1, Helical3/Linker and HEPN2 domains. In our representation, to be consistent with grouping on each side of crRNA, we defined the REC lobe of LbuCas13a as including NTD and Helical-2, whereas the NUC lobe would contain Helical-1, HEPN1, HEPN2, and Helical3/Linker. In EsCas13d, The NTD and Helical1 domains are grouped into the REC lobe, and its NUC lobe contains HEPN1, Helical-2 and HEPN2 domains.
Figure S3. Pre-crRNA and target RNA cleavage properties of Cas13d. Related to Figures 3 and 4
(A) Cas13d pre-crRNA cleavage is enhanced in the presence of Mg2+ . Denaturing gel of EsCas13d pre-crRNA array cleavage in the presence (M, Mg2+ ) or absence (E, EDTA) of 6 mM Mg2+ given an increasing ratio of protein:pre-crRNA (0.5:1, 1:1 and 5:1 molar ratios of EsCas13d to pre-crRNA) at a 1 hour timepoint. Red arrowheads indicate mature crRNA 1, which requires two cleavage events to be generated and is abolished by the addition of EDTA at lower protein ratios. Catalytic HEPN residues are not required for pre-crRNA processing as dEsCas13d exhibits equivalent activity as active EsCas13d.
(B) Possible cleavage products are indicated as i-v.
(C) Denaturing gel depicting timecourse of pre-crRNA processing and sensitivity of mature crRNA generation to EDTA. Pre-crRNA processing is conducted at a limiting protein:effector ratio (1:1). In the absence of Mg2+ , processing of mature crRNA 1 is largely eliminated, while processing of crRNA 2 is only minimally delayed.
(D) Filter binding assays were conducted to determine the binding affinity of Cas13d to mature crRNA 1 in the presence or absence of magnesium. Measured KD (mean ± SEM, n = 3) were 25.6 ± 5.3 (crRNA-Mg2+) and 262.1 ± 76.3 (crRNA-EDTA).
(E-F) Structural rationale for the 3’ PFS requirement in LbuCas13a and EsCas13d. Close-up views of 3’ PFS region of LbuCas13a (E) and EsCas13d (F). Key interactions are shown as dashed lines. Unlike LbuCas13a, whose nt-30 of crRNA DR (magenta) faces toward the HEPN-1 domain , nt-30 of crRNA DR in EsCas13d faces toward the solvent and is free to make base-pairing interactions with incoming target RNA. These would be treated as normal spacer:protospacer interactions or, as in this structure, non-Watson-Crick base-pairs.
(G-H) Point mutations of conserved spacer:target RNA interacting residues in EsCas13d impair ssRNA target cleavage but not pre-crRNA processing. Denaturing gel (G) of ssRNA target (arrowhead) cleavage by EsCas13d wild type (+) and mutant (1–3) proteins, guided by crRNA or pre-cRNA. “1” = N86A/T524A/N641A, “2” = R386A/R679A/Y680A, “3” = K376A/K443A/Y447A. (H) Denaturing gel of pre-crRNA array cleavage by EsCas13d wild type and triple mutant proteins. Full length pre-crRNA (i) is cleaved into smaller products (ii-v) as in B.
Figure S4. Domain movement of EsCas13d from binary to ternary transition. Related to Figure 5
(A) Superposition of individual EsCas13d domains between binary and ternary forms.
(B) Local rearrangement of the hinged region that connects the HEPN1 and Helical-1 domains.
(C) Superposition of the Helical-1 and HEPN1 domains between binary and ternary forms. The structures are aligned to HEPN1.
Figure S5. Cryo-EM data collection, analysis, and modeling of Apo EsCas13d. Related to Figure 6
(A) Representative 2D class averages calculated using Relion. Scale bar, 100 nm.
(B) Ab initio cryo-EM reconstruction colored by local resolution, calculated using “sxlocres.py” implemented within Sparx.
(C) Euler angle distribution plot (left) showing the relative orientation of the particles used in the final 3D reconstruction. 3D FSC isosurfaces (right), thresholded at a cutoff of 0.75 and displayed in three axial orientations, describe the directional resolution (isotropy) of the refined map.
(D) Fourier Shell Correlation (FSC) curves calculated between two half maps.
(E) Comparison of ab initio 2D class averages calculated using cATEM from the raw data with 2D projections from the apo-EsCas13d model, calculated along optimally assigned Euler angles. Densities that are missing in the 2D projections are often seen within ab initio 2D classes averages and are indicated by red arrows.
Figure S6. HDX and electrostatic potential maps for EsCas13d. Related to Figure 6
(A) Percentage of deuterium incorporation was consolidated from peptides and plotted as a function of residue number. The amount of deuterium incorporation for each residue, for apo and binary samples, is colored red and black, respectively. Regions with high Deuterium incorporation are indicated.
(B) The deuterium uptake for individual apo or binary samples is mapped onto the respective structural models. Blue and red colors represent low and high deuterium uptake.
(C) Electrostatic surface maps of &Cas13d in its apo, binary and ternary states. The RNA components are removed for clarity. Red and Blue represent negatively and positively charged regions, respectively.
Figure S7. Multiple sequence alignment and viral packaging schematic of Cas13d. Related to Figure 7
(A) Sequence alignment was conducted for seven Cas13d sequences: Es, Rfx, Adm, P1E0, Ur, Rff and Ra. Residue conservation is indicated by grey-scale shading according to Blosum62. Secondary structural elements observed in EsCas13d binary structure are shown above the sequence. Residues that interact with nucleic acids in the binary and ternary states are labeled with blue and red bars, respectively. The same color scheme as Figure 6 was used to highlight the differential HDX onto EsCas13d protein residues. Magenta triangles, catalytic residues of HEPN domains.
(B) Five CasRx deletion variants from Figure 7 exhibiting minimal cleavage activity were tested on the splicing reporter illustrated in Figure 4F to assay for binding activity. All variants displayed reduced or minimal splicing activity compared to full-length dCasRx. This finding is in agreement with close association of binding and cleavage activities in Cas13d (Figure 4) and the participation of HEPN1 and HEPN2 in target binding.
(C) AAV construct designs with Δ3.3 CasRx and a full-length WPRE post-transcriptional element for enhanced transgene expression with payload size <4.7 kb, the AAV packaging limit.