

Abstract
Interests in the impact of the gastrointestinal microbiota on health and wellbeing have extended from humans to that of companion animals. While relatively fewer studies to date have examined canine and feline gut microbiomes, analysis of the metagenomic DNA from fecal communities using next‐generation sequencing technologies have provided insights into the microbes that are present, their function, and potential to contribute to overall host nutrition and health. As carnivores, healthy dogs and cats possess fecal microbiomes that reflect the generally higher concentrations of protein and fat in their diets, relative to omnivores and herbivores. The phyla Firmicutes and Bacteroidetes are highly abundant, and Fusobacteria, Actinobacteria, and Proteobacteria also feature prominently. Proteobacteria is the most diverse bacterial phylum and commonly features in the fecal microbiota of healthy dogs and cats, although its reputation is often sullied as its members include a number of well‐known opportunistic pathogens, such as Escherichia coli, Salmonella, and Campylobacter, which may impact the health of the host and its owner. Furthermore, in other host species, high abundances of Proteobacteria have been associated with dysbiosis in hosts with metabolic or inflammatory disorders. In this review, we seek to gain further insight into the prevalence and roles of the Proteobacteria within the gastrointestinal microbiomes of healthy dogs and cats. We draw upon the growing number of metagenomic DNA sequence‐based studies which now allow us take a culture‐independent approach to examine the functions that this more minor, yet important, group contribute to normal microbiome function.
Keywords: 16S rRNA gene, canine, fecal microbiome, feline, metagenome, Proteobacteria
Short abstract
The fecal microbiomes of healthy dogs and cats often include Proteobacteria at varying abundances. This phylum can have a sullied reputation as it contains a number of well‐known pathogenic members. We explored the functions of the Proteobacteria in fecal shotgun metagenome datasets from healthy dogs and cats. The Proteobacteria appeared to be enriched for functions that are consistent with a role in helping to maintain the anaerobic environment of the gut for normal microbiome function.
1. INTRODUCTION
Domesticated dogs and cats are popular companion animals, and interest in the importance and impact of their gastrointestinal (GI) microbiota on their health and wellbeing is a growing area (Deng & Swanson, 2015). The GI microbiota is a dense and diverse group of microorganisms that reside in the GI tracts of their hosts, and ferment available substrates derived from both diet and the host. In addition to their more prominent role in digestion, they also provide vitamins and substrates that are required by the host (Leblanc et al., 2013), provide specific energy sources required for intestinal epithelium integrity and contribute to its normal function (Louis & Flint, 2009), modulate the immune system (Kelly et al., 2004), and can protect the GI tract from colonization by pathogens (Ng et al., 2013). Moreover, the contributions of the GI microbiota to signaling between the central and enteric nervous system (the gut–brain axis) are becoming better understood (Perry et al., 2016), and impact on brain development and behavior.
In mammals, both major dietary shifts and host phylogeny have played influential roles in shaping the composition of gut microbiota over evolutionary timescales (Groussin et al., 2017). While modern domestic dogs and cats are generally fed a variety of commercially manufactured pet foods, consistent with the predatory lifestyles of their carnivorous ancestors, their metabolism and digestive system anatomies are adapted to diets that are rich in animal proteins and fat. Carnivore GI tracts are relatively shorter than those of omnivores and herbivores, reflecting the lower retention times required for the digestion of meat. The GI tract walls are typically much thicker to withstand bone fragments in the diet (Bosch, Hagen‐Plantinga, & Hendriks, 2015). Domestic cats are classified as obligate carnivores. However, over the course of domestication, dogs appear to have adapted to eating small amounts of starch and vegetation (Axelsson et al., 2013), thus, are largely considered facultative carnivores (Swanson et al., 2011). Modern domestic dogs and cats are fed diets that vary considerably in both format and nutrient profile. Commercially manufactured diets are typically produced in canned or kibbled formats, and in the case of kibbled diets, often contain considerable amounts of plant‐based carbohydrate, although it is recognized that neither dog nor cat has a nutritional requirement for carbohydrate (AAFCO, 2016). Raw meat‐based diets are also increasing in popularity due to purported benefits associated with their apparent biological fit with nutritional requirements, such as higher macronutrient digestibility (Bermingham, Maclean, Thomas, Cave, & Young, 2017). However, such diets have also come under criticism due to various concerns that include an inherent risk of bacterial and parasite contamination of the diet (van Bree et al., 2018), and shedding of infectious agents to humans, particularly those of high risk such as the elderly, immunocompromised, young, and pregnant (Freeman, Chandler, Hamper, & Weeth, 2013).
2. CHARACTERIZATION OF DOG AND CAT GI MICROBIOTA
Our understanding of the impact of diet on the GI microbiota, and their subsequent impact on host health and wellbeing, has grown in the last decade with the application of high‐throughput sequencing technologies. Traditionally, characterization of the dog and cat GI microbiota has been based on culture‐based approaches, where culture‐based insights into the diversity of microbes of dogs and cats to date have been fairly limited beyond pathogenic agents (Johnston et al., 2001). However, recent developments in systematic culture‐based methodologies have resulted in considerable progress cultivating the previously ‘uncultivable’ species of the human gut microbiota (Lagkouvardos, Overmann, & Clavel, 2017). Similar efforts for the dog and cat GI microbiota are yet to be undertaken.
Our understanding of GI microbiome composition and function in healthy dogs and cats has been vastly enhanced by culture‐independent studies facilitated by molecular approaches, in particular, those enabled by advances in high‐throughput sequencing (Figure 1). The GI microbiomes of companion animals are commonly explored through fecal samples, which are far less invasive to obtain than in situ GI tract content samples. Total DNA extracted from these samples (metagenomic DNA) may be amplified using PCR, commonly of informative marker genes such as the 16S ribosomal RNA (rRNA) genes, and sequenced. These data can provide detailed information on the taxonomic composition of the microbiota (Sogin et al., 2006). Alternatively, metagenomic DNA may be sequenced directly to gain information on both microbiota function, and taxonomic composition, which may be extracted from 16S rRNA or other informative marker genes within the shotgun dataset (Deusch et al., 2015; Swanson et al., 2011; Tun et al., 2012; Young, Moon, Thomas, Cave, & Bermingham, 2016). The fecal microbiota of dogs and cats contain all three domains of life – Archaea, Bacteria, and Eukarya. The Bacteria comprise the vast majority of the community, with recent metagenome‐based estimates indicating that they make up ~98% (Swanson et al., 2011; Tun et al., 2012). Estimates for the minor components of the community, Archaea, Eukarya, and viruses, ranged from ~0.2% to 1.1%, ~0.4% to 1.2%, and ~0.1% to 0.3%, respectively (Deusch et al., 2014; Swanson et al., 2011; Tun et al., 2012). Microbiota composition also varies along the GI tract, where the hindgut compartments contain the highest microbial diversity in comparison to the stomach and small intestine (Honneffer, Steiner, Lidbury, & Suchodolski, 2017; Ritchie, Steiner, & Suchodolski, 2008; Suchodolski, Camacho, & Steiner, 2008). This observation is consistent with the role of the colon and cecum as the main site of fermentation in monogastric mammals, and that the physiological conditions along the GI tract vary in their ability to support microbial life.
Figure 1.
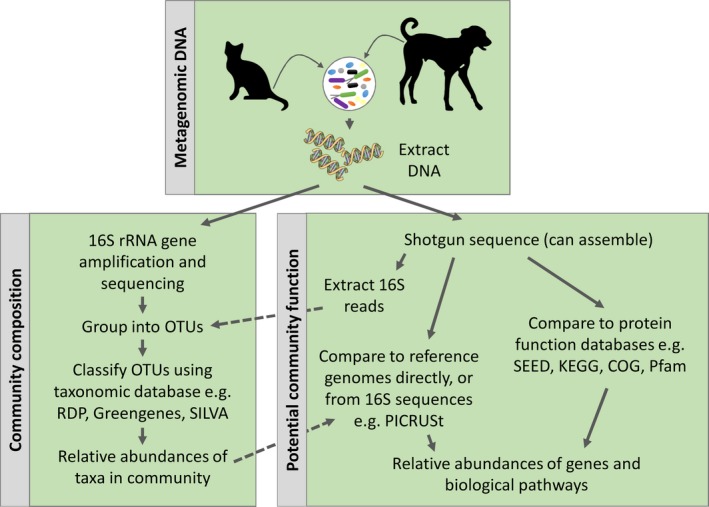
Common metagenomic DNA‐based analyses to define the community composition and function of the microbial communities in the dog and cat GI tract. Metagenomic DNA is extracted from a GI content sample, usually freshly voided fecal material, which contains microbial community members. The microbial community composition (see lower left box) is most commonly determined by amplifying variable regions of the 16S rRNA gene and sequencing the resulting amplicons. Similar 16S rRNA sequences are grouped into Operational Taxonomic Units (OTUs), which can be compared to specialized 16S rRNA sequence‐based taxonomic databases (e.g., RDP, Greengenes, SILVA) to assign taxonomic identities. The community can be described in terms of the relative abundance of the taxa present, and/or their phylogenetic relationships. To enable the potential function of the microbial community to be explored (see lower right box), metagenomic DNA is directly shotgun sequenced. The functional potential of the community can be determined by comparing the sequences to reference genomes, gene catalogs, or functional databases (e.g., SEED, KEGG, and COG). This allows the community to be described in terms of the relative abundances of its genes and pathways. More recently, inferences of community function may be made from 16S rRNA‐based taxonomic profiles using reference genome information implemented in software such as PICRUSt. Moreover, community composition may be deduced from shotgun sequenced DNA by capturing the 16S rRNA gene sequence reads, with classification using dedicated 16S rRNA databases
Bacterial 16S rRNA gene surveys have shown that, like other mammals, dogs and cats harbor complex GI microbial communities whose taxonomic compositions vary not only by diet but also factors such as age (Deusch et al., 2015), incidence of metabolic disorders (e.g., obesity, diabetes) (Bell et al., 2014; Handl et al., 2012; Park et al., 2015) and intestinal issues (e.g., IBD, diarrhea) (Guard et al., 2015; Suchodolski, Markel, et al., 2012; Suchodolski et al., 2015). In clinically healthy dogs and cats, Firmicutes and Bacteroidetes are generally the dominant phyla found in the fecal microbiome, with Fusobacteria, Actinobacteria, and Proteobacteria also featuring prominently (Deng & Swanson, 2015; Garcia‐Mazcorro & Minamoto, 2013). In the dog and cat, the general roles and functions of these bacterial phyla are assumed to be similar to their roles in the gut microbiota of model organisms such as humans and rodents, for which, more information is available. However, there is building evidence that this may not be a viable assumption. For example, Fusobacterium appears to be associated with inflammatory bowel disease (IBD) and colorectal cancer in humans, but not necessarily in dogs (Vázquez‐Baeza, Hyde, Suchodolski, & Knight, 2016), where they have been found in high abundance in healthy dogs fed a raw red meat compared to a kibble diet (Bermingham et al., 2017). Fusobacterium has also generally been found in higher concentrations in healthy carnivore hosts (Ley et al., 2008).
The Proteobacteria are commonly occurring in healthy mammalian GI microbiomes. Proteobacteria are more abundant in dogs and cats fed high protein diets, but are frequently highlighted as a microbial group of particular concern to veterinarians and pet owners as they include a number of clinically important gastrointestinal pathogens, such as diarrheagenic Escherichia coli, Campylobacter jejuni, Klebsiella pneumoniae, Salmonella typhimurium, and Yersenia enterocolitica (Kil & Swanson, 2011) that may affect the health and wellbeing of both the pet and its owner. Moreover, high abundances, or “blooms”, of Proteobacteria in the GI tract, have been suggested as a microbial signature of dysbiosis in humans and mice (Shin, Whon, & Bae, 2015). In dogs and cats, members of the Proteobacteria have significantly increased abundances in individuals with gut inflammation (Minamoto et al., 2015; Suchodolski, Dowd, Wilke, Steiner, & Jergens, 2012; Suchodolski, Markel, et al., 2012; Suchodolski et al.,2015; Vázquez‐Baeza et al., 2016) and metabolic disorders (Park et al., 2015). In addition, companion animals may act as reservoirs for antimicrobial resistant bacteria, further contributing to public health concerns (Rubin & Pitout, 2014).
3. PROTEOBACTERIA
The Proteobacteria are named after Proteus, a Greek god of the sea, capable of assuming many forms in reflection of the broad morphological and metabolic diversity contained within this phylum (Stackebrandt, Murray, & Trüper, 1988). Proteobacteria is the largest bacterial phylum, and six classes and over 116 families are currently recognized (http://www.bacterio.net/). Members are Gram negative, and play a variety of roles in a range of diverse microbial ecosystems, such as in aquatic, soil, plant, and animal niches. Within anaerobic gastrointestinal environments, the Gammaproteobacteria are often the most prevalent class of Proteobacteria present. Unlike the majority of microbes in the GI microbiome that are strict anaerobes, Proteobacteria are often facultatively or obligately anaerobic, thus are able to tolerate a range of oxic conditions. As such, it is postulated that the Proteobacteria contribute to homeostasis of the anaerobic environment of the GI tract, and hence, the stability of the strictly anaerobic microbiota. Moreover, in humans and other mammals, facultative anaerobes including Proteobacteria are among the earliest colonizers and dominant members in the neonatal gut, which is abundant in oxygen immediately post partum. By consuming oxygen, and lowering redox potential, it has been speculated that the Proteobacteria play a key role in preparing the gut for successive colonization by the strict anaerobes required for healthy gut function (Shin et al., 2015). Proteobacteria can also colonize the mucus layer of the acid secreting stomach, where several Helicobacter species have been identified and isolated from dogs and cats (Haesebrouck et al., 2009). Gastric Helicobacter isolates are able to increase the pH in their local external environment via the action of urease to liberate alkaline ammonium salts from urea (Sidebotham, Worku, Karim, Dhir, & Baron, 2003). Many clinically healthy dogs and cats are infected with Helicobacter species, although these species can also associate with inflammation of the gastric mucosa and have been hypothesized to cause gastric lymphoma in cats (Haesebrouck et al., 2009).
