

Abstract
RNA 2′-phosphotransferase Tpt1 converts an internal RNA 2′-monophosphate to a 2′-OH via a two-step NAD+-dependent mechanism in which: (i) the 2′-phosphate attacks the C1″ of NAD+ to expel nicotinamide and form a 2′-phospho-ADP-ribosylated RNA intermediate; and (ii) the ADP-ribose O2″ attacks the phosphate of the RNA 2′-phospho-ADPR intermediate to expel the RNA 2′-OH and generate ADP-ribose 1″–2″ cyclic phosphate. Tpt1 is an essential component of the fungal tRNA splicing pathway that generates a unique 2′-PO4, 3′-5′ phosphodiester splice junction during tRNA ligation. The wide distribution of Tpt1 enzymes in taxa that have no fungal-type RNA ligase raises the prospect that Tpt1 might catalyze reactions other than RNA 2′-phosphate removal. A survey of Tpt1 enzymes from diverse sources reveals that whereas all of the Tpt1 enzymes are capable of NAD+-dependent conversion of an internal RNA 2′-PO4 to a 2′-OH (the canonical Tpt1 reaction), a subset of Tpt1 enzymes also catalyzed NAD+-dependent ADP-ribosylation of an RNA or DNA 5′-monophosphate terminus. Aeropyrum pernix Tpt1 (ApeTpt1) is particularly adept in this respect. One-step synthesis of a 5′-phospho-ADP-ribosylated cap structure by ApeTpt1 (with no subsequent 5′-phosphotransferase step) extends the repertoire of the Tpt1 enzyme family and the catalogue of ADP-ribosylation reactions involving nucleic acid acceptors.
INTRODUCTION
Tpt1 is an enzyme that catalyzes the transfer of an internal RNA 2′-monophosphate (2′-PO4) to NAD+ to form a 2′-OH RNA, ADP-ribose 1″–2″ cyclic phosphate, and nicotinamide (1). Tpt1 was first discovered as an essential component of the fungal tRNA splicing pathway (2–5), which characteristically generates a 2′-PO4, 3′–5′ phosphodiester splice junction during the tRNA ligation reaction (6,7). Tpt1 homologs are distributed widely in eukaryal, archaeal, and bacterial taxa (8). Mammalian, plant, and bacterial Tpt1 homologs can genetically complement an otherwise lethal tpt1Δ deletion in Saccharomyces cerevisiae, signifying that these homologs are capable of removing the tRNA splice junction 2′-phosphate in vivo (8–11). Biochemical studies of yeast and bacterial Tpt1 enzymes have revealed that the Tpt1 mechanism comprises two chemical steps in which: (i) the RNA 2′-phosphate reacts with NAD+ to expel nicotinamide and form a 2′-phospho-ADP-ribosylated RNA intermediate; and (ii) transesterification of the ADP-ribose 2′-OH to the RNA 2′-phosphate displaces the RNA 2′-OH and generates ADP-ribose 1″–2″ cyclic phosphate (10–13) (Supplementary Figure S1). Tpt1 activity in vivo and in vitro depends on a constellation of four conserved amino acids (an Arg-His-Arg-Arg tetrad) that are thought to comprise the enzyme's active site (9,10,14).
Many of the bacterial species with Tpt1 homologs have no known intron-containing tRNAs and/or no known pathways to generate RNAs with internal 2′-PO4 modifications. In metazoa and archaea, the mechanism of tRNA exon ligation is entirely different from that of fungi and does not result in a junction 2′-PO4. Whereas these considerations rationalize the findings that genetic ablation of Tpt1 in Escherichia coli and mouse has no apparent phenoptypic consequences (8,15), we remain in the dark as to what reactions Tpt1 might perform in taxa that lack a fungal-type RNA ligase. One scenario is that bacteria, archaea, or metazoa do have a capacity to install internal RNA 2′-PO4 groups, conceivably during RNA repair pathways that are as yet uncharacterized, and that such RNAs are potential substrates for Tpt1. An alternative is that Tpt1 can catalyze reactions other than RNA 2′-PO4 removal, via its unique NAD+-dependent transferase mechanism.
In the present study, we address the latter scenario, by purifying Tpt1 enzymes from diverse sources and interrogating their reactions with various 5′ 32P-labeled nucleic acid substrates. We report that whereas all of the Tpt1 enzymes are capable of NAD+-dependent conversion of an internal RNA 2′-PO4 to a 2′-OH, a subset of Tpt1 enzymes also formed a novel RNA product. Aeropyrum pernix Tpt1 (ApeTpt1) is particularly adept in this regard. Further characterization of ApeTpt1 revealed that the novel product is formed via NAD+-dependent ADP-ribosylation of the RNA 5′-monophosphate terminus. ApeTpt1 can also quantitatively ADP-ribosylate a DNA 5′-monophosphate end. Enzymatic synthesis of a 5′-phospho-ADP-ribosylated cap structure expands the roster of nucleic acid 5′ end modifications and extends the repertoire of the Tpt1 family. It also extends the spectrum of ADP-ribosylation reactions involving nucleic acid acceptors.
