

Abstract
Self-assembly of proteins into filaments, such as actin and tubulin filaments, underlies essential cellular processes in all three domains of life. The early emergence of filaments in evolutionary history suggests that filament genesis might be a robust process. Here we describe the fortuitous construction of GFP fusion proteins that self-assemble as fluorescent polar filaments in Escherichia coli. Filament formation is achieved by appending as few as 12 residues. Crystal structures reveal that the protomers each donate an appendage to fill a groove between two following protomers along the filament. This exchange of appendages resembles runaway domain swapping but is distinguished by higher efficiency because monomers cannot competitively bind their own appendages. Ample evidence of this “runaway domain coupling” mechanism in nature suggests it could facilitate the evolutionary pathway from globular protein to polar filament, requiring a minimal extension of protein sequence and no significant refolding.
Introduction
The self-association of proteins to form filaments is the basis for the formation of essential cytoskeletal elements such as actin and tubulin in all three domains of life. Cytoskeletal elements participate in a variety of critical cellular processes including DNA segregation, cell division, morphogenesis, cell motility, maintenance of cell shape, and the transport of cellular cargoes. The identification of diverse families of actin and tubulin homologs in prokaryotes over the last 20 years1 has led to the suggestion that these are ancient proteins that predate the split between bacteria, archaea and eukarya2,3,4. Furthermore, the recent discovery that certain metabolic enzymes can polymerize as filaments has provoked the suggestion that an original rudimentary cytoskeleton may have evolved in an ancestral cell through the repurposing of enzyme polymerization5,6,7,8.
The early emergence of filaments in biological history raises the question of what structural mechanisms are capable of establishing filament-competent interfaces, and how facile is their evolution. One mechanism requiring few specific structural features other than a globular fold is runaway 3-dimensional domain swapping. By this mechanism, the intermolecular interface between successive protomers in the filament recapitulates an intramolecular interface that exists within the isolated monomer9,10. Thus, a domain of the protein, usually residing at either the N- or C-terminus and connected to the remainder of the protein by a flexible linker (the so-called hinge-loop), exchanges its intramolecular contacts for otherwise identical intermolecular contacts, an exchange that usually occurs only under non-native conditions. Runaway domain swapping occurs in a unidirectional fashion, with each subunit donating a domain to the next, leading to the formation of an open-ended protofilament. Examples include fibrils of a designed 3-α-helix bundle11, filaments of α1-antitrypsin12, and ribbons of “cab”-type carbonic anhydrase13. Runaway domain swapping has also been evidenced in some amyloid fibrils10,14,15,16,17. (Note, we consider fibrils and filaments to be equivalent in the assembly hierarchy – a level more complex than protofilaments – and we use the term most closely associated with the particular system discussed.)
A simpler, more obvious mechanism for filament formation is the stacking of domains in an end-to-end fashion. Other than runaway domain swapping and end-to-end stacking, no other naturally occurring mechanisms for filament formation have been formally categorized despite the recent rapid increase in the number of filament structures determined.
Here we describe a nested set of GFP fusion proteins with a shared ability to self-assemble as fluorescent filaments in bacterial cells. Remarkably, these filaments typically span the entire length of the cell. The smallest of these fusion proteins bears just 12 non-native amino acids at the C-terminus of GFP. Structure determination by x-ray crystallography revealed that these appended residues form an α-helix that extends away from the GFP domain (a β-barrel) through a hinge loop and docks into a groove formed by the following two β-barrels in the protofilament. We call this filament-forming mechanism “runaway domain coupling” because the protomer-protomer interfaces involve complementation between distinct donor and acceptor domains. It is distinct from end-to-end domain stacking where the protomer-protomer interfaces reside entirely within a single domain type. It is also distinct from runaway domain swapping, because the helix-in-groove complementation cannot be reproduced in the monomer. The fact that runaway domain coupling arose fortuitously suggests that the genesis of coupled interfaces is facile, an inference supported by our discovery of their occurrence in numerous natural filaments. We propose that the chances for such randomly generated surfaces to align in a productive interface are enhanced by a flexible hinge loop that connects the two domains of each protomer. Our findings illustrate a pathway whereby a non-polymerizing protein might evolve the ability to self-assemble as protofilaments and filaments.
Results
GFP-RNase A fusion proteins form fluorescent rods in bacterial cells.
Previous work from the Eisenberg group17,18 demonstrated that RNase A variants containing short amyloidogenic peptide insertions in the C-terminal hinge loop could form amyloid-like fibrils in vitro. We sought to investigate whether or not these RNase A variants could also access the fibrillar form when produced in E. coli cells. To do this, we constructed a plasmid vector that directed the inducible synthesis of RNase A (with or without an inserted amyloidogenic peptide sequence; see Methods) fused to a monomeric variant of GFP (Fig. 1a ). Cells containing these plasmids were examined by fluorescence microscopy at various times after the induction of fusion protein synthesis. Whereas most cells contained diffuse fluorescence, we observed fluorescent rod structures in a small minority (1 to 2%) of cells. Such rod-containing cells typically contained a single rod (Fig. 1b, left). These structures were observed regardless of whether or not the RNase A moiety contained the amyloidogenic peptide insertion (Fig. 1b, left and data not shown). Furthermore, we found that refrigeration dramatically stimulated formation of the rod-like structures. When we grew cells overnight after the induction of fusion protein synthesis and then incubated the cell cultures for 2 hours at 4oC, we detected fluorescent rod structures in essentially all of the cells (Fig. 1b, right).
Figure 1. Fluorescence images of E. coli cells containing GFP-RNase A fusion proteins.