Proteobacteria are able to grow on a range of organic compounds including protein, carbohydrates, and lipids. Recent studies of the human gut microbiome have shown that while the Firmicutes and Bacteroidetes possess many conserved genes that contribute to the functional redundancy of the microbiome; despite their relatively lower abundance, the Proteobacteria contribute to much of the functional variation (Bradley & Pollard, 2017). This observation suggests that the major sources of taxonomic variation in microbiota do not necessarily contribute the most variation in function (Bradley & Pollard, 2017). As many observations regarding the Proteobacteria have been based on human or rodent models, whether these extend to their roles in the microbiomes of the domestic dog and cat requires further investigation. In this study, we review and further analyze the growing number of available community composition and metagenomic datasets based on the GI and fecal microbiota of healthy dogs and cats to better understand the prevalence, diversity, and roles of the Proteobacteria within these hosts.
4. ABUNDANCE OF PROTEOBACTERIAIN DOG AND CAT FECAL MICROBIOMES VIA 16S rRNA GENE ANALYSES
A summary of the prevalence and diversity of Proteobacteria in clinically healthy dog and cat fecal microbiome studies, based on the information reported in 16S rRNA gene survey studies for which key technical information was available, is shown in Tables 1 and 2. These studies have typically analyzed thousands to tens of thousands of bacterial sequences per sample, and compared to traditional culture and clone‐based approaches, have enabled more detailed insight into the Proteobacteria which typically comprise only a few percent of 16S rRNA gene sequences. Within these studies, variations in fecal microbial community profiles, even for cohorts of animals receiving the same diet, could be considerable, and likely impacted by the complex interplay between host genetics and physiology of the individual, with its environment and diet.
Table 1.
Prevalence of Proteobacteria in healthy dog fecal microbiota studies based on 16S rRNA gene sequencing
| Study descriptiona | Subjects/age | Sequencing and analysis methodsb | Proteobacteria abundance and general microbiome descriptionsc | Reference |
|---|---|---|---|---|
| General microbiome characterization | ||||
| Fecal microbiome | Mixed age, breed, and sex (N = 12) | 454, V1‐V3 region, NCBI | Median values for all Proteobacteria genera detected were 0%, and in general, upper end of range was <0.1%. Prevalent genera were Anaerobiospirillum (0%–1.88%), Pseudomomas (0%–0.14%), and Succinivibrio (0%–0.1%) | (Handl et al., 2011) |
| Fecal microbiome | Mixed age, breed, sex NS (N = 6) | 454, V1‐V3 region, NCBI | Proteobacteria ranged from 0% to 17%. Highest abundance in dogs fed the Beneful (10%–17%) and Science Diet (~10%) diets, but negligible in other diets. Alphaproteobacteria (Hyphomicrobiaceae) predominated. Proteobacteria were observed in 4 of the 6 dogs, and was the third‐most abundant phylum after Firmicutes and Actinobacteria | (Garcia‐Mazcorro et al., 2012) |
| Fecal microbiome | Mixed age, miniature schnauzer, mixed sex (N = 11) | 454, V1‐V3 region, RDP | Mean of 11.31% Proteobacteria observed across all dogs. Highest (21.6%) was from dogs fed a “Trial” diet. Gammaproteobacteria (Succinivibrionaceae and Enterobacteriaceae) were predominant. Proteobacteria were observed in all individuals and was generally the fourth‐most abundant phylum after Fusobacteria, Bacteroidetes, and Firmicutes | (Hand et al., 2013) |
| Diet, pre‐ and probiotic trials | ||||
| Dry control (30% CP, 19% fat, 1.4% fiber) vs. beet pulp containing diet (28% CP, 21% fat, 4.5% fiber) | ~20 months, mongrel and hound crosses, female (N = 6) | 454, V3 region, RDP | Proteobacteria ranged ~4%–10.5% (mean 7%), with no significant difference between diet treatments. Proteobacteria were observed in all six dogs, and was generally the fourth‐most abundant phylum after Firmicutes, Bacteroidetes, and Fusobacteria | (Middelbos et al., 2010) |
| Diet (various commercial) with synbiotic administration | Mixed age, breed, and sex (N = 12) | 454, V1‐V3, NCBI | Proteobacteria was the fifth‐most abundant phylum after Firmicutes, Bacteroidetes, Actinobacteria, and Fusobacteria | (Garcia‐Mazcorro et al., 2011) |
| Dry control (31% CP, 14% fat, 3.0% fiber) vs. cooked navy bean diet (30% CP, 14% fat, 3.0% fiber) | Mixed age, breed, and sex (N = 10) | 454, V4‐V6 region, NCBI | Diet did not have a significant effect on Proteobacteria abundance (0.79% in control, 1.68% in navy bean diet). The Enterobacteriaceae were the most abundant family of Proteobacteria. Proteobacteria was the fourth‐most abundant phylum after Firmicutes, Actinobacteria, and Fusobacteria | (Forster et al., 2012; Kerr et al., 2013) |
| Potato fiber at five concentrations in an extruded diet (25% CP, 13%–15% fat, 10.8%–11.4% fiber) | ~6 years, hound, female (N = 10) | 454, V4‐V6 region, Greengenes | Proteobacteria abundance increased with increasing potato fiber concentration (from 1.5% to 2.8%), although this increase was not significant. Sutterella and Succinivibrio were the most abundant Proteobacteria genera, and Proteobacteria were the third‐most abundant phyla after Firmicutes and Fusobacteria | (Panasevich et al., 2015) |
| Beef and chicken raw meat diets with and without 1.4% inulin or 1.4% yeast cell wall (25%–30% CP, 45%–50% fat) | ~5.5 years, beagle, female (N = 6) | 454, V4‐V6 region, NCBI | Proteobacteria ranged from 4.1% to 5.8% in abundance across diets. Beef diets increased the abundance of Escherichia and decreased the abundance of Anaerobiospirillum compared to chicken diets. Inulin reduced the abundance of Enterobacteriaceae and Escherichia. Overall, Proteobacteria were observed in all six dogs, and was generally the fourth‐most abundant phylum after Fusobacteria, Firmicutes, and Bacteroidetes. | (Beloshapka et al., 2013) |
| Raw meat (76% CP, 18% fat, 0.6% fiber) vs. kibble (30% CP, 27% fat, 2.4% fiber) | ~5.8 years, harrier hound, mixed sex (N = 15) | Illumina MiSeq, V4‐V6, Greengenes 13_8 | Dogs fed kibbled diet averaged 1.27% Proteobacteria (0.2%–4.7%), compared to 0.55% (0.08%–1.87%) when fed raw meat diet. Proteobacteria (mainly Gammaproteobacteria) were observed in all dogs, and was the fourth‐most abundant phylum after Firmicutes, Bacteroidetes, and Fusobacteria. | (Bermingham et al., 2017) |
| Diet (various) supplemented with 225 mg FOS+inulin prebiotic for 16 days | Mixed age, breed, and sex (N = 10) | 454, V4‐V5 region, Greengenes, PICRUSt | Sutterella was detected among more dogs after administration of prebiotic. | (Garcia‐Mazcorro et al., 2017) |
| Diet (various) supplemented with FOS+inulin prebiotic at 0.5% of DMI | Mixed age, breed, and sex (N = 10) | 454, V4‐V5 region, Greengenes, PICRUSt | Administration of prebiotic did not significantly impact the relative abundance of Proteobacteria subtaxa. | (Garcia‐Mazcorro et al., 2017) |
| Dry control (27% CP, 11% fat, 2.8% fiber) vs. raw meat based (26% CP, 18% fat, 0.7% fiber) diet | Mixed age, boxer, female (N = 8) | Illumina MiSeq, V3‐V4, RDP | Dogs fed the control diet had an average of 2.4% Proteobacteria compared to 4.4% when fed the raw meat‐based diet. Sutterellaceae (Betaproteobacteria) were prevalent among treatments and Enterobacteriaceae (Gammaproteobacteria) predominated in raw meat fed dogs. Proteobacteria was the fourth‐most abundant phylum after Firmicutes, Bacteroidetes, and Fusobacteria. | (Sandri et al., 2017) |
| Natural (90% raw meat plus vegetables) vs. commercial feed (18%–21% CP, 8%–10% fat) | Mixed age, small breeds, and sex (N = 11) | Illumina MiSeq, V3‐V4, EzBioCloud | Proteobacteria abundance averaged 0.86% in dogs fed the natural diet where it was the fifth‐most abundant phylum, as compared to 8.67% for the commercial diet‐fed dogs, where it was the third‐most abundant. Within the latter treatment, Escherichia coli comprised 7.3% of sequences. | (Kim et al., 2017) |
| Diet shifted from a commercial dry diet to increasing proportions of boiled minced beef added, then reversion to dry diet. | Mixed age, breed, and sex (N = 11) | Illumina MiSeq, V3‐V4, Greengenes | Sutterella and Anaerobiospirillum were the predominant genera present, and like Proteobacteria (~4%–6%), decreased in relative abundance as beef content was increased. Proteobacteria was the third‐most abundant phylum after Firmicutes and Fusobacteria in dogs on all treatments. | (Herstad et al., 2017) |
CP, crude protein; FOS, fructooligosaccharide.
454 refers to 454 GS FLX Titanium sequencing.
If available, descriptions of sub‐phylum level abundance and Proteobacteria prevalence among individuals are given. The level of taxonomic detail provided in each study varied and taxonomy (OTU) tables were not always available.
Table 2.
Prevalence of Proteobacteria in healthy cat fecal microbiota studies based on 16S rRNA gene sequencing
| Study descriptiona | Subjects/age | Sequencing and analysis methodsb | Proteobacteria abundance and general microbiome descriptionsc | Reference |
|---|---|---|---|---|
| General microbiome characterization | ||||
| Fecal microbiome | Mixed age, breed, and sex (N = 12) | 454, V1‐V3 region, NCBI | Median values for all Proteobacteria genera were 0%, and the upper end of range for genus abundance was <0.1%, apart from Succinivibrio, which was 0.51% | (Handl et al., 2011) |
| Diet, pre‐ and probiotic trials | ||||
| Diet (various commercial) with synbiotic administration | Mixed age, breed, and sex (N = 12) | 454, V1‐V3, NCBI | Proteobacteria was the fourth‐most abundant phylum after Firmicutes, Actinobacteria, and Bacteroidetes. Synbiotic did not appear to impact abundance of any class, order, family, or genus in these phyla | (Garcia‐Mazcorro et al., 2011) |
| Kitten fecal microbiome of kittens fed kibble (35% CP, 20% fat, 1.8% fiber) vs. canned (45% CP, 38% fat, 1.5% fiber), from mothers fed canned or kibbled diets (crossover design). | Kittens (8 and 17 weeks), shorthair, mixed sex (N = 20) | 454, V1‐V3 region, RDP | Average Proteobacteria abundance was 1.2%–4.9% across treatments, with 4.9% in the canned‐fed kitten from kibble‐fed mother. Sutterella abundance significantly differed across treatments at 0.79% in canned‐fed kittens from canned‐fed mothers, compared to the three other treatments (0.24%–0.27%). Proteobacteria were observed in 19/20 kittens, and was the fourth‐most abundant phylum after Firmicutes, Bacteroidetes, and Fusobacteria | (Bermingham, Kittelmann, et al., 2013) |
| Dry format diet (33% CP, 11% fat, 1.9% fiber) vs. wet (42% CP, 42% fat, 1.6% fiber) | Mixed age, shorthair, mixed sex (N = 12) | 454, V1‐V3 region, RDP | Average Proteobacteria abundance was 1.1% in the wet vs. 0.4% in the dry diet treatment, where Anaerobiospirillum and Sutterella were significantly more abundant (0.4% and 0.6%, respectively) in the wet treatment. Proteobacteria were detected in all animals, and was the fifth‐most abundant phylum after Firimicutes, Bacteridetes/Fusobacteria, and Actinobacteria | (Bermingham,Young et al., 2013) |
| MPMC diet (34% CP, 19% fat, 6.9% fiber) vs. HPLC diet (53% CP, 24% fat, 2% fiber) | Kittens (8, 12, and 16 weeks), domestic shorthair, mixed sex (N = 14) | 454, V4‐V6 region, NCBI | Proteobacteria abundance, averaged per time point was 3.3%–3.7% in HPLC and 0.1%–1.1% in the MPMC groups | (Hooda et al., 2013) |
| Diet (various) supplemented with 225 mg FOS+inulin prebiotic for 16 days | Mixed age, breed, and sex (N = 10) | 454, V4‐V5 region, Greengenes, PICRUSt | Linear discriminant analysis effect size identified Gammaproteobacteria abundances contribute bacterial community differences between pre‐ and during prebiotic administration. Gammaproteobacteria were detected in 9/10 cats prior to, but only 4 cats after, prebiotic administration suggesting that the prebiotic selected against this class | (Garcia‐Mazcorro et al., 2017) |
| Kibble (composition) vs. raw meat‐based diets (composition); with and without prebiotic. | Mixed age, shorthair, mixed sex (N = 12) | Illumina MiSeq, V3‐V4, Greengenes | Proteobacteria were less abundant in cats fed the raw meat diet (average 2.4% on kibble vs. 0.4% on raw meat), and were dominated by Anaerobiospirillum (0.6%–2.5%) and Succinivibrio (1.2%–2.1%) in the kibble diet‐fed cats. When fed the kibble diet Proteobacteria were the third‐most abundant phylum after Firmicutes and Bacteroidetes. When fed the raw meat diet, Fusobacteria was third‐most abundant and Proteobacteria was fourth | Butowski et al., unpublished |
CP, crude protein; FOS, fructooligosaccharide.
454 refers to 454 GS FLX Titanium sequencing.
If available, descriptions of sub‐phylum level abundance and Proteobacteria prevalence among individuals are given. The level of taxonomic detail provided in each study varied and taxonomy (OTU) tables were not always available.