MATERIALS AND METHODS
Recombinant Tpt1 proteins
The open reading frames encoding Tpt1 homologs from Clostridium thermocellum (NCBI accession ABN54255.1), Pyrococcus horikoshii (NCBI accession WP_010884270.1), and Archaeoglobus fulgidus (NCBI accession WP_010877913.1) were PCR-amplified from genomic DNA with primer pairs designed to introduce an NdeI site at the translation start codon and an XhoI site immediately 3′ of the stop codon. The PCR products were digested with NdeI and XhoI and then inserted into NdeI/XhoI-cut T7 RNA-polymerase-based expression plasmid pET28a so as to fuse the encoded Tpt1 polypeptide to an N-terminal His6 tag. The Tpt1 ORFs from A. pernix (Uniprot: Q9YFP5), Chaetomium thermophilum (NCBI accession XP_006693007.1) and Homo sapiens (Uniprot: Q86TN4) were synthesized by Genscript and then inserted into the NdeI and XhoI sites of pET28a. The Tpt1 ORFs were sequenced completely in the pET-His6Tpt1 plasmids to confirm that no unwanted coding changes were introduced during PCR amplification and cloning. The expression vector for Runella slithyformis Tpt1 was described previously (10).
The pET-His6Tpt1 plasmids were transformed into E. coli BL21(DE3). Cultures (1000 ml) derived from single kanamycin-resistant transformants were grown at 37°C in LB medium containing 50 μg/ml kanamycin until the A600 reached 0.5–0.6, at which time the cultures were adjusted to 0.5 mM isopropyl-β-d-thiogalactoside (IPTG) and then incubated for 16 h at 18°C with continuous shaking. Cells were harvested by centrifugation, and the pellets were resuspended in 25 ml lysis buffer (50 mM Tris–HCl, pH 8.0, 1M NaCl, 20 mM imidazole, 10% glycerol) and stored at −80°C pending purification. All subsequent purification procedures were performed at 4°C for Clostridium, Chaetomium, Archaeoglobus and human Tpt1. Procedures for purification of Pyrococcus and Aeropyrum Tpt1 were performed at room temperature, because these archaeal proteins precipitated at 4°C. The thawed cell suspensions in lysis buffer were adjusted to 1 mg/ml lysozyme and supplemented with one protease inhibitor tablet (Roche) and incubated for 30 min. The lysates were sonicated to reduce viscosity and insoluble material was removed by centrifugation for 45 min at 14 000 rpm. The soluble extract was mixed for 1 h with 3 ml of Ni-NTA agarose (Qiagen) that had been equilibrated in lysis buffer. The resin was collected by centrifugation and resuspended in 50 ml of buffer A (50 mM Tris–HCl, pH 8.0, 300 mM NaCl) containing 20 mM imidazole, then re-collected by centrifugation and suspended in 30 ml of 50 mM Tris–HCl, pH 8.0, 3 M KCl. The resin was recovered again and resuspended in 50 ml of buffer A containing 50 mM imidazole. The resin was then poured into a gravity flow column and the bound material was serially step-eluted with 150 mM, 250 mM, and 500 mM imidazole in buffer A. The elution profiles were monitored by SDS-PAGE. The peak His6Tpt1-containing fractions were pooled and adjusted to 10 mM dithiothreitol (DTT). The Ni-agarose fractions of Pyrococcus and Aeropyrum Tpt1 were dialyzed against 20 mM Tris–HCl, pH 8.0, 300 mM NaCl, 1 mM DTT at room temperature. The other Tpt1 proteins were concentrated by centrifugal ultrafiltration and subjected to gel-filtration through a 120-ml Superdex 200 column that was equilibrated with either 20 mM Tris–HCl, pH 8.0, 300 mM NaCl, 1 mM DTT (for Chaetomium and human Tpt1), 20 mM Tris–HCl, pH 8.0, 300 mM NaCl, 1 mM DTT, 5% glycerol (for Clostridium Tpt1), or 20 mM HEPES–NaOH, pH 7.0, 300 mM NaCl (for Archaeoglobus Tpt1). Peak fractions were pooled and concentrated by centrifugal ultrafiltration and stored at –80°C. Protein concentrations were determined by using the BioRad dye reagent with bovine serum albumin as the standard. Analysis of the polypepide compositions of the Tpt1 preparations by SDS-PAGE is shown in Supplementary Figure S2. The purifications of wild-type Runella Tpt1 and the step 2-defective R64A mutant were performed as described previously (10).
5′ 32P-labeled oligonucleotide substrates
A synthetic 6-mer RNA oligonucleotide 5′-CCAA2′PAU containing an internal 2′-PO4 (10) and standard synthetic 10-mer RNA (5′-CCUGUAUGAU) and DNA (5′-CCTGTATGAT) oligonucleotides were 5′ 32P-labeled by reaction with phosphatase-dead T4 polynucleotide kinase (Pnkp-D167N) in the presence of [γ32P]ATP. The reactions were quenched with 90% formamide, 50 mM EDTA, 0.01% xylene cyanol and the radiolabeled RNAs were purified by electrophoresis through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA. The radiolabeled oligonucleotides were eluted from excised gel slices, recovered by ethanol precipitation, and resuspended in 10 mM Tris–HCl, pH 6.8, 1 mM EDTA (TE) and stored at −20°C.
Assay of Tpt1 activity
Reaction mixtures containing 100 mM Tris–HCl (pH 7.5), 5′ 32P-labeled oligonucleotide substrates, NAD+, and Tpt1 as specified in the figure legends were incubated at 37°C. The reactions were quenched by addition of three volumes of cold 90% formamide, 50 mM EDTA. The products were analyzed by electrophoresis (at 55 W constant power) through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA and visualized by autoradiography and/or scanning the gel with a Fujifilm FLA-7000 imaging device. The products were quantified by analysis of the gel scans in ImageQuant.
Isolation of 5′ 32P-labeled 5′-P-ADPR capped DNA
Reaction mixtures (3 × 20 μl) containing 100 mM Tris–HCl, pH 7.5, 1 mM NAD+, 100 pmol 5′ 32P-labeled 10-mer DNA, and 250 pmol ApeTpt1 were incubated for 60 min at 37°C. The reactions were quenched with 3 volumes of cold 90% formamide, 50 mM EDTA, 0.01% xylene cyanol. The radiolabeled 5′-P-ADPR DNA species was separated from substrate by urea-PAGE, eluted from excised gel slices, ethanol precipitated, resuspended in TE buffer, and stored at −20°C.