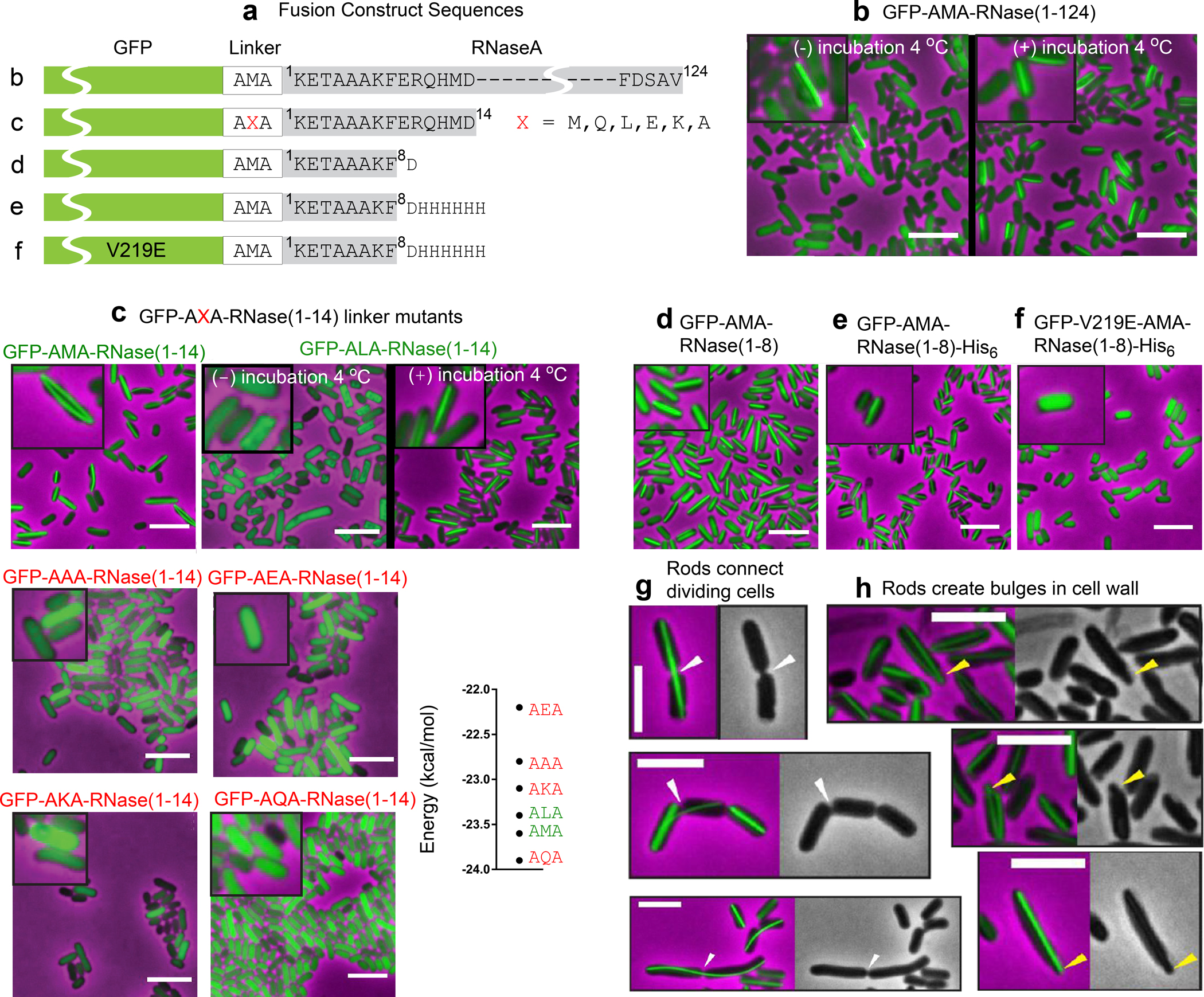
(a) Fusion constructs. RNaseA and truncated segments were fused to the C-terminus of GFP via a 3-residue linker (white box). (b) After overnight induction of GFP-AMA-RNaseA fusion protein, cells were imaged before (left) or after incubation for 2 h at 4oC (right). (c) Variations in the linker of GFP-AMA-RNaseA(1–14) cause variation in rod-forming propensities, as observed after overnight induction before (AMA, ALA [left]) or after incubation for 24 h at 4oC (AQA, ALA [right], AEA, AKA, AAA). The graph indicates the energy of the highest propensity amyloid-forming hexapeptide predicted by ZipperDB19for each of the six variants. Most notably, the AQA variant predicted to have the strongest amyloid propensity produced no rods, suggesting that the rods are not amyloid-like. (d) Eight appended RNase A residues suffice for rod formation. (e) Appending hexa-histidine tag does not interfere with rod formation. Constructs in panels d and e were imaged after overnight induction and incubation for 2 h at 4oC. (f) V219E, a mutation within the GFP moiety, disrupts filament formation. Cells were imaged after overnight induction and incubation for 120 h at 4oC. For all panels, fluorescence images show representative fields, with illustrative portions enlarged (insets). (g) Fluorescence and phase contrast images of dividing cells connected by continuous rod structures. Cell-spanning rods are indicated by white arrowheads. A similar phenomenon was observed in E. coli cells overexpressing an actin homolog.47 (h) Cells exhibiting rod-induced bulges at their poles. Bulges are indicated by yellow arrowheads. Cells in panels g and h contain GFP-RNaseA(1–14) fusion protein, and were imaged after induction for 4 h and incubation for 24 h at 4oC. Scale bars represent 5 μm.
To define the sequence requirements for intracellular rod formation, we constructed a series of truncations from the C-terminus of RNase A (Fig. 1a). We found that as few as eight N-terminal residues of RNase A (separated from the C-terminus of GFP by a three-residue linker) sufficed to permit rod formation. When cell cultures containing this construct were induced overnight and incubated for 2 hours at 4oC, essentially all of the cells contained fluorescent rods (Fig. 1d). In most cases, the rods extended the entire length of the cell.
Rod formation does not depend on the fortuitous presence of an HP segment.
We considered the possibility that the propensities of the constructed GFP fusion proteins to form fluorescent rod structures was due to the fortuitous presence of an amino acid segment with high amyloid fiber-forming propensity (termed an HP segment) either spanning the fusion junction or contained wholly within the appended sequence. We discovered an HP segment comprising the 3-residue linker and the first 3 residues of RNase A (AMAKET) that was shared by our rod-forming constructs (Fig. 1a and Fig S1)19.
Results from mutagenesis experiments were inconsistent with the hypothesis that rod formation was due to the amyloid-forming propensity of the HP segment located at the N-terminal end of the appended peptide sequence. Using a fusion protein containing RNase A residues 1–14 as our reference construct, we compared the rod-forming behaviors of the reference fusion [GFP-AMA-RNase(1–14)] and several variants bearing replacements of the linker methionine residue (residue 240 in the fusion sequence) with A, E, K, L or Q (Fig. 1c, Fig. S1). Whereas the reference construct gave rise to rods in essentially all of the cells after overnight induction of fusion protein synthesis (Fig. 1c), the variant encoding AQAKET, which has a higher predicted amyloid-forming propensity than AMAKET, gave rise to no rod-containing cells even after induced overnight cultures were refrigerated for an additional 24 hours (Fig. 1 c). Among the remaining variants, only that encoding ALAKET gave rise to rods (which formed efficiently only after refrigeration) (Fig. 1c), suggesting that a hydrophobic residue was required at the second linker position (see below). Overall, our analysis of this set of single amino acid substitution variants suggests that rod formation is not due to the amyloid-forming propensity of the appended peptide. Conclusive evidence that the rods lack amyloid architecture follows from diffraction analysis and crystal structure determination presented below.
Crystal structures of GFP fused to eight RNase A residues.
To elucidate the intermolecular interactions that guide assembly of GFP into intracellular fluorescent rods, we crystallized a fusion protein containing RNase A residues 1–8 [GFP-AMA-RNase(1–8)] (Fig. 1d), the shortest fusion protein that efficiently formed rods. To facilitate purification of the fusion we appended a C-terminal His6 tag [GFP-AMA-RNase(1–8)-His6] after determining that the tag did not alter the ability of the fusion protein to form fluorescent rods (Fig. 1e).
We discovered crystals in dozens of conditions, all of which shared the same needle morphology, recapitulating the rods observed in cells (Fig. S2a). The needles were typically over 100 μm long, but seldom greater than 5 μm thick. Among the different conditions, we discovered four different crystal forms (P21212 form1, P212121 form 2, C2, and P21) (Fig. S2b), collected diffraction data, and solved their structures (Table 1).
Table 1.