When comparing the data generated from different studies, care must be exercised as technical differences between the analytical methods employed will generate biases in the data reported. It is well recognized that variation in microbial community composition may arise from differences in factors such as sample storage method, DNA extraction procedure, PCR primer sequences and 16S rRNA gene region amplified, PCR amplification conditions, and sequencing technology (Boers, Jansen, & Hays, 2016). In particular, differences in cell lysis treatments for DNA extraction may bias the representation of Proteobacteria, with lower recovery of intact DNA from Gram‐negative bacteria associated with harsher lysis conditions (Yuan, Cohen, Ravel, Abdo, & Forney, 2012). The choice of bioinformatics procedures used (including sequence data quality filtering, chimera detection, and low‐abundance OTU cut‐offs) and, in particular, how taxonomic assignments are made, also bias the microbiota composition reported. A range of reference databases are commonly used (Cole et al., 2007; Desantis et al., 2006; Pruesse et al., 2007) that are frequently updated as microbial taxonomies are refined. Differences between these databases (Balvočiūtė & Huson, 2017) and updates over time may impact the degree and level of taxonomic detail that assignments have been made. Furthermore, differences between studies in the level of detail that data were reported to (e.g., from taxon frequencies per individual animal to group medians and ranges, and the level of taxonomic rank reported to) were very apparent, as well as the numbers of individuals examined, and these factors also limit the degree to which data can be directly compared between studies. An understanding of the range of microbiota compositions that are ‘normal’ for healthy individuals is only beginning to be established (AlShawaqfeh et al., 2017; Suchodolski, Dowd, et al., 2012). In general, Proteobacteria were detected from almost all individuals examined, which suggests they play a fairly essential role in the GI microbiomes of healthy pets.
4.1. Proteobacteria in dogs
Studies of the fecal microbiomes of clinically healthy adult dogs (Table 1) indicate that the relative abundances of Proteobacteria are generally higher and more variable than in cats (Table 2). In general, Proteobacteria comprised from 0% to 22% of 16S rRNA reads of the fecal microbiomes reported in dogs, although high abundances at the top of this range were seldom observed. In a study of six privately owned dogs of a variety of breeds and ages, the median Proteobacteria relative abundance observed was 1%, although ranged from 0% to 17% across the individuals (Garcia‐Mazcorro, Dowd, Poulsen, Steiner, & Suchodolski, 2012). In a research colony group of 11 adult miniature Schnauzers fed a range of commercial diets, the average abundance of Proteobacteria was 11.3%, with a range from 0.03% to 21.6%, and Sutterella comprised a large majority of the Proteobacteria detected (Hand, Wallis, Colyer, & Penn, 2013). In contrast to these two studies, low proportions of Proteobacteria were detected in a pyrosequencing‐based analysis of the fecal microbiota of 12 privately owned healthy dogs (Handl, Dowd, Garcia‐Mazcorro, Steiner, & Suchodolski, 2011). In this study, data were reported at the genus level, and all Proteobacteria genera had a median percent of sequences as 0%. The most abundant genus was Anaerobiospirillum (family Succinivibrionaceae), which occurred at 0% to 1.88%, although again, the median was 0%. Observations of the healthy subjects in epidemiological studies also show Proteobacteria at a range of abundances. For example, in a study that examined microbial dysbiosis in dogs with acute diarrhea, the control group of 13 healthy dogs displayed a median of 0.1% Proteobacteria (with range 0.0% to 0.3%), where microbiota were heavily dominated by Firmicutes and Bacteroidetes (Guard et al., 2015). Isaiah, Parambeth, Steiner, Lidbury, & Suchodolski (2017) recruited 18 healthy dogs to a study to compare the impact of exocrine pancreatic insufficiency on the fecal microbiome. Proteobacteria comprised 0.2% to 5.2% of the microbiota in this study, with a median abundance of 1.3%. Li, Lauber, Czarnecki‐Maulden, Pan, and Hannah (2017) examined lean and obese dogs fed high protein, low carbohydrate and low protein, high carbohydrate diets. Among the treatment groups, average Proteobacteria relative abundances in lean individuals ranged from 3.9% to 6.7%, with greater abundances in dogs fed the low protein diets (Li et al., 2017). In general, detailed information on diet composition from these studies was insufficient to dissect potential nutritional drivers of the diversity, but it is apparent that when fed a variety of commercial diets, the abundances of Proteobacteria in healthy animals varies considerably (Garcia‐Mazcorro et al., 2012; Hand et al., 2013; Handl et al., 2011).
4.2. Diet‐based studies in dogs
Diet‐related trends in humans have also extended to companion animals and interests in meat‐based, high animal protein and fat diets with minimal carbohydrates, and the use of prebiotics, have been gaining in popularity among pet owners in recent times. A number of studies have investigated the effects of such dietary trends on the dog fecal microbiome. These studies typically involve cohorts of research animals of similar breed, and detailed nutritional information of the diets used is available.
Raw meat diets in particular raise a number of food safety–related concerns, as the potential to carry pathogens such as Escherichia coli, Salmonella, and Listeria, and associated antimicrobial resistance determinants, is greater for the uncooked product (van Bree et al., 2018). Moreover, there are concerns around risks of infection to the pet owner (Schlesinger & Joffe, 2011), thus particular care around food hygiene and storage must be taken. As such, the impact of raw meat diets on the GI microbiome is of interest, and particularly with regard to Proteobacteria.
From studies to date, relative Proteobacteria abundances have varied considerably for raw meat feeding, which has often been contrasted to kibble diets (Bermingham et al., 2017; Kim, An, Kim, Lee, & Cho, 2017; Sandri, Dal Monego, Conte, Sgorlon, & Stefanon, 2017), although differences in study designs and diet formulations confound the ability to directly compare and interpret results. In the study by Sandri et al., a 70% raw beef skeletal muscle–based diet supplemented with carbohydrate fed to adult Boxer dogs resulted in significantly increased proportion (4.4%) of Proteobacteria in the raw meat fed animals compared to those fed a commercial extruded diet (1.3%) (Sandri et al., 2017). A highly significant shift in the abundance of Enterobacteriaceae, from 0.047% to 2.454% (p < 0.01), and Escherichia coli/Shigella was seen, although all dogs were healthy (Sandri et al., 2017). In this study, both diets had similar protein concentrations. However, protein digestibility, and hence, the amount of protein entering the colon, was not measured (Sandri et al., 2017), which is a factor that influences microbiota composition. In contrast, in a trial that involved feeding Harrier Hounds a complete and balanced raw meat diet, Proteobacteria comprised an average of 0.56% (0.08%–1.8%) (Bermingham et al., 2017) after 9 weeks. This was compared to a commercially available kibble diet, where Proteobacteria comprised 1.27% (0.21%–2.01%), although the difference between raw and kibble‐fed animals was not significant (Bermingham et al., 2017). Succinivibrio and an unclassified member of the Burkholderiales were the only genera of Proteobacteria that displayed a significant difference in abundance between the two diets, with both being more abundant in kibble‐fed dogs (Bermingham et al., 2017). Similarly, in a study conducted in Korea (Kim et al., 2017), generally, lower abundances of Proteobacteria were observed in dogs fed a natural diet (>90% raw meat such as kangaroo, beef, and poultry; average 0.86%, range 0.27%–1.84%) than standard commercial kibble fed (8.67%, 0.05%–2.07%) (Students T‐test, p = 0.07), where Proteobacteria were dominated by members of the Enterobacteriaceae. This study examined the fecal microbiomes of 11 mixed‐age, small breed dogs, recruited from a pet owner group based on their existing dietary regimes. Beloshapka and colleagues examined raw chicken‐ and raw beef‐based diets, both with and without inulin and yeast cell wall prebiotics, to beneficially alter the gut microbiota (Beloshapka et al., 2013). In general, the abundances of Proteobacteria averaged from 4.05% to 5.83% when fed each of the diet treatments, and these did not differ significantly with respect to either meat source, or addition of prebiotic. Few taxa significantly differed across treatments at the genus level. Anaerobiospirillum was more abundant when fed the chicken compared to the beef‐based diets. Escherichia abundances differed significantly, both in response to meat type, and prebiotic inclusion, across all diet groups, where they were present at 0.29%–1.69%.
High levels of cooked meat were examined in the study by Herstad and colleagues (Herstad et al., 2017). They observed a gradual decrease in Proteobacteria relative abundance from a kibble control diet through stepwise inclusions of cooked minced beef at 85% of the diet, where beef content strongly negatively correlated with average Proteobacteria abundance per treatment (Pearson's correlation, −0.979). However, in a comparison of Proteobacteria concentrations in the control diet (average 6.24% Proteobacteria, range 1.15%–14.34%) with the 85% beef treatment (3.98%, range 1.11%–11.85%), these differences were not highly significant (Students T‐test, p = 0.10). Sutterella and Anaerobiospirillum dominated the most abundant Proteobacteria genera (Herstad et al., 2017).
A number of studies have investigated the impact of prebiotic and probiotic treatments (Beloshapka et al., 2013; Garcia‐Mazcorro, Barcenas‐Walls, Suchodolski, & Steiner, 2017; Garcia‐Mazcorro et al., 2011; Kerr, Forster, Dowd, Ryan, & Swanson, 2013; Middelbos et al., 2010; Panasevich et al., 2015), although again the impact of these on Proteobacteria levels and composition appears to be minimal. For example, the supplementation of a kibble diet with beet pulp dietary fiber, a common ingredient used in commercial dog food, provided a complex mixture of fermentable and nonfermentable carbohydrates (Middelbos et al., 2010). This resulted in significant increases in Firmicutes and decreases in Fusobacteria, but the relative abundances of Proteobacteria remained the same (Middelbos et al., 2010). A mixture of a fructooligosaccharides (FOS) and inulin was supplemented to dogs fed a range of diets, and led to significant shifts in the abundances of Sutterella among individuals. The responses across animals were highly variable, however, and may have depended on the baseline composition of the microbiota prior to prebiotic administration, and interaction with diet. Inclusion of potato fiber as a prebiotic source appeared to result in a small increase in the relative abundance of Proteobacteria, but this was not significant (Panasevich et al., 2015).
Taken together, the range of Proteobacteria abundances in clinically healthy dogs varies widely, and the influence of diet between studies is not clear cut. Certainly, harmonization of experimental and analytical methods would enable more direct comparisons between studies and overall trends to be deduced such as via a metastudy approach. Moreover, analyses of the microbiota metatranscriptomes would provide much greater insight into the metabolic activities of the Proteobacteria present and their functional contributions under different dietary regimes.
4.3. Proteobacteria in cats
Proteobacteria appeared to be generally less abundant in cats as compared to dogs, being detected at less than 4.3% in clinically healthy adult animals (Table 2), although considerably fewer studies were available to determine if this is a more general trend. However, in one study of the fecal microbiomes of cats with diarrhea (Suchodolski et al., 2015), among the 21 healthy subjects recruited to the control group, 0.26%–27.9% (median 4.79%) Proteobacteria was reported, and this appeared to be underpinned by few individuals that had up to 24% Helicobacter, or up to 7.4% Campylobacter. These results were not consistent with general observations from other studies (Table 2). These cats belonged to staff and students of a veterinary medical teaching hospital, so whether this aspect had contributed to a higher exposure of such microbial taxa remains unknown (Suchodolski et al., 2015).
In kittens, the relative abundance of Proteobacteria in the fecal microbiota generally appeared to be greater when they were fed diets with higher protein and fat content, than kibble, at around 3%–4% (Bermingham, Kittelmann, et al., 2013; Hooda, Vester Boler, Kerr, Dowd, & Swanson, 2013). In adult cats, Proteobacteria were detected at an average abundance of 1.1% when fed a wet (high protein, high fat) diet compared to 0.4% when fed a dry (moderate protein, low fat) commercial diet, with Anaerobiospirillum and Sutterella showing significantly higher abundances in the cats fed a cooked wet diet (Bermingham, Young, et al., 2013). In contrast, however, adults cats fed a raw meat‐based diet had only 0.4% Proteobacteria on average, compared to their kibble‐fed counterparts who averaged 2.4% (Butowski et al., unpubl.). Synbiotic administration did not appear to change the relative abundances of major phyla, although specific information on Proteobacteria abundance was not presented (Garcia‐Mazcorro et al., 2011), while FOS and inulin prebiotic treatments were reported to negatively impact Proteobacteria concentrations (Garcia‐Mazcorro et al., 2017).
4.4. Proteobacteria in the young
Proteobacteria are dominant members in the human neonatal gut, which is abundant in oxygen immediately post partum. The Proteobacteria are thought to play a key role in preparing the gut for colonization by the strict anaerobes required for healthy gut function by consuming oxygen, and lowering redox potential in the gut environment (Shin et al., 2015). Analyses of the GI microbiota of kittens from birth, and hence, an understanding of the dynamics of GI microbiome establishment in dogs and cats, have not yet been reported. Thus, the role of Proteobacteria at this critical stage of life in the dog and cat is yet to be confirmed. However, it would be reasonable to assume that they play similar roles in newborn dogs and cats, as in other mammals.
Two 16S rRNA gene studies examined the fecal microbiomes of newly weaned kittens (Bermingham, Kittelmann, et al., 2013; Hooda et al., 2013). In one study, Proteobacteria comprised, on average, 3% of the 16S rRNA sequences of kittens fed a high protein, low carbohydrate dry diet, compared to 1% or less when fed a moderate protein, moderate carbohydrate dry diet (Hooda et al., 2013). Fecal samples were taken at 8 (weaning), 12, and 16 weeks, where Proteobacteria abundances generally decreased over time (Hooda et al., 2013). In another study, the impact of maternal and postweaning diet was assessed in pre‐ and postweaned kittens at 8 and 17 weeks of age, respectively, using canned (high‐protein, high‐fat concentration) and kibbled (medium‐protein, medium‐fat concentration) diets (Bermingham, Kittelmann, et al., 2013). In this study, the relative abundance of Proteobacteria ranged from ~1% to 5% across the treatments, and although they appeared more abundant in canned‐fed kittens from kibbled‐fed mothers, this difference was not statistically significant. Moreover, Proteobacteria were generally more abundant at 8 weeks than at 17, but again, this was not significant. Sutterella was approximately three times more abundant in canned diet‐fed kittens from canned diet‐fed mothers than from kittens that had been exposed to the kibbled diet, either pre‐ or postweaning, and was the only member of the Proteobacteria whose abundance differed by postweaning diet (Bermingham, Kittelmann, et al., 2013). Shotgun metagenome studies have also examined the fecal microbiomes of kittens, where Escherichia and Desulfovibrio were among predominant bacteria, together with Proteobacteria in general, their relative abundances decreased as the kittens went from 8 to 16 weeks of age (Deusch et al., 2014).