RESULTS
RNA 2′-phosphotransferase activity of recombinant mammalian, fungal and bacterial Tpt1 proteins
Tpt1 proteins from diverse taxa—the fungus Chaetomium thermophilum (Chth), the bacterium Clostridium thermocellum (Clth), the archaea A. pernix (Ape), Pyrococcus horikoshii (Pho), and Archaeoglobus fulgidus (Afu), and the primate Homo sapiens (Hsa)—were produced in E. coli as His6-tagged polypeptides and purified from soluble bacterial lysates (Supplementary Figure S2). These purified proteins were tested for their ability to remove the 2′-PO4 moiety from a 5′ 32P-labeled synthetic 6-mer 2′-PO4 RNA substrate (10). The biochemically well-characterized Runella slithyformis Tpt1 (RslTpt1) was assayed in parallel as a positive control (10). Urea-PAGE analysis revealed that a 30 min reaction of 500 nM Tpt1 with 200 nM RNA and 1 mM NAD+ resulted in each case in the conversion of the input radiolabeled RNA into a more slowly migrating species corresponding to the 2′-OH product (Figure 1A). The reactions with the three archaeal enzymes and ClthTpt1 were distinctive in that they also generated an even more slowly migrating species (Figure 1A, denoted by an asterisk). This novel product is distinct from (and more slowly migrating than) the 2′-phospho-ADP-ribosylated RNA intermediate in the canonical Tpt1 reaction pathway, which accumulates to high levels during reaction of the 6-mer RNA substrate with the RslTpt1-R64A mutant enzyme (10) (Figure 1A). Because the yield of the novel product in our initial experiments was highest for ApeTpt1, we proceeded to characterize the Aeropyrum enzyme in greater detail as described below.
Figure 1.

Formation of a novel RNA product during Tpt1 reaction with 2′-phosphate RNA. (A) Reaction mixtures (10 μl) containing 100 mM Tris–HCl, pH 7.5, 0.2 μM (2 pmol) 5′ 32P-labeled 6-mer 2′-PO4 RNA (shown at bottom), 1 mM NAD+, and 0.5 μM (5 pmol) Tpt1 from the sources specified were incubated at 37°C for 30 min. Tpt1 was omitted from the control reaction mixture in lane –. The sources of enzyme are as follows: Runella slithyformis, Rsl; Clostridium thermocellum, Clth; Chaetomium themophilum, Chth; Homo sapiens, Hsa; Aeropyrum pernix, Ape, Pyrococcus horikoshii, Pho; and Archaeoglobus fulgidus, Afu. The step 2-defective RslTpt1-R64A mutant was assayed in parallel (leftmost lane) in order to demarcate the 2′-P-ADPR RNA intermediate in the canonical Tpt1 reaction pathway. The reactions were quenched by adding three volumes of cold 90% formamide, 50 mM EDTA. (B) A reaction mixture containing 100 mM Tris–HCl, pH 7.5, 0.2 μM 5′ 32P-labeled 6-mer 2′-PO4 RNA, 1 mM NAD+, and 0.5 μM (5 pmol) ApeTpt1 was incubated at 37°C. Aliquots (10 μl) were withdrawn at the times specified and quenched immediately with three volumes of cold 90% formamide, 50 mM EDTA. The time 0 sample was withdrawn prior to adding ApeTpt1. The reaction products were analyzed by urea-PAGE and visualized by autoradiography. The positions of the 6-mer 2′-PO4 RNA substrate (2′-P), 2′-P-ADPR RNA intermediate, and 2′-OH RNA product (2′-OH) of the canonical two-step Tpt1 reaction are indicated on the left and right. The novel RNA product formed by some of the Tpt1 enzymes is denoted by an asterisk. The yields of 2′-OH RNA and novel RNA (expressed as percent of total labeled RNA) are indicated below the lanes.
Parameters affecting formation of the novel RNA product by ApeTpt1
The kinetic profile of the ApeTpt1 reaction showed that that the 2′-phosphate was removed completely within 5 min, at which time the novel species comprised 7% of the total labeled RNA (Figure 1B). The novel species accumulated steadily thereafter, concomitant with a decline in the 2′-OH RNA, such that after 60 min the novel species and 2′-OH RNA comprised 92% and 8% of the total label, respectively (Figure 1B). A finer analysis sampling earlier times indicated that ApeTpt1 completely removed the 2′-PO4 in 10 s, at which point the novel RNA constituted only 0.2% of the total (not shown). ApeTpt1 formed neither the 2′-OH RNA product nor the novel product during a 60 min reaction when NAD+ was omitted from the reaction mixture (Figure 1B). ApeTpt1 titration indicated that 2′-OH product increased with enzyme concentration between 5 and 10 nM (50–100 fmol in Supplementary Figure S3) and was 95% complete at 20 nM enzyme, at which point there was no detectable formation of the novel RNA. The novel product comprised 4% of the total labeled RNA when ApeTpt1 was increased to 50 nM (500 fmol in Supplementary Figure S3). These results imply that the 2′-OH RNA product of the canonical Tpt1 reaction pathway is the substrate for a separate reaction that generates the novel RNA.
