

Abstract
Modern large-scale agricultural practices that incorporate high density farming with sub-therapeutic antibiotic dosing are considered a major contributor to the rise of antibiotic resistant bacterial infections of humans with species of Salmonella being a leading agriculture-based bacterial infection. Microcin J25, a potent and highly stable antimicrobial peptide active against Enterobacteriaceae is a candidate antimicrobial against multiple Salmonella species. Emerging evidence supports the hypothesis that the composition of the microbiota of the gastrointestinal tract prevents a variety of diseases by preventing infectious agents from proliferating. Reducing clearance of off-target bacteria may decrease susceptibility to secondary infection. Of the Enterobacteriaceae susceptible to microcin J25, Escherichia coli are the most abundant within the human gut. To explore the modulation of specificity, a collection of 207 mutants encompassing 12 positions in both the ring and loop of microcin J25 was built and tested for activity against Salmonella and Escherichia coli strain7s. As has been found previously, mutational tolerance of ring residues was lower than loop residues, with 22% and 51% of mutations, respectively, retaining activity towards at least one target within the target organism test panel. The multi-target screening elucidated increased mutational tolerance at position G2, G3, and G14 than previously identified in panels composed of single targets. Multiple mutations conferred differential response between the different targets. Examination of specificity differences between mutants found that 30% showed significant improvements to specificity towards any of the targets. Generation and testing of a combinatorial library designed from the point-mutant study revealed that microcin J25I13T reduces off-target activity towards commensal human-derived E. coli isolates by 81% relative to Salmonella enterica serovar Enteritidis. These in vitro specificity improvements are likely to improve in vivo treatment efficacy by reducing clearance of commensal bacteria in the gastrointestinal tract of hosts.
Keywords: antimicrobial peptide, pathogenic Salmonella, protein engineering, specificity, combinatorial library
Introduction
It is estimated that every year two million people in the United States suffer from antibiotic resistant infections, with at least 23,000 resulting in death (Martens and Demain, 2017). Overuse and decreased development of antibiotics in multiple economic sectors in both developed and emerging countries is projected to result in increased numbers of infections and deaths over time (O’Neill, 2014). One of the largest avenues of infection by antibiotic resistant bacteria is from agricultural products (Van Boeckel et al., 2015). In the United States a leading foodborne infectious agent is Salmonella (Scallan et al., 2011). Due to both concerns over development and spread of antibiotic resistance amongst bacterial communities as well as the decreased antibiotic discovery rate across the pharmaceutical industry (Lewis, 2013), there is growing desire for so-called next generation antimicrobials (The White House Administration, 2015). Among other features, these agents are designed to have improved efficacy and specificity towards target organisms. Improved specificity is important for at least the following two reasons: first, for reducing selective pressure on broader bacterial communities to develop mechanisms of resistance, which can be spread by horizontal gene transfer (Ochman et al., 2000); second for reducing clearance, or modification of the composition, of microbiota within bacterial communities such as in the gastrointestinal tract. These microfloral environments provide protection against primary and opportunistic infection and are a contributor to a wide variety of factors of human health (Guinane and Cotter, 2013).
One potential development platform is the use of antimicrobial peptides (AMPs), either as free proteins (Lopez et al., 2007), or secreted via engineered probiotics at the site of infection (Borrero et al., 2014; Forkus et al., 2017; Geldart et al., 2015; Geldart et al., 2016; Hwang et al., 2017). Microcin J25 (MccJ25) is one such AMP with high activity against multiple species of Salmonella and other enterobacteriaceae such as Escherichia coli (Duquesne and Destoumieux-Garzón, 2007; Hegemann et al., 2015; Li et al., 2015; Maksimov et al., 2012; Sablé et al., 2000; Severinov et al., 2007; Vincent et al., 2004). Mature MccJ25 is a 21 amino acid lasso protoknot with high thermal, chemical, and proteolytic stability (Pan et al., 2011). Its complete maturation and export require the co-expression of two cytoplasmic enzymes and an export protein (Figure 1.A, proteins B, C, D)(Yan et al., 2012). Its primary mode of action is the inhibition of RNA polymerase via competitive binding to the secondary channel (Adelman et al., 2004; Mukhopadhyay et al., 2004).
Figure 1.
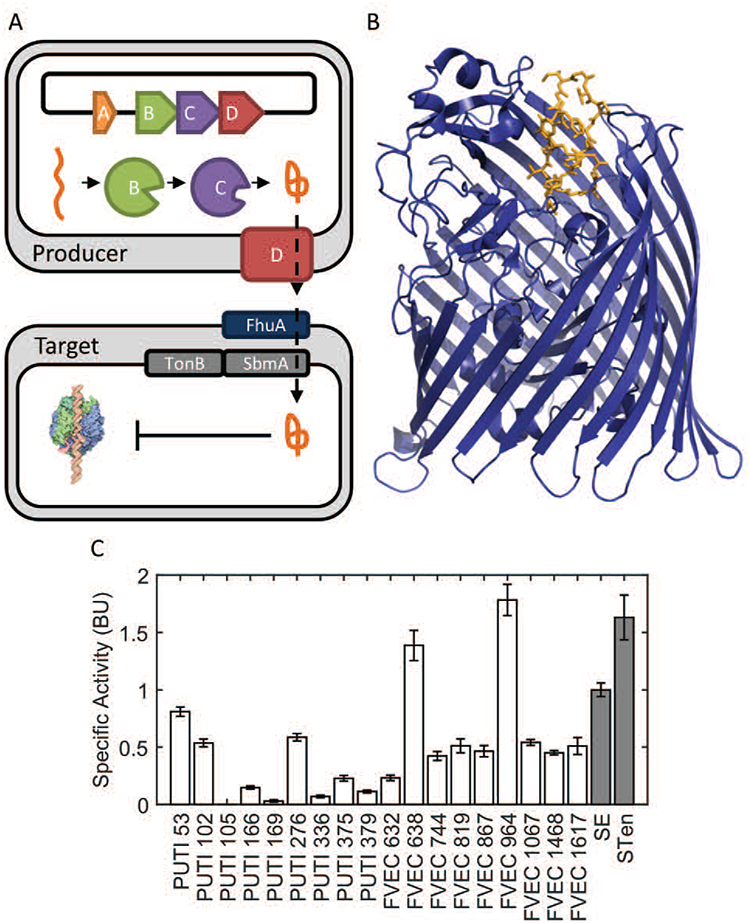
MccJ25 maturation, export, and activity
(A) MccJ25 is the product of a 4-gene cluster composed of A, a 58 amino-acid precursor, B, an ATP-dependent cysteine protease, C, an amidotransferase, and D, an ATP-binding cassette transporter. Following export from a producing bacterium, mature MccJ25 is uptaken through homologs of the iron-siderophore receptor FhuA in a TonB-system mediated transport process. Once intracellular, MccJ25 exerts its more common mode of action, binding to the secondary channel of RNA polymerase thus inhibiting transcription. (B, PDB: 4CU4) Uptake through the FhuA receptor (blue) involves the specific interaction of MccJ25 (orange). (C) Wild-type MccJ25 shows strong activity against pathogenic Salmonella strains (grey) as well as non-pathogenic commensal E. coli (white) isolated from human patients.