5. PROTEOBACTERIA DIVERSITY IN THE DOG AND CAT FECAL MICROBIOME
In the dog, Proteobacteria was generally the fourth‐most abundant phylum behind Firmicutes, Bacteroidetes, and Fusobacteria, while in cats, it generally ranked behind Firmicutes, Bacteroidetes, and Fusobacteria or Actinobacteria (Tables 1 and 2). Gammaproteobacteria and Betaproteobacteria were most commonly observed, while other classes such as the Alphaproteobacteria, Epsilonproteobacteria, and Deltaproteobacteria were generally lower in abundance and not consistently detected. Escherichia, Shigella, Succinivibrio, Anaerobiospirillum, and Sutterella were among the most abundant recognized Proteobacteria genera reported in healthy individuals (Handl et al., 2011). Escherichia and Salmonella are members of the Enterobacteriaceae and while these genera are best known for their prominent pathogenic members, many isolates, even with diarrhea‐related virulence factors, are nonpathogenic and most likely contribute to normal microbiome function. Analysis of fecal samples from 70 diarrheic and 230 nondiarrheic domestic cats identified 15 enteropathogenic E. coli (EPEC) strains from 14 cats, of which only one cat was suffering from diarrheal symptoms (Morato et al., 2009). Additionally, dogs can be colonized by extended‐spectrum beta lactamase producing E. coli for extended periods of time (>6 months) without any clinical signs (Baede et al., 2015). A study in the United States to determine the prevalence of Salmonella in dogs and cats visiting veterinary clinics from 11 geographically dispersed veterinary testing laboratories indicated a low (<1%) overall prevalence in 542 cat fecal samples, and a prevalence of 2.5% (60 of 2422) in dogs, however, almost half the Salmonella‐positive animals were nondiarrheic (Reimschuessel et al., 2017). Similarly, the prevalence of Campylobacter spp. in rectal swab enrichments from 90 healthy dogs and 110 healthy cats was 36% and 16%, respectively (Bojanić et al., 2017).
Succinivibrio and Anaerobiospirillum are both succinate‐producing members of the family Succinivibrionaceae of the Gammaproteobacteria, and are recognized as part of the normal fecal microbiota of dogs and cats. Succinivibrio is commonly found in the rumens of cattle and sheep fed grain‐based diets, where they are involved in the digestion of starch and its breakdown products (Bryant & Small, 1956; Stackebrandt & Hespell, 2006), and it is presumed that they undertake similar roles in the GI microbiomes of dogs and cats. In contrast, Anaerobiospirillum succiniciproducens was first isolated from dog feces (Davis, Cleven, Brown, & Balish, 1976), and A. thomasii, a glucose, galactose, and maltose fermenter, was first isolated from the feces of healthy dogs and cats (Malnick, 1997). While Anaerobiospirillum is known to cause diarrhea and bacteremia in immunocompromised humans, dog ownership (and the potential for zoonotic transmission) is not recognized as an established as a risk factor for infection (Epstein, Ernst, Rogers, Carmody, & Aguero‐Rosenfeld, 2017a,b). Sutterella are members of the Betaproteobacteria (family Alcaligenaceae), where Sutterella stercoricanis, a Gram‐negative anaerobe, was first isolated from healthy canine faeces (Greetham et al., 2004). The asaccharolytic, nitrate‐reducing nature of Sutterella (Greetham et al., 2004) is suggestive of a key role in protein metabolism.
6. THE ROLES OF PROTEOBACTERIA IN DOG AND CAT GI MICROBIOME FUNCTION
An understanding of microbiota function, rather than its community structure alone, is necessary to truly recognize the contributions of the microbiota to host nutrition and wellbeing. Such knowledge will also contribute to developing strategies to improve host health. While the number of microbiome‐based studies in the dog and cat lag behind those of humans and related rodent models, insights into pet gastrointestinal microbiome functions are being pursued using similar molecular methods, revealing considerable similarities in function (Swanson et al., 2011), but also a number of distinct differences (Bermingham et al., 2017; Vázquez‐Baeza et al., 2016). A number of approaches to explore microbiome function from high‐throughput sequencing data have been used by the dog and cat microbiome research community, from inferring community functional gene composition from 16S rRNA gene profiles, to shotgun sequencing of metagenomic DNA. Notable by their current absence are studies that have examined the gene activities in microbiome samples. Metatranscriptomic studies, which sequence the total RNA within the sample, enable an in depth examination of the metabolically active members of the community and their specific contributions to GI metabolic, and other, processes. Although such studies will likely follow in due course, they will be more informative as a more diverse range of dog‐ and cat‐derived microbial isolates are brought into culture, characterized, and microbial reference genome information becomes available. These data will aid the interpretation of metagenomic and metatranscriptomic datasets, and allow more accurate predictions of “who is doing what”.
6.1. Microbiome function inferred from 16S rRNA gene profiles
In the more recent 16S rRNA gene‐based studies, the taxonomic composition of microbiomes has been extended to include predictions of microbiome function via the recruitment of available bacterial reference genome information that matches the 16S rRNA gene profiles (Garcia‐Mazcorro et al., 2017; Guard et al., 2015; Isaiah et al., 2017; Li et al., 2017) (Tables 1 and 2). Such analyses, most commonly implemented in the software, PICRUSt (Langille et al., 2013), have become more prevalent. However, the majority of available GI microbial reference genome data are from human gut (or model rodent) microbiome (Land et al., 2015; Turnbaugh et al., 2007) and rumen microbiome origin (Seshadri et al., 2018). Moreover, the Proteobacteria are such a diverse and heterogeneous group that 16S rRNA gene sequences are rarely sufficiently informative to identify specific strains. Current examination (16 Nov 2017) of the GOLD database of microbial genome sequencing projects (Mukherjee et al., 2017) retrieved 83 entries when the “Isolation host name” field was queried with “Canis”, and 37 entries for “Felis”. Almost all entries were from diseased hosts, with over a quarter of the dog and just under half of the cat entries being of viral origin. The remaining entries appear to be dominated by sequencing projects for pathogenic agents. Thus, it is not known how much the dog and cat commensal gastrointestinal microbial genomes differ to those from the currently available references available, and available reference data are likely to skew functional interpretations toward disease state microbiomes. The predictive accuracy of dog and cat microbiota function, using 16S rRNA gene‐based community data in conjunction with reference genome information, will only improve as more microbial genomes from healthy dog and cat sources become available. However, there is yet to be a systemic effort to sequence the genomes of microbes found in the dog and cat GI tract, and such information will be vital to identify unique functions of microbial taxa that are specific to the dog and cat hosts, and for accurate functional interpretation of metagenomic datasets generated from these sources.
6.2. Microbiome function from shotgun metagenome sequencing
Shotgun metagenome sequence datasets for dog and cat GI microbiota are now becoming more prevalent, and studies that are currently available in the literature are listed in Table 3. To analyze the microbiome to a similar depth as for 16S rRNA studies, the volume of shotgun sequence data required is several orders of magnitude greater, thus such studies are considerably more expensive than community profiling alone. However, advances in sequencing technologies and the decreasing costs of high‐throughput sequencing will likely see an increase in the number of shotgun metagenome sequencing studies for dog and cat GI microbiomes being undertaken in future. Shotgun sequencing not only provides information about the potential functions of the microbiota, but allows simultaneous capture of all major microbial groups (bacteria, archaea, fungi, and viruses), in contrast to amplicon‐based methods that have employed separate amplifications for each group (Kittelmann et al., 2013). Moreover, compared to marker‐dependent amplicon‐based procedures, there are fewer practical steps for potential biases to be incorporated (e.g., via primer‐based amplification biases) which influence data interpretation. However, the more complex bioinformatic pipelines required to analyze metagenomic sequence data may arguably induce more biases in silico. The presence of host DNA sequences, which can comprise a significant proportion of datasets, should be screened and removed before microbiota analysis is performed (Schmieder & Edwards, 2011).
Table 3.
Healthy dog and cat fecal microbiota shotgun metagenome sequencing studies
| Study descriptiona | Subjects/age | Sequencing, analysis methodsb, and accession numbers | Reference |
|---|---|---|---|
| General microbiome characterization | |||
| Cats fed various commercial diets | Cat, 3–16 year, domestic long and shorthair (N = 5) | 454 FLX Titanium (0.15 M reads); Galaxy, MG‐RAST, WebCARMA. NCBI SRA029158.2 | (Tun et al., 2012) |
| Diet trial, treatments | |||
| Dry control (30% CP, 19% fat, 1.4% fiber) vs. beet pulp containing diet (28% CP, 21% fat, 4.5% fiber) | Dog, ~20 month, mongrel and hound crosses, female (N = 6) | 454 FLX Titanium (~0.5 M reads/sample); MG‐RAST. MG‐RAST 4444165 and 4444164; NCBI SRR054690 | (Swanson et al., 2011) |
| Kibble diet with cellulose, FOS, or pectin | Cat, ~20 months, male (N = 4) | 454 FLX Titanium (1.2–1.7 M reads/sample); MG‐RAST | (Barry et al., 2012) |
| Dry diets (HPLC and MPMC) | Kitten, sampled at 8, 12, and 16 weeks (N = 12) | Illumina, TruSeq DNAseq (~96 M reads/sample), MetaCV. ENA PRJEB4391 | (Deusch et al., 2014) |
| Dry diets (HPLC and MPMC) | Kitten, sampled at 18, 30, and 42 weeks (N = 12) | Illumina, Nextera TruSeq SBS (~55 M reads/sample), MetaCV. ENA PRJEB9357 | (Deusch et al., 2015) |
| Canned and kibbled diets | Kitten, 17 weeks (N = 20) | Illumina, TruSeq DNAseq (~4 M reads/sample), MG‐RAST. MG‐RAST 4629274.3–4629293.3 | (Young et al., 2016) |
CP, crude protein; FOS, fructooligosaccharide; HPLC, high protein–low carbohydrate; MPMC, medium protein–medium carbohydrate diet.
454 refers to 454 GS FLX Titanium sequencing, Illumina refers to Illumina HiSeq2000. NCBI, ENA, and MG‐RAST database accession numbers provided where available.
A major bottleneck of metagenome‐based studies is the considerable computational requirements to analyze the volumes of data generated, and the biological interpretation of such analyses. Advances in computing power (Nobile, Cazzaniga, Tangherloni, & Besozzi, 2017) and the development of more efficient analyses methods (Buchfink, Xie, & Huson, 2015) have facilitated shotgun metagenome sequence analyses. Shotgun sequence data may be analyzed unassembled or assembled, where assembled data generate longer contiguous stretches of sequence that can more accurately retrieve hits to against reference databases. Indeed, in deeply sequenced datasets, ‘binning’ together assembled sequences of similar composition and abundance has the potential to recover full or near‐full genomes of highly abundant organisms (Parks et al., 2017; Tyson et al., 2004). However, assembly is likely to be poor for the vast majority of rare organisms (Sogin et al., 2006), and there is the potential to misassemble reads from closely related organisms.
MG‐RAST (Meyer et al., 2008) has been the main metagenome sequence annotation service used by researchers in the field of canine and feline GI microbiology (Barry et al., 2012; Swanson et al., 2011; Tun et al., 2012; Young et al., 2016). Sequences are assessed for quality and annotated for gene function and taxonomy against a number or reference databases. Annotated data can be viewed through the MG‐RAST web application, but more often is downloaded and explored using custom‐based analyses tailored to the focus of the project (Young et al., 2016). The first metagenomic studies of the dog and cat GI microbiome used 454 pyrosequencing technology, and generated from 150 K to 1.6 M reads per dataset (Barry et al., 2012; Swanson et al., 2011; Tun et al., 2012).
6.2.1. Community composition biases revealed by metagenomic data
Shotgun sequence reads associated with rRNA gene sequences (or other marker genes of choice) can be identified from the dataset, and then used to generate a taxonomic profile of the microbial community using clustering techniques and comparison to referenced databases, similar to typical 16S rRNA gene analysis pipelines. From the taxonomic analysis of dog and cat GI shotgun metagenome data, it appears that the relative abundance of Proteobacteria is considerably greater than estimates from 16S rRNA gene amplicon‐based studies. For example, Proteobacteria represented 13%–15% of rRNA gene sequences via shotgun metagenome dataset analyses (Swanson et al., 2011), whereas only 5%–7% of sequences were classified as Proteobacteria in 16S rRNA gene V3 amplicon analysis of the same fecal samples (Middelbos et al., 2010). By comparison, Bacteroidetes/Chlorobi and Firmicutes each represented ~35% of taxa by metagenome analysis, but only comprised 27%–34% and 17%–24% of the amplicon‐based dataset, respectively. In contrast, the amplicon analysis appeared to overestimate Fusobacteria abundance (Swanson et al., 2011). In a separate study, via metagenome sequence analyses Proteobacteria comprised 8.7% of the community of kittens fed a canned diet (from mothers fed a kibbled diet) (Young et al., 2016), whereas 16S rRNA amplicon analyses of the samples placed the estimate for this group at ~4% (Bermingham, Kittelmann, et al., 2013). The Firmicutes also appear to be highly overrepresented by the 16S rRNA data compared to the metagenome sequence (Bermingham, Kittelmann, et al., 2013; Young et al., 2016). These discrepancies likely reflect biases resulting from the choice of amplification primers and conditions used to generate data (Soergel, Dey, Knight, & Brenner, 2012), as well as the different bioinformatics pipelines and reference databases used for each set of analyses. Thus, an additional level of caution is urged in the interpretation of data obtained via these various methods.