The novel product is a phosphatase-resistant 5′ cap structure
Given that the signature step 1 reaction of Tpt1 enzymes is the transfer of ADP-ribose from NAD+ to a phosphomonoester, we suspected that the novel reaction of ApeTpt1 subsequent to removal of the internal 2′-PO4 might entail ADP-ribosylation of the RNA 5′-monophosphate end to form a 5′-phospho-ADPR ‘cap’ structure. Because the 5′-PO4 is 32P-labeled, such an ADP-ribosylation reaction would be expected to render the 5′-PO4 resistant to hydrolysis by alkaline phosphatase (CIP). Indeed, we found that whereas the 32P-label of the 6-mer 2′-PO4 RNA substrate and the 2′-OH product of the canonical ApeTpt1 reaction was effaced completely by treatment with CIP, the 32P-label of the novel reaction product was unaffected by CIP treatment (Figure 2).
Figure 2.
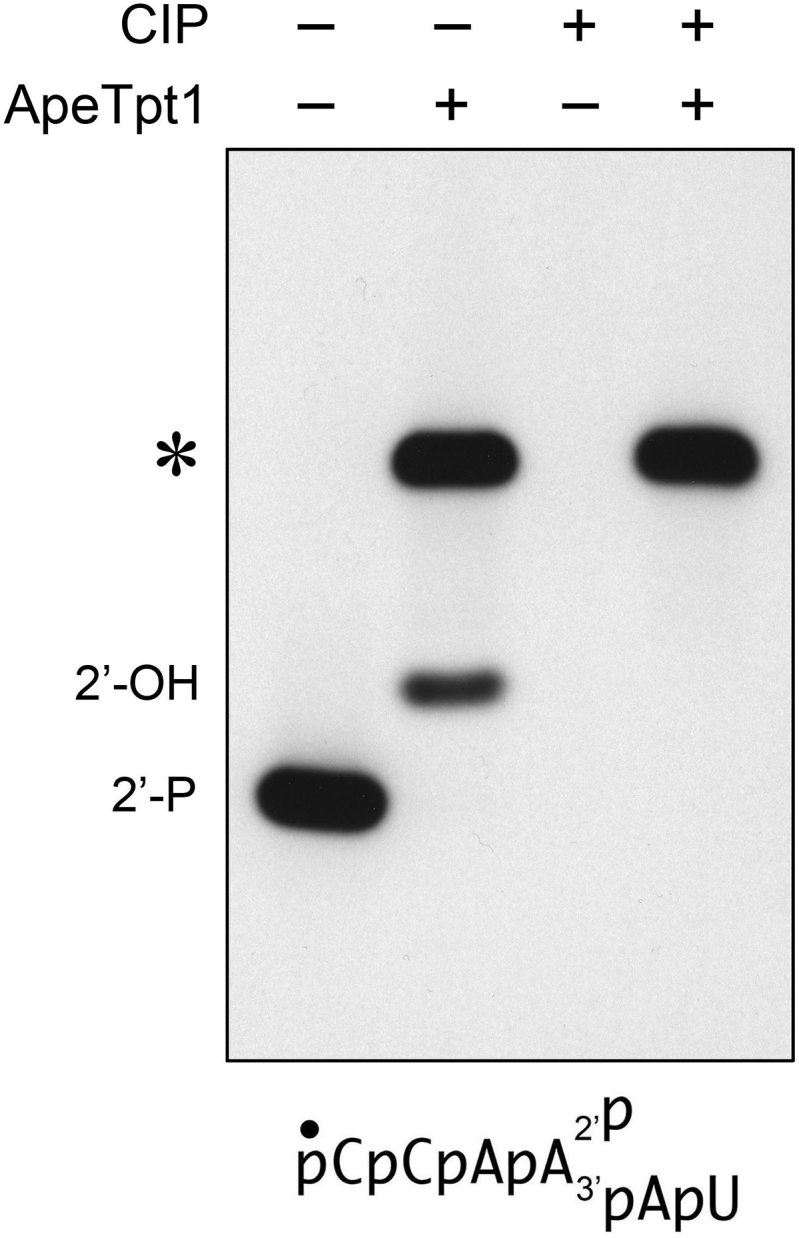
ApeTpt1 synthesizes a phosphatase-resistant 5′ cap structure. Reaction mixtures (10 μl) containing 100 mM Tris–HCl, pH 7.5, 0.2 μM (2 pmol) 5′ 32P-labeled 6-mer 2′-PO4 RNA (shown at bottom), 1 mM NAD+, and 0.5 μM (5 pmol) ApeTpt1 (where indicated by +) were incubated at 37°C for 60 min. The reaction mixtures were heated at 65°C for 5 min and then either treated for 10 min at 37°C with 10 U of calf intestine alkaline phosphatase (CIP; from NEB) in 1× Cutsmart buffer (50 mM potassium acetate, 20 mM Tris-acetate, pH 7.9, 10 mM magnesium acetate, 100 μg/ml BSA, pH 7.9) or mock-incubated without phosphatase. The reactions were quenched with three volumes of cold 90% formamide, 50 mM EDTA and the products were analyzed by urea-PAGE and visualized by autoradiography.
ApeTpt1 caps RNA or DNA 5′-phosphate ends with ADP-ribose
Reaction of 500 nM ApeTpt1 with 200 nM 10-mer 5′ 32P-labeled RNA or DNA oligonucleotides of identical nucleotide sequence (excepting T for U in DNA versus RNA) in the presence of 1 mM NAD+ resulted in the conversion of the substrates into RNA and DNA products of slower electrophoretic mobility (Figure 3). These products were not formed in the absence of NAD+. The 32P label in the RNA and DNA reaction products was resistant to hydrolysis by CIP (Figure 3). An ApeTpt1 titration experiment showed that pDNA and pRNA substrates were capped quantitatively at saturating enzyme (Figure 4). The specific activities, calculated from the slope of the titration curves at sub-saturating enzyme (via linear regression in Prism) were 0.48 ± 0.04 pmol/pmol for pDNA and 0.36 ± 0.015 pmol/pmol for pRNA (Figure 4). Thus, ApeTpt1 is acting in a stoichiometric fashion as a 5′ capping enzyme with the 10-mer substrates under the conditions employed. ApeTpt1 capped the pDNA substrate with high yield (94–100%) at temperatures ranging from 37°C to 65°C (Supplementary Figure S4), and with progressively lower yields at 75°C (68%), 85°C (32%) and 95°C (21%). Retention of capping activity at high temperatures is consistent with the thermophilic character of A. pernix.