Variants of MccJ25 have previously been studied in order to address questions about the flexibility of the maturation machinery, the export mechanism, as well as improved activity (Ducasse et al., 2012; Pan and Link, 2011; Pavlova et al., 2008). As it relates to this work Pavlova et al. previously evaluated nearly the complete set of single mutants of MccJ25 for production, E. coli RNA polymerase inhibition, as well as growth inhibition of E. coli strain DH5α. However, with the exception of one multi-target comparison for a single mutant (MccJ25T15G) the study exclusively focused on a single target organism. Their work revealed that only 45% of MccJ25 point-mutants, which could be produced, exported, and inhibit RNA polymerase in vitro, retained antimicrobial activity. This discrepancy is due to the import process of MccJ25, whose fine details are not yet fully understood (Duquesne and Destoumieux-Garzón, 2007; Severinov et al., 2007). Differences among proteins involved in the import of MccJ25 from different bacterial targets could potentially enable the specificity of MccJ25 to be modulated. The lack of a detailed biophysical description of MccJ25’s interaction with import machinery necessitates high-throughput activity assays to develop MccJ25 variants with improved target specificity. This modulation is critical, as E. coli isolates from humans show high susceptibility to MccJ25 (Figure 1.C).
Though there has been considerable effort to study AMP mutants for improved activity (Avram et al., 2011; Boakes et al., 2012; Field et al., 2013; Healy et al., 2013; Liu and Hansen, 1992; McClintock et al., 2016; Molloy et al., 2013; Tominaga and Hatakeyama, 2006; Tominaga and Hatakeyama, 2007) there are few examples of studies focusing on the modulation of AMP specificity (Haugen et al., 2011; Kazazic et al., 2002). To our knowledge an evaluation of specificity modulation of MccJ25 has not been undertaken and herein we present specificity differences between single-mutant variants of MccJ25 between a collection of Salmonella and E. coli targets (Supplemental Table I). We also explore the ability of a combinatorial library incorporating the most promising mutants to generate functional variants of MccJ25.
Materials and Methods
Bacterial culture and strains
All bacterial growth was performed in either liquid or solid (1% agar) lysogeny broth (LB) with or without supplements. For propagation and maintenance of NEB Iq cells transformed with pJP4 and derivatives, 100 μg/mL ampicillin were used. For induction of MccJ25 variant production, in liquid or solid, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to final concentration of 0.5 mM. For a complete list of plasmids, Escherichia coli production strains, as well as bacterial indicator strains including human commensal E. coli isolates (generously provided by University of Minnesota students and Veterans Affairs Hospital patients and/or their families via Dr. Johnson of the VA Medical Center of Minneapolis), pathogenic E. coli, and Salmonella see Supplemental Table I.
Generation of pJP4 for MccJ25 variant expression
Plasmid containing MccJ25 and maturation operon (pJP3), provided by the Link lab from Princeton (Pan et al., 2010), was digested with XhoI and HindIII (New England Biolabs). Modified MccJ25 insert (Supplemental Table II) containing the sequence to move XhoI downstream as well as an optimized ribosomal binding site (Espah Borujeni et al., 2014) were Gibson assembled (HiFi Assembly Kit, New England Biolabs) with digested plasmid to generate pJP4. This was done to both reduce the size of library inserts used by 30%, as well as remove high redundancy present in the promoter region, simplifying library assembly. This plasmid possesses the T5 promoter under regulation with LacO/LacI to enable inducible production of MccJ25.
Generation of single-site saturation mutagenic libraries of MccJ25
For single-site saturation mutagenesis, oligonucleotides (Supplemental Table II) with NNK degeneracy (all amino acids + stop) at positions 2–7 and 9–14 (each site independently randomized on an independent oligonucleotide) were synthesized and assembled via polymerase chain reaction (PCR, all PCRs conducted with NEB Q5 Polymerase) with additional oligonucleotides to construct MccJ25 variant inserts. Inserts and pJP4 were digested with XhoI and HindIII, and subsequently ligated together with T4 DNA ligase (New England Biolabs) overnight at 16°C. Subsequently, each of the 12 ligations was transformed separately into NEB Express Iq and plated on LB with ampicillin. Once grown, random colonies from each transformation were inoculated into 100 μL LB with ampicillin in 12 sterile 96-well plates and grown overnight. Once grown, 5 μL from each well on each plate were added to a new sterile 96-well plate and mixed to preserve well locations in the final mixed plate. Whole-cell PCR was then conducted to amplify each well separately with unique 4-base pair row and column indices adjacent to the coding region of mature MccJ25. PCR products were then mixed, purified (Qiagen spin column), and amplified with a subsequent PCR to append appropriate adapters for Illumina Sequencing.
Illumina MiSeq sequencing (1/8th lane) generated 1.3 million reads. Sequences lacking full-length MccJ25 with indices with E>33 at all positions were discarded. Unique sequences with >100 reads were isolated and analyzed by a custom MATLAB script. The identity of each mutant within the 96-well plates was processed excluding out-of-library mutants as well as wells with multiple entries identified. For each amino acid mutant, the optimal codon sampled was selected based on that codon’s usage in E. coli (Nakamura et al., 1999).
Generation of multi-site saturation mutagenesis libraries of MccJ25
Oligonucleotides (Supplemental Table II) were synthesized with the introduction of degenerate codons at positions 2, 3, 6, 11, 13, and 14. Degenerate codons were selected that included the desired set of mutant amino acids as well as wild-type. Oligonucleotides were assembled via PCR, restriction digested with XhoI and HindIII, ligated into linearized pJP4 as described above and transformed into NEB Express Iq cells. Random colonies were selected from transformation plates and used directly for screening as described in the following section.
After screening, active clones from the multi-mutant library were indexed in a 96-well plate format using the same indexing primers described previously. To sequence all variants screened, screened plates were scraped for bacteria and plasmid DNA was purified. Plasmid DNA recovered was used for amplification in the same aforementioned indexed primer set. Illumina MiSeq sequencing (1/8th lane) was then used to determine sequence identities for active variants as well as the total pool of variants screened.