6.3. Proteobacteria contributions to microbioTA function
Metagenome studies have revealed primary functions that are abundant in the healthy dog and cat GI microbiomes, which include carbohydrate metabolism; protein metabolism; DNA metabolism, cofactors vitamins, prosthetic groups and pigments; amino acids and derivatives; cell wall and capsule; and virulence (Barry et al., 2012; Swanson et al., 2011; Tun et al., 2012; Young et al., 2016; Deusch et al., 2014, 2015).
The functional contributions that the Proteobacteria contribute to the microbiome cannot be readily deduced from the published data, as the reported functions for individual sequence reads are decoupled from taxonomic information (Barry et al., 2012; Deusch et al., 2014, 2015; Swanson et al., 2011; Tun et al., 2012; Young et al., 2016). We have therefore reanalyzed a representative metagenomic sequence dataset for each of the dog (Swanson et al., 2011), cat (Tun et al., 2012), and kitten (Young et al., 2016) fecal microbiota for which data were readily available (details of these datasets in Table 3) using MEGAN (Huson et al., 2016). MEGAN captures both functional and taxonomic information for each sequence read based on homology searches using DIAMOND (Buchfink et al., 2015) against the nr database. The resulting. daa files were meganized and visualized in MEGAN V6.10.2 Community Edition (Huson et al., 2016). This allowed direct relationships between the function and taxonomic origin to be examined, and thus, the functional roles of specific microbial taxa within complex communities, such as the Proteobacteria, to be explored. A comparison of the SEED subsystem profiles from each of representative dog, cat, and kitten pooled fecal microbiota, with those extracted from the reads that relate to Proteobacteria only, is presented in Figure 2.
Figure 2.
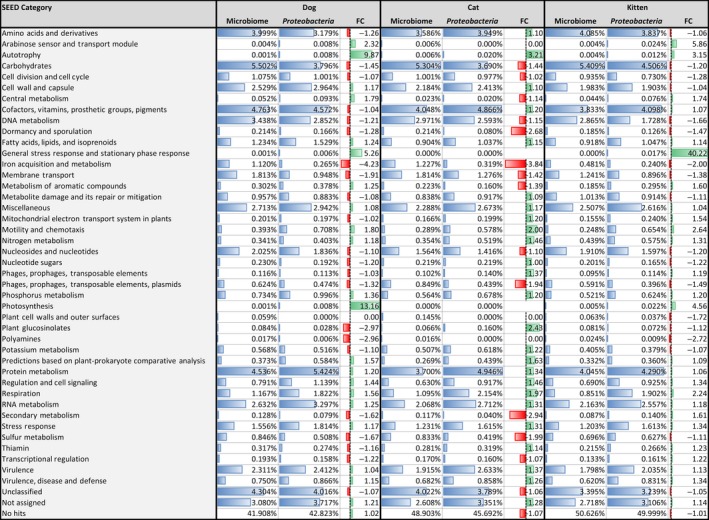
Functional profiles of representative healthy dog (Swanson et al., 2011), cat (Tun et al., 2012), and kitten (Young et al., 2016) fecal microbiota, compared to profiles from the subset of Proteobacteria‐associated reads within these datasets. Metagenomic shotgun sequence data were analyzed in MEGAN6 CE. The relative abundances of sequence hits for each SEED subsystem are shown as percentages with the length of the blue bar indicating their relative magnitude within the dataset, apart from the “No hits” category. Fold changes (FC) for Proteobacteria relative to microbiota abundances are shown, where fold increases are represented with green bars, and fold decreases (negative values) are represented by red bars.
At Level 1 of the SEED classification system, the most abundant Proteobacteria functions included “Protein Metabolism” (5.4%, 4.9%, and 4.3% of Proteobacteria reads in the dog, cat, and kitten datasets, respectively), “Cofactors, Vitamins, Prosthetic Groups, Pigments” (4.6%, 4.9%, and 4.1%), “Carbohydrates” (3.8%, 3.7%, and 4.5%), “RNA Metabolism” (3.3%, 2.7%, and 2.6%), and “Amino Acids and Derivatives” (3.2%, 3.9%, and 3.8%) (Figure 2). A considerable proportion of reads were to “Unclassified” sequences (4.0%, 3.8%, and 3.2%), “Not assigned” (3.7%, 3.4%, and 3.1%), or “No hits” (42.8%, 45.7%, and 50.0%) were observed (Figure 2), which reflect the extent to which gene and sequence identities are known for the pet gastrointestinal microbiota. The relative abundances of SEED subsystems for the whole community generally reflected those for the Proteobacteria. However, several classes appeared to have a considerably greater relative abundance in the Proteobacteria as compared to the whole microbiome: “Autotrophy” (3.2‐ to 9.8‐fold greater in Proteobacteria), which may reflect the metabolic versatility of the Proteobacteria as a whole and their ability to produce complex organic compounds from simpler substances; “General Stress Response and Stationary Phase Response” (ca. 40‐fold greater in the kitten, 5.2‐fold greater in the dog) and “Respiration” (1.6‐ to 2.2‐fold greater), which may reflect their respiratory abilities and ability to respond to stresses associated with aerobic respiration; and “Nitrogen Metabolism” (ca. 1.8‐ to 2.6‐fold greater) which is consistent with their contribution to protein metabolism (Figure 2). In contrast, the “Polyamines” (from undetected to ~3‐fold lower in Proteobacteria), “Iron Acquisition and Metabolism” (2‐ to 4.2‐fold lower), and “Carbohydrates” (1.2‐ to 1.5‐fold lower) are among SEED subsystems that appeared to be consistently underrepresented among the Proteobacteria in the datasets examined (Figure 2).
To better understand the specific functions that the Proteobacteria contributed to within their roles as protein degraders, sugar and oxygen utilizers within the gut, and their potential roles in pathogenicity, we examined the relative abundances of Level 2 SEED classification sequence hits for Proteobacteria, as compared to the whole microbiome. Due to the small number of datasets available, it was not possible to statistically determine the significance of apparent differences between datasets. As such we highlight subsystems that appeared to be consistently enriched for in both the dog and cat fecal datasets, or which had particularly large fold‐change differences.
6.3.1. Proteobacteria contributions to protein metabolism
Among the “Protein Metabolism” Level 2 subsystems (Table S1), the “Putative TldE‐TldD proteolytic complex” subsystem had a 13‐fold greater relative abundance in Proteobacteria in the dog, and was 29‐fold greater in the cat dataset than the general microbiome. While best characterized in the capacity for biosynthesis of microcin B17, a peptide antibiotic (Ghilarov et al., 2017), tldD and tldE genes are highly conserved and common in prokaryotic genomes (Allali, Afif, Couturier, & van Melderen, 2002). They have recently been shown to encode a heterodimeric metalloprotease with unusual regulation of substrate specificity by directing unfolded polypeptides through a narrow channel with cleavage in a processive manner (Ghilarov et al., 2017). Rather than contributing to a general role in protein degradation, tld genes may contribute to important functions in maintaining protein quality control through the activation and degradation of specific products across a range of bacteria (Ghilarov et al., 2017).
“Peptide methionine sulfoxide reductase” was fivefold more abundant in the dog dataset, but was not among Proteobacteria sequences in the cat dataset. Moreover, “Periplasmic disulphide interchange” displayed 2.5‐fold greater abundance in the dog, and 2.1‐fold greater relative abundance in the cat datasets. These subsystems are associated with the repair of oxidatively damaged of proteins, where sulfur‐containing amino acids such as methionine and cysteine are particularly susceptible to damage by reactive oxygen species, which are normal byproducts of aerobic respiration. Methionine sulfoxide reductase reverses oxidative damage to methionine (Weissbach et al., 2002). Cysteine‐mediated disulfide bridges help maintain the tertiary structure of secreted proteins, and are introduced into proteins within an oxidative environment such as the periplasm of Gram‐negative bacteria (Lasica & Jagusztyn‐Krynicka, 2007). Hence, these functions are consistent to the role of Proteobacteria as facultative anaerobes, and are likely to contribute to normal protein function during aerobic respiration where additional pressures imposed by the generation of reactive oxygen species are higher.
Within the “Amino Acids and Derivatives” a number of subsystems were considerably more abundant in the Proteobacteria than in the general microbiome for both the dog and cat microbiome datasets (Table S2), including, “Putrescine utilization pathways” (13.2‐fold more abundant in the dog and 28.9‐fold more in the cat) and “Ketoisovalerate oxidoreductase” (3.8‐fold dog and 9.6‐fold cat). The putrescine utilization pathway (Puu pathway) gene cluster may have evolved to allow utilization of polyamines, which exist at relatively high concentrations in the gut, where it is found in E. coli and closely related enterobacteria, but is uncommon in other bacterial groups (Nemoto et al., 2012). The ketoisovalerate oxidoreductase catalytic domain (IPR019752; (Finn et al., 2017)) is generally involved in carbohydrate fermentation processes, but this subsystem was also likely also classified within the “Amino Acids and Derivatives” as the family includes pyruvate flavodoxin oxidoreductase, which in cyanobacterium, is required for growth on molecular oxygen when iron is limited (Bauer, Scappino, & Haselkorn, 1993). In addition, tyrosine and phenylalanine are both essential amino acids in the dog and cat (Council, 2006). The Proteobacteria may contribute to the supply of tyrosine and phenylalanine as the relative abundance of the “Tyrosine and phenylalanine metabolism in plants” subsystem in Proteobacteria was 2.1‐fold greater than the microbiome in general in the dog, and 1.8‐fold greater in the cat dataset. The relevance of the dog and cat fecal microbiome functions to those in plants is presumed to be due to general sequence similarities rather than specific plant functions, however.
6.3.2. Proteobacteria contributions to carbohydrate metabolism
Within “Carbohydrate” (Table S3), there was little consistency among the top Level 2 classes that were overrepresented within the dog and cat microbiome. In common, the “Methylcitrate cycle” had Proteobacteria 12.5‐fold more highly represented than the general microbiota in the dog, and 22.4‐fold in the cat dataset. In E. coli, propionate, a major end‐product of gut fermentation, can be used as the sole source of carbon and energy using the methylcitrate cycle, to producing pyruvate, which can then be metabolized aerobically (Textor et al., 1997). Moreover, the dog dataset appeared to be highly enriched for the “CitAB” subsystem (13.1‐fold greater in Proteobacteria than the general microbiota) and a number of other Citrate Metabolism subsystems (Table S3). CitAB comprises a two‐component system involved in citrate fermentation (Scheu et al., 2012), although this subsystem was not detected in the cat dataset. Subsystems relating to the metabolism of a variety of carbohydrates and organic acids, such as d‐allose, d‐galactonate, “unknown carbohydrates”, and lactate utilization, were also more highly prevalent in the Proteobacteria in the dog microbiome dataset as compared to the cat. These differences may reflect differences in carbohydrate content of diets, where the dog study focused on beet pulp supplementation of a basal diet (Swanson et al., 2011), although the diets of the cats examined were not explicitly defined (Tun et al., 2012).
In the gut, mucins represent a major carbohydrate source for the microbiota (Pereira & Berry, 2017). Members such as E. coli colonize the mucus layer, and although they generally cannot degrade oligosaccharides or polysaccharides, they acquire mono‐ and disaccharides that result from extracellular hydrolysis of mucus and dietary polysaccharides by other microbial community members (Conway & Cohen, 2015). The ability of E. coli to colonize the mucus layer has been shown to depend on the ability to use a variety of mucin‐derived sugars, including gluconate, N‐acetylglucosamine, N‐acetylneuraminic acid, glucoronate, mannose, fucose, and ribose (Chang et al., 2004). However, the contributions of Proteobacteria relative to the general microbiota for subsystems related to the metabolism of these sugars did not show specific enrichment (Table S3). Thus, while mucins represent an important carbohydrate source in the gut, Proteobacteria do not appear to differ, more than the microbiota in general, in the level of their genetic potential to utilize these substrates.
6.3.3. Proteobacteria contributions to aerobic respiration
Among the “Respiration” Level 2 subsystems (Table S4) a notably high proportion of categories were overrepresented in Proteobacteria relative to the whole microbiome for both dog and cat, with a number of subsystems associated with cytochrome C oxidase being among the most highly differentially abundant (~13‐ and 28‐fold in dog and cat microbiota, respectively). Cytochrome C oxidase is the last enzyme in the respiratory electron transport chain, and is critical for maintaining the electrochemical potential gradient across the cell membrane to facilitate ATP synthesis, while converting molecular oxygen into water (Ludwig, 1987). A number of general respiration subsystems related to the human gut microbiome were also relatively more abundant in the Proteobacteria (3.5‐ and 5.1‐fold, in dogs and cat, respectively), consistent with the Proteobacteria filling a role in aerobic respiration within the gut, and contributing to the redox homeostasis of the gut required for normal function of the strictly anaerobic microbial community members.
Subsystems involving formate dehydrogenases and formate hydrogenases were also more prevalent among the Proteobacteria as compared to the microbiome in general. In E. coli, the extracellular accumulation of formate at low pH induces biosynthesis of the formate hydrogen lyase complex, which enables formate transport into the cell and oxidation to CO2 and H2 (McDowall et al., 2014; Sawers, 1994). This process may generally contribute to the utilization of formate resulting from anaerobic fermentation by other gut microbes.