Figure 3.

ApeTpt1 caps RNA or DNA 5′-phosphate ends with ADP-ribose. Reaction mixtures (10 μl) containing 100 mM Tris–HCl (pH 7.5), 0.2 μM (2 pmol) 5′ 32P-labeled 10-mer pDNA or pRNA substrates (shown at bottom), 1 mM NAD+ (where indicated by +), and 0.5 μM (5 pmol) ApeTpt1 (where indicated by +) were incubated at 37°C for 60 min. The reaction mixtures were heated at 65°C for 5 min and then either treated with calf intestine alkaline phosphatase (CIP +) or mock- treated (CIP –) as described in Figure 2. The products were analyzed by urea-PAGE and visualized by autoradiography.
Figure 4.

Dependence of pDNA and pRNA capping on ApeTpt1 concentration. Reaction mixtures (10 μl) containing 100 mM Tris–HCl (pH 7.5), 0.2 μM (2 pmol) 5′ 32P-labeled 10-mer pDNA or pRNA substrates, 1 mM NAD+, and ApeTpt1 as specified were incubated at 37°C for 60 min. The extents of pDNA and pRNA capping are plotted as a function of input ApeTpt1. Each datum in the graph is the average of three separate enzyme titration experiments ±SEM.
Although the preceding experiments show that the 5′-PO4 DNA and RNA ends are capped, they do not reveal the nature of the capping moiety. We reasoned that if the novel capping reaction is ADP-ribosylation, then the electrophoretic mobility of the capped oligonucleotide should be affected by a bulky modification of the adenine base of NAD+. To test this idea, we performed ApeTpt1 reactions with the pRNA and pDNA substrates in parallel with 50 μM NAD+ or 6-Biotin-17-NAD+ (an NAD+ analog in which biotin is attached to the adenine N6 position via a 17-mer spacer chain) and found that the biotin modification did indeed slow the mobility of the capped RNA and DNA oligonucleotides (Figure 5). Additional evidence that the cap moiety was ADP-ribose was provided by an experiment in which the 32P-labeled capped oligonucleotides were gel-purified and reacted with ApeTpt1 in the presence of 1 mM nicotinamide (the leaving group of the Tpt1 ADP-ribosyl-transferase reaction). This resulted in de-capping of 20% of the ADPR-pDNA substrate to re-generate the pDNA strand (Supplementary Figure S5).
Figure 5.

pDNA and pRNA capping by ApeTpt1 using biotin-NAD+ as substrate. Reaction mixtures (10 μl) containing 100 mM Tris–HCl (pH 7.5), 0.2 μM (2 pmol) 5′ 32P-labeled 10-mer pDNA or pRNA substrates (shown at bottom), 50 μM NAD+ or biotin-NAD+ (where specified by +), and 0.5 μM (5 pmol) ApeTpt1 (where indicated by +) were incubated at 37°C for 60 min. The products were analyzed by urea-PAGE and visualized by autoradiography. 6-Biotin-17-NAD+ was from BPS Bioscience (catalog # 80610).
Test of pDNA capping activity of other Tpt1 enzymes
In the experiment in Figure 6, we surveyed the collection of seven Tpt1 enzymes for their ability to ADP-ribosylate the 5′-PO4 end of the pDNA substrate (i.e. without a competing reaction at a 2′-PO4 that applied in the initial experiment in Figure 1A). Again, ApeTpt1 was clearly the ‘winner’ as the most effective 5′-phospho-ADP-ribosyltransferase (98% yield of capped product). Pyrococcus and Chaetomium Tpt1 were less active (20–21%), followed by Archaeoglobus (13% yield), and Clostridium Tpt1 (9%). By contrast, the Runella and human Tpt1 enzymes were inactive in DNA capping.
Figure 6.

Assay of pDNA capping by Tpt1 enzymes from diverse taxa. Reaction mixtures (10 μl) containing 100 mM Tris–HCl (pH 7.5), 0.2 μM (2 pmol) 5′ 32P-labeled 10-mer pDNA substrate (shown at bottom), 1 mM NAD+, and 0.5 μM (5 pmol) Tpt1 from the indicated sources (as per Figure 1) were incubated at 37°C for 30 min. The products were analyzed by urea-PAGE and visualized by autoradiography. The extents of capping are indicated below the lanes.
DISCUSSION
The present study sheds light on a puzzling feature of the Tpt1 enzyme family, viz. that Tpt1 homologs are widely prevalent in diverse taxa, many of which have no obvious need for an RNA 2′-phosphotransferase because: (i) they lack tRNA introns; and/or (ii) they are not known to have an enzymatic pathway that generates 2′-PO4, 3′–5′ phosphodiester RNA structures. Despite this lack of existential rationale, it is clear by the criteria of genetic complementation of yeast tpt1Δ (8–10) and direct biochemical assay of recombinant Tpt1 proteins ((10,11) and the present study) that Tpt1 enzymes from widely divergent organisms all possess NAD+-dependent RNA 2′-phosphotransferase activity. The salient discovery here is that a subset of Tpt1 enzymes have an additional capacity to catalyze the transfer of ADP-ribose from NAD+ to a 5′-monophosphate end of RNA or DNA to install a 5′-phospho-ADP-ribose cap structure. This distinctive 5′ capping reaction has important implications for how we think about the Tpt1 mechanism and the prospect that 5′ phospho-ADP-ribose capping is just the tip of the iceberg of what Tpt1′s catalytic repertoire might embrace.
Is Tpt1 fundamentally an ADP-ribosyltransferase or a phosphotransferase?
The experiments presented here indicate that ApeTpt1 catalyzes 5′-PO4 capping of RNA and DNA via a single-step ADP-ribosyltransferase reaction (as depicted in Figure 7) in which a 5′-PO4 oxygen is the nucleophile that attacks the C1″ carbon of NAD+ and displaces nicotinamide, resulting in the covalent attachment of ADP-ribose to the 5′-PO4 RNA or DNA terminus. The capping reaction is chemically analogous to the first step in the RNA 2′-phosphotransferase pathway (Supplementary Figure S1), wherein a 2′-PO4 oxygen is the nucleophile that displaces nicotinamide during formation of the 2′-phospho-ADPR RNA intermediate. The crucial distinction between the two ADP-ribosyltransferase reactions is that Tpt1 is apparently unable to catalyze a second chemical 5′-phosphoryltransferase step once the 5′-phospho-ADPR cap structure is formed in situ. Several lines of evidence underscore this point. First, there was no appreciable decrement in 32P-label in the 5′ capped product that would be expected if ApeTpt1 did catalyze attack of the ADP-ribose O2″ on the 5′-PO4, which would release 32P-labeled ADP-ribose 1″–2″ cyclic phosphate as a second-step end product. Second, whereas 32P-labeled ADP-ribose 1″–2″ cyclic phosphate would be CIP-resistant, the slower mobility of the 32P-labeled capping reaction product versus the 10-mer pRNA or pDNA substrates (Figure 3) weighs strongly against this CIP-resistant species being ADP-ribose 1″–2″ cyclic phosphate. Indeed, we detected no radiolabeled capping reaction product migrating more rapidly than the 10-mer pRNA or pDNA substrate, as would be expected of ADP-ribose 1″–2″ cyclic phosphate. Third, the fact that the mobility of the 32P-labeled capping reaction product varied according to whether the susbtrate was 10-mer pRNA or pDNA (Figure 3) is incompatible with the product being 32P-labeled ADP-ribose 1″–2″ cyclic phosphate (which should be indifferent to whether the source of the 32P-labeled phosphate group was DNA or RNA) and instead diagnostic of the pRNA or pDNA being a constituent of the reaction end product. Fourth, reaction of the isolated 5′-phospho-ADPR capped DNA reaction product with ApeTpt1 in the absence of NAD+ resulted in no detectable decrement in 32P-label in this species (Supplementary Figure S5).
Figure 7.

Proposed mechanism of 5′-phosphate capping via transfer of ADP-ribose from NAD+.
By contrast, previous studies of the canonical Tpt1 pathway showed that: (i) the second 2′-phosphotransferase reaction is significantly faster than the prior (rate-limiting) ADP-ribosyltransferase step; and (ii) reaction of the isolated 2′-phospho-ADPR RNA intermediate with Tpt1 in the absence of NAD+ results in the rapid and quantitative removal of the RNA 2′-phosphate to yield a 2′-OH RNA product (10). We hypothesize that the different reaction endpoints with a 5′-PO4 versus 2′-PO4 nucleophile reflects variations in the geometry of the 5′-PO4 and 2′-PO4 linked ADP-ribose moieties in the Tpt1 active site, such that the 2′-phospho-ADP-ribosylated RNA is oriented so that the RNA ribose O2′ is positioned apically relative to the ADP-ribose O2″ in a manner conducive to step 2 cyclization and explusion of the RNA 2′-OH product, whereas the orientation of the terminal O5′ and the ADP-ribose O2″ in the capped nucleic acid is inimical to cyclization and displacement of the 5′-OH.
In sum, these considerations focus attention on ADP-ribosyl transfer to a phosphorylated substrate as the universal mechanistic feature of Tpt1-catalyzed reactions, in addition to (or in lieu of) the 2′-phosphotransfer feature that was central to the discovery of Tpt1 and its role in healing the 2′-PO4, 3′-5′ phosphodiester RNA splice junction formed during fungal and plant tRNA splicing (1–5).
5′-Phospho-ADP-ribose: a novel enzymatically synthesized RNA cap structure
5′-Phospho-ADP-ribosylation of RNA by ApeTpt1 adds to the roster of enzymatic RNA capping reactions, each of which entails a distinct biochemical mechanism. The classic m7GpppN cap structure of eukaryal mRNAs is formed by transfer of GMP from GTP to a 5′-diphosphate RNA end, followed by AdoMet-dependent guanine-N7 methylation (16). Eukaryal snRNAs undergo two more sequential methyl additions to the guanine-N2 atom of the m7GpppN monomethyl cap (catalyzed by the enzyme Tgs1) to form a trimethylguanosine cap structure m2,2,7GpppN (17). AdoMet-dependent methylation of an RNA terminal phosphate oxygen (either the γ-phosphate of a 5′-triphosphate-terminated RNA or a 5′-monophosphate RNA terminus) by BCDIN3-family enzymes yields a monomethyl-phosphate cap structure on certain non-coding eukaryal RNAs (18–20). A subset of bacterial and eukaryal RNAs acquire non-canonical 5′ caps when RNA polymerase initiates transcription with NAD+, NADH, desphospho-CoA, FAD, or UDP-glucose instead of a standard NTP (21,22). The enzyme RtcA synthesizes a 5′ AppN RNA cap via transfer of AMP from ATP to an RNA 5′-monophosphate terminus (23).