Agar-diffusion assay to measure growth inhibition of MccJ25 and variants
For single-mutant evaluation selected MccJ25 variants were inoculated from fresh colonies into 100 μL of LB in 96 well sterile polystyrene plates and incubated overnight shaking at 250 rpm at 37°C. Separately, pathogens to be evaluated were inoculated into 3 mL of LB and grown overnight at 250 rpm at 37°C. The following day, optical densities were determined, and wells were diluted to a final OD600 value of 1.0 (pathlength corrected to 1.0 cm). In addition, plates containing pathogenic target were prepared. This was done by first spreading 50 mL of LB agar supplemented with IPTG. Once solidified, 25 mL of LB agar (cooled to 42°C) supplemented with IPTG as well as 1000x dilution of pathogenic target grown overnight was spread as a thin layer on the plate. Once solidified, 2 μL of cell suspension was then deposited on target pathogen plates using a multi-channel pipette, allowed to dry, and placed at 37°C for overnight growth. After growth, the zone of inhibition was measured using a custom MATLAB script (see Supplemental Figure 1 for example output).
For initial multi-mutant library panning, plates containing Salmonella enterica serovar Enteritidis were prepared as described above. Fresh, randomly selected colonies from the multi-mutant library were transferred onto the pathogen plate and grown overnight at 37°C. Following growth, colonies with any sized zone of growth inhibition surrounding them were classified as active.
For evaluation of active candidates from the multi-mutant screen, fresh colonies of mutants were inoculated into 1 mL of LB supplemented with IPTG in sterile deep 96-well plates and grown at 250 rpm at 37°C for 20 hours. Following this, cells were pelleted at 1000g for 15 minutes. The top 0.5 mL were removed from each well and stored in 1.5 mL sterilized microcentrifuge tubes (supernatant). Supernatant was then sterilized by heating to 98°C for 15 minutes. Plates containing Salmonella targets as well as the subset of commensal E. coli susceptible to MccJ25 were prepared as described above. 5 μL of sterilized supernatant of each variant were then applied using a multi-channel pipette. Once supernatants had dried, plates were grown overnight at 37°C.
For determination of activity of wild-type MccJ25 and MccJ25I13T against Salmonella and commensal E. coli, supernatants from producers as prepared before were diluted with spent LB (prepared the same as supernatants described above but with a non-expressing producer cell line). 5 μL of dilutions were plated on Salmonella as well as commensal E. coli plates as described above, allowed to dry, and grown overnight at 37°C. Minimum inhibitory concentration was determined as a linear fit of the dilution number to diameter of zone of inhibited target growth.
Proteolytic stability assay
Supernatants of MccJ25 and variants, whose production was described previously, were incubated at 60°C for 10 minutes as a 1:1 mixture with phosphate buffered saline (PBS) and varying concentrations of proteinase K (New Englad Biolabs). The samples were then heated to 98°C for 20 minutes to inactive proteinase K. Residual activity of treated supernatants was then determined as described previously using indicator strain Salmonella enterica serovar Enteritidis.
Acid stability assay
Supernatants of MccJ25 and variants, whose production was described previously, had pH adjusted to 1.5 using 1 M HCl and were incubated at 37°C for 30 minutes. Following incubation pH was normalized to 7.0 using 1 M NaOH. Residual activity of treated supernatants was then determined as described previously using indicator strain Salmonella enterica serovar Enteritidis.
Bactericidal/bacteriostatic assay
Supernatants of MccJ25 and variants, whose production was described previously, were applied to indicator strains, in exponential phase growth at 106 colony forming units (cfu)/mL in LB, to a final volume fraction of 20% MccJ25 supernatant. Indicator strains were incubated for one hour at 37°C. Following incubation, cell suspensions were dilution-plated to determine cfu after treatment.
MccJ25 purification
Following (Pan et al., 2010), supernatants of MccJ25 and variants, whose production was described previously, were vigorously mixed with two volumes of n-butanol. The n-butanol phase was removed and dried under vacuum; dried solute was then resuspended in 200 μL of ultrapure (MilliQ) water. Samples were then analyzed by reverse-phase high-performance liquid chromatography (RP-HPLC) with a XBridge Peptide BEH C18 300 Å column with a 10–90% gradient of acetonitrile in water and 0.1% trifluoroacetic acid. All peaks were isolated and evaluated for activity against indicator Salmonella strain. Active peaks were freeze-dried under vacuum, resuspended in ultrapure water and analyzed for identity via matrix-assisted laser desorption/ionization time-of-flight mass spectrometry using an AB Sciex TOF/TOF 5800. Active peaks were re-assessed for activity in ultrapure water against indicator Salmonella strain.
Normalized activity analysis
Throughout this text, normalized activity (Ni,j) for a particular mutant (j) on a particular target organism (i) refers to the normalization of activity data to the activity data of wild-type MccJ25. In the case of the agar-plate assay presented, these wild-type data come from the same plate; thus, this is referred to as intra-plate normalization.
Specific normalized activity analysis
Throughout this text, specific normalized activity refers to the ratio of normalized activity between some organism and the global reference organism (i.e. Salmonella enterica serovar Enteritidis in this text). The principle for this normalization is to evaluate the relative effectiveness of a mutant of MccJ25 compared with wild-type MccJ25 in the context of the assays conducted.
Specificity analysis
In the context of the four-pathogen screening, a mutation (j) is deemed to be specific if it causes a statistically significant deviation in the specificity metric compared to wild type. This metric is defined to be:
There exists no universal metric to evaluate specificity of a process. To enable quantitative analysis of specificity the above metric was generated with the following property: range from zero (all equal normalized activity) to one (only one target having activity greater than zero). The significance of this metric was determined by bootstrap sampling wild-type measurements that were normalized to the intra-plate average activity of two wild-type MccJ25 measurements and performing the above calculation. Based upon this null distribution, values of Sj > 0.3127 were deemed significant (p<0.001).
Statistical analysis of specific normalized activity of MccJ25I13T
For statistical analysis of specific normalized activity of MccJ25I13T against commensal E. coli relative to Salmonella enteritidis a two-sample t-test with unequal variance with the Bonferri correction was utilized. Isolates were deemed significant with p<0.05.
Results
Library Construction and Validation
A collection of degenerate oligonucleotides was used to synthesize point-mutants of MccJ25 at 12 residues. Six of these sites were in the ring region (positions 2–7) which has been previously identified to be highly sensitive to mutation (Pavlova et al., 2008) and form critical interactions with the plug domain of FhuA (Figure 1.B) (Lai and Kaznessis, 2017; Mathavan et al., 2014). Six other sites from the loop region were selected to incorporate positions immediately following the lactam linked E8 as well as several sites previously identified (Pan and Link, 2011; Pavlova et al., 2008) to have high mutational tolerance (positions 9–14). Randomly selected colonies from each sub-library were grown in 96-well plates. The following day, aliquots from each plate were pooled with maintained plate indices into a master 96-well plate. Whole-cell PCR was conducted on each of the wells in isolation (see Supplemental Table II for primers). This process appended row and column indices to each construct which were then used to identify mutant identity via deep sequencing.