Crystallographic data collection and refinement statistics
| GFP-RNase(1–8)- His6 (PDB 5HGE) |
GFP-RNase(1–8)- His6 (PDB 6AS9) |
GFP-RNase(1–8)- His6 (PDB 5HBD) |
GFP-RNase(1- 8)-His6 (PDB 5HW9) |
|
|---|---|---|---|---|
| Data collection | ||||
| Space group | P212121 form 1 | P212121 form 2 | C2 | P21 |
| Cell dimensions | ||||
| a, b, c (Å) | 51.2, 62.9, 69.3 | 51.1, 54.5, 85.5 | 112.7, 51.3, 55.1 | 45.7, 51.8, 54.8 |
| α, β, γ (°) | 90.0,90.0,90.0 | 90.0,90.0,90.0 | 90.0,99.6,90.0 | 90.0, 96.0, 90.0 |
| Resolution (Å) | 46.0–1.86(1.96– 1.86)a |
46.0–1.75(1.80– 1.75) |
46.0–1.65(1.75– 1.65) |
46.0–3.0(3.08– 3.00) |
| Rmerge | 0.106 (0.556) | 0.117 (0.585) | 0.094 (0.534) | 0.189 (0.750) |
| I/σ(I) | 9.4 (2.3) | 14.1 (3.8) | 13.7 (2.3) | 5.8 (1.2) |
| CC1/2 | 0.997(0.836) | 0.996 (0.630) | 0.998(0.765) | 0.975 (0.554) |
| Completeness (%) | 96.7 (98.2) | 99.4 (98.5) | 91.2 (63.8) | 95.1 (84.4) |
| Redundancy | 3.7 (3.7) | 7.1(7.0) | 6.0 (2.9) | 3.2 (2.1) |
| Refinement | ||||
| Resolution (Å) | 46.0–1.86 | 46.0–1.75 | 46.0–1.65 | 46.0–3.00 |
| No. reflections | 18,492 | 24,416 | 30,817 | 4,963 |
| Rwork / Rfree | 0.169/0.211 | 0.156/0.156 | 0.174/0.206 | 0.186/0.232 |
| No. atoms | ||||
| Protein | 1937 | 1952 | 1925 | 1932 |
| Ligands | ||||
| 2-Methyl-2,4-pentanediol | 8 | 16 | 0 | 0 |
| Acetate | 0 | 8 | 0 | 0 |
| Water | 172 | 237 | 312 | 6 |
| B factors | ||||
| Protein | 24.4 | 14.5 | 19.1 | 50.0 |
| Ligands | ||||
| 2-Methyl-2,4-pentanediol | 46.7 | 36.7 | ||
| Acetate | 24.3 | |||
| Water | 30.6 | 25.9 | 30.9 | 39.2 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.010 | 0.010 | 0.013 | 0.010 |
| Bond angles (°) | 1.1 | 1.1 | 1.7 | 1.2 |
Values in parentheses are for highest-resolution shell.
Remarkably, the structures obtained from the four crystal forms revealed essentially identical protofilament architectures, suggesting this architecture is likely to persist under many other conditions, including those within the cell. In each crystal form, the protofilament is assembled with 21 screw symmetry, meaning that each protomer is related to its closest neighbor protomer by a 180° rotation around the protofilament axis and translation along the axis by half the pitch length (i.e. rise per turn along the protofilament axis) (Fig 2a and c). In each crystal form, this pitch length is approximately 51.4 Å, (less than 1 Å variation) and corresponds to one of the unit cell dimensions (a=51.2 Å in P212121 form 1, a=51.1 Å in P212121 form 2, b=51.3 Å in C2, and b=51.8 Å in P21) (Fig. 3b). There is no significant structural variation in protofilament architecture among crystal forms (Fig. 2b). Indeed, the root mean square deviation is less than 0.8 Å between protofilament segments from different crystals (composed of three consecutive GFP-AMA-RNase(1–8)-His6 protomers totaling 5667 atoms). Thus, the robust nature of this protofilament assembly is evidenced by the high degree of similarity in protofilament structures (Fig. 2b) despite clear differences in packing between protofilaments (Fig. 3b).
Fig. 2. Structure of the GFP-RNase(1–8) protofilament determined by x-ray crystallography.

(a and c) Two orientations of the protofilament related by a 45o rotation around the vertical axis. The protofilament is represented as a schematic on the left (large cylinders represent the GFP β-barrel) and cartoon ribbons on the right. The protofilament axis (with 2-fold screw symmetry) is marked by the vertical black line. (b) Superposition of GFP protofilaments from four crystal forms, each shaded according to the color key. The three center protomers (blue, brown and green) were superposed as unified trimers, rather than monomers. The four protofilaments superimpose nearly identically, indicating the protofilament architecture is highly conserved and robust. (d, e, f) Molecular interfaces responsible for robust assembly. (d) A close-up view of the fortuitously formed C-terminal helix of protomer i (cartoon ribbon) fitting in a groove between i+1 and i+2, represented as solvent accessible surfaces. The sequence of the C-terminal fusion and its origins are diagrammed. Arrows connect the individual sequence components to the structure. (e) Details of the helix-groove interface contacts. (f) Barrel-to-barrel stacking contacts between protomers i+1 and i+2 that facilitate fibril formation. The barrel-barrel interface has been observed as a crystal packing interaction previously (for example, see PDB ID 1EMA48).
Fig. 3. Protofilament architecture and bundling in vivo.

(a) Four independent crystal structures of the GFP-RNase fusion reveal essentially identical protofilament assemblies (b) Remarkably, this consensus protofilament structure (one is highlighted in green, blue, brown) is unperturbed despite differences in its alignment with neighboring protofilaments (parallel in C2, and P21, and antiparallel in P212121 forms 1 and 2) and different spacings between neighboring protofilaments. Only one of the unit cell dimensions is constant among the four crystal forms, and this corresponds to the filament pitch (brown scale bar). Crystallographic 21 symmetry axes are denoted by black symbols and half-arrows. (c) An X-ray diffraction pattern from dried E. coli cells carrying GFP-RNase(1–8) filaments reveals six low resolution reflections (labeled 55 to 25 Å). (d) Diffraction radial profiles calculated for each crystal form (orange traces) are overlaid with the observed radial profile of E. coli cells (blue dashed traces). Remarkably, one of the calculated patterns (space group C2) matches the observed pattern, implying that protofilament bundling in vivo resembles the packing observed in the C2 crystal. Correlation coefficient between observed and simulated diffraction profiles are noted in the lower right corner of each subpanel. (e) Calculated fiber diffraction pattern of the C2 crystal structure. (f) Overlay of observed and calculated (space group C2) diffraction patterns. Bands of dark green color highlight the coincidence of the most intense reflections. Similarity is evident in the Bragg spacings, relative intensities of the reflections, and orientation of the reflections with respect to the fibril axis (fibril axis is vertical). Labels indicate Miller indices and Bragg spacings.
A closer view of the protofilament reveals that one structural feature in particular stabilizes protofilament assembly--a new, fusion-created, C-terminal helix which fastens together neighboring pairs of protomers by binding in a groove between them (Fig. 2a, c, and d). This new C-terminal helix comprises seventeen residues which span a sequence derived from three different origins: six residues from the natural GFP C-terminus (233-MDELYK-238), a three-residue linker (239-AMA-241), and eight residues from RNase (242-KETAAAKF-249) (Fig. 2d). The six residues contributed by GFP are normally disordered in crystal structures of natural-length GFP. The propensity of this segment to form a helix was evidently enhanced by its fusion with the RNase residues 1–8, which also form a helix in native RNase.
The new, fusion-created helix extends up to 40 Å away from its attachment to the barrel’s last strand, enabling the helix to reach a binding groove that is formed by the following two protomers in the protofilament. This 40 Å reach is thus directed by the relative positions of three successive protomers in the protofilament; the helix originates from protomer i, and the groove is formed between protomers i+1 and i+2. The ability of the helix to reach this groove is facilitated by the extended conformation adopted by GFP residues 228-GITHG-232, which connect the helix to strand 11, the last strand in the GFP barrel (Fig. 2d).
Protomer i+1 forms one wall of the groove (Fig. 2e, brown surface). This wall comprises the surfaces of strands 3, 10, and 11 near the waist of the barrel, and the helix docks tangentially across these. The helix-in-groove interface is snug and defined almost entirely by side-chain-to-side-chain contacts. A stripe of hydrophobic side-chains (M233, L236, Y237, M240, and T244) lines the length of the α-helix and these embed between side-chains on the surface of the barrel (T43, K45, A206, S208, V219, and L221). Loss of this hydrophobic interface upon substituting M240 with polar, charged, or small residues, such as Q, E, K, or A, would likely explain our observation that these substitutions interfere with efficient rod formation (Fig. 1c) (Fig. S3). This interface between helix i and protomer i+1 buries a total of 727 Å2 on the two surfaces (Fig. S4).