Having demonstrated enzymatic synthesis of a 5′-phospho-ADP-ribosyl RNA cap, it now remains to be seen whether such cap structures exist in nature and how widely prevalent they are among taxa. Needless to say, this will require the development and implementation of sensitive analytical methods for 5′-phospho-ADP-ribosyl RNA cap detection and their application to non-standard organisms like A. pernix that are good candidates to have such a cap synthetic capacity. Potential roles for 5′-phospho-ADP-ribosyl RNA caps might be to protect RNA ends from exonucleolytic decay (à la the m7GpppN mRNA cap) and recruit proteins that recognize this cap structure in service of particular RNA transactions (akin to how eIF4E recognizes the m7GpppN RNA cap to promote translation initiation). 5′-phospho-ADP-ribosyl caps might also come into play in host-virus dynamics, e.g., as a means of distinguishing self from non-self nucleic acids. In that vein, it is noteworthy that Aeropyrum pernix is susceptible to infection by thermophilic viruses (24,25).
Tpt1 expands the scope of ADP-ribosylation reactions that modify DNA
We show here that ApeTpt1 efficiently caps DNA 5′-PO4 ends with ADP-ribose. A similar mono-ADP-ribosylation reaction at DNA 5′-monophosphate ends was described recently for mammalian PARP3 (26,27). As discussed for PARPs, the capping of DNA 5′-phosphate ends with ADPR catalyzed by Tpt1 enzymes might play a role in cellular responses to DNA damage, e.g. by protecting the capped 5′ ends from digestion by exonucleases and/or providing a covalent ‘mark’ that is recognized by a cellular protein that signals the presence of the ADPR DNA cap. ADP-ribosylation of terminal monophosphates is but one of several modes of DNA covalent modification by ADPR. Other examples include: (i) the attachment of ADPR to the N2 atom of deoxyguanosine nucleobases catalyzed by the ADP-ribosyltransferase cytotoxins Pierisin-1, Scabin and CARP-1 (28–30); and (ii) attachment of ADPR to the deoxythymidine nucleobases by the ADP-ribosyltransferase cytotoxin DarT (31).
How broad is the catalytic repertoire of Tpt1 enzymes?
The capacity for 5′-phospho-ADPR capping is exemplified and characterized for the case of ApeTpt1, but is not restricted to this member of the Tpt1 family. Among the enzymes surveyed here, two other archaeal Tpt1s (from Pyrococcus and Archaeoglobus), one bacterial Tpt1 (from Clostridium) and one fungal Tpt1 (from Chaetomium) exhibit DNA capping activity in vitro, albeit less vigorously than ApeTpt1. By contrast, human and Runella Tpt1 are inactive in DNA capping. Although we cannot exclude the possibility that the DNA substrate and reaction conditions that are conducive to 5′-phospho-ADPR capping by ApeTpt1 are not permissive for capping by human Tpt1, we prefer to think that different Tpt1 orthologs are truly differentially adept when it comes to ADP-ribosyltransfer to non-canonical phosphoacceptor substrates. A natural extension of this idea is that Tpt1 enzymes might catalyze other novel ADP-ribosylation reactions, including ADP-ribosylation of non-nucleic acid phosphoacceptors, with or without subsequent removal of the phosphate moiety. To fortify these notions, it will be important to correlate Tpt1 activity with the detection of 5′-phospho-ADP-ribose caps in vivo and to survey Tpt1 homologs for activity in vitro with a broad spectrum of potential substrates.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
U.S. National Institutes of Health [R35GM122575]; Geoffrey Beene Cancer Research Center. Funding for open access charge: U.S. National Institutes of Health [R35GM122575].
Conflict of interest statement. None declared.
REFERENCES
- 1. Culver G.M., McCraith S.M., Zillman M., Kierzek R., Michaud N., LaReau R.D., Turner D.H., Phizicky E.M.. An NAD derivative produced during transfer RNA splicing: ADP-ribose 1′′-2′′ cyclic phosphate. Science. 1993; 261:206–208. [DOI] [PubMed] [Google Scholar]
- 2. McCraith S.M., Phizicky E.M.. A highly specific phosphatase from Saccharomyces cerevisiae implicated in tRNA splicing. Mol. Cell. Biol. 1990; 10:1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCraith S.M., Phizicky E.M.. An enzyme from Saccharomyces cerevisiae uses NAD+ to transfer the splice junction 2′-phosphate from ligated tRNA to an acceptor molecule. J. Biol. Chem. 1991; 266:11986–11992. [PubMed] [Google Scholar]
- 4. Culver G.M., McCraith S.M., Consaul S.A., Stanford D.R., Phizicky E.M.. A 2′-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae.. J. Biol. Chem. 1997; 272:13203–13210. [DOI] [PubMed] [Google Scholar]
- 5. Spinelli S.L., Consaul S.A., Phizicky E.M.. A conditional lethal yeast phosphotransferase mutant accumulates tRNA with a 2′-phosphate and an unmodified base at the splice junction. RNA. 1997; 3:1388–1400. [PMC free article] [PubMed] [Google Scholar]
- 6. Greer C.L., Peebles C.L., Gegenheimer P., Abelson J.. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell. 1983; 32:537–546. [DOI] [PubMed] [Google Scholar]