6.3.4. Proteobacteria virulence
Within “Virulence” and “Virulence, Disease and Defence” (Table S5), the most prominent function that was enriched for in Proteobacteria was “Type 4 secretion and conjugative transfer” (7.5‐ and 18.4‐fold in the dog and cat, respectively), followed by “Mycobacterial MmpL2 membrane protein cluster” (4.1‐ and 5.8‐fold greater), “Mycobacterial MmpL6 membrane protein cluster” (1.7‐ and 2.9‐fold greater), and “Multidrug efflux pump in Campylobacter jejuni (CmeABC operon)” (2.4‐ and 1.4‐fold greater). Type 4 secretion systems are found in many bacterial species and represent significant functional diversity through conjugative transfer of genetic material, effector translocation, DNA exchange with the outside environment, biofilm formation, and lethal toxin delivery to bacterial neighbors (Grohmann, Christie, Waksman, & Backert, 2018). Short‐read polymorphisms associated with the CmeABC operon of C. jejuni have been associated with increased resistance to a number of quinolones and other antibiotics (Yang et al., 2017). Proteobacteria protein sequences with hits to Mycobacterial MmpL2 and MmpL6 membrane protein cluster SEED subsystems suggest that Proteobacteria are able to contribute related transporter functions to the microbiome. However, any potential role in virulence as for Mycobacteria (Domenech, Reed, & Barry, 2005), which are generally not detected in the healthy dog and cat, remains unclear. These proteins instead may simply be related to lipid transport in Proteobacteria taxa. Noteworthy, in the cat were the greater abundance of functions related to the resistance to heavy metals “Cadmium resistance” (9.6‐fold) “Copper homeostasis: copper tolerance” (2.6‐fold cat), although these were not as pronounced in the dog.
6.3.5. Highly correlated taxa and gene functions involve Proteobacteria
Correlation analyses between microbial taxa and gene abundances can be undertaken using annotated shotgun sequencing data to explore functions that are potentially uniquely contributed to by specific taxa. The kitten fecal metagenome study (Young et al., 2016) is comprised of 20 shotgun sequence datasets, which has allowed correlation analyses to be performed (Figure 3). Interestingly, the highest correlations observed (R ≥ 0.9) involved many members of the Proteobacteria including Escherichia, Salmonella, and Shigella, which correlated with a wide variety of genes predominantly involved in amino acid, sugar, and sulfate transport systems, secretion systems, and lipopolysaccharide biosynthesis (Figure 3). These genes are in keeping with known functions of these taxa, but whether or not they are from these taxa would require further validation. Given the tight network of genes with these closely related taxa, there is good likelihood that this was the case.
Figure 3.
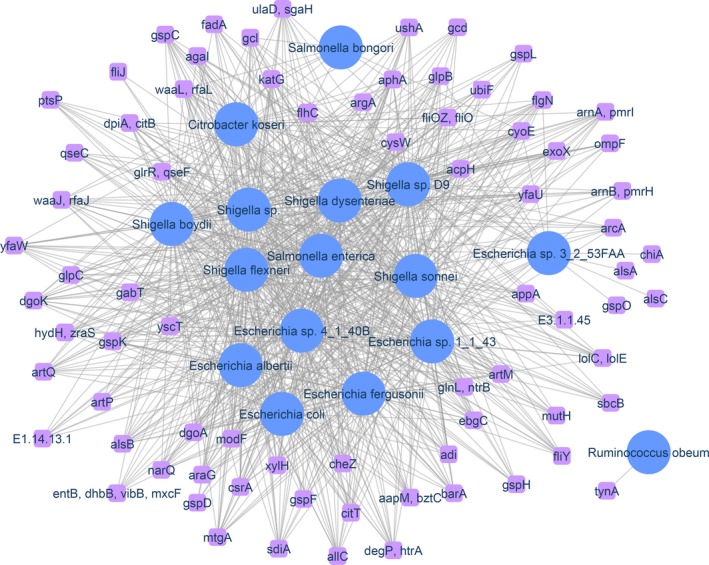
Network of the most highly correlated taxa (blue circles) and genes (purple squares) among the fecal microbiomes of 17‐week‐old kittens fed canned and kibbled diets. Kitten fecal microbiome sequence data were previously annotated in MG‐RAST (Meyer et al., 2008) for taxonomy and COG and KO functions as described (Young et al., 2016). Gene and taxon abundance network generated by sparse partial least squares regression using the spls function in the mixOmics package (Lê Cao & González, 2009) for R, using a correlation cut off of > |0.9|. The networks were viewed in Cytoscape 3.5.1 (Shannon et al., 2003)
Ruminococcus also featured in this analysis, but its abundance only highly correlated with one gene, tynA, which encodes a primary amine oxidase.
7. SUMMARY AND FUTURE DIRECTIONS
In the fecal microbiomes of healthy dogs and cats, Proteobacteria were detected in almost all individuals examined, where they were generally the third‐ to fifth‐most abundant bacterial phylum present. Proteobacteria abundances and diversity were variable, however, and 16S rRNA gene‐based surveys showed that the gene copy abundance ranged widely in dogs, from 0% to ~22%, while in cats, average abundances were generally <5%. Commonly observed genera include Escherichia, Shigella, Succinivibrio, Anaerobiospirillum, and Sutterella, although the relative abundances of each varied between individuals and studies. It is difficult to discern the key factors that influence Proteobacteria abundance and diversity, given the confounding factors between studies, however, that diet is a key driver of microbiota composition is clearly demonstrated within controlled studies. Metagenomic sequence data show that the Proteobacteria encode a variety of functions, which most prominently include protein and amino acid metabolism, carbohydrate metabolism, and cofactor/vitamin/prosthetic group/pigment metabolism. Compared to the fecal microbiota in general, the Proteobacteria appear to confer more abundantly to a number of functions that relate to their ability to grow aerobically such as respiration, and help maintain energy efficiency and integrity in the high redox potential environment generated while doing so, such as the utilization of propionate as a carbon source, and repair of protein from oxidative damage. These insights have been obtained from datasets based on relative gene abundances, and using reference genome and sequence data whose relevance to the dog and cat GI microbiota remains unclear. However, the functional contributions of Proteobacteria to microbiome function will become clearer with (1) greater efforts to cultivate and characterize diverse microbial representatives from dog and cat GI sources, (2) greater availability of reference genomes from dog‐ and cat‐derived strains, (3) greater depth of metagenomic sequencing, given the lower representation of Proteobacteria within the microbiome, and (4) metatranscriptome studies to identify metabolically active members of the microbiome and the pathways they utilize. Such information will allow us to further determine the roles and contributions of Proteobacteria to microbiome function in healthy dogs and cats, including their interactions with other members of the microbiota, as well as during dysbiosis and disease. As in other mammalian hosts, these observations point to the Proteobacteria occupying a unique ecological niche in the microbiome, where they broadly contribute to protein and carbohydrate metabolism, and also maintaining oxygen homeostasis in the gastrointestinal tract of the healthy dog and cat.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
The authors would like to acknowledge Christina Butowski for access to unpublished data on the cat fecal microbiome, and Jane Mullaney and two anonymous reviewers for critically reviewing this manuscript. This review was funded by the New Zealand Ministry of Business, Innovation and Employment (C10X1501) including financial contributions from Bombay Petfoods Ltd, K9 Natural Ltd, and ZiwiPeak Ltd. The funders did not contribute to the content of this review, or the interpretation of the information presented.
Moon C, Young W, Maclean P, Cookson A, Bermingham E. Metagenomic insights into the roles of Proteobacteria in the gastrointestinal microbiomes of healthy dogs and cats. MicrobiologyOpen. 2018;7:e677 10.1002/mbo3.677
REFERENCES
- AAFCO . (2016). Association of American Feed Control Officials 2016 Official Publication.
- Allali, N. , Afif, H. , Couturier, M. , & van Melderen, L. (2002). The highly conserved TldD and TldE proteins of Escherichia coli are involved in microcin B17 processing and in CcdA degradation. Journal of Bacteriology, 184, 3224–3231. 10.1128/JB.184.12.3224-3231.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlShawaqfeh, M. K. , Wajid, B. , Minamoto, Y. , Markel, M. , Lidbury, J. A. , Steiner, J. M. , … Suchodolski, J. S . 2017. A dysbiosis index to assess microbial changes in fecal samples of dogs with chronic inflammatory enteropathy. FEMS Microbiology Ecology, 93: fix136 10.1093/femsec/fix136. [DOI] [PubMed] [Google Scholar]
- Axelsson, E. , Ratnakumar, A. , Arendt, M. L. , Maqbool, K. , Webster, M. T. , Perloski, M. , … Lindblad‐Toh, K. (2013). The genomic signature of dog domestication reveals adaptation to a starch‐rich diet. Nature, 495, 364. [DOI] [PubMed] [Google Scholar]
- Baede, V. O. , Wagenaar, J. A. , Broens, E. M. , Duim, B. , Dohmen, W. , Nijsse, R. , … Hordijk, J. (2015). Longitudinal study of extended‐spectrum‐beta‐lactamase‐ and AmpC‐producing Enterobacteriaceae in household dogs. Antimicrobial Agents and Chemotherapy, 59, 3117–3124. 10.1128/AAC.04576-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balvočiūtė, M. , & Huson, D. H. (2017). SILVA, RDP, Greengenes, NCBI and OTT — how do these taxonomies compare? BMC Genomics, 18, 114 10.1186/s12864-017-3501-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry, K. A. , Middelbos, I. S. , Vester Boler, B. M. , Dowd, S. E. , Suchodolski, J. S. , Henrissat, B. , … Swanson, K. S. (2012). Effects of dietary fiber on the feline gastrointestinal metagenome. Journal of Proteome Research, 11, 5924–5933. 10.1021/pr3006809 [DOI] [PubMed] [Google Scholar]
- Bauer, C. C. , Scappino, L. , & Haselkorn, R. (1993). Growth of the cyanobacterium Anabaena on molecular nitrogen: NifJ is required when iron is limited. Proceedings of the National Academy of Sciences of the United States of America, 90, 8812–8816. 10.1073/pnas.90.19.8812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, E. T. , Suchodolski, J. S. , Isaiah, A. , Fleeman, L. M. , Cook, A. K. , Steiner, J. M. , & Mansfield, C. S. (2014). Faecal microbiota of cats with insulin‐treated diabetes mellitus. PLoS ONE, 9, e108729 10.1371/journal.pone.0108729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloshapka, A. N. , Dowd, S. E. , Suchodolski, J. S. , Steiner, J. M. , Duclos, L. , & Swanson, K. S. (2013). Fecal microbial communities of healthy adult dogs fed raw meat‐based diets with or without inulin or yeast cell wall extracts as assessed by 454 pyrosequencing. FEMS Microbiology Ecology, 84, 532–541. 10.1111/1574-6941.12081 [DOI] [PubMed] [Google Scholar]
- Bermingham, E. N. , Kittelmann, S. , Young, W. , Kerr, K. R. , Swanson, K. S. , Roy, N. C. , & Thomas, D. G. (2013). Post‐weaning diet affects faecal microbial composition but not selected adipose gene expression in the cat (Felis catus). PLoS ONE, 8, e80992 10.1371/journal.pone.0080992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham, E. N. , Maclean, P. , Thomas, D. G. , Cave, N. J. , & Young, W. (2017). Key bacterial families (Clostridiaceae, Erysipelotrichaceae and Bacteroidaceae) are related to the digestion of protein and energy in dogs. PeerJ, 5, e3019 10.7717/peerj.3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham, E. N. , Young, W. , Kittelmann, S. , Kerr, K. R. , Swanson, K. S. , Roy, N. C. , & Thomas, D. G. (2013). Dietary format alters fecal bacterial populations in the domestic cat (Felis catus). MicrobiologyOpen, 2, 173–181. 10.1002/mbo3.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boers, S. A. , Jansen, R. , & Hays, J. P. (2016). Suddenly everyone is a microbiota specialist. Clinical Microbiology and Infection, 22, 581–582. 10.1016/j.cmi.2016.05.002 [DOI] [PubMed] [Google Scholar]
- Bojanić, K. , Midwinter, A. C. , Marshall, J. C. , Rogers, L. E. , Biggs, P. J. , & Acke, E. (2017). Isolation of Campylobacter spp. from client‐owned dogs and cats, and retail raw meat pet food in the Manawatu, New Zealand. Zoonoses Public Health, 64, 438–449. 10.1111/zph.12323 [DOI] [PubMed] [Google Scholar]
- Bosch, G. , Hagen‐Plantinga, E. A. , & Hendriks, W. H. (2015). Dietary nutrient profiles of wild wolves: Insights for optimal dog nutrition? British Journal of Nutrition, 113, S40–S54. 10.1017/S0007114514002311 [DOI] [PubMed] [Google Scholar]
- Bradley, P. H. , & Pollard, K. S. (2017). Proteobacteria explain significant functional variability in the human gut microbiome. Microbiome, 5, 36 10.1186/s40168-017-0244-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bree, F. P. J. , Bokken, G. C. A. M. , Mineur, R. , Franssen, F. , Opsteegh, M. , van der Giessen, J. W. B. , … Overgaauw, P. A. M. (2018). Zoonotic bacteria and parasites found in raw meat‐based diets for cats and dogs. Veterinary Record, 182, 50 10.1136/vr.104535 [DOI] [PubMed] [Google Scholar]
- Bryant, M. P. , & Small, N. (1956). Characteristics of two new genera of anaerobic curved rods isolated from the rumen of cattle. Journal of Bacteriology, 72, 22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink, B. , Xie, C. , & Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nature Methods, 12, 59–60. 10.1038/nmeth.3176 [DOI] [PubMed] [Google Scholar]
