

In the terminal steps of methane formation in freshwater lake sediments, fermenting bacteria cooperate syntrophically with methanogens and homoacetogens at minimum energy increments via interspecies electron transfer. The energy yields of the partner organisms in these cooperations have so far been calculated based mainly on in situ hydrogen partial pressures. In the present study, we also analyzed pools of formate as an alternative electron carrier in sediment cores of an oligotrophic lake. The formate and hydrogen pools appeared to be energetically nearly equivalent and are likely to be used simultaneously for interspecies electron transfer. Calculations of reaction energies of the partners involved suggest that propionate degradation may also proceed through the Smithella pathway, which converts propionate via butyrate and acetate to three acetate residues, thus circumventing one energetically difficult fatty acid oxidation step.
KEYWORDS: secondary fermentations, energetics, fatty acids, methanogenesis, pool sizes, syntrophy
ABSTRACT
The energetic situation of terminal fermentations in methanogenesis was analyzed by pool size determinations in sediment cores taken in the oligotrophic Lake Constance, Germany. Distribution profiles of fermentation intermediates and products were measured at three different water depths (2, 10, and 80 m). Methane concentrations were constant below 10 cm of sediment depth. Within the methanogenic zone, concentrations of formate, acetate, propionate, and butyrate varied between 1 and 40 μM, and hydrogen was between 0.5 and 5 Pa. From the distribution profiles of the fermentation intermediates, Gibbs free energy changes for their interconversion were calculated. Pool sizes of formate and hydrogen were energetically nearly equivalent, with −5 ± 5 kJ per mol difference of free energy change (ΔG) for a hypothetical conversion of formate to hydrogen plus CO2. The ΔG values for conversion of fatty acids to methanogenic substrates and their further conversion to methane and CO2 were calculated with hydrogen and with formate as intermediates. Syntrophic propionate oxidation reached energetic equilibrium with formate as the sole electron carrier but was sufficiently exergonic if at least some of the electrons were transferred via hydrogen. The energetic consequences of formate versus hydrogen transfer in secondary and methanogenic fermentations indicate that both carrier systems are probably used simultaneously to optimize the energy yields for the partners involved.
IMPORTANCE In the terminal steps of methane formation in freshwater lake sediments, fermenting bacteria cooperate syntrophically with methanogens and homoacetogens at minimum energy increments via interspecies electron transfer. The energy yields of the partner organisms in these cooperations have so far been calculated based mainly on in situ hydrogen partial pressures. In the present study, we also analyzed pools of formate as an alternative electron carrier in sediment cores of an oligotrophic lake. The formate and hydrogen pools appeared to be energetically nearly equivalent and are likely to be used simultaneously for interspecies electron transfer. Calculations of reaction energies of the partners involved suggest that propionate degradation may also proceed through the Smithella pathway, which converts propionate via butyrate and acetate to three acetate residues, thus circumventing one energetically difficult fatty acid oxidation step.
INTRODUCTION
Methanogenesis is the terminal process of anaerobic digestion of organic matter in deeper layers of freshwater sediments that contributes to the global atmospheric methane budget to a minor but significant extent (6 to 16%) (1). Fermentation of biomass to methane and CO2 proceeds through a network of primary fermentations, secondary fermentations, homoacetogenesis, and methanogenesis (2–5), which all are carried out by different microbes that have to share the small amount of energy available in the overall process. In particular, the last players in this concerted action, i.e., secondary (“syntrophic”) fermenters, homoacetogens, and methanogens, have to run their energy metabolism at energy increments that barely provide sufficient energy for ATP synthesis. The minimum amount of metabolic energy that is needed for synthesis of a fraction of an ATP unit has been defined to be equivalent to the energy needed to transport a proton or a sodium ion across the charged cytoplasmic membrane, i.e., equivalent to −20 kJ per mol reaction (4, 6). Considering that starving cells may have a lower energy charge and that higher proton-to-ATP stoichiometries are possible due to unusual ATPase architectures, this calculated minimum value was later decreased to about −10 to −12 kJ per mol reaction (7–9).
In secondary fermentations, fatty acids and alcohols that are released as products of primary fermentations are converted to compounds that can be used by methanogens, i.e., C1 compounds, hydrogen, and acetate. The electrons released in these fermentations are transferred to methanogenic partners either as hydrogen gas or as formate at low concentrations to render these fermentations energetically feasible (2, 4, 10–13). For many years, hydrogen was considered the more important electron carrier, mostly because hydrogen partial pressures could easily be measured in the headspace of culture bottles or digesters. Nonetheless, formate was already discussed as an alternative electron shuttle with the descriptions of the very first defined syntrophic cocultures (4, 10, 12–14). Later work has shown that several secondary (“syntrophically”) fermenting bacteria may release formate rather than hydrogen as an electron sink (15–22), and most hydrogen-oxidizing methanogenic partners can use both hydrogen and formate as an electron source. Homoacetogenic bacteria can also use these electron carriers and, with this, interfere with the interspecies electron flow from secondary fermenters to their methanogenic partners (23). Sulfate reducers compete for available hydrogen or formate as well, but only if sufficient sulfate is available (24, 25). Beyond experiments with isolated cultures, several studies have tried to assess the energetic situation of secondary fermentations and methane formation in the past by measuring pool sizes of fermentation intermediates, including hydrogen partial pressures in situ, both in natural sediments and in technical settings, such as sewage sludge digesters or biogas reactors (26–32).
Only recently have techniques been developed that allow measurement of formate in the micromolar range in environmental samples, even in the presence of large amounts of complex organic matter such as, e.g., humic compounds (26, 27). In the present study, the importance of formate as an alternative electron shuttle in methanogenic biomass degradation was investigated in sediments of the oligotrophic Lake Constance in southern Germany, focusing on the energetic limitations of the transformation processes involved.
RESULTS
Stratification of sediment cores.