- 7. Remus B.S., Shuman S.. A kinetic framework for tRNA ligase and enforcement of a 2′-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA. 2013; 19:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spinelli S.L., Malik H.S., Consaul S.A., Phizicky E.M.. A functional homolog of a yeast tRNA splicing enzyme is conserved in higher eukaryotes and in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14136–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sawaya R., Schwer B., Shuman S.. Structure-function analysis of the yeast NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA. 2005; 11:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Munir A., Abdullahu L., Damha M.J., Shuman S.. Two-step mechanism and step-arrest mutants of Runella slithyformis NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA. 2018; 24:1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spinelli S.L., Kierzek R., Turner D.H., Phizicky E.M.. Transient ADP-ribosylation of a 2′-phosphate implicated in its removal from ligated tRNA during splicing in yeast. J. Biol. Chem. 1999; 274:2637–2644. [DOI] [PubMed] [Google Scholar]
- 12. Steiger M.A., Kierzek R., Turner D.H., Phizicky E.M.. Substrate recognition by a yeast 2′-phosphotransferase involved in tRNA splicing and its Escherichia coli homolog. Biochemistry. 2001; 40:14098–14105. [DOI] [PubMed] [Google Scholar]
- 13. Steiger M.A., Jackman J.E., Phizicky E.M.. Analysis of 2′-phosphotransferase (Tpt1p) from Saccharomyces cerevisiae: evidence for a conserved two-step reaction mechanism. RNA. 2005; 11:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato-Murayama M., Bessho Y., Shirouzu M., Yokoyama S.. Crystal structure of the RNA 2′-phosphotransferase from Aeropyrum pernix K1. J. Mol. Biol. 2005; 348:295–305. [DOI] [PubMed] [Google Scholar]
- 15. Harding H.P., Lackey J.G., Hsu H.C., Zhang Y., Deng J., Xu R.M., Damha M.J., Ron D.. An intact unfolded protein response in Trpt1 knockout mice reveals phylogenetic divergence in pathways for RNA ligation. RNA. 2008; 14:225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shuman S. What messenger RNA capping tells us about eukaryotic evolution. Nat. Rev. Mol. Cell. Biol. 2002; 3:619–625. [DOI] [PubMed] [Google Scholar]
- 17. Hausmann S., Shuman S.. Specificity and mechanism of RNA cap guanine-N2 methyltransferase (Tgs1). J. Biol. Chem. 2005; 280:4021–4024. [DOI] [PubMed] [Google Scholar]
- 18. Cosgrove M.S., Ding Y., Rennie W.A., Lane M.J., Hanes S.D.. The Bin3 RNA methyltransferase targets 7SK RNA to control transcription and translation. Wiley Interdiscip. Rev. RNA. 2012; 3:633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xhemalce B., Robson S.C., Kouzarides T.. Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell. 2012; 151:278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martinez A., Yamashita S., Nagaike T., Sakaguchi Y., Suzuki T., Tomita K.. Human BCDIN3D monomethylates cytoplasmic histidine transfer RNA. Nucleic Acids Res. 2017; 45:5423–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bird J.G., Zhang Y., Tian Y., Panova N., Barvík I., Greene L., Liu M., Buckley B., Krásný L., Lee J.K. et al. The mechanism of RNA 5′ capping with NAD+, NADH and desphospho-CoA. Nature. 2016; 535:444–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Julius C., Yuzenkova Y.. Bacterial RNA polymerase caps RNA with various cofactors and cell wall precursors. Nucleic Acids Res. 2017; 45:8282–8290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chakravarty A.K., Shuman S.. RNA 3′-phosphate cyclase (RtcA) catalyzes ligase-like adenylylation of DNA and RNA 5′-monophosphate ends. J. Biol. Chem. 2011; 286:4117–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mochizuki T., Yoshida T., Tanaka R., Forterre P., Sako Y., Prangishvili D.. Diversity of viruses of the hyperthermophilic archaeal genus Aeropyrum, and isolation of the Aeropyrum pernix bacilliform virus 1, APBV1, the first representative of the family Clavaviridae. Virology. 2010; 402:347–354. [DOI] [PubMed] [Google Scholar]
- 25. Mochizuki T., Krupovic M., Pehau-Arnaudet G., Sako Y., Forterre P., Prangishvili D.. Archaeal virus with exceptional virion architecture and the largest single-stranded DNA genome. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:13386–13391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Munnur D., Ahel I.. Reversible mono-ADP-ribosylation of DNA breaks. FEBS J. 2017; 284:4002–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zarkovic G., Belousova E.A., Talhaoui I., Saint-Pierre C., Kutuzov M.M., Matkarimov B.T., Biard D., Gasparutto D., Lavrik O.I, Ishchenko A.A.. Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic Acids Res. 2018; 46:2417–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takamura-Enya T., Watanabe M., Totsuka Y., Kanazawa T., Matsushima-Hibiya Y., Koyama K., Sugimura T., Wakabayashi K.. Mono(ADP-ribosyl)ation of 2′-deoxyguanosine residue in DNA by an apoptosis-inducing protein, pierisin-1, from cabbage butterfly. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:12414–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakano T., Matsushima-Hibiya Y., Yamamoto M., Enomoto S., Matsumoto Y., Totsuka Y., Watanabe M., Sugimura T., Wakabayashi K.. Purification and molecular cloning of a DNA ADP-ribosylating protein, CARP-1, from the edible clam Meretrix lamarckii.. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13652–13657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lyons B., Ravulapalli R., Lanoue J., Lugo M.R., Dutta D., Carlin S., Merrill A.R.. Scabin, a novel DNA-acting ADP-ribosyltransferase from Streptomyces scabies. J. Biol. Chem. 2016; 291:11198–11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jankevicius G., Ariza A., Ahel M., Ahel I.. The toxin-antitoxin system DarTG catalyzes reversible ADP-ribosylation of DNA. Mol. Cell. 2016; 64:1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.