Following colony isolation and high-throughput sequencing it was determined that 207 of the 228 (91%) possible point mutations across MccJ25 positions 2–7 and 9–14 were generated via the site saturation mutagenesis. This compares reasonably with the 221 variants (97%) that would be expected by completely random sampling with three-fold coverage of the NNK diversity, as some sampling efficiency was lost due to wells containing multiple mutants.
Single Mutant Activity Analysis
Point mutants were evaluated for growth inhibition, upon recombinant production and secretion, via an agar diffusion assay against two strains of Salmonella enterica (serovar Enteritidis (SE) and serovar Tennessee (STen)) and two strains of E. coli (JJ1887 and O157:H7). These strains were selected due to their known susceptibility to MccJ25, prevalence in multiple domains as pathogens, and high level of laboratory characterization including available genomic data. Following overnight growth, the sizes of the zones of growth inhibition were measured and normalized to the sizes of wild-type MccJ25 zones on each plate (see Materials and Methods for analysis). These data, summarized in Figure 2.A, reveal a range of mutational tolerance. Against all targets, mutations at positions G4 and F10 had no detectable activity, while tolerance was strongly limited at H5 (only R mutant tolerated), P7 (only A mutant tolerated and with notably weaker activity), and F9 (only Y mutant tolerated and with notably weaker activity) (Figure 2.B). Conversely, sites 3 and 12–14 exhibit high tolerance, with moderate mutational tolerance observed for sites 2, 6, and 11. In the context of the assay, ring and loop positions showed an average per-residue mutational tolerance of 22% and 51%, respectively (Supplemental Figure 2).
Figure 2.
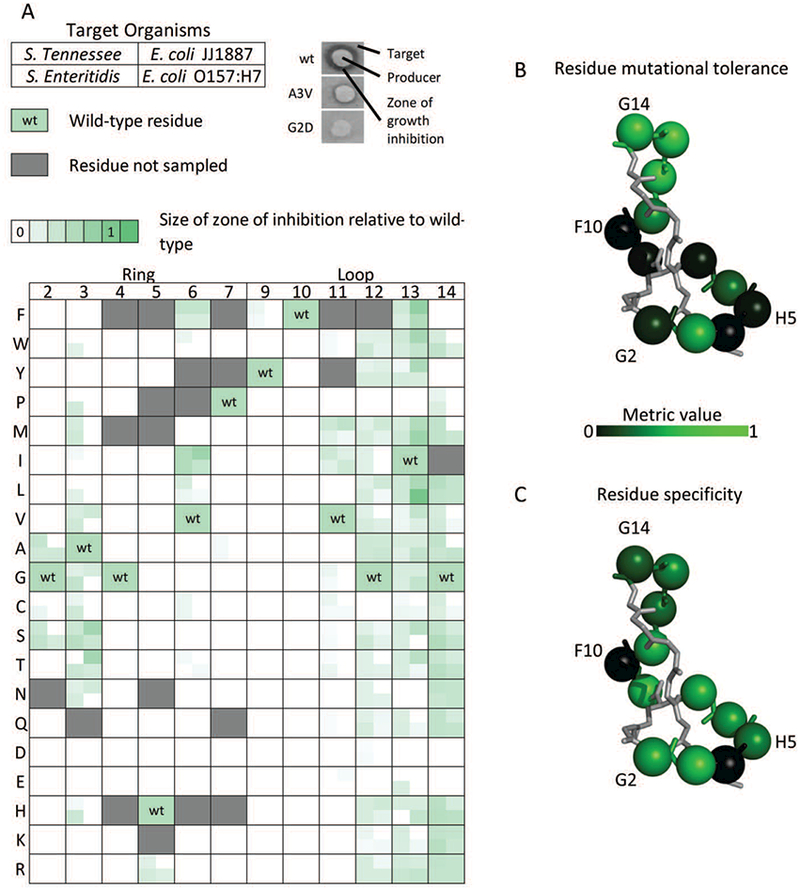
Normalized activity of MccJ25 single-site mutants against four targets
(A) The normalized activities of MccJ25 mutants against two strains of Salmonella as well as two pathogenic E. coli were determined via the size of zones of growth inhibition surrounding colonies of producer cells in solid culture. Each box corresponds to a MccJ25 site and particular amino acid variant (or wild-type as indicated) with four quadrants each that correspond to activity against the four indicated strains. (B-C) The main chain structure of mature MccJ25 with residues highlighted across the mutated position in the ring (2–7), and loop (9–14) as spheres. (B) Mutated positions are colored according to their mutational tolerance, defined as the fraction of mutations showing activity against at least one target. (C) Mutated positions colored according to the average of the specificity metric for mutants at that position which had activity against at least one target.
Negatively charged mutations are broadly unsupported (active in only 4% of test pairs), whereas positively charged mutations show higher broad tolerance (31%), with near-complete tolerance in positions 12–14 (92%).
Chemical homology between wild-type residues and mutants was predictive of activity differences at positions G2, A3, V6, V11, and I13 (p < 0.05; Supplemental Figure 3). Activities of mutants of G12 or G14 were not predictable by homology, despite having high mutational tolerance (84% and 78%, respectively). Tolerated mutations at G2 and A3 were mostly substitutions of small amino acids (A, G, C, S). Against Salmonella targets, partial activity was observed for several hydrophobic mutations (W, P, M, I, L, V) of A3.
This multi-target activity evaluation also enables the determination of how mutations impact the normalized specificity. Across the entirety of MccJ25, 63/207 (30%) mutations show statistically significant specificity modulation (p < 0.001; Figure 3.A). Specificity was negatively correlated with the maximum activity against the four targets (p < 0.001; Figure 3.B). Mutants in the ring region active against at least one target showed higher specificity than mutants in the loop (p < 0.01; Figure 2.C). Chemical homology was not found to be predictive of specificity (Supplemental Figure 4).
Figure 3.
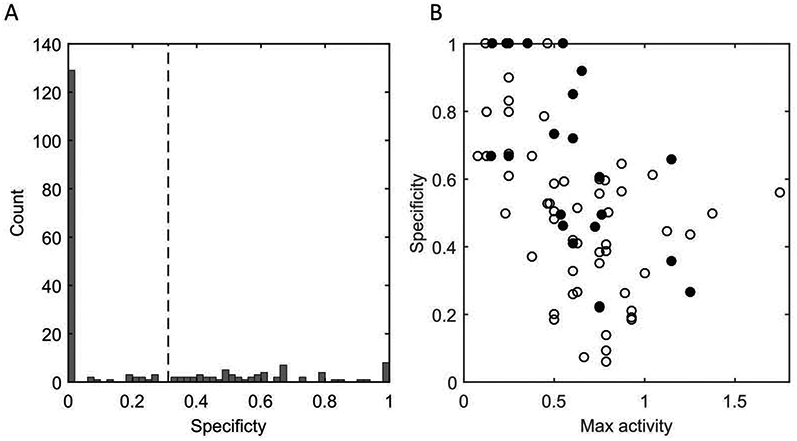
Mutant specificity and correlation with maximum activity
(A) The computed specificity metric across all mutants (including non-active, which have specificity of zero) is presented. The dashed line indicates the 99.999 percentile of the bootstrap sampled wild-type distribution which was defined as the null. Mutants with specificity greater than this were deemed statistically significant from 0. (B) The specificity of active mutants is plotted versus the maximum activity and specificity for mutations with a max activity above 0 for ring (closed circle), and loop (open circle) positions. Spearman’s rho: −0.58 (p = 2.3×10–8). When comparing the specificities of the ring and loop positions, it is found that the mutations in the ring confer greater specificity than those of the loop when considering active mutants (p = 0.0016, Wilcoxon rank-sum right-tailed statistic, μring>μloop).
Multiple Mutant Activity Analysis
Utilizing the single mutant activity data, a collection of mutations across both the ring and loop regions were combined in a multi-mutant library. To design the library, the set of mutants with an average activity against SE and STen of at least 0.5 and a significant specificity metric were identified. A3T was selected as an exception due to differential response between the two Salmonella strains. Three positions in each region were selected: residues 2, 3, and 6 from the ring and residues 11, 13, and 14 from the loop. Due to higher specificity metrics, ring positions were preferentially given more phenotypic variation (4, 5, and 3 options) relative to loop positions (2, 4, and 2 options). Repertoire for each residue was selected with preference for SE and STen as targets with reduced activity against one or both pathogenic E. coli strains. G2T and A3D were included because of codon degeneracy and not selected for attributes of interest. G12 remained fixed in contrast to 13 and 14 to reduce library diversity and give preference to the top of the loop region (Table I). In addition to aforementioned mutational options, library degeneracy included all wild-type residues.
Table I.
Multi-mutant saturation mutagenic library of MccJ25
The composition of the multi-mutant library. Each of the listed positions was encoded using a set of degenerate oligonucleotides to include all combinations of the wild-type and mutant amino acids. The genetic diversity as well as screening results for activity are included.
| Position | Wild-Type | Mutants |
|---|---|---|
| 2 | G | T,S,A |
| 3 | A | M,N,D,T |
| 6 | V | F,L |
| 11 | V | M |
| 13 | I | Y,S,T |
| 14 | G | A |
| Theoretical Diversity | 960 | |
| Sampled Colonies | 2000 | |
| Unique In-library Sequences | 919 | |
| Active Sequences | 32 |
From this library of 960 possible unique sequences, 2000 randomly selected colonies were screened for activity against SE with 64 colonies showing some level of activity (Table I). The relative rarity of active clones was surprising given the library was composed of combinations of active single-mutants (extended observation provided in Discussion). Following Illumina deep sequencing of active and inactive colonies, it was found that the screened set contained 919 unique in-library sequences covering 96% of the designed library sequence space. There was a total of 32 unique sequences (31 mutants and wild-type, Table II) which showed functional activity against SE in the assay. Six mutants of the 12 sampled active single-mutants did not show activity in this assay whereas they did in the first (G2S, A3N, V6F, V6L, I13S, I14A). Codon usage is unable to explain these differences (p > 0.1; Supplemental Table III). Though unidentified as single mutants, each of these single mutants was represented in variants with 2 or more mutations.
Table II.
Active mutants from multi-mutant saturation mutagenesis library
Sequences of active variants from the multi-mutant initial screening with at least two mutations are listed. Blank cells are wild-type amino acids. The activities of these variants against Salmonella enterica serovar enteritidis (SE) are listed.
| 21 | Activity against SE† | ||||||||||||||||||||
| G | +++++ | ||||||||||||||||||||
| Active multi-mutant | |||||||||||||||||||||
| A | Y | ++ | |||||||||||||||||||
| F | T | + | |||||||||||||||||||
| F | Y | ++ | |||||||||||||||||||
| L | Y | + | |||||||||||||||||||
| S | A | + | |||||||||||||||||||
| N | F | ++ | |||||||||||||||||||
| N | M | + | |||||||||||||||||||
| N | A | + | |||||||||||||||||||
| N | S | + | |||||||||||||||||||
| T | F | + | |||||||||||||||||||
| T | A | +++ | |||||||||||||||||||
| T | T | ++++ | |||||||||||||||||||
| T | Y | +++ | |||||||||||||||||||
| S | F | ++++ | |||||||||||||||||||
| S | T | + | |||||||||||||||||||
| S | N | + | |||||||||||||||||||
| A | N | Y | + | ||||||||||||||||||
| A | T | F | + | ||||||||||||||||||
| F | T | A | + | ||||||||||||||||||
| T | M | Y | + | ||||||||||||||||||
| S | M | Y | + | ||||||||||||||||||
| S | N | Y | + | ||||||||||||||||||
| S | T | Y | + | ||||||||||||||||||
| A | M | L | T | + | |||||||||||||||||
Activity against SE was determined by zone-of-inhibition size for each mutant and indicated in one of five bins <20%, <40%, <60%, <80%, and 100% of wild-type MccJ25 indicated by +, ++, +++, ++++, and +++++, respectively.
The activity of MccJ25 and all active multi-mutants were tested against randomly selected human commensal E. coli isolates. These isolates are identified via genetic screening to be probable non-extraintestinal pathogenic E. coli (Johnson et al., 2003), representing non-pathogenic commensal E. coli and are gathered from both urine and fecal samples. Of these 20 isolates, 18 were found to have susceptibility to wild-type MccJ25 in a liquid-culture growth inhibition assay (Supplemental Figure 5).
Those isolates which showed susceptibility were tested against heat-sterilized supernatant derived from active variants from the multi-mutant MccJ25 library (examples from multi-mutant screening stages displayed in Supplemental Figure 6). It was found that the I13T single mutation significantly reduces the specific normalized activity against 15 of 18 of the strains, susceptible to MccJ25 in liquid culture, relative to SE (p<0.05; Figure 4.A–C). Assessment of MccJ25 and I13T for bactericidal/bacteriostatic action demonstrated similar responses against three commensal E. coli strains with a range of MccJ25 responses as well as the indicator Salmonella strains (Figure 4.D). On average, this mutation reduced the specific normalized activity against the 18 evaluated commensals by 81%. This mutation also maintained >50% supernatant activity compared to wild-type MccJ25 against SE. This contrasted with the majority of multi-mutants which had considerable lower normalized activity against SE (Table II).
Figure 4.
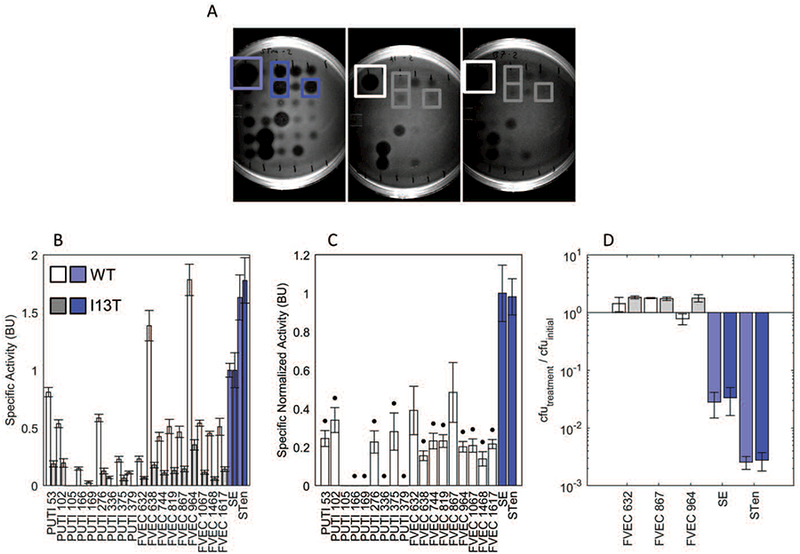
MccJ25I13T demonstrates improved specificity
For pre-purified activity quantification MccJ25 mutants were first grown and induced in liquid culture, then the supernatants were isolated and sterilized by heating to 98 ºC for 15 min. and then plated on agar containing target organisms yielding a spectrum of responses (A, left: STen, middle: PUTI 53, right: FVEC 964). The specific activity, assessed via dilution plating, of wild-type MccJ25 (B, light bars) and MccJ25I13T (B, dark bars), as well as the specific normalized activity (C) are shown against 18 commensal E. coli human isolates (grey bars) as well as well as two strains of Salmonella (blue bars). Data use Salmonella enterica serovar enteritidis (SE) as a reference point. These data demonstrate significant reduction of specific normalized activity for 15 of the 18 E. coli isolates (p<0.05). (D) To differentiate bactericidal from only bacteriostatic response, a subset of commensal E. coli and both Salmonella were incubated for one hour in exponential phase growth with MccJ25 or MccJ25I13T and colony forming units (cfu) determined. A value below unity (100) is indicative of bactericidal activity.
MccJ25 and MccJ25I13T exhibit good thermal stability as evidenced by their activity after heat sterilization (98ºC for 15 min) in the previous assay. Stabilities in the presence of protease and acidic conditions were also evaluated. MccJ25I13T and MccJ25 are essentially equivalent in protease tolerance up to the highly stringent condition of 0.25 U/mL proteinase K at 60ºC for 10 minutes. At more extreme conditions, MccJ25I13T does lose activity, dropping to zero with 1 U/mL protease whereas MccJ25 retained 50% residual activity up to 4 U/mL protease. To assess stability in acid conditions, supernatants of both MccJ25 and MccJ25I13T were incubated at pH 1.5 for 30 min via HCl addition and returned to pH 7.0 via NaOH addition. Both peptides retained full activity (Figure 5.B). The activities of purified MccJ25 and MccJ25I13T, extracted and purified from supernatant, were assessed against SE, demonstrating activity of the pure form of both (Supplemental Figure 7).
Figure 5.
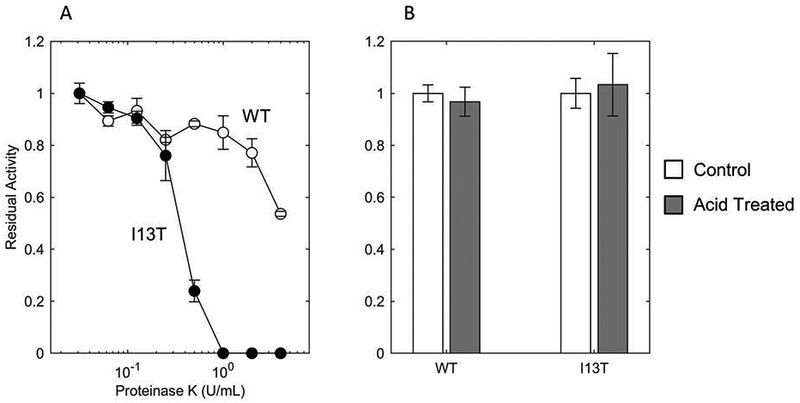
Proteolytic and acid stability of MccJ25 and MccJ25I13T
(A) Supernatant of wild-type MccJ25 (WT, open circles) and MccJ25I13T (I13T, filled circles) were incubated at 60°C for 10 minutes in the presence of varying concentrations of Proteinase K followed by Proteinase K inactivation by incubating at 98°C for 20 minutes. These processed supernatants were then deposited on SE-seeded LB agar plates. Following growth, the size of the zones of growth inhibition were measured to determine residual activity. (B) Supernatant of WT and I13T were incubated at 37°C for 30 minutes at pH 1.5 (via HCl, grey bars) followed by normalization to pH 7.0 (via NaOH). Residual activity was then determined using SE as an indicator strain.
Discussion
Significant effort remains to develop platforms for next generation antimicrobials. The use of ribosomally synthesized AMPs offers the advantage to explore functional sequence space using straightforward genetic manipulation toolkits. Though many efforts have explored the functional sequence landscape of ribosomally synthesized AMPs, few studies have focused on the modulation of specificity of these proteins. Improved specificity for target pathogens can reduce the pressure for AMP resistance development against off-target bacteria as well as improve patient standard of care by reducing microfloral disruption.
Through the evaluation of a collection of single- and multi-mutants of MccJ25, an I13T mutation was identified which significantly reduces activity towards 15 of 18 randomly selected E. coli isolates from humans relative to activity against SE. Though wild-type MccJ25 specificity and efficacy is an improvement over tradition broad-spectrum antibiotics, its high efficacy towards commensal E. coli could pose an issue for human application. Of the E. coli isolates tested, 90% have susceptibility to MccJ25, 55% have susceptibility at least half that of SE and 10% show higher susceptibility. In contrast, 83% of MccJ25-susceptible isolates have reduced susceptibility to MccJ25I13T relative to SE, with an average reduction of 81%.
All testing was done using assays that measure total activity, the combination of production rate and per-molecule activity. The high specificity modulation of ring-position mutants, possibly driven by interactions during binding to the plug domain of FhuA, lost out in the screening performed likely because of the significant losses to activity these mutations acquired. Future studies could explore promising ring-position mutations further to evaluate per-molecule activity.
In addition to the specificity modulation, it should be noted that though the mutational tolerance data provided by singletons is complementary of work done by Pavlova et al., there are several deviations. Several mutations at position G2 (A, C, S) were found to be active in the agar diffusion assay used here, however they were not detected for production, maturation, export, and stability by Pavlova et al. It is possible that the expression system in this work may produce larger quantities of MccJ25 and variants than the naturally-occurring gene cluster utilized in Pavlova et al.’s work. The gene cluster used in this work is derived from the cluster developed by Pan and co-workers (Pan et al., 2010).
As demonstrated in Figure 3.B, there is an inverse relationship between specificity and activity for single-mutants of MccJ25. Though unexplored in AMP research, this trade-off is well known in other protein classes, most notably enzymes (Tawfik, 2014). This tradeoff is due to the large combinatorial landscape of proteins, resulting in multi-function optima being rare. A direct consequence of this property is the lack of multi-mutants retaining high levels of activity, further highlighting the necessity of high-throughput screening to discover mutants with both specificity as well as high activity.
MccJ25 and MccJ25I13T exhibit high stability under thermal, acidic, and proteolytic stresses. Though MccJ25I13T showed higher susceptibility to proteinase K, the conditions of the assay (60°C for 10 minutes, high concentration of nonspecific protease) were elevated in comparison to physiological conditions in order to stress the peptides.
In this work it was demonstrated that mutagenic libraries can be used to identify variants of MccJ25 with improved specificity towards pathogenic targets over commensal organisms. This study resonates with previous work regarding the flexibility of MccJ25’s loop region to functional modification (Knappe et al., 2011; Pan and Link, 2011; Pomares et al., 2009) suggesting a capacity of MccJ25 to be tailored to applications of interest. Though these methods are laborious, scaling linearly with target screening, they offer tremendous opportunity to tune AMPs for use in treating human infections. For many AMPs, in particular bacteriocins produced by gram-negative bacteria, the lack of information provided by homologous sequences, solved structures, or descriptions of uptake and mode of action, necessitates studies such as this to provide insight which can be utilized to design sequences with desired properties.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (GM121777 and GM111358) and a grant from the National Science Foundation (CBET-1412283). S.C.R was also supported by a predoctoral National Institutes of Health traineeship (T32GM008347).
We thank Dr. Timothy Johnson and Dr. Michael Sadowsky of the University of Minnesota for their donations of pathogenic E. coli and Salmonella strains used in this study. In addition, we thank Dr. James Johnson of the Veterans Affairs Hospital of Minneapolis for his donation of pathogenic and commensal E. coli strains used in this study. We thank Dr. James Link of Princeton for providing the pJP3 vector. Support from the University of Minnesota Genomics Center, the Digital Technology Center, and the Biotechnology Institute is gratefully acknowledged.
Grant numbers: National Institutes of Health (GM121777 and GM111358) and the National Science Foundation (CBET-1412283). S.C.R was also supported by a predoctoral National Institutes of Health traineeship (T32GM008347).
Footnotes
Competing financial interests: Y. Kaznessis is the founder and president of General Probiotics, Inc., a startup company that plans to commercialize technologies based on antimicrobial probiotics. This interest has been reviewed and managed by the University of Minnesota in accordance with its Conflict of Interest policies.
References
- Adelman K, Yuzenkova J, La Porta A, Zenkin N, Lee J, Lis JT, Borukhov S, Wang MD, Severinov K. 2004. Molecular mechanism of transcription inhibition by peptide antibiotic Microcin J25. Mol. Cell 14:753–762. [DOI] [PubMed] [Google Scholar]
- Avram S, Duda-Seiman D, Borcan F, Radu B, Duda-Seiman C, Mihailescu D. 2011. Evaluation of antimicrobial activity of new mastoparan derivatives using QSAR and computational mutagenesis. Int. J. Pept. Res. Ther 17:7–17. [Google Scholar]
- Boakes S, Ayala T, Herman M, Appleyard AN, Dawson MJ, Cortés J. 2012. Generation of an actagardine A variant library through saturation mutagenesis. Appl. Microbiol. Biotechnol 95:1509–1517. [DOI] [PubMed] [Google Scholar]
- Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin S a., Robinson TP, Teillant A, Laxminarayan R. 2015. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci 112:5649–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrero J, Chen Y, Dunny GM, Kaznessis YN. 2014. Modified Lactic Acid Bacteria Detect and Inhibit Multiresistant Enterococci. ACS Synth. Biol 4:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducasse R, Yan KP, Goulard C, Blond A, Li Y, Lescop E, Guittet E, Rebuffat S, Zirah S. 2012. Sequence determinants governing the topology and biological activity of a lasso peptide, microcin J25. ChemBioChem 13:371–380. [DOI] [PubMed] [Google Scholar]
- Duquesne S, Destoumieux-Garzón D. 2007. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod … 24:75005. [DOI] [PubMed] [Google Scholar]
- Espah Borujeni a., Channarasappa a. S, Salis HM. 2014. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res 42:2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field D, Molloy EM, Iancu C, Draper LA, O’ Connor PM, Cotter PD, Hill C, Ross RP. 2013. Saturation mutagenesis of selected residues of the α-peptide of the lantibiotic lacticin 3147 yields a derivative with enhanced antimicrobial activity. Microb. Biotechnol 6:564–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forkus B, Ritter S, Vlysidis M, Geldart K, Kaznessis YN. 2017. Antimicrobial Probiotics Reduce Salmonella enterica in Turkey Gastrointestinal Tracts. Sci. Rep 7:40695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldart K, Borrero J, Kaznessis YN. 2015. A Chloride-Inducible Expression Vector for Delivery of Antimicrobial Peptides Against Antibiotic-Resistant Enterococcus faecium. Appl. Environ. Microbiol 81:3889–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geldart K, Forkus B, McChesney E, McCue M, Kaznessis YN. 2016. pMPES: A Modular Peptide Expression Syste for the delivery of Antimicrobial Peptides. Pharmaceuticals 9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinane CM, Cotter PD. 2013. Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic organ. Therap. Adv. Gastroenterol 6:295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen HS, Fimland G, Nissen-Meyer J, Pa- BP, Haugen S, Fimland G, Nissen-Meyer J, Haugen HS, Fimland G, Nissen-Meyer J. 2011. Mutational Analysis of Residues in the Helical Region of the Class IIa. Appl. Environ. Microbiol 77:1966–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy B, Field D, O’Connor PM, Hill C, Cotter PD, Ross RP. 2013. Intensive mutagenesis of the nisin hinge leads to the rational design of enhanced derivatives. PLoS One 8:e79563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegemann JD, Zimmermann M, Xie X, Marahiel MA. 2015. Lasso Peptides: An Intriguing Class of Bacterial Natural Products. Acc. Chem. Res 48:1909–1919. [DOI] [PubMed] [Google Scholar]