Protomer i+2 forms the opposite wall of the groove. This wall comprises the end of strand 5 and residues at one cap of the barrel (Fig. 2e, blue surface). Specifically, residues L7, P89, and F114 create a small hydrophobic pocket into which docks the side chain of A245 of the helix of protomer i. This contact is flanked on both sides by salt bridges (protomer i+2 residues E90 and D117 with helix i residues K242 and K248, respectively). This interface between protomer i+2 and helix i buries a total of 426 Å2 on the two surfaces (Fig. S4). Overall, the shape complementarity between the helix and groove (0.78) indicates that the fit is even tighter than typically observed among antibody-antigen complexes20.
Direct contact between the sides of neighboring GFP barrels strengthens and rigidifies the contiguous helix-in-groove interaction (Fig. 2f). The interface is approximately flat. Side-chains from strands 1, 2, 5, and 6 of protomer i+1 nestle with side-chains from strands 3, 10 and 11 of protomer i+2. The interactions are primarily polar rather than non-polar. The area buried by the two surfaces of this interface is 901 Å2 (Fig. S4). This patch of direct contact between barrels appears to be adventitious; it does not correspond in any way to the natural dimer interface of wild-type GFP21.
The combined surface area buried by protomers i, i+1, and i+2 appears large enough to maintain protofilament architecture in solution, free of the crystal lattice. Coordination of neighboring protomers by the C-terminal helix buries a total of 1153 Å2. Direct contact between protomers buries an additional 901 Å2 of surface area. Either interface by itself would likely be insufficient to support protofilament assembly. But, together, these total to 2054 Å2, which exceeds the 1712 Å2 threshold value that discriminates between biological and artificial dimers22. The division of the protomer-protomer interface over three surfaces implies cooperativity in protofilament assembly.
Single amino acid substitution disrupts rod formation.
As a further test of whether or not the intermolecular interface observed in the crystals is relevant to rod formation in the cell, we mutated a solvent exposed residue on the GFP barrel that was predicted to contribute significantly to the interface formed by the binding of the C-terminal alpha helix of the neighboring protomer. Specifically, we introduced a charged residue (E) in place of V219, which should disfavor the docking of M233, L236, and M240 in the C-terminal alpha helix (Fig. S3). We then induced the synthesis of GFP-AMA-RNase(1–8)-His6 with or without the V219E substitution in the GFP moiety. Whereas the unmutated fusion protein formed rod structures in essentially all of the cells after overnight induction followed by incubation for 2 hours at 4oC, the mutant fusion protein failed to form rods even after overnight cultures were refrigerated for several days, and instead gave rise to diffuse fluorescence (Fig. 1f and Fig. S5). These findings provide strong support for the hypothesis that the intermolecular interface observed in the four crystal forms is also the basis for fluorescent rod formation in bacterial cells.
The pattern of protofilament bundling in intracellular rods resembles that observed in the C2 crystal form.
We performed in cellulo diffraction experiments to evaluate the pattern of protofilament bundling in fluorescent rods produced in E. coli. X-ray diffraction patterns from E. coli producing GFP-AMA-RNase A(1–8)-His6 revealed six reflections with Bragg spacings between 55 Å and 25 Å (Fig. 3c). Remarkably, these spacings observed from intracellular filaments coincide with those of the crystal structure of purified GFP-AMA-RNase(1–8)-His6 in space group C2 (Fig. 3e). Moreover, the relative intensities of the six observed reflections correlate between the observed and calculated patterns, as well as the geometric disposition with respect to the meridional and equatorial directions (Fig. 3f). Structurally, we find that the Bragg planes for these six reflections coincide well with the spacings between protomers in the crystal (Fig. S6a-g). The sum of these six Fourier terms produces a low-resolution image of the protofilament in close agreement with the crystal structure (Fig. S6h). These correlations, like matching fingerprints, indicate that the protofilaments bundle together in the fluorescent rods in the same pattern as observed in the C2 crystal (Supplementary note 1).
Poor correlation is observed between the in cellulo diffraction and the remaining three crystal forms (P21, P212121 form 1 and P212121 form 2; Fig. 3d). This poor correlation is not an indication of dissimilarity among the protofilament structures. Recall that all four crystal forms are composed of the same protofilament structure (Fig. 2b and 3a). Rather, the differences among the simulated diffraction patterns arise from alternative packing interactions between structurally identical protofilaments (Fig. 3b). Apparently, the cellular milieu favors the same packing of protofilaments as the C2 crystal form.
The symmetry of the protofilaments themselves likely makes them prone to crystal-like bundling, because it corresponds to one of the most prevalent symmetry elements in crystallography—a two-fold screw axis. Fibers of hemoglobin E6V are also rigidified by crystal-like bundling23, and also feature a two-fold screw protofilament symmetry24. Conversely, many functional protofilaments, such as actin and tubulin, have resisted crystallization because their symmetry is imprecise or does not coincide with common crystallographic symmetry elements.
Discussion
We found that appending as few as twelve residues to the C-terminus of GFP enabled the fortuitous assembly of GFP filaments both in vivo and in vitro. These filaments were detectable as fluorescent rods that spanned the length of bacterial cells harboring the fusion construct. The purified fusion protein readily formed needle-like crystals and structure determination revealed the basis for filament formation. Specifically a fusion-created C-terminal α-helix extends away from the barrel to which it is attached and nestles into a groove that is formed by the barrels of two successive protomers along the protofilament axis.
This helix-in-groove linkage between protomers resembles runaway domain swapping10, a mechanism proposed for the assembly of disease-related fibrils. By this mechanism, each protomer swaps a part of its structure into an identical protomer along the protofilament (Fig. 4e). The swapped part is termed the “swapping domain” and may be as small as a single α-helix or β-strand; the receiving part is termed the “complementary domain”. The swapping event begins when a folded or “closed” monomer (Fig. 4c) opens up to expose its own swapping domain to another protomer (Fig. 4d). The flexibility required to open the monomer and execute the swap is afforded by a hinge loop that links the two domains. In GFP-RNase(1–8) protofilaments, the C-terminal helix acts as the swapping domain, the GFP barrel acts as the complementary domain, and intervening residues 229–232 form the hinge loop.
Figure 4. Four basic protofilament-forming mechanisms and their observed combinations.
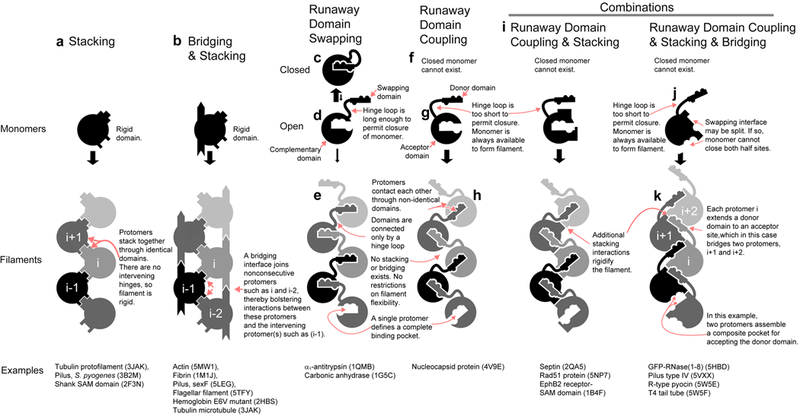
(a) In domain stacked filaments, protomers stack together through rigid domains, with no intervening hinges to confer flexibility. (b) Bridging interfaces join nonconsecutive protomers, creating a mutually supportive closed network that includes the intervening “bridged” protomer(s) (c-e) Runaway domains swapped filaments are characterized by the exchange of domains between neighboring protomers. Hinge loops, which link the domains, confer flexibility to the filament. Nucleation usually requires lengthy or harsh conditions because the closed, self-complemented state (c) is far more stable than the open state (d) in which the swapping element (in this case indicated by a bumpy rectangle) is exposed and the complementing surface (indicated by a cavity) is available for swapping. (f-h) Protofilament assembly through runaway domain coupling features a similar exchange of domains between protomers, but is more easily achieved because the donor domain is permanently open (g), geometrically restricted from self-complementing (for example, by a hinge loop that is too short). Thus no closed monomer exists (f) to compete for protofilament assembly. (i-k)Combinations of stacking, bridging, and coupling elements are commonly observed. For example, stacking, bridging and coupling elements are consolidated in the GFP fusion (j-k). In this case, the complementation site is divided over two surfaces (j). That is, two (or more) protomers cooperate to complement the swapping domain (k), rather than just one (h). Cooperativity implies a larger entropic barrier to nucleation because the nucleus is composed of more protomers, but this limit may be offset by the absence of closed monomer to compete for protofilament assembly. Conceivably, stacking, bridging, and swapping could also coexist if the complementation site resided entirely within protomer i+2.
Other properties of the GFP-RNase filaments, such as facile nucleation and mechanical rigidity, seem incongruous with runaway domain swapping. Indeed, GFP-RNase(1–8) filaments assemble spontaneously in vivo within hours, unlike runaway domain swapped filaments which may take years to assemble in vivo, or require high concentrations, harsh conditions, proteolytic cleavage, or multiple days to nucleate in vitro14, 25 Furthermore, GFP-RNase filaments are mechanically strong and rigid. In some instances, the rods connect two deeply constricted cells, implying that septum formation was incapable of severing a preexisting filament (Fig. 1g). In other instances, the ends of growing GFP-RNase rods appear to impose bulges at the cell poles (Fig. 1h). These distortions of the cell shape are reminiscent of those observed in red blood cells upon aggregation of hemoglobin bearing a single glutamate-to-valine substitution, E6V, associated with sickle cell anemia. This mutation causes hemoglobin tetramers to assemble into rigid fibrils that distort the normally disk-shaped cell to a sickle shape26. In contrast, runaway domain-swapped filaments are connected by flexible hinge loops which typically confer the appearance of “beads-on-a-string.”27. These incongruities prompted us to examine the GFP-RNase protofilament structure for attributes that differ from runaway domain swapping and facilitate nucleation and mechanical rigidity.
Consideration of the monomeric state illuminates the defining attribute that distinguishes GFP-RNase filament formation from runaway domain swapping and explains why GFP-RNase filaments grow more robustly than runaway domain swapped filaments; that is, the GFP-RNase monomer cannot competitively bind its own helical appendage. A defining feature of runaway domain swapping is the high affinity for binding the swapping domain, whether nested within its own protomer (monomeric state) or swapped into a neighboring protomer (filamentous state). The equally deep energetic stability of both the monomeric and filamentous states impedes filament nucleation and growth in two ways: (1) it imposes a large kinetic barrier to the conversion from monomer to filament because the intramolecular domain-domain interface in the monomer must be disrupted to form the equivalent intermolecular domain-domain interface in the filament28, and (2) it renders the monomeric state an energetically viable competitor of filament nucleation and growth since the two states contain equally stabilizing domain-domain interfaces.
In violation of a defining principle of runaway domain swapping, the helix-in-groove interface that stabilizes GFP-RNase protofilaments is unattainable within individual monomers. Indeed, the C-terminal helix of GFP-RNase (donor domain) cannot bind its complementary site within the same monomer (acceptor domain) because the acceptor site is out of reach; the hinge loop is too short (Fig. 4g). Self-complementation is further precluded by that fact that the acceptor site itself is not a single surface, but assembled from two protomers (i+1 and i+2), which respectively contribute residues from opposite faces of the barrel (Fig. 2d and Fig. 4j).
The unattainability of a self-complementary donor-acceptor interface within GFP-RNase monomers explains why this protein forms filaments more readily than proteins that undergo runaway domain swapping. Without a means of self-complementation, protomers in the monomeric state cannot attain the same low energy state as do the protomers in the filament, thereby tilting the energy landscape in favor of filament growth (Fig. 4f-h). The absence of stable self-complementation also has the advantage of eliminating the kinetic barrier to filament formation—with no donor-acceptor interface in the monomer there is no barrier imposed by its disruption as there would be in runaway domain swapped filaments.
We discovered that many natural filaments display donor-acceptor linkages analogous to those observed in the GFP-RNase filaments (Table S1). For example, in filaments of nucleocapsid protein from phlebovirus, each protomer donates a helix to an acceptor domain in the neighboring protomer29. Similarly in pilus rods of fimA protein, each protomer donates an N-terminal strand to its neighboring protomer30. Moreover, GFP-RNase, nucleocapsid protein, and fimA are all incapable of self-complementation. The mechanism of pilus rod formation has been named “donor-strand exchange”, but a more general name is needed to encompass those filaments assembled by donation of elements other than strands. We refrain from using the previously coined term “obligate domain swapping” 31,32, because we have shown the mechanism is not a true domain swap. Instead, we coin the term “runaway domain coupling” to describe all filaments like GFP-RNase in which protomers interface through distinct donor and acceptor domains--interfaces which are unattainable in individual monomers.
Runaway domain coupling describes many protofilament assemblies, but it does not explain why some, like GFP-RNase, are mechanically rigid. Indeed, a flexible linker is characteristic of this mechanism. Rigidity, on the other hand, is a defining characteristic of domain stacking, in which copies of a single rigid domain stack together in uninterrupted succession (Fig. 4a). In GFP-RNase protofilaments, protomers stack through patches on the rigid barrel domains, thus rigidifying the otherwise flexible domain coupled linkages (Fig 2f). This combination of domain coupling and stacking is also evident in naturally occurring RecA nucleoprotein filaments; even in the absence of nucleic acid, RecA forms filaments with a persistence length of about 100 nm (Table S1)(Fig. 4i)33. In contrast, the runaway domain coupled filaments of phlebovirus nucleocapsid protein have no supplementary connection through domain stacking (Fig. 4h). Accordingly, the filaments give the appearance of a random walk in electron micrographs29. Hence, domain stacking imparts rigidity to a protofilaments, acting alone or in combination with other mechanisms.
Bridging is yet another mechanism of filament formation that contributes to the mechanical rigidity of GFP-RNase protofilaments. Bridging interfaces join nonconsecutive protomers in the protofilament, creating a mutually supportive closed network that includes the intervening “bridged” protomer(s) (Fig 4b). In GFP-RNase protofilaments, a bridging interface exists between protomers i and i+2, as part of the helix-in-groove interaction (Fig 4k). This interface in turn, bolsters contacts between these protomers and the intervening protomer i+1. Bridging is also observed in tropomyosin, which forms a coiled coil bridge across protomers in actin filaments34, thereby reinforcing stacking between actin domains and increasing the persistence length by two-fold (Table S1)35. Bridging also helps assemble soluble E6V hemolgobin tetramers into cell-deforming rods24 and tubulin protofilaments into microtubules. Cooperativity is implied by bridging because assembly of two protomers enhances binding of a third protomer.
Genesis of protofilaments by either runaway domain coupling or domain stacking appears relatively difficult to achieve through a single, de novo interface alone. The interface would have to be sufficiently large22 and snug for the protofilament to have a significant lifetime, presumably requiring many iterations of genetic selection. Circular permutation may offer a shortcut by exploiting a preexisting intramolecular interface. For example, circular permutation may shift an N-terminal element to the C-terminus. From its new location, the element may not reach its own complementary site near the N-terminus, but it might easily reach the complementary site of another protomer—a perfect setup for runaway domain coupling (Fig. S7). Circular permutation occurs with sufficient frequency in nature to be a plausible step in protofilament genesis; according to one study, as many as 47% of all domains examined have a circularly permuted homolog36. Simpler still, runaway domain coupled protofilaments might also arise from insertion of a stop codon in a multi-domain oligomeric protein, as engineered in the T7 primase-helicase gene37. Evidently, the removal of some structural elements disrupted cyclic oligomerization in favor of a runaway polymerization.
Surprisingly, protofilaments might be generated faster when protomer-protomer interfaces are divided over multiple surfaces, as observed in GFP-RNase protofilaments (Fig 4k), rather than collected in a single surface. Contrary to the elevated level of complexity inferred by the presence of three distinct interfaces in GFP-RNase protofilaments (runaway domain coupling, stacking, and bridging), this combination allows each of the interfaces to be relatively smaller, and therefore relatively easier to achieve. In fact, we found that a majority of crystal structures of monomeric proteins demonstrate intermolecular interfaces as large as the individual interfaces observed in GFP-RNase protofilaments (Supplementary Note 2).
Flexible linkage between domains, such as occurs in runaway domain coupling, seems advantageous for consolidating these smaller interfaces into a network stable enough to support filament genesis. Consider an early stage of protofilament evolution in which domains can stack, but the stacking interface is too small to be stable. Suppose that this stacking domain is linked to an accessory domain with the potential to evolve a second, stabilizing interface in parallel with the first. If the linker between domains is rigid, only a small portion of the accessory domain may be in proximity of establishing a new interface without disturbing the first. However, if the linker is flexible, as is the case with domain coupling, additional residues may be brought in proximity, improving chances of encountering a snug interface. Indeed, one third of all the naturally occurring filament structures we surveyed involve combinations of runaway domain coupling with stacking (Table S1). Notably, runaway domain coupling, stacking, and bridging were revealed recently in CTP synthase38, a representative of a class of metabolic enzymes that evolved filamentation as a survival response to certain cellular conditions5,6,8,39,40.
The advantage of flexibility for consolidating stacking interfaces comes with an entropic penalty--the immobilization of the interdomain loop. This penalty partially diminishes the stability of the protofilament, perhaps explaining the temperature sensitivity of filament formation by some of the GFP-RNase constructs (Fig. 1b, c) and the sensitivity of CTP synthase filamentation to cellular conditions. These two filaments also score lower values of interface area and solvation free energy than the majority of naturally occurring filaments (Table S1, Fig. S8), suggesting another reason for their sensitivity.
A strategy of combining runaway domain coupling with stacking may offer new advantages for filament design. Notably, flexible linkers were not included in the few examples where globular or helical proteins were successfully engineered to form filaments11, 41,42,43,44,45,46. In all these cases, alignment between protomers was designed computationally, thereby eliminating the need for flexibility to attain alignment. However, the success of most of these designs depended on the presence of a naturally evolved dimerization interface. We propose that a design strategy of combining runaway domain coupling with stacking could open up filamentation to the majority of monomeric proteins, employing interfaces as small as the average crystal packing interface.
Online Methods
Plasmids, Strains, and Cell Growth.
GFP fusions were produced from pBR322-derived plasmids under the control of the arabinose-inducible promoter pBAD. Specifically, the constructs depicted in Fig. 1 (top to bottom) were produced from plasmids pLM163, pLM168, pLM180, pLM179, pLM177, pLM178, pLM176, pLM187, pLM192, and pLM198. Plasmid pLM136 encodes the full-length RNaseA fusion with the amyloidogenic peptide GGVVIA (derived from Aβ) inserted between RNaseA residues 113 and 11418. Constructs with only eight RNaseA residues included an additional aspartate (D) residue with or without a C-terminal hexa-histidine tag. Overnight cultures of E. coli BW2778549,50 transformed with the appropriate GFP-fusion construct were diluted to an OD600 of 0.02 in 50 mL LB supplemented with carbenicillin (100 μg/mL), grown for 30 min at 30 °C, and induced with L-arabinose (0.2% wt/vol). For experiments in which early stationary phase cultures were examined, cells were harvested after 4 h induction at 30 ºC. Otherwise, cultures were induced overnight (~20 h) at 30 ºC. Where indicated, both early stationary phase and overnight cultures were refrigerated at 4 ºC for 2 h or ≥24 h in order to enhance formation of GFP rods.
Fluorescence Microscopy.
Cells were imaged after overnight induction of fusion protein synthesis either without any subsequent incubation or following incubation for the time specified at 4oC. Cells were harvested at 3,000 × g, resuspended in PBS, and spotted onto agarose pads (1% wt/vol in PBS; Seakem LE Agarose, Lonza) for visualization with a UplanFLN 100× phase contrast objective on an Olympus BX61 microscope as described elsewhere51. Images were captured with a CoolSnapHQ camera (Photometrics) and the Metamorph software package, version 6.1 (Universal Imaging). Exposures were typically 50–100 ms. Images were cropped and adjusted using Metamorph or ImageJ52.
Bacterial Extract Preparation and Western Blotting.
Cultures (50 mL) were grown as described above, and the cell densities were recorded (OD600). The cultures were centrifuged 10 min at 3,000 × g at 4 ºC, and the pellets were resuspended in BugBuster Protein Extraction Reagent (EMD Millipore) to normalize for cell density (800μL of lysis buffer for an OD600 of 0.8.). rLysozme (EMD Millipore) was added to 3,000 units/mL and Omnicleave endonuclease (Epicentre) was added to 200 units/mL; the mixture was incubated, rocking, at room temperature (RT) for 30 min. For SDS-PAGE and Western blot analysis, samples were diluted 1:25 in standard SDS-PAGE loading buffer and equal volumes were loaded. Blots prepared in duplicate were probed with either an anti-His mouse monoclonal antibody (Genscript A00612), to detect His6-tagged GFP-fusion protein, or with an antibody specific for the RpoA (α) subunit of E. coli RNA polymerase (Neoclone, clone 4RA2), which served as a loading control. Blots were detected using an ECLplus Western Blotting Detection System (GE Heathcare).
Purification of GFP-RNase(1–8) for crystallization.
E. coli strain BW27785 was transformed with pLM192 (pBAD-gfp-AMA-KETAAAKFD-his6) to produce the GFP fusion, GFP-AMA-KETAAAKFD-His6, for crystallization. We term this fusion protein GFP-RNase(1–8). A culture of LB supplemented with100 μg/mL carbenicillin was inoculated with a single transformed colony and grown at 30 ºC overnight. This overnight culture was back diluted to an OD600 of 0.03 in 1 L of fresh LB medium supplemented with 100 μg/mL carbenicillin and grown at 30 ºC 220 rpm until the culture reached mid-log phase (OD600 0.5). L-arabinose was added to a final concentration of 0.2%, and the culture was grown at 30 ºC at 220 rpm for an additional 5 h. Cells were harvested from a 500 mL aliquot of this culture by centrifugation and stored at −80 ºC until used. On the day of purification, the cell pellet was thawed on ice, washed once in cold, sterile PBS, and lysed in 8.5 mL BugBuster supplemented with Santa Cruz Complete™ Protease Inhibitor Cocktail Tablet (1 tablet per 50 mL lysis buffer), 5 μL rLysozyme, and 5 μL Omnicleave. The lysate was centrifuged for 20 min at 12,600 rpm to remove cell debris, and the supernatant fraction was incubated with 3 mL pre-washed Ni-NTA resin (Qiagen) at 4 ºC for 1 h with rocking. After this incubation step, the resin/supernatant mixture was added to a gravity column and drained. The column was washed three times with a buffer containing 50 mM Hepes pH 7.9, 300 mM NaCl, 5 mM BME, and increasing concentrations of imidazole (0, 20 mM, and 60 mM, respectively). Bound protein was eluted in 8 mL elution buffer (50 mM Hepes pH 7.9, 300 mM NaCl, 5 mM BME, 200 mM imidazole), and 1 mL aliquots were frozen on dry ice. Purification protocol was adapted from Ormö et al.48
Crystallization of GFP-RNase(1–8).
The protein was concentrated to approximately 40 mg/mL in a centrifugal concentrator. The buffer was exchanged three times with 20 mM HEPES pH 7.5 and 10 mM NaCl. Crystals were grown by the hanging-drop vapor diffusion method. The drops were set up using a Mosquito robot, dispensing drops composed of 200 nL of protein and 200 nL reservoir. The reservoir volume was 100 μL. Crystals typically grew within a few hours. Crystals in space group P21 and P212121 were cryoprotected by the addition of glycerol to a concentration of 35%.
Crystallization of GFP-RNase(1–8) in space group P212121 form 1.
The reservoir contained 20% ethanol and 30% (v/v) 2-methyl-2,4-pentanediol (Qiagen MPD Suite condition 90 [H6]). The crystal grew at 20°C.
Crystallization of GFP-RNase(1–8) in space group P212121 form 2.
The reservoir contained 0.1 M sodium acetate, pH 5.0 and 65% (v/v) 2-methyl-2,4-pentanediol (Qiagen MPD Suite condition 68 [F8]). The crystal grew at 4°C.
Crystallization of GFP-RNase(1–8) in space group C2.
The reservoir contained 0.1 M sodium citrate, 0.1 M HEPES pH 7.5, and 10% (v/v) 2-methyl-2,4-pentanediol (Qiagen MPD Suite condition 73 [G1]). The crystal grew at 4°C.
Crystallization of GFP-RNase(1–8) in space group P21.
The reservoir contained 0.2 M KCl, 0.005M MgCl2, 0.05 M sodium cacodylate pH 6.5, and 10% (w/v) 1,6-hexanediol (Qiagen Nucleix Suite condition 21 [B9]). The crystal grew at 4°C.
Data collection and processing.
Diffraction data from GFP-RNase(1–8) crystals in space group P212121 (form 1), C2, and P21 were collected at the Advanced Photon Source beamline 24-ID-C using a Dectris Pilatus 6M-F pixel detector. The data set from crystal form 2 of space group P212121 was collected at the beamline 24-ID-E using an ADSC Quantum 315 CCD detector. Each data set was collected from a single crystal at a temperature of 100 K. A cryoprotectant mixture of 35% glycerol and 65% reservoir was used for crystal form P21. The remaining crystal forms required no additional cryoprotectant. The following X-ray wavelengths were used for crystal forms P212121 (form 1), P212121 (form 2), C2, and P21, respectively: 0.9795 Å, 0.9791 Å, 0.9795 Å, and 1.4760 Å. Data was processed using the XDS package53 for all crystals. In addition, autoPROC54 was used for processing data sets in P212121 (form 1), and C2; aimless55 was used for scaling in P212121 (form 1). Data collection statistics are reported in supplemental Table S1.
Structure determination and refinement.
The first crystal form we obtained of GFP-RNase(1–8), P212121 form 1, was isomorphous with a previously determined GFP structure, PDB ID code 4P1Q56. We used these coordinates as a starting model in the refinement of the GFP-RNase(1–8) Buster57. Difference density maps revealed the position of the N-terminal fusion, and these atoms were built using the graphics program Coot58. The remaining structures were determined by molecular replacement using the program Phaser59. The search model for molecular replacement in space group C2 was the refined structure from space group P212121 form 1. The search model for molecular replacement in space groups P21 and P212121 form 2 was the refined structure from space group C2. All models were built and refined using the same software as described above for P212121 form 1. Structures were illustrated using the program Pymol60.
Fiber diffraction simulation.
Crystal diffraction intensities were cylindrically averaged around the filament axis to simulate the radial disorder of a filament using a custom-written Fortran code.
Identification and analysis of potential protofilament-forming interfaces in the Protein Data Bank.
To identify how many globular, monomeric proteins are poised to form protofilaments, we began by screening 130,446 PDB entry headers to identify monomers. We defined “monomers” as entries that contained only a single protein chain, were determined by crystallography, had resolution better than 3.0 Å, and had no biological assembly operators other than the identity matrix. We also excluded redundant entries defined as having the same MOLECULE record and unit cell lengths identical to ± 3Å and unit cell angles identical to ±5°. We found 12,933 entries that passed these criteria for monomers.
To determine how many of these monomers were poised to form screw-related protofilaments, we performed the following steps. First, we applied crystallographic operations to bring the molecule as close as possible to the origin, generating a reference molecule. Then, we applied crystallographic screw operations to the reference molecules, allowing for up to 3 unit cell translations in any combination along positive and negative a, b, and c. Screw operators were accepted as potential protofilament generators only if the reference molecule and the operated molecule were close enough to touch. We defined touching distance as any distance less than twice the distance of the furthest atom of the reference molecule from its center of mass. These criteria produced 73,968 screw-related pairs. Surface areas buried by these interfaces were calculated using the CCP4 program, areaimol55. Of these, 26,375 screw-related pairs meet the criterion of a quasi-stable interface as defined by equaling or surpassing the 901 Å2 of buried surface area (450 Å2 per surface) observed between barrel domains of our GFP fusion protein protofilament. That is, 9119 PDB entries, or 74% of the monomer entries. Including a constraint that one of the termini be located within 12 Å of the intermolecular interface (a threshold defined by our GFP fusion protein) leaves 53% of monomeric PDB entries meeting these combined criteria. That is, over half the monomer entries in the PDB (6816 entries) are poised for filament formation.
Simulation of helix-forming propensity for randomly generated 13-mers.
We used a random number generator and probability weighted amino acid frequencies in vertebrates to calculate one million peptide sequences. We scored the sequences for helix propensity using Chou & Fasman rules61. Using two different seeds for the random number generator, we found 127,952 and 128,171 sequences passed the criteria for helix. That is approximately 13%.
Supplementary Material
Acknowledgments
We thank the Howard Hughes Medical Institute and National Science Foundation (grant 1616265) for support to DSE, and National Institutes of Health (grants OD003806 and GM115941) for support to AH. Diffraction data were collected at the Northeastern Collaborative Access Team beamlines 24-ID-E and C, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. We thank M. Collazo and D. Cascio of the UCLA-DOE X-ray Crystallization and Crystallography Core Facilities for their assistance with crystallization and data collection, J. Abraham for helpful discussion, and S. Dove for critical reading of the manuscript.
Footnotes
Data availability. Atomic coordinates of the X-ray structures of GFP-RNase(1–8) in space groups P212121 forms 1 and 2, C2, and P21 were deposited in the Protein Data Bank (PDB) under accession codes PDB 5HGE, 6AS9, 5HBD, and 5HW9, respectively.
Life Sciences Reporting Summary for this article is available.
Code availability. The custom written Fortran program used to calculate the fibril diffraction pattern displayed in Fig 3e is available upon request.
Competing interests
The authors declare no competing financial interests.
References
- 1.Shih Y-L & Rothfield L The Bacterial Cytoskeleton. Microbiol. Mol. Biol. Rev 70, 729–754 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jockusch BM & Graumann PL The Long Journey: Actin on the Road to Pro- and Eukaryotic Cells. in Reviews of Physiology, Biochemistry and Pharmacology 161 (eds. Amara SG et al. ) 67–85 (Springer Berlin; Heidelberg, 2011). doi: 10.1007/112_2011_1 [DOI] [PubMed] [Google Scholar]
- 3.Bernander R, Lind AE & Ettema TJG An archaeal origin for the actin cytoskeleton: Implications for eukaryogenesis. Commun. Integr. Biol 4, 664–667 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wickstead B & Gull K The evolution of the cytoskeleton. J. Cell Biol 194, 513–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Connell JD, Zhao A, Ellington AD & Marcotte EM Dynamic Reorganization of Metabolic Enzymes into Intracellular Bodies. Annu. Rev. Cell Dev. Biol 28, 89–111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrovska I et al. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. eLife (2014). doi: 10.7554/eLife.02409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aughey GN & Liu J-L Metabolic regulation via enzyme filamentation. Crit. Rev. Biochem. Mol. Biol 51, 282–293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barry RM et al. Large-scale filament formation inhibits the activity of CTP synthetase. eLife 3, e03638 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y & Eisenberg D 3D domain swapping: as domains continue to swap. Protein Sci. Publ. Protein Soc 11, 1285–1299 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett MJ, Sawaya MR & Eisenberg D Deposition Diseases and 3D Domain Swapping. Structure 14, 811–824 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Ogihara NL et al. Design of three-dimensional domain-swapped dimers and fibrous oligomers. Proc. Natl. Acad. Sci. U. S. A 98, 1404–1409 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huntington JA et al. A 2.6 A structure of a serpin polymer and implications for conformational disease. J. Mol. Biol 293, 449–455 (1999). [DOI] [PubMed] [Google Scholar]
- 13.Strop P, Smith KS, Iverson TM, Ferry JG & Rees DC Crystal structure of the ‘cab’-type beta class carbonic anhydrase from the archaeon Methanobacterium thermoautotrophicum. J. Biol. Chem 276, 10299–10305 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Guo Z & Eisenberg D Runaway domain swapping in amyloid-like fibrils of T7 endonuclease I. Proc. Natl. Acad. Sci 103, 8042–8047 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahlbom M et al. Fibrillogenic oligomers of human cystatin C are formed by propagated domain swapping. J. Biol. Chem 282, 18318–18326 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Jaskólski M 3D domain swapping, protein oligomerization, and amyloid formation. Acta Biochim. Pol 48, 807–827 (2001). [PubMed] [Google Scholar]
- 17.Sambashivan S, Liu Y, Sawaya MR, Gingery M & Eisenberg D Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature 437, 266–269 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Teng PK & Eisenberg D Short protein segments can drive a non-fibrillizing protein into the amyloid state. Protein Eng. Des. Sel. PEDS 22, 531–536 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldschmidt L, Teng PK, Riek R & Eisenberg D Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci 107, 3487–3492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawrence MC & Colman PM Shape complementarity at protein/protein interfaces. J. Mol. Biol 234, 946–950 (1993). [DOI] [PubMed] [Google Scholar]
- 21.Yang F, Moss LG & Phillips GN The molecular structure of green fluorescent protein. Nat. Biotechnol 14, 1246–1251 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Ponstingl H, Henrick K & Thornton JM Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins 41, 47–57 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Wellems TE & Josephs R Crystallization of deoxyhemoglobin S by fiber alignment and fusion. J. Mol. Biol 135, 651–674 (1979). [DOI] [PubMed] [Google Scholar]
- 24.Harrington DJ, Adachi K & Royer WE Jr The high resolution crystal structure of deoxyhemoglobin S1. J. Mol. Biol 272, 398–407 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Gotte G, Libonati M & Eisenberg D Structures of the two 3D domain-swapped RNase A trimers. Protein Sci 11, 371–380 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, Kishchenko G, Chen Y & Josephs R Polymerization of deoxy-sickle cell hemoglobin in high-phosphate buffer. J. Struct. Biol 131, 197–209 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Lomas DA, Evans DL, Finch JT & Carrell RW The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 357, 605–607 (1992). [DOI] [PubMed] [Google Scholar]
- 28.Bennett MJ, Choe S & Eisenberg D Domain swapping: entangling alliances between proteins. Proc. Natl. Acad. Sci 91, 3127–3131 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raymond DD, Piper ME, Gerrard SR, Skiniotis G & Smith JL Phleboviruses encapsidate their genomes by sequestering RNA bases. Proc. Natl. Acad. Sci. U. S. A 109, 19208–19213 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hospenthal MK et al. The Cryoelectron Microscopy Structure of the Type 1 Chaperone-Usher Pilus Rod. Structure 25, 1829–1838.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhlman B, O’Neill JW, Kim DE, Zhang KY & Baker D Conversion of monomeric protein L to an obligate dimer by computational protein design. Proc. Natl. Acad. Sci. U. S. A 98, 10687–10691 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelley BS, Chang LC & Bewley CA Engineering an obligate domain-swapped dimer of cyanovirin-N with enhanced anti-HIV activity. J. Am. Chem. Soc 124, 3210–3211 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Ruigrok RWH & DiCapua E On the polymerization state of recA in the absence of DNA. Biochimie 73, 191–198 (1991). [DOI] [PubMed] [Google Scholar]
- 34.von der Ecken J et al. Structure of the F-actin-tropomyosin complex. Nature 519, 114–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenberg MJ, Wang C-LA, Lehman W & Moore JR Modulation of Actin Mechanics by Caldesmon and Tropomyosin. Cell Motil. Cytoskeleton 65, 156–164 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung J & Lee B Circularly permuted proteins in the protein structure database. Protein Sci. Publ. Protein Soc 10, 1881–1886 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawaya MR, Guo S, Tabor S, Richardson CC & Ellenberger T Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell 99, 167–177 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Lynch EM et al. Human CTP synthase filament structure reveals the active enzyme conformation. Nat. Struct. Mol. Biol 24, 507–514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barry RM & Gitai Z Self-assembling enzymes and the origins of the cytoskeleton. Curr. Opin. Microbiol 14, 704–711 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen Q-J et al. Filamentation of Metabolic Enzymes in Saccharomyces cerevisiae. J. Genet. Genomics Yi Chuan Xue Bao 43, 393–404 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandya MJ et al. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry (Mosc.) 39, 8728–8734 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Potekhin SA et al. De novo design of fibrils made of short alpha-helical coiled coil peptides. Chem. Biol 8, 1025–1032 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Padilla JE, Colovos C & Yeates TO Nanohedra: using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc. Natl. Acad. Sci. U. S. A 98, 2217–2221 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeates TO & Padilla JE Designing supramolecular protein assemblies. Curr. Opin. Struct. Biol 12, 464–470 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Mou Y, Yu J-Y, Wannier TM, Guo C-L & Mayo SL Computational design of co-assembling protein-DNA nanowires. Nature 525, 230–233 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Sayre TC, Lee TM, King NP & Yeates TO Protein stabilization in a highly knotted protein polymer. Protein Eng. Des. Sel 24, 627–630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bharat TAM, Murshudov GN, Sachse C & Löwe J Structures of actin-like ParM filaments show architecture of plasmid-segregating spindles. Nature 523, 106–110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ormö M et al. Crystal structure of the Aequorea victoria green fluorescent protein. Science 273, 1392–1395 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Garrity SJ, Sivanathan V, Dong J, Lindquist S & Hochschild A Conversion of a yeast prion protein to an infectious form in bacteria. Proc. Natl. Acad. Sci. U. S. A 107, 10596–10601 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khlebnikov A, Datsenko KA, Skaug T, Wanner BL & Keasling JD Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiol. Read. Engl 147, 3241–3247 (2001). [DOI] [PubMed] [Google Scholar]
- 51.Doan T, Marquis KA & Rudner DZ Subcellular localization of a sporulation membrane protein is achieved through a network of interactions along and across the septum. Mol. Microbiol 55, 1767–1781 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabsch W XDS. Acta Crystallogr. D Biol. Crystallogr 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vonrhein C et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr 67, 293–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Auerbach D et al. Replacement of highly conserved E222 by the photostable non-photoconvertible histidine in GFP. Chembiochem Eur. J. Chem. Biol 15, 1404–1408 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Bricogne G et al. Buster version 1.10.0 (Global Phasing Ltd, 2016). [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delano W The PyMOL Molecular Graphics System (Schrödinger LLC; ). [Google Scholar]
- 61.Chou PY & Fasman, and G. D. Empirical Predictions of Protein Conformation. Annu. Rev. Biochem 47, 251–276 (1978). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.