- Chang, D. E. , Smalley, D. J. , Tucker, D. L. , Leatham, M. P. , Norris, W. E. , Stevenson, S. J. , … Conway, T. (2004). Carbon nutrition of Escherichia coli in the mouse intestine. Proceedings of the National Academy of Sciences of the United States of America, 101, 7427–7432. 10.1073/pnas.0307888101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, J. R. , Chai, B. , Farris, R. J. , Wang, Q. , Kulam‐Syed‐mohideen, A. S. , McGarrell, D. M. , … Tiedje, J. M. (2007). The ribosomal database project (RDP‐II): Introducing myRDP space and quality controlled public data. Nucleic Acids Research, 35, D169–D172. 10.1093/nar/gkl889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, T. , & Cohen, P. S. (2015). Commensal and pathogenic Escherichia coli metabolism in the gut. Microbiology Spectrum, 3, MBP‐0006‐2014. 10.1128/microbiolspec.MBP-0006-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Council, N. R. (2006). Proteins and amino acids. Nutrient requirements of dogs and cats. Washington DC: The National Academies Press. [Google Scholar]
- Davis, C. P. , Cleven, D. , Brown, J. , & Balish, E. (1976). Anaerobiospirillum, a new genus of spiral‐shaped bacteria. International Journal of Systematic Bacteriology, 26, 498–504. 10.1099/00207713-26-4-498 [DOI] [Google Scholar]
- Deng, P. , & Swanson, K. S. (2015). Gut microbiota of humans, dogs and cats: Current knowledge and future opportunities and challenges. British Journal of Nutrition, 113, S6–S17. 10.1017/S0007114514002943 [DOI] [PubMed] [Google Scholar]
- Desantis, T. Z. , Hugenholtz, P. , Larsen, N. , Rojas, M. , Brodie, E. L. , Keller, K. , … Andersen, G. L. (2006). Greengenes, a chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Applied and Environmental Microbiology, 72, 5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deusch, O. , O'Flynn, C. , Colyer, A. , Morris, P. , Allaway, D. , Jones, P. G. , & Swanson, K. S. (2014). Deep Illumina‐based shotgun sequencing reveals dietary effects on the structure and function of the fecal microbiome of growing kittens. PLoS ONE, 9, e101021 10.1371/journal.pone.0101021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deusch, O. , O'Flynn, C. , Colyer, A. , Swanson, K. S. , Allaway, D. , & Morris, P. (2015). A longitudinal study of the feline faecal microbiome identifies changes into early adulthood irrespective of sexual development. PLoS ONE, 10, e0144881 10.1371/journal.pone.0144881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech, P. , Reed, M. B. , & Barry, C. E. (2005). Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infection and Immunity, 73, 3492–3501. 10.1128/IAI.73.6.3492-3501.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, D. J. , Ernst, K. , Rogers, R. , Carmody, E. , & Aguero‐Rosenfeld, M. (2017a). The brief case: Anaerobiospirillum succiniciproducens bacteremia and pyomyositis. Journal of Clincal Microbiology, 55, 665–669. 10.1128/JCM.01351-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein, D. J. , Ernst, K. , Rogers, R. , Carmody, E. , & Aguero‐Rosenfeld, M. (2017b). Closing the brief case: Anaerobiospirillum succiniciproducens bacteremia and pyomyositis. Journal of Clinical Microbiology, 55, 986–987. 10.1128/JCM.01358-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, R. D. , Attwood, T. K. , Babbitt, P. C. , Bateman, A. , Bork, P. , Bridge, A. J. , … Mitchell, A. L. (2017). InterPro in 2017‐beyond protein family and domain annotations. Nucleic Acids Research, 45, D190–D199. 10.1093/nar/gkw1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster, G. M. , Hill, D. , Gregory, G. , Weishaar, K. M. , Lana, S. , Bauer, J. E. , & Ryan, E. P. (2012). Effects of cooked navy bean powder on apparent total tract nutrient digestibility and safety in healthy adult dogs. Journal of Animal Science, 90, 2631–2638. 10.2527/jas.2011-4324 [DOI] [PubMed] [Google Scholar]
- Freeman, L. M. , Chandler, M. L. , Hamper, B. A. , & Weeth, L. P. (2013). Current knowledge about the risks and benefits of raw meat–based diets for dogs and cats. Journal of the American Veterinary Medical Association, 243, 1549–1558. 10.2460/javma.243.11.1549 [DOI] [PubMed] [Google Scholar]
- Garcia‐Mazcorro, J. F. , Barcenas‐Walls, J. R. , Suchodolski, J. S. , & Steiner, J. M. (2017). Molecular assessment of the fecal microbiota in healthy cats and dogs before and during supplementation with fructo‐oligosaccharides (FOS) and inulin using high‐throughput 454‐pyrosequencing. PeerJ, 5, e3184 10.7717/peerj.3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Mazcorro, J. F. , Dowd, S. E. , Poulsen, J. , Steiner, J. M. , & Suchodolski, J. S. (2012). Abundance and short‐term temporal variability of fecal microbiota in healthy dogs. MicrobiologyOpen, 1, 340–347. 10.1002/mbo3.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Mazcorro, J. F. , Lanerie, D. J. , Dowd, S. E. , Paddock, C. G. , Grützner, N. , Steiner, J. M. , … Suchodolski, J. S. (2011). Effect of a multi‐species synbiotic formulation on fecal bacterial microbiota of healthy cats and dogs as evaluated by pyrosequencing. FEMS Microbiology Ecology, 78, 542–554. 10.1111/j.1574-6941.2011.01185.x [DOI] [PubMed] [Google Scholar]
- Garcia‐Mazcorro, J. F. , & Minamoto, Y. (2013). Gastrointestinal microorganisms in cats and dogs: A brief review. Archivos de Medicina Veterinaria, 45, 111–124. 10.4067/S0301-732X2013000200002 [DOI] [Google Scholar]
- Ghilarov, D. , Serebryakova, M. , Stevenson, C. E. M. , Hearnshaw, S. J. , Volkov, D. S. , Maxwell, A. , … Severinov, K. (2017). The origins of specificity in the microcin‐processing protease TldD/E. Structure, 25, 1549–1561. 10.1016/j.str.2017.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greetham, H. L. , Collins, M. D. , Gibson, G. R. , Giffard, C. , Falsen, E. , & Lawson, P. A. (2004). Sutterella stercoricanis sp. nov., isolated from canine faeces. International Journal of Systematic and Evolutionary Microbiology, 54, 1581–1584. 10.1099/ijs.0.63098-0 [DOI] [PubMed] [Google Scholar]
- Grohmann, E. , Christie, P. J. , Waksman, G. , & Backert, S. (2018). Type IV secretion in Gram‐negative and Gram‐positive bacteria. Molecular Microbiology, 107, 455–471. 10.1111/mmi.13896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groussin, M. , Mazel, F. , Sanders, J. G. , Smillie, C. S. , Lavergne, S. , Thuiller, W. , & Alm, E. J. (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nature Communications, 8, 14319 10.1038/ncomms14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guard, B. C. , Barr, J. W. , Reddivari, L. , Klemashevich, C. , Jayaraman, A. , Steiner, J. M. , … Suchodolski, J. S. (2015). Characterization of microbial dysbiosis and metabolomic changes in dogs with acute diarrhea. PLoS ONE, 10, e0127259 10.1371/journal.pone.0127259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haesebrouck, F. , Pasmans, F. , Flahou, B. , Chiers, K. , Baele, M. , Meyns, T. , … Ducatelle, R. (2009). Gastric Helicobacters in domestic animals and nonhuman primates and their significance for human health. Clinical Microbiology Reviews, 22, 202–223. 10.1128/CMR.00041-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand, D. , Wallis, C. , Colyer, A. , & Penn, C. W. (2013). Pyrosequencing the canine faecal microbiota: Breadth and depth of biodiversity. PLoS ONE, 8, e53115 10.1371/journal.pone.0053115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handl, S. , Dowd, S. E. , Garcia‐Mazcorro, J. F. , Steiner, J. M. , & Suchodolski, J. S. (2011). Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiology Ecology, 76, 301–310. 10.1111/j.1574-6941.2011.01058.x [DOI] [PubMed] [Google Scholar]
- Handl, S. , German, A. J. , Holden, S. L. , Dowd, S. E. , Steiner, J. M. , & Heilmann, R. M. (2012). Faecal microbiota in lean and obese dogs. FEMS Microbiology Ecology, 84, 332–343. 10.1111/1574-6941.12067 [DOI] [PubMed] [Google Scholar]
- Herstad, K. M. V. , Gajardo, K. , Bakke, A. M. , Moe, L. , Ludvigsen, J. , Rudi, K. , … Skancke, E. (2017). A diet change from dry food to beef induces reversible changes on the faecal microbiota in healthy, adult client‐owned dogs. BMC Veterinary Research, 13, 147 10.1186/s12917-017-1073-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honneffer, J. B. , Steiner, J. M. , Lidbury, J. A. , & Suchodolski, J. S. (2017). Variation of the microbiota and metabolome along the canine gastrointestinal tract. Metabolomics, 13, 26 10.1007/s11306-017-1165-3 [DOI] [Google Scholar]
- Hooda, S. , Vester Boler, B. M. , Kerr, K. R. , Dowd, S. E. , & Swanson, K. S. (2013). The gut microbiome of kittens is affected by dietary protein: carbohydrate ratio and associated with blood metabolite and hormone concentrations. British Journal of Nutrition, 109, 1637–1646. 10.1017/S0007114512003479 [DOI] [PubMed] [Google Scholar]
- Huson, D. H. , Beier, S. , Flade, I. , Gorska, A. , El‐Hadidi, M. , Mitra, S. , … Tappu, R. (2016). MEGAN Community Edition ‐ interactive exploration and analysis of large‐scale microbiome sequencing data. PLoS Computational Biology, 12, e1004957 10.1371/journal.pcbi.1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaiah, A. , Parambeth, J. C. , Steiner, J. M. , Lidbury, J. A. , & Suchodolski, J. S. (2017). The fecal microbiome of dogs with exocrine pancreatic insufficiency. Anaerobe, 45, 50–58. 10.1016/j.anaerobe.2017.02.010 [DOI] [PubMed] [Google Scholar]
- Johnston, K. L. , Swift, N. C. , Forster‐Van Hijfte, M. , Rutgers, H. C. , Lamport, A. , Ballàvre, O. , & Batt, R. M. (2001). Comparison of the bacterial flora of the duodenum in healthy cats and cats with signs of gastrointestinal tract disease. Journal of the American Veterinary Medical Association, 218, 48–51. 10.2460/javma.2001.218.48 [DOI] [PubMed] [Google Scholar]
- Kelly, D. , Campbell, J. I. , King, T. P. , Grant, G. , Jansson, E. A. , Coutts, A. G. , … Conway, S. (2004). Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear‐cytoplasmic shuttling of PPAR‐γ and RelA. Nature Immunology, 5, 104–112. 10.1038/ni1018 [DOI] [PubMed] [Google Scholar]
- Kerr, K. R. , Forster, G. , Dowd, S. E. , Ryan, E. P. , & Swanson, K. S. (2013). Effects of dietary cooked navy bean on the fecal microbiome of healthy companion dogs. PLoS ONE, 8, e74998 10.1371/journal.pone.0074998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kil, D. Y. , & Swanson, K. S. (2011). Companion animals symposium: Role of microbes in canine and feline health. Journal of Animal Science, 89, 1498–1505. 10.2527/jas.2010-3498 [DOI] [PubMed] [Google Scholar]
- Kim, J. , An, J.‐U. , Kim, W. , Lee, S. , & Cho, S. (2017). Differences in the gut microbiota of dogs (Canis lupus familiaris) fed a natural diet or a commercial feed revealed by the Illumina MiSeq platform. Gut Pathogens, 9, 68 10.1186/s13099-017-0218-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittelmann, S. , Seedorf, H. , Walters, W. A. , Clemente, J. C. , Knight, R. , Gordon, J. I. , & Janssen, P. H. (2013). Simultaneous amplicon sequencing to explore co‐occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE, 8, e47879 10.1371/journal.pone.0047879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagkouvardos, I. , Overmann, J. , & Clavel, T. (2017). Cultured microbes represent a substantial fraction of the human and mouse gut microbiota. Gut Microbes, 8, 493–503. 10.1080/19490976.2017.1320468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land, M. , Hauser, L. , Jun, S.‐R. , Nookaew, I. , Leuze, M. R. , Ahn, T.‐H. , … Ussery, D. W. (2015). Insights from 20 years of bacterial genome sequencing. Functional & Integrative Genomics, 15, 141–161. 10.1007/s10142-015-0433-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille, M. G. I. , Zaneveld, J. , Caporaso, J. G. , Mcdonald, D. , Knights, D. , Reyes, J. A. , … Huttenhower, C. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31, 814–821. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasica, A. M. , & Jagusztyn‐Krynicka, E. K. (2007). The role of Dsb proteins of Gram‐negative bacteria in the process of pathogenesis. FEMS Microbiology Reviews, 31, 626–636. 10.1111/j.1574-6976.2007.00081.x [DOI] [PubMed] [Google Scholar]
- Lê Cao, K. A. , & González, I. S. D. (2009). Integromics: An R package to unravel relationships between two omics data sets. Bioinformatics, 25, 2855–2856. 10.1093/bioinformatics/btp515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc, J. G. , Milani, C. , de Giori, G. S. , Sesma, F. , van Sinderen, D. , & Ventura, M. (2013). Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Current Opinion in Biotechnology, 24, 160–168. 10.1016/j.copbio.2012.08.005 [DOI] [PubMed] [Google Scholar]
- Ley, R. E. , Hamady, M. , Lozupone, C. , Turnbaugh, P. , Ramey, R. R. , Bircher, J. S. , … Gordon, J. I. (2008). Evolution of mammals and their gut microbes. Science, 320, 1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , Lauber, C. L. , Czarnecki‐Maulden, G. , Pan, Y. , & Hannah, S. S . (2017). Effects of the dietary protein and carbohydrate ratio on gut microbiomes in dogs of different body conditions. mBio, 8, e01703–e01716. 10.1128/mBio.01703-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis, P. , & Flint, H. J. (2009). Diversity, metabolism and microbial ecology of butyrate‐producing bacteria from the human large intestine. FEMS Microbiology Letters, 294, 1–8. 10.1111/j.1574-6968.2009.01514.x [DOI] [PubMed] [Google Scholar]