- Hwang IY, Koh E, Wong A, March JC, Bentley WE, Lee YS, Chang MW. 2017. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat. Commun 8:15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JR, Gajewski A, Lesse AJ, Russo T a. 2003. Extraintestinal Pathogenic Escherichia coli as a Cause of Invasive Nonurinary Infections Extraintestinal Pathogenic Escherichia coli as a Cause of Invasive Nonurinary Infections. Society 41:5798–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazic M, Nissen-Meyer J, Fimland G. 2002. Mutational analysis of the role of charged residues in target-cell binding, potency and specificity of the pediocin-like bacteriocin sakacin P. Microbiology 148:2019–2027. [DOI] [PubMed] [Google Scholar]
- Knappe TA, Manzenrieder F, Mas-Moruno C, Linne U, Sasse F, Kessler H, Xie X, Marahiel MA. 2011. Introducing lasso peptides as molecular scaffolds for drug design: Engineering of an integrin antagonist. Angew. Chemie - Int. Ed 50:8714–8717. [DOI] [PubMed] [Google Scholar]
- Lai PK, Kaznessis YN. 2017. Free Energy Calculations of Microcin J25 Variants Binding to the FhuA Receptor. J. Chem. Theory Comput 13:3413–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis K 2013. Platforms for antibiotic discovery. Nat. Rev. Drug Discov 12:371–87. [DOI] [PubMed] [Google Scholar]
- Li Y, Zirah S, Rebuffat S. 2015. Lasso Peptides: Bacterial Strategies to Make and Maintain Bioactive Entangled Scaffolds 1st ed New York: Springer-Verlag New York. [Google Scholar]
- Liu W, Hansen JN. 1992. Enhancement of the chemical and antimicrobial properties of subtilin by site-directed mutagenesis. J. Biol. Chem 267:25078–25085. [PubMed] [Google Scholar]
- Lopez FE, Vincent PA, Zenoff AM, Salomón RA, Farías RN. 2007. Efficacy of microcin J25 in biomatrices and in a mouse model of Salmonella infection. J. Antimicrob. Chemother 59:676–680. [DOI] [PubMed] [Google Scholar]
- Maksimov MO, Pan SJ, James Link a. 2012. Lasso peptides: structure, function, biosynthesis, and engineering. Nat. Prod. Rep 29:996. [DOI] [PubMed] [Google Scholar]
- Martens E, Demain AL. 2017. The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. (Tokyo) 70:520–526. [DOI] [PubMed] [Google Scholar]
- Mathavan I, Zirah S, Mehmood S, Choudhury HG, Goulard C, Li Y, Robinson CV, Rebuffat S, Beis K. 2014. Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nat. Chem. Biol 10:340–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock MK, Kaznessis YN, Hackel BJ. 2016. Enterocin A mutants identified by saturation mutagenesis enhance potency towards vancomycin-resistant Enterococci. Biotechnol. Bioeng 113:414–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy EM, Field D, O’Connor PM, Cotter PD, Hill C, Ross RP. 2013. Saturation Mutagenesis of Lysine 12 Leads to the Identification of Derivatives of Nisin A with Enhanced Antimicrobial Activity. PLoS One 8:e58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay J, Sineva E, Knight J, Levy RM, Ebright RH. 2004. Antibacterial peptide Microcin J25 inhibits transcription by binding within and obstructing the RNA polymerase secondary channel. Mol. Cell 14:739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Gojobori T, Ikemura T. 1999. Codon usage tabulated from the international DNA sequence databases; its status 1999. Nucleic Acids Res 27:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J 2014. Antimicrobial Resistance : Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist [Google Scholar]
- Ochman H, Lawrence JG, Groisman E a. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. [DOI] [PubMed] [Google Scholar]
- Pan SJ, Cheung WL, Fung HK, Floudas CA, Link AJ. 2011. Computational design of the lasso peptide antibiotic microcin J25. Protein Eng. Des. Sel 24:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan SJ, Cheung WL, Link AJ. 2010. Engineered gene clusters for the production of the antimicrobial peptide microcin J25. Protein Expr. Purif 71:200–206. [DOI] [PubMed] [Google Scholar]
- Pan SJ, Link a. J. 2011. Sequence diversity in the lasso peptide framework: Discovery of functional microcin J25 variants with multiple amino acid substitutions. J. Am. Chem. Soc 133:5016–5023. [DOI] [PubMed] [Google Scholar]
- Pavlova O, Mukhopadhyay J, Sineva E, Ebright RH, Severinov K. 2008. Systematic structure-activity analysis of microcin J25. J. Biol. Chem 283:25589–25595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomares MF, Salomón R a., Pavlova O, Severinov K, Farías R, Vincent P a. 2009. Potential applicability of chymotrypsin-susceptible microcin J25 derivatives to food preservation. Appl. Environ. Microbiol 75:5734–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablé S, Pons AM, Gendron-Gaillard S, Cottenceau G. 2000. Antibacterial activity evaluation of microcin J25 against diarrheagenic Escherichia coli. Appl Env. Microbiol 66:4595–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV., Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States-Major pathogens. Emerg. Infect. Dis 17:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K, Semenova E, Kazakov A, Kazakov T, Gelfand MS. 2007. Low-molecular-weight post-translationally modified microcins. Mol. Microbiol 65:1380–1394. [DOI] [PubMed] [Google Scholar]
- Tawfik DS. 2014. Accuracy-rate tradeoffs: How do enzymes meet demands of selectivity and catalytic efficiency? Curr. Opin. Chem. Biol 21:73–80. [DOI] [PubMed] [Google Scholar]
- The White House Administration. 2015. National Action Plan for Combating Antibiotic-Resistant Bacteria. Open Gov. Natl. Action Plans:1–63. [Google Scholar]
- Tominaga T, Hatakeyama Y. 2006. Determination of essential and variable residues in pediocin PA-1 by NNK scanning. Appl. Environ. Microbiol 72:1141–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga T, Hatakeyama Y. 2007. Development of innovative pediocin PA-1 by DNA shuffling among class IIa bacteriocins. Appl. Environ. Microbiol 73:5292–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent PA, Delgado MA, Farías RN, Salomón RA. 2004. Inhibition of Salmonella enterica serovars by microcin J25. FEMS Microbiol. Lett 236:103–107. [DOI] [PubMed] [Google Scholar]
- Yan KP, Li Y, Zirah S, Goulard C, Knappe TA, Marahiel MA, Rebuffat S. 2012. Dissecting the Maturation Steps of the Lasso Peptide Microcin J25 in vitro. ChemBioChem 13:1046–1052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.