Profiles of physicochemical data and pool sizes of microbial metabolites obtained in the present study are shown in Fig. 1. The porosity in the core taken at 2 m water depth was rather homogeneous, between 60 and 80% (Fig. 1a). In the core from 10 m water depth, the porosity decreased from 60 to 40% and reached a steady value (40%) at 22.5 cm sediment depth. In the sediment core taken at 80 m depth, the porosity decreased between 10 and 20 cm sediment depth, from around 70% to 55%. The pH value (Fig. 1b) was rather constant (about 7.6) in all three sediment profiles and was in general slightly more alkaline than the sediment surface. The sediment core taken at 10 m water depth was in total slightly more acidic (pH 7.0).
FIG 1.

Physicochemical properties and pool sizes of fermentation intermediates in sediment cores taken in Lake Constance at three different water depths (2 m, 10 m, and 80 m).
Oxygen was not measured in the present study. It penetrates into Lake Constance sediments down to about 4 to 5 mm depth (33). Sulfate declined from a maximum of 270 μM in the water column to values of 20 to 40 μM at 10 to 15 cm sediment depth in the sediment cores taken at 2 m and 80 m depth, and to 50 to 60 μM at 20 cm depth in the core taken at 10 m depth (Fig. 1c).
Methane showed high concentrations up to saturation below 10 cm sediment depth in the cores taken at 2 m and 10 m depth (1.8 to 4.3 kPa; Fig. 1d). In the profundal core taken at 80 m, the methane concentration was in general lower and decreased steadily from 30 cm depth upwards, indicating that methane was mainly formed further down in the sediment. In all three cores, the methane concentrations decreased distinctly within the uppermost 5 to 8 cm depth toward the sediment surface. The majority of the methane produced in the sediment is reoxidized within the top 1 to 2 cm depth, either by aerobic or by nitrite-dependent oxidation (33–35).
Pool sizes of reaction intermediates in sediment cores.
The concentrations of dissolved compounds and gases were analyzed in all layers of the studied sediments in triplicate. The data shown in Fig. 1e through i are representative of similar data (within ± 40% error) obtained in parallel measurements on replicate cores. In all cores, the concentrations of short-chain fatty acids were rather similar (formate, 5 to 15 μM; acetate, 5 to 15 μM; propionate, 5 to 40 μM; and butyrate, 0.5 to 2 μM; Fig. 1e through h). The concentrations of dissolved hydrogen showed maximal and rather constant values at greater sediment depth. Below 20 cm sediment depth, dissolved hydrogen reached about 0.5 Pa in the core taken at 2 m and about 2 Pa in the core taken at 80 m, and it oscillated between these values in the 10-m core (Fig. 1i). The hydrogen partial pressure decreased distinctly from about 10 cm depth toward the sediment surface in the cores taken at 2 m and the 10 m, and from 15 cm depth downwards in the core taken at 80 m. We also detected carbon monoxide at partial pressures of 8.8 to 430 Pa in all core samples (see Fig. S1 in the supplemental material).
Free reaction energies of terminal fermentation processes in sediment cores.
From the pool sizes of the fermentation intermediates shown in Fig. 1, we calculated via the respective activities the free energy changes of the respective conversion reactions listed in Table 1. The alternative electron carriers formate and hydrogen turned out to be energetically not exactly equivalent, as Fig. 2 shows. A (hypothetical) conversion of formate to hydrogen plus CO2 yielded free energy changes in the range of −5 ± 5 kJ per mol reaction.
TABLE 1.
Terminal transformation reactions in biomass conversion to methane and CO2a
| Reaction | Conversion reaction | ΔG5 | Figure |
|---|---|---|---|
| Formate lysis | HCOO− + H+ → CO2 + H2 | +0.69 | 2 |
| Methanogenic reactions | |||
| Acetate to methane | CH3COO− + H+ → CO2 + CH4 | −30.3 | 3a |
| Hydrogen to methane | CO2 + 4H2 → CH4 + 2H2O | −139.0 | 3b |
| Formate to methane | 4HCOO− + 4H+ → CH4 + 2H2O + 3CO2 | −136.2 | 3c |
| Acetogenic reactions | |||
| Hydrogen to acetate | 2CO2 + 4H2 → CH3COO− + H+ + 2H2O | −108.7 | 3d |
| Formate to acetate | 4HCOO− + 3H+ → CH3COO− + 2H2O + 2CO2 | −105.9 | 3e |
| Syntrophic oxidation reactions | |||
| Propionate to hydrogen | CH3CH2COO− + 2H2O → CH3COO− + CO2 + 3H2 | +82.7 | 3f |
| Propionate to formate | CH3CH2COO− + 2CO2 + 2H2O → CH3COO− + 3HCOO− + 3H+ | +80.6 | 3g |
| Propionate to hydrogen (Smithella pathway) | 2CH3CH2COO− + 2H2O → 3CH3COO− + H+ + 2H2 | +56.7 | 3f |
| Propionate to formate (Smithella pathway) | 2CH3CH2COO− + 2H2O + 2CO2 → 3CH3COO− + 2HCOO− + 3H+ | +55.3 | 3g |
| Butyrate to H2 | CH3CH2CH2COO− + 2H2O → 2CH3COO− + H+ + 2H2 | +54.0 | 3h |
| Butyrate to formate | CH3CH2CH2COO− + 2CO2 + 2H2O → 2CH3COO− + 2HCOO− + 3H+ | +52.7 | 3i |
FIG 2.
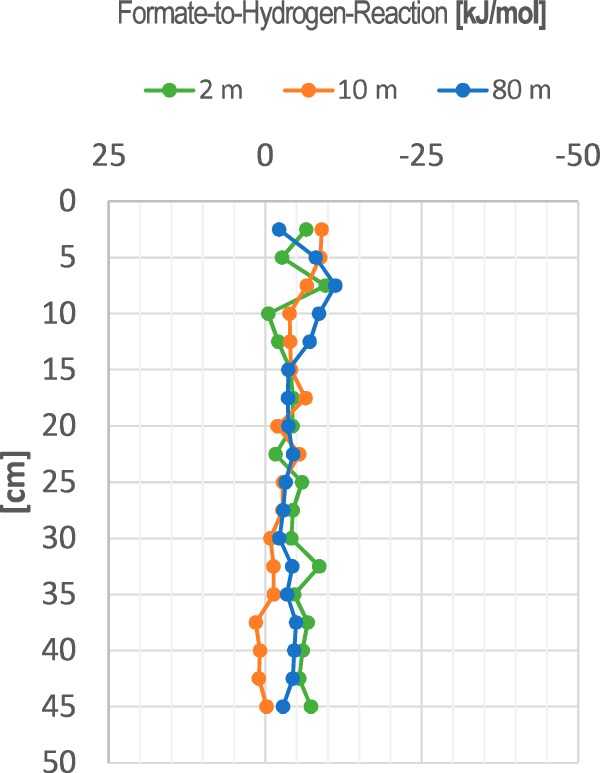
Gibbs free energy changes of (hypothetical) conversion of formate to hydrogen plus CO2 in sediment cores taken in Lake Constance at three different water depths (2 m, 10 m, and 80 m).
As shown in Fig. 3a, the conversion of acetate to methane plus CO2 released energy in the range of −10 kJ per mol in all sediment cores below 10 cm sediment depth, where net methane formation was observed. As a consequence of the slight energetic differences between formate and hydrogen shown in Fig. 2, electron consumptions in methanogenesis or homoacetogenesis and electron releases in secondary fermentations yield slightly different free energy changes, as Fig. 3b through i exemplify. While methanogenesis from hydrogen and bicarbonate was exergonic, with −14 to −40 kJ per mol in all cores below 10 to 15 cm depth (Fig. 3b), the available free energy increased to −30 to−50 kJ per mol if formate, rather than hydrogen, was used as the electron donor (Fig. 3c). Homoacetogenesis in the methanogenic sediment layers with hydrogen as the electron donor was exergonic, with only about −10 kJ per mol in the cores from 2 and 10 m water depth, and it yielded more than −30 kJ per mol in the core from 10 m water depth (Fig. 3d). If formate, rather than hydrogen, was used for homoacetate fermentation, the available free energy changes shifted to −20 to −40 kJ per mol reaction. (Fig. 3e).
FIG 3.

Gibbs free energy changes of secondary fermentation reactions as specified in Table 1, based on interspecies hydrogen or formate transfer in sediment cores taken in Lake Constance at three different water depths (2 m, 10 m, and 80 m). In panels f and g, energetics of fermentations according to Syntrophobacter are shown with solid lines, and those according to Smithella are shown with dashed lines.
The situation is quite the opposite for the electron-releasing secondary fermentations. Syntrophic propionate oxidation according to the Syntrophobacter-catalyzed reaction (Table 1), leading to acetate and three molecules of H2, released about −10 kJ per mol in most depth layers of sediment cores from 2 m and 10 m water depth and no energy at all in nearly all methanogenic layers of the 80-m sediment core (Fig. 3f). If formate was used as an electron carrier, the oxidation of propionate no longer yielded free energy, or even became slightly endergonic in several sediment layers examined (Fig. 3g). The energetic situation improves substantially if propionate is fermented through an alternative fermentation pathway converting two propionates to three acetates and two hydrogens, as Smithella propionica does (36) (Table 1; Fig. 3f and g). In a similar manner, syntrophic oxidation of butyrate yields −15 to −35 kJ per mol with hydrogen as the coproduct (Fig. 3h) but only −10 to −30 kJ per mol if electrons are released as formate (Fig. 3i). The mentioned deviations of the measured pool sizes (±40%, see above) cause differences in the Gibbs free reaction energies in the range of 1.5 to 4 kJ per mol reaction, depending on the number of electron pairs transferred, and are therefore insignificant for our energetical considerations.
DISCUSSION
Stratification of sediments.
The sediment cores examined in the present study were taken in areas of different water depths of a large oligotrophic lake. Although the littoral sediments taken at 2 m water depth were coarser than those taken from deeper sediments, the porosity of all three sediments was roughly similar, with a tendency to lower porosity and higher compaction with increasing depth. In addition, the pH values of the sediments were similar, with slightly higher values toward the sediment surface. Sulfate content decreased from the surface to 10 cm sediment depth from nearly 300 μM to values of 10 to 50 μM, most clearly exemplified with the core taken at 80 m water depth. In the other two cores, sulfate penetrated down to 15 to 20 cm depth, where low values similar to those in the core taken at 80 m were reached. These uneven distributions may be attributed to bioturbation activities in these sediments from shallow waters, unlike the distributions in the deep-water core, where bioturbation appears to be irrelevant (37). Methane distributions showed very stable values close to saturation below 10 cm sediment depth in the cores from 2 and 10 m water depth. In the upper 10 cm of sediment, methane concentrations decreased upwards, indicating diffusive methane transport toward the sediment surface (38), where methane is consumed nearly quantitatively by either aerobic (33, 34) or nitrate-dependent microbial oxidation (33, 39). In the sediment core from 80 m water depth, the methane concentration decreased continuously from about 20 cm depth upwards, indicating a diffusive flux from the layers below and lower methanogenic activity in the layers of 10 to 20 cm sediment depth, compared to those of the other two cores. These lower methane formation activities may be due to lower biomass import into these deep-lying sediments, as opposed to that into shallow-water sediments. We can state that oxidation of organic matter with oxygen, nitrate, Fe(III), or sulfate may take place in our sediments within the uppermost 10 cm depth, and that below this depth, methanogenesis is the dominant process of organic matter degradation, even if sulfate reduction may play a marginal role based on the small sulfate pool detected in the lower sediment layers as well. Reoxidation of sulfide in the sense of a “hidden sulfur cycle” can be ruled out due to lack of alternative oxidants in these deep sediment layers.
Pool sizes of fermentation intermediates and energy gains of terminal fermentation processes.
Pool sizes of fatty acids in the methanogenic zones of the sediments studied (below 10 cm sediment depth or below 15 cm depth in the core taken at 80 m water depth) were stable over more than 30 cm of core depth, with values of about 10 μM for acetate, 1 μM for butyrate, and 5 to 50 μM for propionate. Previous studies on Lake Constance sediments at times when this lake was eutrophic due to high phosphate contents (up to 90 mg phosphate-P per liter of water) and corresponding high productivity reported pool sizes of 50 to 250 μM acetate, 50 to 80 μM butyrate, and 5 to 30 μM propionate (40–42). It appears that the higher trophic state in those days caused increased acetate and butyrate pools, whereas propionate pools were less affected, compared to today's oligotrophic state of this lake, with phosphate contents of 8 to 10 mg phosphate-P per liter of water (37).
In the present study, formate pools were measured in freshwater sediment profiles to compare them with the profiles of hydrogen distribution as an alternative interspecies electron carrier. The measured formate pool sizes were rather similar in all three cores, with concentrations of 2 to 15 μM over the entire core lengths and a slight tendency to increase from the surface toward greater depth. With a similar tendency, pool sizes of molecular hydrogen increased with depth between 10 cm and 45 cm sediment depth from 0.5 to 5 Pa. Our formate and acetate pools were similar to those measured recently in an oligotrophic sulfate-reducing marine sediment (4 to 6 μM [27]).
Simultaneously with hydrogen measurements, we also detected carbon monoxide (CO) at rather high concentrations (10 to 400 Pa) in all samples. CO is known to be formed, at concentrations similar to those we observed, when anoxic sediment material or paddy soil is handled under air and daylight (43–45). We therefore assume that this CO was formed as an artifact during the sample preparation process and do not discuss it any further here. The measured CO profiles are shown in the supplemental material (Fig. S1).
The Gibbs free energy changes of acetate conversion to methane and CO2 as calculated from the pool sizes measured in our study were in the range of −8 to −13 kJ per mol acetate in all three sediment cores over the entire length below 10 cm sediment depth. With this, these reactions were at the lowermost limit of energy potentials that can just support microbial life, according to our present-day understanding of microbial energy metabolism (7, 8). In paddy soil microcosms that were well supplied with organic matter, minimum energy yields in the range of −17 to −20 kJ per mol acetate were calculated for this reaction (46, 47). It appears that the poor substrate supply situation in our oligotrophic lake sediment drives the energy supply of the acetate-cleaving methanogens down to its lowermost limits, where they operate with optimal kinetic efficiency.
Hydrogen partial pressures increased from the sediment surface downwards from 0.5 Pa to a maximum of 5 Pa and stayed stable at about 10 Pa (10 ppm) in the methanogenic zones. These values exactly match the hydrogen concentrations that were measured earlier in iron- or sulfate-reducing and methanogenic sediments (0.5 to 10 nM H2, corresponding to 0.5 to 10 ppm equivalent in the gas phase [29]). Similar values were measured in marine sediments (2 to 15 nM [30]), and slightly higher values (10 to 60 nM) were measured by a hydrogen diffusion probe in Lake Constance sediments when it was still eutrophic (28). These concentrations are close to the threshold concentrations for hydrogen uptake by pure cultures of methanogens (20 to 100 ppm [48]), meaning that they are controlled by the uptake efficiency of the hydrogen consumers and cannot go much further down in a methanogenic environment.
The pool sizes of formate and hydrogen measured in our study allowed calculations of a (hypothetical) conversion of formate to H2 plus CO2 in the range of −5 ± 5 kJ per mol, a range mostly below the minimal energy quantum needed for ATP synthesis. Thus, this small energy potential will hardly be sufficient to drive microbial growth. Nonetheless, microbial energy conservation based on this reaction has been postulated before (49) and has been biochemically proven for a specialized marine archaeon living in a formate-rich environment (50). Since many strict anaerobes contain enzymes which equilibrate the formate and the hydrogen pool without concomitant energy conservation (21, 51), it appears unlikely that other organisms in the same environment can exploit this reaction for energy conservation.
Nonetheless, the small energy potential between formate and hydrogen in our sediments may have energetic consequences in reactions in which two, three, or four electron pairs are transferred in syntrophic associations. Thus, it is energetically more favorable to release electrons as hydrogen than as formate, e.g., in butyrate or propionate oxidation. Both processes can release electrons either as formate or as hydrogen (21, 52). According to our measurements, butyrate oxidation with electron release as hydrogen is sufficiently exergonic at all three sampling sites but comes to energetic limits with formate as the electron sink in the shallow-water sediment core. The same difference applies, of course, to beta oxidation of long-chain fatty acids from lipid degradation, which has to face the same energetic problems as those of butyrate oxidation.
The energetic difference between hydrogen and formate release is even more pronounced in propionate oxidation by bacteria similar to Syntrophobacter spp., which comes to thermodynamic equilibrium when coupled to formate release (see Fig. 3g). This is surprising because just for this syntrophic oxidation, a release of formate as electron carrier has been indicated from physiological and biochemical studies in defined cocultures (14, 52–55). Our calculated energy gains in this step are in the same range as values published earlier for paddy soil and laboratory cocultures (56, 57). Boone et al. (14) measured formate concentrations of 16 μM in a propionate-degrading defined methanogenic coculture. Perhaps especially in propionate oxidation, both electron transfer channels are used simultaneously; i.e., some of the electrons are released via hydrogen and others via formate, and both balance out to a situation that attributes just sufficient energy to the propionate oxidizer. The choice of electron carrier might as well depend on the availability of suitable partner methanogens available in the immediate surroundings (13). Although earlier radiolabeling experiments have shown that the majority of methanogenic propionate degradation in lake sediments proceeds through syntrophic oxidation via succinate (58), one may consider also the alternative fermentation path via transformation of two propionates to three acetates that Smithella propionica catalyzes (36, 59, 60). This pathway turns out to be an elegant alternative for transferring the “difficult” electrons of propionate oxidation via acetate directly to methane, thus avoiding the thermodynamically difficult oxidation of succinate with protons or CO2 as the electron acceptor. Stable isotope-probing experiments with rice paddy soil showed that Smithella-like organisms contributed a significant part to propionate turnover in this environment (61). Nonetheless, the comparably high propionate concentrations we found indicate that propionate degradation runs only at limited efficiency, perhaps because Syntrophobacter-like organisms operate at their energetic limit and the alternative path via the Smithella fermentation is not being used, although it is energetically more favorable.
Of course, the energetic gain on the one hand is the energetic loss on the other one; methane formation or homoacetogenic acetate formation in our sediment is energetically easier with formate than with hydrogen electrons. Both processes are sufficiently exergonic to support microbial growth with either electron source, with changes of Gibbs free energy in the range of −10 to −50 kJ per mol. Especially in homoacetogenesis, formate utilization and interspecies formate transfer could be involved in a mixed-carrier strategy. Beyond that, one has to keep in mind that homoacetogenic bacteria are metabolically very versatile and can also use, e.g., sugars, methanol from pectin degradation (62), and methyl groups from phenyl methyl ethers (63) as the substrates for acetate formation.
On the other hand, our calculations clearly show that the opposite of homoacetogenesis, i.e., syntrophic acetate oxidation, will hardly be possible in our lake sediment. This process has been observed so far only at high substrate supply in thermal biogas reactors (64) or in mesothermal reactors stressed with high ammonia contents (65). Slightly alkaline and cold oligotrophic sediments such as ours are obviously unsuitable environments for this type of metabolism.
MATERIALS AND METHODS
Harvesting of sediment cores.
Sediment cores (3 at each sampling site) were taken in August and September 2015, in Lake Constance littoral (light zone and transition zone) (2 m depth; 18°C; 47°42′9.86″ N, 9°11′32.82″ E; and 10 m depth; 8°C; 47°41′51.27″ N, 9°11′32.94″ E) and profundal (80 m depth; 4°C; 47°41′44.0″ N, 9°12′51.9″ E) zones. Lake Constance water stays nearly air saturated through the entire water column down to the sediment surface. Preliminary sampling and analysis series in the summers of 2013 and 2014 yielded similar results. The corer used was 8 cm in diameter (equating to 50 ml volume per cm depth). It could extract cores up to 100 cm in length. The harvested sediment cores were typically 50 to 60 cm in length. At 2 and 10 m water depth, the corer was pushed into the sediment by a modular, air-tight aluminum tube system (3 m per component). The floatable rod and the corer were disconnected, and the core was pulled out by a rope. Samples at 80 m depth were taken with a multicorer with lead ballast and deployed by a steel rope on a crane. Core tubes were pushed into the sediment by gravitational acceleration 10 m over the ground and pulled out with the heaver. The cylinders were sealed from the lower side with a butyl rubber stopper and from the upper side with a polyetheretherketone (PEEK) screw plug.
Sediment preparation.
Core tubes were equipped with two rows of bore holes at an angle of 90° to subsample slices of 2.5 cm each. Subsamples from each core were taken with a tip-cut syringe (30 ml, 2.4-cm inner diameter). This method allows quick sample preparation to minimize losses of gas and volatile organic acids. For each sampling site, the entire extraction and analysis experiment was run twice with two separate cores, with similar results. Because of differences in core stratification, it does not make sense to average these results. Therefore, representative results from doublet cores of each single sampling are shown.
Analysis of pool sizes.
For analysis of gaseous compounds, 30-ml sediment subcores were transferred into 150-ml infusion bottles and sealed with butyl rubber stoppers. Headspaces were flushed with nitrogen gas for 3 min. A total of 30 ml of saturated NaCl solution was added to the sediment by a syringe to force all gases from the pore water into the headspace. After mixing on a shaker and sonication for 10 min, gas concentrations were measured. Gaseous compounds were analyzed in the headspace by gas chromatography. H2 and CO were measured with a reductive gas chromatograph (Peak Performer 1, Peak Laboratories, Mountain View, CA) (66). The system was equipped with an automatic sampling device (100-μl sample loop) to enhance injection accuracy of replicates. Methane was measured with a gas chromatograph (Shimadzu GC-9A) equipped with a zeolite molecular sieve column and flame ionization detector. The CO2 partial pressure was calculated with 100 mM bicarbonate in the interstitial water (based on experimentally determined 0.4 M total carbonate + bicarbonate in the sediment), a temperature of 5°C, and the measured pH value. For high-performance liquid chromatography (HPLC) analyses, subsamples were transferred into 50-ml Falcon tubes and centrifuged at 10°C at 15,000 rpm for 10 min. The supernatant was used directly without acidification for analysis of soluble compounds. Short-chain fatty acids were measured using a Shimadzu Prominence high-performance liquid chromatograph equipped with an Aminex HPX87H column, a photo diode array detector, and a trace sensitive method for organic acids (26). pH measurement was done with a potentiometric electrode in the supernatant of the centrifugation step.
Porosity was determined as weight loss of sediment samples during 24 h of drying at 150°C.
Calculation of Gibbs free energies of reactions.
Temperature-corrected standard Gibbs free energies of reactions were calculated from standard Gibbs free energy changes of formation taken from (67) at 5°C by polynomic interpolation of the given data. Volatile fatty acids were used at molar concentrations, and H2, CH4, and CO2 were calculated as partial pressures in the headspace. The measured pool sizes were normalized to activities, which were used to calculate the Gibbs free energies of reactions via the Nernst equation.
Chemicals.
All chemicals were of analytical grade or higher quality and were obtained from Boehringer (Mannheim, Germany), Eastman Kodak (Rochester, NY), Fluka (Neu-Ulm, Germany), Merck (Darmstadt, Germany), Pharmacia (Freiburg, Germany), Serva (Heidelberg, Germany), and Sigma (Deisenhofen, Germany). Gases were purchased from Messer-Griesheim (Darmstadt, Germany) and Sauerstoffwerke Friedrichshafen (Friedrichshafen, Germany).
Supplementary Material
ACKNOWLEDGMENTS
Technical help with specific experiments in the lab by Antje Wiese and Joana Thiel and in the field by Maren Bornemann is highly appreciated.
This study was funded by the German Federal Ministry for Education and Research, project BioPara (project number 03SF0421E).
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01572-18.
REFERENCES
- 1.Bastviken D, Cole J, Pace M, Tranvik L. 2004. Methane emissions from lakes: dependence of lake characteristics, two regional assessments, and a global estimate. Global Biogeochem Cycles 18:GB4009. [Google Scholar]
- 2.Bryant MP. 1979. Microbial methane production—theoretical aspects. J Animal Sci 48:193–201. [Google Scholar]
- 3.Gujer W, Zehnder A. 1983. Conversion processes in anaerobic digestion. Water Sci Technol 15:127–167. [Google Scholar]
- 4.Schink B. 1997. Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61:262–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schink B, Stams AJM. 2013. Syntrophism among prokaryotes, p 471–493. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes—prokaryotic communities and ecophysiology. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 6.Thauer RK, Morris JG. 1984. Metabolism of chemotrophic anaerobes: old views and new aspects, p 123–168. In Kelly DP, Carr NG (ed), The microbe: part II: prokaryotes and eukaryotes. Cambridge University Press, Cambridge, UK. [Google Scholar]
- 7.Spahn S, Brandt K, Müller V. 2015. A low phosphorylation potential in the acetogen Acetobacterium woodii reflects its lifestyle at the thermodynamic edge of life. Arch Microbiol 197:745–751. doi: 10.1007/s00203-015-1107-2. [DOI] [PubMed] [Google Scholar]
- 8.Lever MA, Rogers K, Lloyd KG, Overmann J, Schink B, Thauer RK, Hoehler TM, Jørgensen BB. 2015. Life under extreme energy limitation: a synthesis of laboratory- and field-based investigations. FEMS Microbiol Ecol 39:688–728. doi: 10.1093/femsre/fuv020. [DOI] [PubMed] [Google Scholar]
- 9.Müller V. 2015. Microbial life at the thermodynamic limit: how much energy is required to sustain life? Environ Microbiol Rep 7:31–32. doi: 10.1111/1758-2229.12232. [DOI] [PubMed] [Google Scholar]
- 10.McInerney MJ, Bryant MP, Pfennig N. 1979. Anaerobic bacterium that degrades fatty acids in syntrophic association with methanogens. Arch Microbiol 122:129–135. doi: 10.1007/BF00411351. [DOI] [PubMed] [Google Scholar]
- 11.Zehnder AJB, Ingvorsen K, Marti T. 1982. Microbiology of methane bacteria, p 45–68. In Hughes DE, Stafford DA, Wheatley BI, Baader W, Lettinga G, Nyns EJ, Verstraete W (ed), Anaerobic digestion. Elsevier Biomedical Press, Amsterdam, The Netherlands. [Google Scholar]
- 12.Stams AJ, Plugge CM. 2009. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol 7:568–577. doi: 10.1038/nrmicro2166. [DOI] [PubMed] [Google Scholar]
- 13.Schink B, Montag D, Keller A, Müller N. 2017. Hydrogen or formate – alternative key players in methanogenic degradation. Environ Microbiol Rep 9:189–202. doi: 10.1111/1758-2229.12524. [DOI] [PubMed] [Google Scholar]
- 14.Boone DR, Bryant MP. 1980. Propionate-degrading bacterium, Syntrophobacter wolinii sp. nov. gen. nov., from methanogenic ecosystems. Appl Environ Microbiol 40:626–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boone DR, Johnson RL, Liu Y. 1989. Diffusion of the interspecies electron carriers H2 and formate in methanogenic ecosystems and its implications in the measurement of Km for H2 or formate uptake. Appl Environ Microbiol 55:1735–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiele JH, Zeikus JG. 1988. Control of interspecies electron flow during anaerobic digestion: significance of formate transfer versus hydrogen transfer during syntrophic methanogenesis in flocs. Appl Environ Microbiol 54:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong X, Cheng G, Stams AJM. 1994. Butyrate oxidation by Syntrophospora bryantii in coculture with different methanogens and in pure culture with pentenoate as electron acceptor. Appl Microbiol Biotechnol 42:647–652. doi: 10.1007/BF00173934. [DOI] [Google Scholar]
- 18.Dong X, Plugge CM, Stams AJM. 1994. Anaerobic degradation of propionate by a mesophilic acetogenic bacterium in co- and triculture with different methanogens. Appl Environ Microbiol 60:2834–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller N, Schleheck D, Schink B. 2009. Involvement of NADH: acceptor oxidoreductase and butyryl coenzyme A dehydrogenase in reversed electron transport during syntrophic butyrate oxidation by Syntrophomonas wolfei. J Bacteriol 191:6167–6177. doi: 10.1128/JB.01605-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Müller N, Worm P, Schink B, Stams AJ, Plugge CM. 2010. Syntrophic butyrate and propionate oxidation processes: from genomes to reaction mechanisms. Environ Microbiol Rep 2:489–499. doi: 10.1111/j.1758-2229.2010.00147.x. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt A, Müller N, Schink B, Schleheck D. 2013. A proteomic view at the biochemistry of syntrophic butyrate oxidation in Syntrophomonas wolfei. PLoS One 8:e56905. doi: 10.1371/journal.pone.0056905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sieber JR, Le HM, McInerney MJ. 2014. The importance of hydrogen and formate transfer for syntrophic fatty, aromatic and alicyclic metabolism. Environ Microbiol 16:177–188. doi: 10.1111/1462-2920.12269. [DOI] [PubMed] [Google Scholar]
- 23.Drake HL. 1994. Acetogenesis, acetogenic bacteria, and the acetyl-CoA “Wood/Ljungdahl” pathway: past and current perspectives, p 3–60. In Drake HL. (ed) Acetogenesis. Chapman and Hall, New York, NY. [Google Scholar]
- 24.Winfrey M, Zeikus JG. 1977. Effect of sulfate on carbon and electron flow during microbial methanogenesis in freshwater sediments. Appl Environ Microbiol 33:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kristjansson JK, Schönheit P, Thauer RK. 1982. Different Ks values for hydrogen of methanogenic bacteria and sulfate reducing bacteria—an explanation for the apparent inhibition of methanogenesis by sulfate. Arch Microbiol 131:278–282. doi: 10.1007/BF00405893. [DOI] [Google Scholar]
- 26.Montag D, Schink B. 2015. Biogas process parameters—energetics and kinetics of secondary fermentations in methanogenic biomass degradation. Appl Microbiol Biotech 100:1019–1026. doi: 10.1007/s00253-015-7069-0. [DOI] [PubMed] [Google Scholar]
- 27.Glombitza C, Jaussi M, Roy H, Seidenkrantz M-S, Lomstein BA, Jorgensen BB. 2015. Formate, acetate, and propionate as substrates for sulfate reduction in sub-arctic sediments of Southwest Greenland. Front Microbiol 6:846. doi: 10.3389/fmicb.2015.00846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kramer H, Conrad R. 1993. Measurement of dissolved H2 concentrations in methanogenic environments with a gas diffusion probe. FEMS Microbiol Ecol 12:148–158. [Google Scholar]
- 29.Lovley DR, Goodwin S. 1988. Hydrogen concentrations as an indicator of the predominant terminal electron-accepting reactions in aquatic sediments. Geochim Cosmochim Acta 52:2993–3003. doi: 10.1016/0016-7037(88)90163-9. [DOI] [Google Scholar]
- 30.Hoehler TM, Alperin MJ, Albert DB, Martens CS. 1998. Thermodynamic control on hydrogen concentrations in anoxic sediments. Geochim Cosmochim Acta 62:1745–1756. doi: 10.1016/S0016-7037(98)00106-9. [DOI] [Google Scholar]
- 31.Conrad R, Goodwin S, Zeikus JG. 1987. Hydrogen metabolism in a midly acidic lake sediment (Knaack Lake). FEMS Microbiol Ecol 45:243–249. doi: 10.1111/j.1574-6968.1987.tb02362.x. [DOI] [Google Scholar]
- 32.Conrad R, Schink B, Phelps TJ. 1986. Thermodynamics of H2-consuming and H2-producing metabolic reactions in diverse methanogenic environments under in situ conditions. FEMS Microbiol Ecol 38:353–360. doi: 10.1111/j.1574-6968.1986.tb01748.x. [DOI] [Google Scholar]
- 33.Rahalkar M, Deutzmann J, Schink B, Bussmann I. 2009. Abundance and activity of methanotrophic bacteria in littoral and profundal sediments of Lake Constance. Appl Environ Microbiol 75:119–126. doi: 10.1128/AEM.01350-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rahalkar M, Schink B. 2007. Comparison of aerobic methanotrophic communities in littoral and profundal sediments of Lake Constance by a molecular approach. Appl Environ Microbiol 73:4389–4394. doi: 10.1128/AEM.02602-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deutzmann JS, Stief P, Brandes J, Schink B. 2014. Anaerobic methane oxidation coupled to denitrification is the dominant methane sink in a deep lake. Proc Natl Acad Sci U S A 111:18273–18278. doi: 10.1073/pnas.1411617111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Bok FA, Stams AJ, Dijkema C, Boone DR. 2001. Pathway of propionate oxidation by a syntrophic culture of Smithella propionica and Methanospirillum hungatei. Appl Environ Microbiol 67:1800–1804. doi: 10.1128/AEM.67.4.1800-1804.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Güde H, Straile D. 2016. Bodensee. Ökologie und anthropogene Belastungen eines tiefen Voralpensees. Limnologie aktuell vol. 15 Schweizerbart, Stuttgart, Germany. [Google Scholar]
- 38.Chanton JP, Martens CS, Kelley CA. 1989. Gas transport from methane-saturated, tidal freshwater and wetland sediments. Limnol Oceanogr 34:807–819. doi: 10.4319/lo.1989.34.5.0807. [DOI] [Google Scholar]
- 39.Deutzmann JS, Schink B. 2011. Anaerobic oxidation of methane in sediments of Lake Constance, an oligotrophic freshwater lake. Appl Environ Microbiol 77:4429–4436. doi: 10.1128/AEM.00340-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thebrath B, Rothfuss F, Whiticar MJ, Conrad R. 1993. Methane production in littoral sediment of lake constance. FEMS Microbiol Lett 102:279–289. doi: 10.1111/j.1574-6968.1993.tb05819.x. [DOI] [Google Scholar]
- 41.Rothfuss F, Conrad R. 1993. Thermodynamics of methanogenic intermediary metabolism in littoral sediment of Lake Constance. FEMS Microbiol Ecol 12:265–276. doi: 10.1111/j.1574-6941.1993.tb00039.x. [DOI] [Google Scholar]
- 42.Schulz S, Conrad R. 1996. Influence of temperature on pathways to methane production in the permanently cold profundal sediment of Lake Constance. FEMS Microbiol Ecol 20:1–14. doi: 10.1111/j.1574-6941.1996.tb00299.x. [DOI] [Google Scholar]
- 43.Conrad R, Seiler W. 1980. Role of microorganisms in the consumption and production of atmospheric carbon monoxide by soil. Appl Environ Microbiol 40:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conrad R, Seiler W. 1980. Photooxidative production and microbial consumption of carbon monoxide in seawater. FEMS Microbiol Lett 9:61–64. doi: 10.1111/j.1574-6968.1980.tb05606.x. [DOI] [Google Scholar]
- 45.Conrad R, Seiler W. 1985. Characteristics of abiological carbon monoxide formation from soil organic matter, humic acids, and phenolic compounds. Environ Sci Technol 19:1165–1169. doi: 10.1021/es00142a004. [DOI] [PubMed] [Google Scholar]
- 46.Fey A, Conrad R. 2000. Effect of temperature on carbon and electron flow and on the archaeal community in methanogenic rice field soil. Appl Environ Microbiol 66:4790–4797. doi: 10.1128/AEM.66.11.4790-4797.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Penning H, Conrad R. 2006. Effect of inhibition of acetoclastic methanogenesis on growth of archaeal populations in an anoxic model environment. Appl Environ Microbiol 72:178–184. doi: 10.1128/AEM.72.1.178-184.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cord-Ruwisch R, Seitz HJ, Conrad R. 1988. The capacity of hydrogenotrophic anaerobic bacteria to compete for traces of hydrogen depends on the redox potential of the terminal electron acceptor. Arch Microbiol 149:350–357. doi: 10.1007/BF00411655. [DOI] [Google Scholar]
- 49.Dolfing J, Jiang B, Henstra AM, Stams AJ, Plugge CM. 2008. Syntrophic growth on formate: a new microbial niche in anoxic environments. Appl Environ Microbiol 74:6126–6131. doi: 10.1128/AEM.01428-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim JK, Mayer F, Kang SG, Müller V. 2014. Energy conservation by oxidation of formate to carbon dioxide and hydrogen via a sodium ion current in a hyperthermophilic archaeon. Proc Nat Acad Sci U S A 111:11497–11502. doi: 10.1073/pnas.1407056111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuchmann K, Müller V. 2013. Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 342:1382–1385. doi: 10.1126/science.1244758. [DOI] [PubMed] [Google Scholar]
- 52.Stams AJM, Dong X. 1995. Role of formate and hydrogen in the degradation of propionate and butyrate by defined suspended cocultures of acetogenic and methanogenic bacteria. Antonie Van Leeuwenhoek 68:281–284. doi: 10.1007/BF00874137. [DOI] [PubMed] [Google Scholar]
- 53.Dong X, Stams AJM. 1995. Evidence for H2 and formate formation during syntrophic butyrate and propionate degradation. Anaerobe 1:35–39. doi: 10.1016/S1075-9964(95)80405-6. [DOI] [PubMed] [Google Scholar]
- 54.de Bok FAM, Hagedoorn PL, Silva PJ, Hagen WR, Schiltz E, Fritsche K, Stams AJ. 2003. Two W-containing formate dehydrogenases (CO2-reductases) involved in syntrophic propionate oxidation by Syntrophobacter fumaroxidans. Eur J Biochem 270:2476–2485. doi: 10.1046/j.1432-1033.2003.03619.x. [DOI] [PubMed] [Google Scholar]
- 55.Worm P, Fermoso FG, Stams AJ, Lens PN, Plugge CM. 2011. Transcription of fdh and hyd in Syntrophobacter spp. and Methanospirillum spp. as a diagnostic tool for monitoring anaerobic sludge deprived of molybdenum, tungsten and selenium. Environ Microbiol 13:1228–1235. doi: 10.1111/j.1462-2920.2011.02423.x. [DOI] [PubMed] [Google Scholar]
- 56.Krylova NI, Conrad R. 1998. Thermodynamics of propionate degradation in methanogenic paddy soil. FEMS Microbiol Ecol 26:281–288. doi: 10.1111/j.1574-6941.1998.tb00512.x. [DOI] [Google Scholar]
- 57.Scholten JC, Conrad R. 2000. Energetics of syntrophic propionate oxidation in defined batch and chemostat cocultures. Appl Environ Microbiol 66:2934–2942. doi: 10.1128/AEM.66.7.2934-2942.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schink B. 1985. Mechanism and kinetics of succinate and propionate degradation in anoxic freshwater sediments and sewage sludge. J Gen Microbiol 131:643–650. [Google Scholar]
- 59.Dolfing J. 2013. Syntrophic propionate oxidation via butyrate: a novel window of opportunity under methanogenic conditions. Appl Environ Microbiol 79:4515–4516. doi: 10.1128/AEM.00111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leng L, Yang P, Singh S, Zhuang H, Xu L, Chen WH, Dolfing J, Li D, Zhang Y, Zeng H, Chu W, Lee PH. 2018. A review on the bioenergetics of anaerobic microbial metabolism close to the thermodynamic limits and its implications for digestion applications. Bioresour Technol 247:1095–1106. doi: 10.1016/j.biortech.2017.09.103. [DOI] [PubMed] [Google Scholar]
- 61.Gan YL, Qiu QF, Liu PF, Rui JP, Lu YH. 2012. Syntrophic oxidation of propionate in rice field soil at 15 and 30 degrees C under methanogenic conditions. Appl Environ Microbiol 78:4923–4932. doi: 10.1128/AEM.00688-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schink B, Zeikus JG. 1980. Microbial methanol formation: A major end product of pectin metabolism. Curr Microbiol 4:387–389. doi: 10.1007/BF02605383. [DOI] [Google Scholar]
- 63.Bache R, Pfennig N. 1981. Selective isolation of Acetobacterium woodii on methoxylated aromatic acids and determination of growth yields. Arch Microbiol 130:255–261. doi: 10.1007/BF00459530. [DOI] [Google Scholar]
- 64.Zinder SH, Koch M. 1984. Non-aceticlastic methanogenesis from acetate: acetate oxidation by a thermophilic syntrophic coculture. Arch Microbiol 138:263–272. doi: 10.1007/BF00402133. [DOI] [Google Scholar]
- 65.Moestedt J, Müller B, Westerholm M, Schnürer A. 2016. Ammonia threshold for inhibition of anaerobic digestion of thin stillage and the importance of organic load rate. Microb Biotechnol 9:180–194. doi: 10.1111/1751-7915.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seiler W, Giehl H, Roggendorf P. 1980. Detection of carbon monoxide and hydrogen by conversion of mercury oxide to mercury vapor. Atmos Technol 12:40–45. [Google Scholar]
- 67.Amend JP, Shock EL. 2001. Energetics of overall metabolic reactions of thermophilic and hyperthermophilic archaea and bacteria. FEMS Microbiol Rev 25:175–243. doi: 10.1111/j.1574-6976.2001.tb00576.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.