- Ludwig, B. (1987). Cytochrome c oxidase in prokaryotes. FEMS Microbiology Reviews, 3, 41–56. 10.1111/j.1574-6968.1987.tb02451.x [DOI] [Google Scholar]
- Malnick, H. (1997). Anaerobiospirillum thomasii sp. nov., an anaerobic spiral bacterium isolated from the feces of cats and dogs and from diarrheal feces of humans, and emendation of the genus Anaerobiospirillum . International Journal of Systematic Bacteriology, 47, 381–384. 10.1099/00207713-47-2-381 [DOI] [PubMed] [Google Scholar]
- McDowall, J. S. , Murphy, B. J. , Haumann, M. , Palmer, T. , Armstrong, F. A. , & Sargent, F. (2014). Bacterial formate hydrogenlyase complex. Proceedings of the National Academy of Sciences of the United States of America, 111, E3948–E3956. 10.1073/pnas.1407927111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, F. , Paarmann, D. , D'Souza, M. , Olson, R. , Glass, E. M. , Kubal, M. , … Edwards, R. A. (2008). The metagenomics RAST server ‐ a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics, 9, 386 10.1186/1471-2105-9-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelbos, I. S. , Vester Boler, B. M. , Qu, A. , White, B. A. , Swanson, K. S. , & Fahey, G. C.,JR. (2010). Phylogenetic characterization of fecal microbial communities of dogs fed diets with or without supplemental dietary fiber using 454 pyrosequencing. PLoS ONE, 5, e9768 10.1371/journal.pone.0009768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamoto, Y. , Otoni, C. C. , Steelman, S. M. , Büyükleblebici, O. , Steiner, J. M. , Jergens, A. E. , & Suchodolski, J. S. (2015). Alteration of the fecal microbiota and serum metabolite profiles in dogs with idiopathic inflammatory bowel disease. Gut Microbes, 6, 33–47. 10.1080/19490976.2014.997612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morato, E. P. , Leomil, L. , Beutin, L. , Krause, G. , Moura, R. A. , & Pestana de Castro, A. F. (2009). Domestic cats constitute a natural reservoir of human enteropathogenic Escherichia coli types. Zoonoses Public Health, 56, 229–237. 10.1111/j.1863-2378.2008.01190.x [DOI] [PubMed] [Google Scholar]
- Mukherjee, S. , Stamatis, D. , Bertsch, J. , Ovchinnikova, G. , Verezemska, O. , Isbandi, M. , … Reddy, T. B. K. (2017). Genomes OnLine Database (GOLD) v. 6: Data updates and feature enhancements. Nucleic Acids Research, 45, D446–D456. 10.1093/nar/gkw992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto, N. , Kurihara, S. , Kitahara, Y. , Asada, K. , Kato, K. , & Suzuki, H. (2012). Mechanism for regulation of the putrescine utilization pathway by the transcription factor PuuR in Escherichia coli K‐12. Journal of Bacteriology, 194, 3437–3447. 10.1128/JB.00097-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, K. M. , Ferreyra, J. A. , Higginbottom, S. K. , Lynch, J. B. , Kashyap, P. C. , Gopinath, S. , … Sonnenburg, J. L. (2013). Microbiota‐liberated host sugars facilitate post‐antibiotic expansion of enteric pathogens. Nature, 502, 96–99. 10.1038/nature12503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile, M. S. , Cazzaniga, P. , Tangherloni, A. , & Besozzi, D. (2017). Graphics processing units in bioinformatics, computational biology and systems biology. Briefings in Bioinformatics, 18, 870–885. 10.1093/bib/bbw058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasevich, M. R. , Kerr, K. R. , Dilger, R. N. , Fahey, G. C. , Guérin‐Deremaux, L. , Lynch, G. L. , … Swanson, K. S. (2015). Modulation of the faecal microbiome of healthy adult dogs by inclusion of potato fibre in the diet. British Journal of Nutrition, 113, 125–133. 10.1017/S0007114514003274 [DOI] [PubMed] [Google Scholar]
- Park, H. J. , Lee, S. E. , Kim, H. B. , Isaacson, R. E. , Seo, K. W. , & Song, K. H. (2015). Association of obesity with serum leptin, adiponectin, and serotonin and gut microflora in beagle dogs. Journal of Veterinary Internal Medicine, 29, 43–50. 10.1111/jvim.12455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D. H. , Rinke, C. , Chuvochina, M. , Chaumeil, P.‐A. , Woodcroft, B. J. , Evans, P. N. , … Tyson, G. W. (2017). Recovery of nearly 8,000 metagenome‐assembled genomes substantially expands the tree of life. Nature Microbiology, 2, 1533–1542. 10.1038/s41564-017-0012-7 [DOI] [PubMed] [Google Scholar]
- Pereira, F. C. , & Berry, D. (2017). Microbial nutrient niches in the gut. Environmental Microbiology, 19, 1366–1378. 10.1111/1462-2920.13659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. J. , Peng, L. , Barry, N. A. , Cline, G. W. , Zhang, D. , Cardone, R. L. , … Shulman, G. I. (2016). Acetate mediates a microbiome‐brain‐β cell axis promoting metabolic syndrome. Nature, 534, 213–217. 10.1038/nature18309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse, E. , Quast, C. , Knittel, K. , Fuchs, B. M. , Ludwig, W. , Peplies, J. , & Glöckner, F. O. (2007). SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research, 35, 7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimschuessel, R. , Grabenstein, M. , Guag, J. , Nemser, S. M. , Song, K. , Qiu, J. , … Okwumabua, O. (2017). Multilaboratory survey to evaluate Salmonella prevalence in diarrheic and nondiarrheic dogs and cats in the United States between 2012 and 2014. Journal of Clinical Microbiology, 55, 1350–1368. 10.1128/JCM.02137-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, L. E. , Steiner, J. M. , & Suchodolski, J. S. (2008). Assessment of microbial diversity along the feline intestinal tract using 16S rRNA gene analysis. FEMS Microbiology Ecology, 66, 590–598. 10.1111/j.1574-6941.2008.00609.x [DOI] [PubMed] [Google Scholar]
- Rubin, J. E. , & Pitout, J. D. (2014). Extended‐spectrum beta‐lactamase, carbapenemase and AmpC producing Enterobacteriaceae in companion animals. Veterinary Microbiology, 170, 10–18. 10.1016/j.vetmic.2014.01.017 [DOI] [PubMed] [Google Scholar]
- Sandri, M. , Dal Monego, S. , Conte, G. , Sgorlon, S. , & Stefanon, B. (2017). Raw meat based diet influences faecal microbiome and end products of fermentation in healthy dogs. BMC Veterinary Research, 13, 65 10.1186/s12917-017-0981-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawers, G. (1994). The hydrogenases and formate dehydrogenases of Escherichia coli . Antonie van Leeuwenhoek, 66, 57–88. 10.1007/BF00871633 [DOI] [PubMed] [Google Scholar]
- Scheu, P. D. , Witan, J. , Rauschmeier, M. , Graf, S. , Liao, Y. F. , Ebert‐Jung, A. , … Unden, G. (2012). CitA/CitB two‐component system regulating citrate fermentation in Escherichia coli and its relation to the DcuS/DcuR system in vivo . Journal of Bacteriology, 194, 636–645. 10.1128/JB.06345-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger, D. P. , & Joffe, D. J. (2011). Raw food diets in companion animals: A critical review. The Canadian Veterinary Journal, 52, 50–54. [PMC free article] [PubMed] [Google Scholar]
- Schmieder, R. , & Edwards, R. (2011). Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE, 6, e17288 10.1371/journal.pone.0017288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri, R. , Leahy, S. C. , Attwood, G. T. , Teh, K. H. , Lambie, S. C. , Cookson, A. L. , … Kelly, W. J. (2018). Cultivation and sequencing of rumen microbiome members from the Hungate 1000 Collection. Nature Biotechnology, 36, 359–367. 10.1038/nbt.4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , … Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13, 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, N.‐R. , Whon, T. W. , & Bae, J.‐W. (2015). Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends in Biotechnology, 33, 496–503. 10.1016/j.tibtech.2015.06.011 [DOI] [PubMed] [Google Scholar]
- Sidebotham, R. L. , Worku, M. L. , Karim, Q. N. , Dhir, N. K. , & Baron, J. H. (2003). How Helicobacter pylori urease may affect external pH and influence growth and motility in the mucus environment: Evidence from in‐vitro studies. European Journal of Gastroenterology & Hepatology, 15, 395–401. 10.1097/00042737-200304000-00010 [DOI] [PubMed] [Google Scholar]
- Soergel, D. A. W. , Dey, N. , Knight, R. , & Brenner, S. E. (2012). Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. The ISME Journal, 6, 1440 10.1038/ismej.2011.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogin, M. L. , Morrison, H. G. , Huber, J. A. , Mark Welch, D. , Huse, S. M. , Neal, P. R. , … Herndl, G. J. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences of the United States of America, 103, 12115–12120. 10.1073/pnas.0605127103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackebrandt, E. , & Hespell, R. B . (2006). The family Succinivibrionaceae In: Dworkin M., Falkow S., Rosenberg E., Schleifer K.‐H. & Stackebrandt E. (Eds.,) The Prokaryotes: Volume 3: Archaea. Bacteria: Firmicutes, Actinomycetes. New York, NY: Springer New York. [Google Scholar]
- Stackebrandt, E. , Murray, R. G. E. , & Trüper, H. G. (1988). Proteobacteria classis nov., a name for the phylogenetic taxon that includes the “Purple bacteria and their relatives”. International Journal of Systematic Bacteriology, 38, 321–325. 10.1099/00207713-38-3-321 [DOI] [Google Scholar]
- Suchodolski, J. S. , Camacho, J. , & Steiner, J. M. (2008). Analysis of bacterial diversity in the canine duodenum, jejunum, ileum, and colon by comparative 16S rRNA gene analysis. FEMS Microbiology Ecology, 66, 567–578. 10.1111/j.1574-6941.2008.00521.x [DOI] [PubMed] [Google Scholar]
- Suchodolski, J. S. , Dowd, S. E. , Wilke, V. , Steiner, J. M. , & Jergens, A. E. (2012). 16S rRNA gene pyrosequencing reveals bacterial dysbiosis in the duodenum of dogs with idiopathic inflammatory bowel disease. PLoS ONE, 7, e39333 10.1371/journal.pone.0039333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchodolski, J. S. , Foster, M. L. , Sohail, M. U. , Leutenegger, C. , Queen, E. V. , Steiner, J. M. , & Marks, S. L. (2015). The fecal microbiome in cats with diarrhea. PLoS ONE, 10, e0127378 10.1371/journal.pone.0127378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchodolski, J. S. , Markel, M. E. , Garcia‐Mazcorro, J. F. , Unterer, S. , Heilmann, R. M. , Dowd, S. E. , … Toresson, L. (2012). The fecal microbiome in dogs with acute diarrhea and idiopathic inflammatory bowel disease. PLoS ONE, 7, e51907 10.1371/journal.pone.0051907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson, K. S. , Dowd, S. E. , Suchodolski, J. S. , Middelbos, I. S. , Vester, B. M. , Barry, K. A. , … Fahey, G. C. (2011). Phylogenetic and gene‐centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. The ISME Journal, 5, 639–649. 10.1038/ismej.2010.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Textor, S. , Wendisch, V. F. , de Graaf, A. A. , Muller, U. , Linder, M. I. , Linder, D. , & Buckel, W. (1997). Propionate oxidation in Escherichia coli: Evidence for operation of a methylcitrate cycle in bacteria. Archives of Microbiology, 168, 428–436. 10.1007/s002030050518 [DOI] [PubMed] [Google Scholar]
- Tun, H. M. , Brar, M. S. , Khin, N. , Jun, L. , Hui, R. K.‐H. , Dowd, S. E. , & Leung, F. C.‐C. (2012). Gene‐centric metagenomics analysis of feline intestinal microbiome using 454 junior pyrosequencing. Journal of Microbiological Methods, 88, 369–376. 10.1016/j.mimet.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh, P. J. , Ley, R. E. , Hamady, M. , Fraser‐Liggett, C. M. , Knight, R. , & Gordon, J. I. (2007). The human microbiome project. Nature, 449, 804–810. 10.1038/nature06244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson, G. W. , Chapman, J. , Hugenholtz, P. , Allen, E. E. , Ram, R. J. , Richardson, P. M. , … Banfield, J. F. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature, 428, 37–43. 10.1038/nature02340 [DOI] [PubMed] [Google Scholar]
- Vázquez‐Baeza, Y. , Hyde, E. R. , Suchodolski, J. S. , & Knight, R. (2016). Dog and human inflammatory bowel disease rely on overlapping yet distinct dysbiosis networks. Nature Microbiology, 1, 16177 10.1038/nmicrobiol.2016.177 [DOI] [PubMed] [Google Scholar]
- Weissbach, H. , Etienne, F. , Hoshi, T. , Heinemann, S. H. , Lowther, W. T. , Matthews, B. , … Brot, N. (2002). Peptide methionine sulfoxide reductase: Structure, mechanism of action, and biological function. Archives of Biochemistry and Biophysics, 397, 172–178. 10.1006/abbi.2001.2664 [DOI] [PubMed] [Google Scholar]
- Yang, W. , Zhang, M. , Zhou, J. , Pang, L. , Wang, G. , & Hou, F. (2017). The molecular mechanisms of ciprofloxacin resistance in clinical Campylobacter jejuni and their genotyping characteristics in Beijing, China. Foodborne Pathogens and Disease, 14, 386–392. 10.1089/fpd.2016.2223 [DOI] [PubMed] [Google Scholar]
- Young, W. , Moon, C. D. , Thomas, D. G. , Cave, N. J. , & Bermingham, E. N. (2016). Pre‐ and post‐weaning diet alters the faecal metagenome in the cat with differences vitamin and carbohydrate metabolism gene abundances. Scientific Reports, 6, 34668 10.1038/srep34668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, S. , Cohen, D. B. , Ravel, J. , Abdo, Z. , & Forney, L. J. (2012). Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS ONE, 7, e33865 10.1371/journal.pone.0033865 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials