

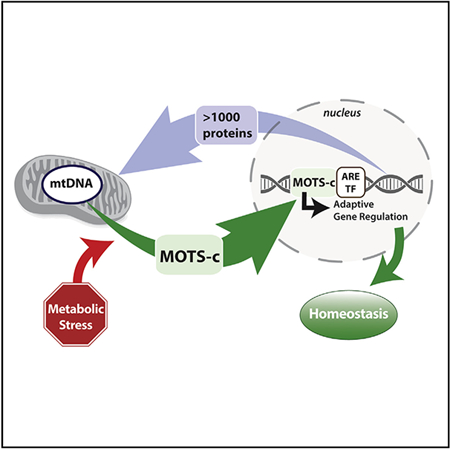
SUMMARY
Cellular homeostasis is coordinated through communication between mitochondria and the nucleus, organelles that each possess their own genomes. Whereas the mitochondrial genome is regulated by factors encoded in the nucleus, the nuclear genome is currently not known to be actively controlled by factors encoded in the mitochondrial DNA. Here, we show that MOTS-c, a peptide encoded in the mitochondrial genome, translocates to the nucleus and regulates nuclear gene expression following metabolic stress in a 5′-adenosine mono-phosphate-activated protein kinase (AMPK)-dependent manner. In the nucleus, MOTS-c regulated a broad range of genes in response to glucose restriction, including those with antioxidant response elements (ARE), and interacted with ARE-regulating stress-responsive transcription factors, such as nuclear factor erythroid 2-related factor 2 (NFE2L2/NRF2). Our findings indicate that the mitochondrial and nuclear genomes co-evolved to independently encode for factors to cross-regulate each other, suggesting that mitonuclear communication is genetically integrated.
In Brief
The mitochondrial genome is regulated by factors encoded in the nucleus. Kim et al. now show that, reciprocally, MOTS-c, a mitochondrial-encoded peptide, can dynamically translocate to the nucleus in response to metabolic stress and regulate adaptive nuclear gene expression. Their findings suggest that mitonuclear communication is genetically integrated.
Graphical Abstract
INTRODUCTION
Mitochondria coordinate complex cellular functions, such as metabolism and stress response, by communicating to the rest of the cell through diverse pathways. Many of the known mitochondrial communication mechanisms include those that respond to cellular stress so as to maintain cellular homeostasis (Chandel, 2015; Quirós et al., 2016). Traditionally, the mediators of mitochondrial communication have been known to be nuclear-encoded proteins, secondary metabolites, transient molecules, and damaged mitochondrial components, but not factors encoded in the mtDNA (Lee et al., 2013).
Mitochondria possess an independent circular genome that is considered a remnant of their symbiotic bacterial ancestry. Thus, the mitochondrial and the nuclear genomes have co-evolved to coordinate complex cellular functions, including metabolism and stress response (Chandel, 2015). To function effectively, mitochondria host over 1,000 proteins that are encoded in the nuclear genome (Hodel et al., 2001). In contrast, factors encoded in the mitochondrial genome that reside in the nucleus with defined functions are currently unknown. In fact, all 13 proteins encoded in the mitochondrial DNA (mtDNA) are structural components of the electron transport chain with critical roles in oxidative phosphorylation with no evident regulatory roles (Gustafsson et al., 2016). Recently, regulatory peptides encoded as short open reading frames (sORFs) in the mitochondrial genome have been identified (Kim et al., 2017; Lee et al., 2013). Such mitochondrial-derived peptides (MDPs) have diverse biological roles, including stress response and metabolic regulation, and add another layer to mitochondrial communication. However, it is unclear whether MDPs can directly and actively regulate nuclear gene expression in response to cellular stress.
MOTS-c is an MDP encoded as a sORF within the mitochondrial 12S rRNA that can regulate cellular metabolism in an AMPK-dependent manner (Lee et al., 2015). MOTS-c treatment prevented diet-induced obesity and insulin resistance and also reversed age-dependent muscle insulin resistance in mice, suggesting a role as a regulator of metabolic homeostasis (Lee et al., 2015).
Of the many organelles that mitochondria communicate with (Murley and Nunnari, 2016), the nucleus is of special interest because it involves adaptive gene regulation. Recent studies have shown that UPRmt can epigenetically regulate the nuclear genome through nuclear-encoded proteins for longevity (Merkwirth et al., 2016; Tian et al., 2016). Further, there are multiple nuclear-encoded mitochondrial-targeted proteins that can also reside in the nucleus as a means to transmit mitochondrial signals (Monaghan and Whitmarsh, 2015). On the contrary, no factors inherently encoded in the mtDNA are currently known to actively migrate to the nucleus. Here, we show that MOTS-c is a mitochondrial-encoded peptide that can dynamically translocate to the nucleus in response to metabolic stress and regulate adaptive nuclear gene expression.
RESULTS
MOTS-c Is a Mitochondrial-Encoded Peptide that Can Reside in the Nucleus
Under resting conditions, endogenous MOTS-c exhibited a predominantly extra-nuclear localization pattern with a certain degree of mitochondrial association (Lee et al., 2015) (Figures 1A and S1A). However, low levels of endogenous MOTS-c were detected in the nucleus under resting conditions by immunofluorescence microscopy (Figures 1B and S1B). Also, we detected MOTS-c by immunoblotting increasing levels of nuclear extracts from resting cells; antibody specificity to MOTS-c was tested by competing out with synthetic MOTS-c peptide (Figures 1C and S1C). We next examined the cellular localization of exogenously treated MOTS-c peptide conjugated with a FITC fluorophore (FITC-MOTS-c; 1 μM) relative to mitochondria and the nucleus. Within 30 min of treatment, FITC-MOTS-c showed mitochondrial and nuclear localization, with a notable punctate pattern throughout the nucleus (Figures 1D and S1D). This suggests that the biological effects of exogenously treated MOTS-c peptide (Lee et al., 2015; Ming et al., 2016) may be, in part, mediated by its actions within the nucleus. To obtain additional spatial information on intracellular MOTS-c, we expressed MOTS-c tagged with an enhanced green fluorescent protein (EGFP-MOTS-c), a widely used technique to determine subcellular protein localization that is complementary to immunofluorescence (Stadler et al., 2013). Similar to nuclear localization signals, which are very short basic peptide motifs that direct large proteins to the nucleus, we found that EGFP-MOTS-c localized not only to mitochondria, but also to the nucleus, using subcellular fractionation and confocal microscopy (Figures 1E and1F). MOTS-c does not possess a known nuclear localization sequence (NLS) but has a cluster of basic residues (13RKLR16), which is typical of a classical NLS (Hodel et al., 2001). Removing the charged 13RKLR16 sequence by substituting with the non-charged alanine residues (i.e., 13AAAA16) did not block the nuclear localization of EGFP-MOTS-c (Figures 1E and1F). MOTS-c also has a hydrophobic core (8YIFY11) that could facilitate interaction with other proteins (Keskin et al., 2008). Substituting the hydrophobic core residues of MOTS-c to alanines (8YIFY11 to 8AAAA11) prevented EGFP-MOTS-c from entering the nucleus (Figures 1E and1F), suggesting that the nuclear translocation of MOTS-c may require interaction with other proteins. Further, the fact that EGFP-MOTS-c only enters in a sequence-specific manner, despite high ectopic expression, suggests that increased concentration alone does not trigger its nuclear translocation. In agreement with our previous report (Lee et al., 2015), wild-type MOTS-c overexpression reduced oxygen consumption rate (OCR) by approximately 40% (Figure S1E). Notably, both nuclear loss-of-function mutants also reduced OCR (Figure S1E), suggesting that the effect of MOTS-c on cellular respiration may involve non-nuclear targets, such as mitochondria. Together, these data indicate that, unlike traditional views, the nuclear proteome includes a peptide that is encoded in the mitochondrial DNA (mtDNA).
Figure 1. MOTS-c Is a Mitochondrial-Encoded Peptide that Can Reside in the Nucleus.

(A) Immunoblots of endogenous MOTS-c from different subcellular fractions of resting HEK293 cells. W, whole-cell lysate; C, cytoplasm; N, nucleus; and M, mitochondria.
(B) Endogenous MOTS-c detection by immunofluorescence microscopy in resting HEK293 cells. The nucleus is outlined by white dashed lines.
(C) Immunoblot detection of endogenous MOTS-c in increasing levels of nuclear extracts from resting HEK293 cells. Antibody specificity to MOTS-c was confirmed by neutralizing peptide competition. Nuclear proteins such as lamin B1 and histone H2B were used as nuclear loading controls.
(D) Localization of exogenously treated FITC-MOTS-c peptide (1 μM; 30 min) in HEK293 cells by confocal microscopy.
(E and F) Subcellular distribution pattern of wild-type (WT) and mutant (8YIFY11 → 8AAAA11 and 13RKLR16 → 13AAAA16) MOTS-c tagged with EGFP in resting HEK293 cells by (E) confocal microscopy (EGFP-MOTS-cWT, EGFP-MOTS-cYIFY, and EGFP-MOTS-cRKLR, respectively) and (F) immunoblotting of subcellular fractions (MWT, MYIFY, and MRKLR, respectively). Representative images are shown (n = 3).
Cells were co-stained with DAPI (nucleus) and MitoTracker Red (mitochondria). Scale bar, 10 μm. See also Figure S1.
Metabolic Stress Triggers MOTS-c to Dynamically Translocate into the Nucleus
Adaptive nuclear translocation of proteins and peptides can occur upon cellular stress (Jovaisaite and Auwerx, 2015; Monaghan and Whitmarsh, 2015). Based on the role of MOTS-c as a metabolic regulator (Lee et al., 2015), we tested if the nuclear trafficking of MOTS-c can be induced by metabolic stress, which would represent a novel nuclear stress-response mechanism that is actively and directly regulated by a mitochondrial-encoded peptide. HEK293 cells were challenged with three different types of metabolic stress: glucose restriction (GR; 0.5 g/L glucose), serum deprivation (SD; 1% fetal bovine serum [FBS]), and oxidative stress (tert-butyl hydrogen peroxide [tBHP]; 100 mM). In response to each of these three stressors, MOTS-c rapidly translocated into the nucleus, as early as 30 min, as determined by subcellular fractionation (Figures 2A–2C) and immunofluorescence imaging (Figures 2D–2G). Such nuclear translocation was transient and shifted back to a largely extra-nuclear state within 24 hr (Figures 2A–2C), reflecting a rapid and progressive stress-response process. Further, paraquat (PQ), another pro-oxidant, also led to an increase in nuclear MOTS-c levels in a dose-dependent manner (Figure S2A). Mitonuclear communication can be mediated by several nuclear-encoded proteins that exhibit dual distribution in mitochondria and the nucleus with differential roles in cellular stress-response. The level of MOTS-c, which showed strong extra-nuclear and mitochondrial-associated localization under resting conditions (Figures 1, 2A–2G, S1, and S2), rapidly declined in the mitochondrial fraction concomitantly with its accumulation in the nuclear compartment upon stress (Figures 2A–2C), suggesting MOTS-c as an adaptive interorganellar communication factor. Interestingly, cytosolic MOTS-c levels decreased in response to GR and SD (1 hr) in HEK293 cells (Figure S2B), which may reflect a transient state of peptide trafficking. Similar effects were observed in HepG2 cells (Figures S2C–S2I). These stressors all increased intracellular reactive oxygen species (ROS) levels (Figures 2H and2I), and its inhibition by pre-treating with N-acetylcysteine (NAC; 10 mM) for 2 hr prevented MOTS-c from translocating to the nucleus upon tBHP (100 μM) treatment (Figure 2J). Further, considering that NAC treatment exhibited elevated MOTS-c levels in whole-cell and mitochondrial extracts, simply increasing total level of MOTS-c alone doesn’t seem to drive the nuclear translocation of MOTS-c. These results collectively indicate that metabolic stress can induce the mitochondrial-encoded peptide MOTS-c to rapidly translocate to the nucleus.
Figure 2. Metabolic Stress Triggers MOTS-c to Dynamically Translocate into the Nucleus.

(A–G) Spatial and temporal assessment of MOTS-c localization in HEK293 after glucose restriction (GR; 0.5 g/L), serum deprivation (SD; 1% fetal bovine serum), and tert−butyl hydrogen peroxide (tBHP; 100 μM) by (A–C) subcellular fraction immunoblots and (D–G) immunofluorescence microscopy. Subcellular fractions were purified at 0, 0.5, 1, 3, 6, and 24 hr post-stress, and confocal microscopy images were acquired 3 hr post-stress. Representative images shown (n = 3). Cells were co-stained with DAPI (nucleus) and MitoTracker Red (mitochondria). Scale bar, 10 μm.
(H) Flow cytometry based on DHE to assess time-dependent reactive oxygen species (ROS) production in response to GR (0.5 g/L), SD (1% fetal bovine serum), and tBHP (100 μM) in HEK293 cells (n = 4).
(I) Fluorescence-activated cell sorting based on MitoSOX staining to assess mitochondrial ROS production following control (DMSO; 0.05%), rotenone (Rot; 10 μM), GR (0.5 g/L), SD (1% fetal bovine serum), and tBHP (100 μM) in HEK293 cells (n = 4).
(J) Subcellular fraction immunoblots following tBHP (100 μM) treatment with and without pre-treatment with NAC (10 mM) for 2 hr. Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by Student’s t test. See also Figure S2.
Stress-Induced Nuclear Translocation of MOTS-c Is Dependent on AMPK
We next set out to identify stress-responsive pathways that intersect with MOTS-c. AMP-activated protein kinase (AMPK) is a key energy-sensing kinase and a master metabolic regulator that is activated by metabolic stress (Hardie et al., 2016) and also mediates the metabolic actions of MOTS-c (Lee et al., 2015; Ming et al., 2016). To that end, we investigated if AMPK was also required for the stress-responsive nuclear translocation of MOTS-c. Pharmacological and genetic interventions that inhibit AMPK activity (compound C [C.C.] and siRNA against AMPKα, respectively) prevented GR-, SD-, and tBHP-induced nuclear translocation of MOTS-c, as determined by subcellular fractionation (Figures 3A and3B) and immunofluorescence imaging (Figures S3A–S3D). Thereafter, we tested if AMPK activation, using metformin and 5-aminomidazole-4-carboxamide ribonucleotide (AICAR), would mimic a stress-like cellular response and cause MOTS-c to translocate to the nucleus. MOTS-c rapidly entered the nucleus within 1 hr of metformin and AICAR treatment in HEK293 cells (Figures 3C–3E). Metformin is thought to inhibit mitochondrial complex I (Willyard, 2017) and increase AMP/ATP ratio (Stephenne et al., 2011), whereas AICAR acts as an AMP mimetic that does not perturb cellular ATP, ADP, or AMP levels (Corton et al., 1995). We confirmed, using C.C. or siRNA against AMPKa, that the effect of metformin and AICAR on MOTS-c translocation was mediated by AMPK (Figures 3D and3E). Notably, both metformin and AICAR did not cause significant ROS production (Figure S3E), indicating that AMPK may regulate the nuclear transport of MOTS-c downstream of ROS (Hardie et al., 2012). These data indicate that MOTS-c translocates to the nucleus in an AMPK-dependent manner.
Figure 3. Stress-Induced Nuclear Translocation of MOTS-c Is Dependent on AMPK.

(A and B) The level of nuclear MOTS-c after 1 hr of GR, SD, and/or tBHP (100 μM) treatment with and without (A) compound C (10 μM), an AMPK inhibitor, for 30 min prior to stress and (B) siRNA against AMPKα or a non-specific sequence (cont) for 48 hr prior to stress, determined by subcellular fractionation immunoblots.
(C) Immunofluorescence staining of MOTS-c after metformin (5 mM; left) or AICAR treatment (2 mM; right).
(D and E) Immunoblots of nuclear fractions from cells treated with metformin or AICAR with and without (D) compound C (10 μM) or (E) siRNA against AMPKα or a non-specific sequence (cont).
Representative images are shown (n = 3). Cells were co-stained with DAPI (nucleus). Scale bar, 10 μm. See also Figure S3.
MOTS-c Binds to Nuclear DNA and Interacts with ARE-Regulating Transcription Factors to Regulate Gene Expression and Increase Cellular Resistance to Metabolic Stress
Within the nucleus of resting cells, MOTS-c could be detected in chromatin extracts under resting conditions (Figure 4A), suggesting that MOTS-c can bind to nuclear DNA. In fact, chromatin-associated MOTS-c levels increased significantly after GR and SD (1 hr) (Figure 4B). Multiple factors translocate to the nucleus upon cellular stress to regulate adaptive gene expression to maintain cellular homeostasis (de Nadal et al., 2011). Nuclear factor erythroid 2-related factor 2 (NFE2L2/NRF2) is a stress-responsive transcription factor that responds to ROS under oxidative stress (Ma, 2013), which was involved in the nuclear translocation of MOTS-c (i.e., GR, SD, and tBHP) (Figures 2H–2J). Notably, NRF2 intersects with AMPK (Joo et al., 2016) and can regulate MOTS-c-related metabolic pathways (Hayes and Dinkova-Kostova, 2014). To start elucidating the role of MOTS-c within the nucleus, we tested whether it interacted with NRF2. Because nuclear MOTS-c levels were abundant 3 hr after stress and returned to baseline after 24 hr (Figures 2A–2G), we studied the MOTS-c/NRF2 interaction at 0, 3, and 24 hr after stress. First, NRF2 co-immunoprecipitated with MOTS-c only from nuclear extracts of cells treated with GR and tBHP for 3 hr; interaction was low under resting states (i.e., 0 and 24 hr) (Figure 4C). Further, we confirmed the interaction between MOTS-c and NRF2 by co-immunoprecipitating EGFP-MOTS-c and Myc-NRF2 (Figure 4D). Interestingly, EGFP-MOTS-cRKLR (13RKLR16 → 13AAAA16 mutant that can translocate to the nucleus) was still capable of interacting with Myc-NRF2 (Figure S4A). MOTS-c was able to translocate to the nucleus under NRF2 knockdown, induced by siRNA (Figure S4B). Conversely, NRF2 was able to translocate to the nucleus under MOTS-c depletion, achieved by actinonin (Lee et al., 2015) (Figure S4C). This indicates that their interaction likely takes place once in the nucleus and that their translocation to the nucleus occurs independently of each other. Second, using electrophoretic mobility shift assays (EMSA), we found that MOTS-c directly bound DNA sequences of ARE-containing promoter regions of NRF2 target genes, including HO-1, NQO1, UGT1A1, UGT1A6, TXN, FTL, and GPX2, in a concentration-dependent manner (Figures 4E and S4D–S4F; Table S1). The binding of MOTS-c to such DNA fragments was lost upon substituting the core hydrophobic or basic residues, which are critical for protein-DNA interactions (Rohs et al., 2010), to alanines (i.e., 8YIFY11 → 8AAAA11 and 13RKLR16 → 13AAAA16, respectively; Figures 4E and S4). Therefore, the interaction with NRF2 and binding to DNA may occur independently of each other. Third, NRF2 generally binds to the antioxidant response element (ARE) sequences (5′-TGACNNNGC-3′) in the promoter of its target genes, such as HO-1 and NQO1, in coordination with other DNA-binding proteins (e.g., small basic leucine zipper transcriptional co-factor Maf proteins) (Hirotsu et al., 2012). Therefore, to test if MOTS-c could also bind to ARE sites in response to stress, we performed chromatin immunoprecipitation followed by quantitative PCR (ChIP-qPCR). The binding of MOTS-c to promoter regions of HO-1 and NQO1 containing ARE sites significantly increased 3 hr after being challenged with GR and tBHP (Figure 4F), then returned to baseline after 24 hr. Further, the binding of NRF2 to the promoter region of HO-1 was significantly increased upon MOTS-c overexpression, determined by ChIP-qPCR (Figure 4G), indicating that regulating the DNA binding of transcriptional factors may be a possible role of nuclear MOTS-c. Fourth, using a luciferase reporter coupled with four copies of NRF2-responsive ARE sequences, we confirmed that the overexpression of MOTS-c significantly increased ARE-dependent transcription in an NRF2-dependent manner (Figures 4H and S4G). Fifth, the over-expression of MOTS-c, but not the 8YIFY11 / 8AAAA11 and 13RKLR16 → 13AAAA16 mutants, significantly increased the transcriptional activity of HO-1 and NQO1 (Figure 4I). However, sulforaphane was able to trigger NRF2-dependent expression of HO-1 in the absence of MOTS-c, achieved using actinonin (Figure S4H). This could be explained by the existence of other functionally redundant NRF2 co-factors (Hirotsu et al., 2012). In fact, actinonin treatment, in addition to depleting MOTS-c levels, causes metabolic stress (Biswas and Chan, 2010), which caused NRF2 to translocate into the nucleus (Figure S4C) as well as increased HO-1 expression (Figure S4H). Because NRF2 shares 73% homology with NFE2L1/NRF1 (OrthoDB entry EOG0906037N [Willyard, 2017]), and considering that they are both involved in ARE-dependent stress-response (Biswas and Chan, 2010), we tested if MOTS-c also interacted with NRF1. Unlike NRF2, the interaction between MOTS-c and NRF1 was relatively unaltered after GR (3 hr) in nuclear extracts but was decreased in chromatin extracts (Figure S4I). This may be explained, in part, by the competitive nature of NRF1 and NRF2 (Stephenne et al., 2011).
Figure 4. MOTS-c Binds to Nuclear DNA and Interacts with Nrf2 to Regulate Gene Expression.

(A and B) Immunoblots of endogenous MOTS-c from chromatin extracts of (A) resting HEK293 cells and (B) HEK293 and HepG2 cells with and without GR (3 hr).
(C) Co-immunoprecipitation of NRF2 by anti-MOTS-c antibody from HEK293 cell nuclear extracts at 0, 3, and 24 hr after GR (top panel) or tBHP (100 μM; bottom panel) determined by immunoblotting.
(D) Co-immunoprecipitation of EGFP-MOTS-c and Myc-tagged NRF2 (Myc- NRF2) in HEK293 cells determined by immunoblotting.
(E) MOTS-cYIFY: 8YIFY11 → 8AAAA11 and pMOTS-cRKLR : 13RKLR16 → 13AAAA16 mutant MOTS-c peptides. Direct MOTS-c/DNA interactions were examined by electrophoretic mobility shift assay (EMSA).
(F) ChIP-qPCR analysis of ARE-containing promoter regions of NQO1 (left) and HO-1 (right) bound to MOTS-c at 0, 3, and 24 hr after GR (top panels) and tBHP (100 μM; bottom panels) (n = 3).
(G) ChIP-qPCR analysis of ARE-containing promoter regions of HO-1 bound to NRF2 in HEK293 cells transfected with empty vector (pEV) or MOTS-c (pMOTS-c) plasmid (n = 3).
(H) ARE-luciferase reporter activity on cells transfected with pEV or pMOTS-c in HEK293 cells in combination with siRNA against NRF2 or a non-specific control (NS) (n = 15).
(I) qRT-PCR analysis of HO-1 and NQO1 expression in response to the overexpression of WT or mutant MOTS-c in HEK293 cells (n = 3).
(J and K) HEK293 cells were stably transfected with pEV, pMOTS-cWT, pMOTS-cYIFY, and pMOTS-cRKLR and subjected to 96 hr of GR + SD. Survival was assessed by (J) phase contrast microscopy images and (K) flow cytometry (n = 3).
(L–N) RNA-seq analyses on HEK293 cells that were transfected with MOTS-c (or empty vector) and subjected to glucose restriction (GR) for 3 hr (n = 6).
(L) Heatmap of significantly differentially regulated genes by MOTS-c upon GR at false discovery rate (FDR) < 5%.
(M) Overlap of genes upregulated by MOTS-c during GR and bona fide NRF2 target genes.
(N) Significantly enriched transcription factor DNA binding motifs in the promoters of genes upregulated (top) and downregulated (bottom) by MOTS-c.
(O) Co-immunoprecipitation of MOTS-c and ATF1 from nuclear extracts of HEK293 cells after GR (3 hr) determined by immunoblotting.
(P) A schematic illustration of the nuclear role of the mitochondrial-encoded peptide MOTS-c in response to metabolic stress.
Error bars represent mean ± SEM. **p < 0.01, ***p < 0.001 by Student’s t test. See also Figure S4 and Tables S1 and S2.
We previously reported that exogenous MOTS-c peptide treatment causes a broad range of gene expression changes in HEK293 under basal conditions using a DNA microarray (Lee et al., 2015). Here, we performed MOTS-c-dependent gene expression profiling under GR by RNA-seq on HEK293 cells that were transfected with a MOTS-c overexpression vector (or empty vector) and subjected to GR for 3 hr. We identified 802 genes that were significantly regulated upon MOTS-c overexpression during GR (412 downregulated and 390 upregulated genes; FDR < 5%) (Figure 4L). Using NRF2 public ChIP-seq datasets in several human cell lines to identify robust NRF2 target genes, we found that a portion of genes upregulated by MOTS-c overlapped with bona fide NRF2 target genes (Figure 4M). In addition, we also found significant enrichment of transcription factor binding motifs in promoters of genes regulated by MOTS-c (Figures 4N). In particular, some of the enriched motifs belonged to activating transcription factor 1 and 7 (ATF1, ATF7) and JUND, which are related to NRF2 and are known to cross-regulate AREs (Chepelev et al., 2013; Stephenne et al., 2011; Willyard, 2017). Further, using ATF1 and JUND public ChIP-seq datasets in several human cell lines, we found that their bona fide targets (without directionality) partially overlap with MOTS-c-regulated genes (Figure S4J–S4N). Interestingly, the overlap between NRF2, ATF1, and JUND targets, although they can all regulate AREs, was also limited, indicating a complex mechanism of target gene selection (Figure S4O). Notably, another member of the ATF family, ATF5, and its C. elegans homolog ATFS-1, can translocate between mitochondria and the nucleus to mediate mitonuclear communication in response to mitochondrial unfolded protein response (UPRmt) (Fiorese et al., 2016). Thus, we tested if the enrichment of ATF1 motifs in MOTS-c-regulated genes under GR was paralleled by increased interaction between MOTS-c and ATF1. We found that MOTS-c co-immunoprecipitated with ATF1 in the nucleus and that their interaction was strengthened after GR (3 hr) (Figure 4O). In contrast to upregulated genes, the overwhelming majority of motifs enriched in promoters of genes downregulated by MOTS-c upon GR were associated to transcription factors involved in immunity, which is in accord with our previously published microarray data (Figure 4N) (Lee et al., 2015).
On a functional level, nuclear MOTS-c increased cellular resistance against metabolic stress. HEK293 cells that stably overexpressed wild-type MOTS-c exhibited significant protection against glucose/serum restriction compared to cells transfected with empty vector control (Figures 4J, 4K, and S4P–S4R). Moreover, cells overexpressing the nuclear loss-of-function MOTS-c mutants (i.e., 8YIFY11 → 8AAAA11 and 13RKLR16 → 13AAAA16) did not confer such protection (Figures 4J and4K). Based on the data presented here, the loss of function of the RKLR mutant may result from a deficient DNA-interaction (Figures 4E and S4F) rather than from an inability to interact with NRF2 (Figure S4A). These data indicate that MOTS-c promotes resistance to metabolic stress by regulating the nuclear genome.
DISCUSSION
It is likely that the endosymbiotic proto-mitochondrial bacteria used peptides encoded in their genome to communicate with our ancestral cells, which is a communication system that is still used by bacteria (Waters and Bassler, 2005). It is plausible that the two genomes co-evolved to cross-regulate each other to coordinate cellular functions. Mitochondria-to-nucleus (retrograde) communication mechanisms that respond to cellular stress, including UPRmt (Jovaisaite and Auwerx, 2015) and damage-associated molecular patterns (DAMPs) (Galluzzi et al., 2012), have been well described, yet are known to be mediated by nuclear-encoded proteins/peptides. To date, factors encoded in the mitochondrial genome that directly regulate the nuclear genome are unknown. We have identified a peptide encoded in the mitochondrial genome that (1) actively translocates into the nucleus in coordination with nuclear-encoded AMPK in response to metabolic stress and (2) directly regulates ARE-containing target genes in the nuclear genome, in part, by interacting with NRF2 (Figure 4P).
In line with the increasing interest in mitonuclear communication in aging, the role of MOTS-c as a mitochondrial-encoded regulator of the nuclear genome may have implications in organismal aging. MOTS-c treatment reversed age-dependent skeletal muscle insulin resistance in mice (Lee et al., 2015), and a MOTS-c polymorphism has been associated with human longevity (Fuku et al., 2015). Thus, considering that glucose depletion, AMPK activation, and increased ROS formation have all been linked to C. elegans longevity (Schulz et al., 2007), MOTS-c may hold key implications for aging and age-related diseases by promoting cellular homeostasis in response to metabolic stress (Bratic and Larsson, 2013; Fuku et al., 2015; Quirós et al., 2016) and maintaining mitonuclear genomic compatibility (Hamilton, 2015; Latorre-Pellicer et al., 2016).
Limitations to Study
Our data are consistent with MOTS-c-dependent mitochondria-to-nucleus communication. However, as discussed above, MOTS-c may also interact with other organelles, suggesting that it may be involved in a more complex interorganellar connectome. Indeed, our RNA-seq data (Figures 4L–4N and S4J–S4O) and microarray data (Lee et al., 2015) indicate that MOTS-c regulates a range of genes involved in various biological processes, including ARE-regulated targets. In addition, that nuclear-defective MOTS-c mutants also reduced OCR suggests that the multifaceted roles of MOTS-c should be studied with defined organellar context. Also, the use of primary human cells and tissues would provide invaluable physiological context and scope of MOTS-c actions. Further work on the mechanistic details of nuclear genome regulation by MOTS-c are required and ongoing in our laboratories.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MOTS-c | This paper | N/A |
| Anti-ATF1 Polyclonal | Thermo Fisher Scientific | Cat# PA5–24788; RRID: AB_2542288 |
| Anti-NFE2L1 Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5–34837; RRID: AB_2552189 |
| Anti-NFE2L2-human (IP) | Abeam | Cat# ab62352 ; RRID: AB_944418 |
| Anti-NRF2 (D1Z9C) XP Rabbit monoclonal | Cell Signaling Technology | Cat# PA5–24788; RRID: AB_2542288 |
| phospho-AMPKα antibody | Cell Signaling Technology | Cat#2535S; RRID: AB_331250 |
| AMPKα antibody | Cell Signaling Technology | Cat#2532S; RRID: AB_330331 |
| Anti-Lamin B1 (D4Q4Z) Rabbit mAb | Cell Signaling Technology | Cat#12586S; RRID: AB_2650517 |
| Anti-Histone H2B (D2H6) Rabbit mAb | Cell Signaling Technology | Cat# 12364; RRID: AB_2714167 |
| Histone H4 antibody | Abeam | Cat#ab10158; RRID: AB_296888 |
| MT-COII antibody | Abeam | Cat#ab79393; RRID: AB_1603751 |
| VDAC antibody | Cell Signaling Technology | Cat#4661S; RRID: AB_10557420 |
| Anti-GAPDH (D16H11) XP Rabbit mAb | Cell Signaling Technology | Cat#5174S; RRID: AB_10622025 |
| Anti-GFP (D5.1) XP Rabbit mAb | Cell Signaling Technology | Cat#2956S; RRID: AB_1196615 |
| Anti-GFP (FL) | Santa Cruz Biotechnology | Cat#sc-8334; RRID: AB_641123 |
| Anti-Myc | Cell Signaling Technology | Cat#5605S; RRID: AB_1903938 |
| Anti-Normal Rabbit IgG | Cell Signaling Technology | Cat#2729S; RRID: ABJ031062 |
| Anti-Rabbit IgG, HRP-linked | Cell Signaling Technology | Cat#7074S; RRID: AB_2099233 |
| Alexa Fluor 488 AffiniPure Goat Anti-Rabbit IgG (H+L) | Thermo Fisher Sceintific | Cat#A-11034; RRID:AB_2576217 |
| Bacterial and Virus Strains | ||
| DH5α Competent Cells | Thermo Fisher Scientific | Cat#18258012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinonin | Enzo Life Sciences | Cat#ALX-260–128 |
| DL-Sulforaphane | Sigma-Aldrich | Cat#S4441 |
| tert-Butyl hydroperoxide solution | Sigma-Aldrich | Cat#458139 |
| Methyl viologen dichloride hydrate | Sigma-Aldrich | Cat#856177 |
| Metformin | Sigma-Aldrich | Cat#317240 |
| AICAR | AdipoGen | Cat#AGCR 10061 |
| MitoTracker Deep Red FM | Thermo Fisher Scientific | Cat#M22426 |
| MOTS-c peptide | GenScript | N/A |
| DAPI | Sigma | Cat#D9542 |
| N-Acetyl Cysteine | Sigma | Cat#A9165 |
| tert-Butyl Hydroperoxide | Sigma | Cat#B2633 |
| Rotenone | Sigma | Cat#8875 |
| Dimethyl Sulfoxide | Sigma | Cat#D8418 |
| Clarity ECL Substrate | Bio-Rad | Cat#1705060 |
| Proteinase K | Thermo Fisher Scientific | Cat#26160 |
| SYBR Green Master Mix | Thermo Fisher Scientific | Cat#4309155 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L3000015 |
| MitoSOX Red | Thermo Fisher Scientific | Cat#M36008 |
| Critical Commercial Assays | ||
| SMARTer Stranded RNA-Seq Kit | Takara/Clontech | Cat#634839 |
| Quant-iT dsDNA HS Kit | Thermo Fisher Scientific | Cat#Q33120 |
| ChIP DNA Clean & Concentrator Kits | Zymo | Cat#D5205 |
| LightShift Chemiluminescent EMSA Kit | Thermo Fisher Scientific | Cat#20148 |
| Dual-Luciferase Reporter Assay System | Promega Corporation | Cat#E1910 |
| NEBNext rRNA Depletion Kit | New England Biolabs | Cat#E6310L |
| Deposited Data | ||
| MOTS-c overexpression RNA-seq | This paper | SRA:SRP136364 |
| Human transcript sequence data | Biomart | Ensv91 |
| ATF1 ChlP-seq in HepG2 cells (and Input controls) | ENCODE DCC; Biswas and Chan, 2010; Brunmair et al., 2004 | SRR2184485; SRR2184486; SRR2184487; SRR2184488; SRR2184489; SRR2184490; SRR2184491; SRR2184492; ENCFF190EPQO; ENCFF950AXC |
| ATF1 ChlP-seq in K562 cells (and Input controls) | ENCODE DCC; Brunmair et al., 2004 | ENCFF000YFC; ENCFF000YFE; SRR351543; SRR351542; SRR351541; SRR351540 |
| ATF1 ChlP-seq in LoVo cells (and Input controls) | Stephenne et al., 2011 | SRR952507; SRR952608 |
| JUND ChlP-seq in HCT-116 cells (and Input controls) | Brunmair et al., 2004 | SRR577903; SRR577904 |
| JUND ChlP-seq in HepG2 cells (and Input controls) | Brunmair et al., 2004 | SRR351817;SRR351818 |
| JUND ChlP-seq in SK-N-SH cells (and Input controls) | Brunmair et al., 2004 | SRR578017;SRR578018 |
| JUND ChlP-seq in MCF-7 cells (and Input controls) | Brunmair et al., 2004 | SRR577997; SRR577998 |
| JUND ChlP-seq in T-47D cells (and Input controls) | Brunmair et al., 2004 | SRR577650; SRR577651 |
| NRF2 ChlP-seq in A549 cells (and Input controls) | ENCODE DCC; Brunmair et al., 2004 | ENCFF003TGD; ENCFF004JMG |
| NRF2 ChlP-seq in HepG2 cells (and Input controls) | ENCODE DCC; Brunmair et al., 2004 | ENCFF063AFU; ENCFF421XBU |
| NRF2 ChlP-seq in Hel_a-S3 cells (and Input controls) | ENCODE DCC; Brunmair et al., 2004 | ENCFF686TXK; ENCFF068LXE |
| NRF2 ChlP-seq in IMR-90 cells (and Input controls) | ENCODE DCC; Brunmair et al., 2004 | ENCFF461GBT; ENCFF467POD |
| NRF2 ChlP-seq in Lymphoblastoid cells cells (and Input controls) | Chepelev et al., 2013 | SRR491140; SRR491141; SRR491142; SRR491143 |
| Experimental Models: Cell Lines | ||
| HEK293 | American Type Culture Collection | CRL-1573 |
| HepG2 | American Type Culture Collection | HB-8065 |
| C2C12 | American Type Culture Collection | CRL-1772 |
| Oligonucleotides | ||
| NFE2L2 Human siRNA Oligo Duplex | Origene | Cat#SR321100 |
| AMPKα2siRNAII | Cell Signaling Technology | Cat# 6630S |
| Please refer to Table S1 for oligonucleotide information | N/A | N/A |
| Recombinant DNA | ||
| pcDNA3.1/NT-eGFP | Gen Script | N/A |
| pcDNA3.1 | Thermo Fisher Scientific | Cat# V79520 |
| Renilla luciferase | Promega Corporation | Cat#E2231 |
| pGL4.37[luc2P/ARE/Hygm]) | Promega Corporation | Cat#E3641 |
| Software and Algorithms | ||
| kallisto 0.43.0 | Bray et al., 2016 | https://pachterlab.github.io/kallisto/download |
| DESeq2 1.16.1 | Corton et al., 1995 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Pheatmap 1.0.8 | Kim et al., 2016 | https://cran.r-project.org/web/packages/pheatmap/index.html |
| HOMER software suite (v4.9.1) | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| GOrilla enrichment tool | Eden et al., 2007, 2009 | http://cbl-gorilla.cs.technion.ac.il/ |
| bowtie 0.12.7 | Langmead et al., 2009 | N/A |
| MACS2 V2.0.8 | Zhang et al., 2008 | N/A |
| bedtools V2.26.0 | Rabinovitch et al., 2017 | https://github.com/arq5x/bedtools2/releases |
| QuantStudio 6 Flex Real-Time PCR software | Thermo Fisher Scientific | N/A |
| GraphPad Prism v6 | GraphPad Software | N/A |
| Other | ||
| In house scripts | This paper | https://github.com/Benayounl_aboratory/MOTSC_nuclear_project |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Changhan David Lee (changhan.lee@usc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293 (source: female), HepG2 (source: male), and C2C12 (source: mouse) were grown in Dulbecco’s modified Eagles’ medium (DMEM) (Corning) supplemented with 10% fetal bovine serum (FBS). All cells were incubated in a humidity-controlled environment at 37 C, 5% CO2 (Heracell VIOS 160i). All cell lines were purchased from ATCC and did not undergo further validation. All cells were utilized up to 20 passages.
METHOD DETAILS
Transfection
Cells were transfected at 60%–80% confluency using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. After 48 hr, cells were used for subcellular fractionation, total cell lysate preparation, immunofluorescence staining, reporter assay, and real-time PCR. pcDNA3.1(+)-MOTS-c construct was described previously (Lee et al., 2015). MOTS-c mutants with quadruple alanine substitutions for residues 8–11 (8YIFY11 → 8AAAA11) and 13–16 (13RKLR16 → 13AAAA16) were constructed in pcDNA3.1(+) by site-directed PCR mutagenesis as previously described (Kadowaki et al., 1989).
For siRNA-mediated knockdown, cells were transfected when they reached 60%–80% confluence. siRNA and Lipofectamine RNAiMAX (Thermo Fisher Scientific) were prepared as instructed in Opti-MEM medium transfection media (GIBCO). A master mix of siRNA (30 pmol per dish) and lipofectamine (9 μL per dish) were individual diluted in 150 μL Opti-MEM medium per dish. These solutions were then combined, mixed gently, and allowed to incubate for 5 min and 250 μL of siRNA-lipid complex were added directly to cells in culture medium. The cells were exposed to the transfection medium for 72 hr. Further experiments were conducted as described above. Knockdown in each experiment was confirmed by western blot.
Treatments
Cells were cultured to reach 70%–80% confluency and washed with phosphate buffered saline (PBS) prior to treatments. Glucose restriction (GR) was carried out using glucose-free DMEM (Thermo Fisher Scientific) supplemented with low glucose (0.5 g/L) and 10% FBS. Serum deprivation (SD) was done by incubating cells in high-glucose DMEM (4.5 g/L) with 1% FBS. Oxidative stress was induced by tert-Butyl Hydroperoxide (tBHP; Sigma) or paraquat (Sigma-Aldrich). Actinonin (Enzo Life Sciences) was treated to deplete MOTS-c levels, (Lee et al., 2015). To activate NRF2, 10 uM sulforaphane was treated for 16 hr.
Subcellular Fractionation
For nuclei isolation, a non-ionic detergent-based purification method was carried out as previously described with some modifications (Gagnon et al., 2014). Briefly, cells were harvested, washed in ice-cold PBS, and resuspended in hypotonic lysis buffer (10 mM Tris, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.3% NP-40, and 10% glycerol) in the presence of a protease/phosphatase inhibitor cocktail (Thermo Fisher Scientific). The suspension was incubated for 30 min on ice, passed five times through a 28-gauge blunt-ended needle and centrifuged at 400 g for 5 min at 4°C. The nuclear pellet was washed three times with hypotonic lysis buffer and collected as a total nuclear fraction.
For chromatin isolation, Wuarin-Schibler buffer (MWS) (10 mM Tris pH 7.0, 4 mM EDTA, 300 mM NaCl, 1 M urea, and 1% NP-40) was used as previously described with minor modifications (Gagnon et al., 2014). Briefly, the washed nuclear pellet was resuspended with MWS containing a protease/phosphatase inhibitor cocktail, incubated for 30 min on ice, and vortexed every 5 min. The suspension was centrifuged at 1,000 g for 5 min at 4°C and the resulting pellets containing chromatin were washed three times with MWS buffer and saved the pellets as chromatin fraction. Genomic DNA was removed by treatment with DNase.
For isolation of mitochondria-enriched cellular fractions, a crude mitochondrial fraction was first obtained as described previously with minor modifications (Frezza et al., 2007). In brief, cells were washed with ice-cold PBS and suspended in chilled mitochondria isolation buffer (IBc) (10 mM Tris-MOPS, pH 7.4, 10 mM EGTA-Tris, 200 mM sucrose, and 5 mM MgCl2) with a protease/phosphatase inhibitor cocktail. The cells were homogenized in IBc buffer using a Teflon pestle (about 50 strokes) and centrifuged at 500 g for 10 min at 4°C to remove unbroken cells. The supernatant was further centrifuged at 2,000 g for 10 min at 4°C to remove cell debris and nucleus. Then the supernatant was collected and centrifuged at 7,500 g for 10 min at 4°C. The pellets containing mitochondria were washed twice with IBc buffer and saved the pellets as mitochondria-enriched fraction. Mitochondria-free cytosolic fraction was collected from the supernatant and further centrifuged at 12,000 g for 10 min at 4°C. The supernatant was saved as cytosolic fraction. Protein levels were determined using a Pierce BCA protein assay (Thermo Fisher Scientific), and each fraction was loaded for western blotting to confirm purity.
Western Blotting
Cellular lysates were extracted with modified RIPA (50 mM Tris, pH 8.0, 75 mM NaCl, 0.3% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing a protease/phosphatase inhibitor cocktail, sonicated, and centrifuged at 12,000 g for 10 min at 4°C to remove debris. The supernatants were subjected to electrophoresis using 8%–16% pre-cast SDS-PAGE gels (Bio-Rad). The resolved gels from pre-cast SDS-PAGE were transferred to PVDF membranes, blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline containing 0.05% Tween-20 (TBS-T) and incubated with primary antibody at 4°C overnight. The membranes were washed three times with TBS-T and incubated with HRP-conjugated secondary antibodies at room temperature for 1 hr. The membrane was washed again three times, developed using Clarity Western ECL substrates (Bio-Rad) and imaged using the ChemiDoc XRS+ system (Bio-Rad).
ChIP-qPCR
ChIP was performed as described previously with some modifications (Nelson et al., 2006). Briefly, cells were cross-linked with 1% formaldehyde for 15 min at room temperature and subsequently quenched by glycine (125 mM) for 5 min at room temperature. Nuclei were isolated as described above and the extracts were resuspended in lysis buffer (50 mM Tris, pH 7.5, 10 mM EDTA, 1% SDS) supplemented with a protease/phosphatase inhibitor cocktail. The chromatin fraction was mechanically sheared at high sonication intensity in cycles of 30 s-on/30 s-off for 30 min. Sonication efficiency was monitored by microscopy and analyzed by agarose gel electrophoresis. Then, 10% Triton-x100 (final concentration of 1%) was added to sheared chromatin extracts before centrifuging at 12,000 g for 10 min at 4°C. The supernatant containing chromatin was immunoprecipitated as input as described above. Protein/DNA complexes were pelleted and washed once each with low salt wash buffer (0.1% SDS, 0.1% Triton X-100, 150 mM NaCl, 2 mM EDTA, 20 mM Tris. pH 8), high salt wash buffer (0.1% SDS, 0.1% Triton X-100, 500 mM NaCl, 2 mM EDTA, 20 mM Tris. pH 8), LiCl wash buffer (0.5 M LiCl, 1% NP-40, 1% Deoxycholic acid, 100 mM Tris. pH 8), and TE buffer (1 mM EDTA, 10 mM Tris. pH 8). After the final wash, the complexes were eluted with elution buffer (50 mM Tris. pH 7.5, 10 mM EDTA, 1% SDS) at 50°C for 30 min. Proteinase K (final concentration of 10 μg/μL; Thermo Fisher Scientific) was added to the eluted chromatin and cross-linking was reversed by incubation at 65°C overnight. The DNA was purified with ChIP DNA Clean & Concentration kit (Zymo Research) and quantified with Quant-iT dsDNA HS kit (Thermo Fisher Scientific) on Qubit 3.0 Fluorometer (Thermo Fisher Scientific). 5 ng of ChIP DNA was amplified using SYBR Green Master Mix (Thermo Fisher Scientific) on QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific). Data were normalized over input as % input. The primer sequences are listed in Table S1.
Luciferase Reporter Assay
HEK293 and HepG2 cells were plated in 12-well plates at 0.5 3 106 cells/well. 24 hr after seeding, cells were transfected with firefly luciferase reporter vectors encoding the ARE (pGL4.37[luc2P/ARE/Hygro]) and renilla luciferase encoding plasmids (pRL-TK) (Promega Corporation) using Lipofectamine 3000 (Thermo Fisher Scientific) per manufacturer’s instruction. Cells were equilibrated for 24 hr, then transfected with these luciferase plasmids in combination with empty pcDNA 3.1 or pcDNA3.1-MOTS-cWT. Dual luciferase assays were performed according to the manufacturer’s recommended protocol with minor modifications using a Dual-luciferase Reporter assay system (Promega Corporation). In brief, 48 hr after transfection, the cells were washed once with PBS, lysed in 100 μL of passive lysis buffer (Promega Corporation) with agitation for 10 min at room temperature, and subjected to one freeze-thaw cycle (−80°C). After centrifugation at 12,000 rpm for 5 min at 4°C, 20 μL of the lysate was mixed with 100 μL of luciferase assay substrate (Promega Corporation). The luciferase activity was normalized against the activity of the control Renilla luciferase reporter and recorded as the mean of three independent biological replicates.
Survival Assay
HEK293 cells were stably transfected with pEV, pMOTS-cWT, pMOTS-cYIFY, and pMOTS-cRKLR and incubated in glucose-free DMEM supplemented with low glucose (0.5 g/L) and 1% FBS (glucose and serum restriction: GR+SD) for 96 hr. Thereafter, cells were allowed to recover for 7 days in complete media (DMEM (4.5 g/L) and 10% FBS). Survival was assessed by trypan blue exclusion using the Contessa Automated Cell Counter (Thermo Fisher Scientific), crystal violet staining, and flow cytometry using the Muse Cell Viability Kit (Millipore).
Immunofluorescence Imaging
To improve the adherence of HEK293 cells, glass slides pretreated with 1 mg/mL collagen (Corning) for 2 hr at room temperature were used. The cells were washed three times with PBS and treated or transiently transfected with expression vectors as described above. Live cells were labeled for 15 min with MitoTracker Deep Red FM (Thermo Fisher Scientific) at a final concentration of 250 nM in cell culture medium to stain mitochondria. Then, the cells were fixed with 4% paraformaldehyde for 10 min at room temperature, washed with PBS, permeabilized for 10 min at room temperature with 0.2% Triton X-100 in PBS. Samples were blocked with 3% BSA in PBS with 0.1% Tween 20 (PBS-T), following incubation with primary antibody at 4°C overnight, and then incubated with secondary antibody for 1 hr. Samples were washed three times with PBS-T and incubated with DAPI (Sigma) for 20 min at room temperature to visualize nuclei. After three washes in PBS-T, the coverslips were mounted in PVA (polyvinyl alcohol) mounting medium as described previously (Khoo et al., 2015). The cellular images were obtained using a Zeiss LSM700 confocal microscope system and software (magnification ×63).
Immunoprecipitation
The isolated nuclei were lysed in modified RIPA buffer in the presence of a protease/phosphatase inhibitor cocktail as described above. 150 μg of nuclear extracts were precleared with 1 μg of IgG antibody bound to protein G Dynabeads (Thermo Fisher Scientific) for 1 hr at 4°C according to the manufacturer’s protocol. The beads were coated with 6 μg of specific antibodies or 2 μg of normal IgG at 4°C overnight. The precleared nuclear extract was incubated with antibody-conjugated Dynabeads and rotated at 4°C overnight. The immunoprecipitation complex was washed three times with PBS containing 0.05% Tween-20, eluted with glycine buffer (0.1 M, pH 2.8). The eluted samples were suspended with SDS sample buffer and subjected to western blotting.
Real-time PCR
Total RNA was purified using the Direct-zol RNA MiniPrep kit (Zymo Research) as per manufacturer’s protocol. cDNA was synthesized by reverse transcription of 1 μg of total RNA using iScript cDNA synthesis Kit (Bio-Rad) following manufacturer’s instructions. Quantitative real-time PCR was performed in 20 μL of reaction mixture containing 1 μL of cDNA, 200 nM of each primer, and 10 μL of SYBR Select Master Mix (Thermo Fisher Scientific) using QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific). Products were amplified with the primers listed in Table S1 and ribosomal protein L27 (RPL27) was used as a reference control for each reaction. The cycle to threshold (Ct) value was determined for each primer pair using QuantStudio 6 Flex Real-Time PCR software (Thermo Fisher Scientific) following recommended guidelines. Three biological replicates reactions were carried for each time point and primer pair.
Oxygen Consumption Measurement
Real-time oxygen consumption rates (OCR) in HEK293 cells that were stably transfected with empty vector (EV) or MOTS-c vectors (WT, YIFY mutant, RKLR mutant) were measured using the XF96 Analyzer (Seahorse Bioscience) at the USC Leonard Davis School of Gerontology Seahorse Core. All readings were normalized to relative protein concentration.
ROS Measurement
Total cellular ROS was measured using the Muse Oxidative Stress Kit (EMD Millipore) according to the manufacturer’s protocol. Briefly, HEK293 cells were seeded onto 6-well at 0.5 × 106 cells/well. After 24 hr, the cells were treated with GR, SD, tBHP as described above or 5 mM metformin, 2 mM AICAR. Then, live cells that are negative for ROS and positive for ROS were distinguished and counted using the Muse Cell Analyzer (EMD Millipore) per manufacturer’s instructions.
Electrophoretic Mobility Shift Assay (EMSA)
For detection of DNA-binding affinity of MOTS-c, wild-type or quadruple alanine mutant MOTS-c (8YIFY11 → 8AAAA11 and 13RKLR16 → 13AAAA16) peptide was synthesized (> 95% purity; New England Peptide). The oligonucleotide probes containing ARE sites were biotinylated and synthesized (Integrated DNA Technologies) as listed in Table S1. The biotinylated oligonucleotides were annealed with their respective complementary strands in annealing buffer (10 mM Tris, pH. 7.5, 1 mM EDTA, 50 mM NaCl) for 2 min at 90°C and allowed to cool slowly to room temperature. MOTS-c peptides were incubated for 30 min on ice in PBS with biotinylated DNA probes and the binding affinity was assessed by LightShift Chemiluminescent EMSA kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
RNA-seq Library Preparation
1 μg of total RNA was subjected to rRNA depletion using the NEBNext rRNA Depletion Kit (New England Biolabs), according to the manufacturer’s protocol. Strand-specific RNA-seq libraries were then constructed using the SMARTer Stranded RNA-Seq Kit (Clontech # 634839), according to the manufacturer’s protocol. Based on rRNA-depleted input amount, 11 cycles of amplification were performed to generate RNA-seq libraries. Paired-end 100bp reads were generated on the Illumina HiSeq2500 platform at the USC UPC Genome and Cytometry Core. The raw sequencing data were deposited to the NCBI Sequence Read Archive (accession number SRA: SRP136364). The resulting data were then analyzed with a standardized RNA-seq data analysis pipeline (described below).
RNA-seq Analysis Pipeline
cDNA sequences of protein coding and lincRNA genes were obtained through Biomart from the latest annotation of the human genome (Ensemble release v91; accessed 2018-03-22). Paired-end 100bp reads were mapped to this reference using kallisto 0.43.0 and the –fr-stranded option (Bray et al., 2016). DEseq2 normalized fold-changes were then used to estimate differential gene expression between control and MOTS-C expressing cells using the ‘DESeq2’ R package (DESeq2 1.16.1). We identified 802 significantly differentially expressed genes (412 downregulated, 390 upregulated) at FDR < 5%. The heatmap of expression across samples for significant genes (Figure 4L) was plotted using the R package ‘pheatmap’ 1.0.8 (Raivo Kolde, 2015-12-11; https://cran.r-project.org/web/packages/pheatmap/index.html).
Promoter Motif Analysis
Motif enrichment analysis was performed on the promoters of the genes significantly regulated by MOTS-C overexpression after 3 hr of glucose restriction using the HOMER software suite (v4.9.1) (Heinz et al., 2010). Briefly, we considered regions from −500 bp to +100 bp of annotated TSS in the hg19 human genome assembly (HOMER annotation package v5.10). Promoter sequences of MOTS-C regulated genes were scanned for enrichment of known motifs relative to all annotated hg19 promoters. Enriched known motifs were considered to be enriched and are reported if Benjamini q value < 0.1.
Gene Ontology Enrichment Analysis
To perform functional enrichment analysis with Gene Ontology (GO), we leveraged the minimum HyperGeometric test through the use of the GOrilla enrichment tool (Eden et al., 2007, 2009). GOrilla is a powerful tool for identifying enriched GO terms in ranked lists of genes. For our analysis, genes were ranked by the t-statistic computed by the DESeq2 analysis. The analysis was run on 2018-03-28 and will reflect the state of the Gorilla GO database at that date.
ChIP-Seq Data Processing
We obtained raw ChIP-seq data derived from human cells for putative MOTS-C transcription factor partners ATF1, JUND and NRF2 from public repositories (see below). Peaks were mapped to the UCSC hg19 assembly using bowtie 0.12.7 (Langmead et al., 2009). Regions of significant enrichment were determined using MACS2 v2.0.8 (Zhang et al., 2008) with default options. We identified high confidence ChIP target sites by selecting peaks calling in 3 out 5 cell lines (JUND, NRF2), or 2 out of 3 cell lines (ATF1). Significant high confidence ChIP peaks of interest were annotated using the HOMER software suite (v4.9.1) to the gene with the closest transcription start site in the hg19 assembly (v5.10) (Heinz et al., 2010).
| Factor | Cell Line | Accession Numbers (ChIP) | Accession Numbers (Input for Peak Calling) |
|---|---|---|---|
| ATF1 | HepG2 | SRR2184485; SRR2184486; SRR2184487; SRR2184488; SRR2184489; SRR2184490; SRR2184491; SRR2184492 | ENCFF190EPQO; ENCFF950AXC |
| ATF1 | K562 | ENCFF000YFC; NCFFOOOYFE | SRR351543; SRR351542; SRR351541; SRR351540 |
| ATF1 | LoVo | SRR952507 | SRR952608 |
| JUND | HCT-116 | SRR577903; SRR577904 | SRR351775 |
| JUND | HepG2 | SRR351817; SRR351818 | SRR351751 |
| JUND | HCT-116 | SRR577903; SRR577904 | SRR351775 |
| JUND | SK-N-SH | SRR578017; SRR578018 | SRR351874 |
| JUND | MCF-7 | SRR577997; SRR577998 | SRR577916 |
| JUND | T-47D | SRR577650; SRR577651 | SRR351697 |
| NRF2 (NFE2L2) | A549 | ENCFF003TGD; ENCFF004JMG | ENCFF918AJW; ENCFF949XNJ |
| NRF2 (NFE2L2) | HepG2 | ENCFF063AFU; ENCFF421XBU | ENCFF190EPQO; ENCFF950AXC |
| NRF2 (NFE2L2) | Hel_a-S3 | ENCFF686TXK; ENCFF068LXE | ENCFF459QXO; ENCFF714ZPY |
| NRF2 (NFE2L2) | IMR-90 | ENCFF461GBT; ENCFF467POD | ENCFF841PLU |
| NRF2 (NFE2L2) | Lymphoblastoid cells | SRR491140; SRR491141; SRR491142; SRR491143 | SRR491147 |
All codes will be made available on the Benayoun lab github (https://github.com/BenayounLaboratory/MOTSC_nuclear_project).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was tested using Student’s t test. Statistical tests were performed using GraphPad Prism. P scores greater than 0.05 were considered not significant. All statistical tests and n values are indicated in the figure legends. t test results are indicated consistently in all figures as *p < 0.05, ** p < 0.01, ***p < 0.001 and ns for not significant (p > 0.05).
DATA AND SOFTWARE AVAILABILITY
Custom scripts used in this paper are available at https://github.com/BenayounLaboratory/MOTSC_nuclear_project. The raw sequencing data have been deposited to the NCBI Sequence Read Archive (SRA: SRP136364).
Supplementary Material
Figure S1, Related to Figure 1. MOTS-c is a nuclear peptide encoded in the mitochondrial genome. (A) Subcellular distribution pattern of MOTS-c by immunoblotting (equal amounts of total protein loaded for C, N, M) in resting HepG2 cells. (B) Immunofluorescence staining for endogenous MOTS-c in resting HepG2 cells. Nucleus is outlined by white dashed lines. Representative images shown (n=3). Cells were co-stained with DAPI (nucleus) and mitotracker red (mitochondria). W: whole cell lysate, C: cytosol, N: nucleus, M: mitochondria. (C) Immunoblot of endogenous MOTS-c from nuclear extracts of resting HepG2 cells. Antibody specificity to MOTS-c was confirmed by neutralizing peptide competition. Lamin B1 and histone H2B were used as nuclear loading controls. (D) Localization of exogenously treated FITC-MOTS-c peptide (1 ¼M) in C2C12 myoblasts by confocal microscopy. (E) Oxygen consumption rate (OCR), normalized to relative protein concentration, of HEK293 cells that were stably transfected with empty vector (EV) or MOTS-c vectors (WT, YIFY mutant, RKLR mutant) (n=15). WT: wild-type MOTS-c, YIFY: 8YIFY11>8AAAA11 mutant MOTS-c, and RKLR: 13RKLR16>13AAAA16 mutant MOTS-c. Scale bar, 10 ¼m. Error bars represent mean ± s.e.m. ***p<0.001 by Student's t−test.
Figure S2, Related to Figure 2. MOTS-c translocates to the nucleus in response to metabolic stress. (A) MOTS-c levels in the nucleus with and without tBHP (150 μM) or paraquat (PQ) at the indicated concentrations. (B) Cytosolic MOTS-c levels detected by immunoblotting with and without GR or SD (1 hr). 50 ug of protein was loaded and the membrane was blotted with 2 ng/uL of MOTS-c antibody (final concentration) and exposed for 4 minutes to detect cytosolic MOTS-c. (C-I) Spatial and temporal assessment of MOTS-c localization in HepG2 cells after glucose restriction (GR; 0.5 g/L), serum deprivation (1% FBS), and tert−butyl hydrogen peroxide (tBHP; 100 μM) by (C-E) subcellular fraction immunoblots and (F-I) immunofluorescence microscopy. Subcellular fractions were purified at 0, 3, and 24 hours post-stress, and confocal microscopy images were acquired 3 hours post-stress. Representative images shown (n=3). Cells were co-stained with DAPI (nucleus) and mitotracker red (mitochondria). Scale bar, white: 10 μm, yellow: 2 μm.
Figure S3, Related to Figure 3. AMPK inhibition prevents stress-induced nuclear translocation of MOTS-c. (A) Flow cytometry based on DHE to assess time-dependent reactive oxygen species (ROS) production in response to a 1-hour treatment with tBHP (100 μM), metformin (5mM), and AICAR (2mM) in HEK293 cells (n=4). (B-E) The level of nuclear MOTS-c by immunofluorescence microscopy in (B) resting HEK293 cells and after one hour of (C) GR, (D) SD, (E) tBHP (100 μM) co-treated with compound C (10 μM), an AMPK inhibitor, for 30 minutes prior to stress. Representative images shown (n=3). Cells were co-stained with DAPI (nucleus). Error bars represent mean ± s.e.m. ***p<0.001 by Student's t−test. Scale bar, 10 μm.
Figure S4, Related to Figure 4. MOTS-c interacts with NRF2 and binds to DNA fragments containing consensus ARE sequences. (A) Co-immunoprecipitation of EGFP-MOTS-c (WT and RKLR mutant) and Myc-tagged Nrf2 (Myc-Nrf2) in HEK293 cells determined by immunoblotting. (B) MOTS-c levels in nuclear extracts in response to GR (3 hrs) with and without NRF2 knockdown using siRNA. (C) NRF2 levels in nuclear extracts with and without actinonin treatment (100 μM; 72 hours), which depletes MOTS-c (Lee et al., 2015). (D-F) EMSA analysis of ARE motifs in several Nrf2 target genes by incubation with (D) WT, (E) YIFY mutant MOTS-c, or (F) RKLR mutant MOTS-c peptide at 0, 0.5, 1.5, 3, and 6 μg. WT: wild-type MOTS-c, YIFY: 8YIFY11>8AAAA11 mutant MOTS-c, and RKLR: 13RKLR16>13AAAA16 mutant MOTS-c. Representative images are shown (n=3). (G) ARE luciferase reporter activity on cells transfected with pEV or pMOTS-c in HepG2 cells. Firefly luciferase activity was normalized to Renilla luciferase activity (n=4). (H) qRT-PCR analysis of HO-1 expression in response to the sulforaphane (SFN; 10 μM) and actinonin (150 μM) in HEK293 cells (n=3). (I) Co-immunoprecipitation of MOTS-c and NRF1 from nuclear extracts and chromatin extracts derived thereof of HEK293 cells after GR (3 hrs) determined by immunoblotting. (J-O) RNA-seq-derived Venn diagrams of genes upregulated by MOTS-c and bona fide target genes as indicated (n=6). (P-R) HEK293 cells stably transfected with MOTS-c plasmid (or EV) underwent GR+SD for 4 days and allowed to recover for 7 days in complete media. Survival was assessed by (P) live cell counts using trypan blue exclusion, (Q) phase contrast images, and (R) crystal violet staining of cells (n=6). Error bars represent mean ± s.e.m. **p<0.01, ***p<0.001 by Student's t−test.
Real-time PCR primer sequences
ChIP-qPCR primer sequences
EMSA oligonucleotide sequences
Table. S1, Related to Figure 4. List of primers and oligonucleotides used for real-time PCR, ChIP-qPCR, and EMSA.
Table. S2, Related to Figure 4. Gene ontology (GO) analysis on RNA-seq performed on HEK293 cells that overexpressed MOTS-c (vs EV) under GR (3 hrs).
Highlights.
MOTS-c, a peptide encoded by the mitochondrial DNA, can localize to the nucleus
Metabolic stress triggers AMPK-dependent translocation of MOTS-c to the nucleus
MOTS-c can bind to chromatin and regulate nuclear gene expression
MOTS-c can promote cellular stress resistance against metabolic stress
ACKNOWLEDGMENTS
We thank Daniel Campo and Suchi P. Patel at the USC UPC Genome & Cytometry Core for help with Next-Generation Sequencing, Hemal Mehta at the USC Gerontology Seahorse Core, Conscience Bwiza for generating stable cell lines, and Angelina Holcom for her help on ChIP optimization. This work was funded by grants from NIH (R01AG052558), the Ellison Medical Foundation, the American Federation for Aging Research (AFAR), and the Hanson-Thorell family to C.L. and by NIH grant R00AG049934 and the Hanson-Thorell family to B.A.B.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and two tables and can be found with this article online at https://doi.org/10.1016/j.cmet.2018.06.008.
DECLARATION OF INTERESTS
C.L. is a consultant for and a shareholder of CohBar, Inc. The remaining authors declare no competing interests.
REFERENCES
- Biswas M, and Chan JY (2010). Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol 244, 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratic A, and Larsson NG (2013). The role of mitochondria in aging. J. Clin. Invest 123, 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhäusl W, and Fürnsinn C (2004). Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 53, 1052–1059. [DOI] [PubMed] [Google Scholar]
- Chandel NS (2015). Evolution of mitochondria as signaling organelles. Cell Metab. 22, 204–206. [DOI] [PubMed] [Google Scholar]
- Chepelev NL, Zhang H, Liu H, McBride S, Seal AJ, Morgan TE, Finch CE, Willmore WG, Davies KJ, and Forman HJ (2013). Competition of nuclear factor-erythroid 2 factors related transcription factor isoforms, Nrf1 and Nrf2, in antioxidant enzyme induction. Redox Biol. 1, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, and Hardie DG (1995). 5-amino-imidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem 229, 558–565. [DOI] [PubMed] [Google Scholar]
- de Nadal E, Ammerer G, and Posas F (2011). Controlling gene expression in response to stress. Nat. Rev. Genet 12, 833–845. [DOI] [PubMed] [Google Scholar]
- Eden E, Lipson D, Yogev S, and Yakhini Z (2007). Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol 3, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, and Haynes CM (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol 26, 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Fuku N, Pareja-Galeano H, Zempo H, Alis R, Arai Y, Lucia A, and Hirose N (2015). The mitochondrial-derived peptide MOTS-c: a player in exceptional longevity? Aging Cell 14, 921–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KT, Li L, Janowski BA, and Corey DR (2014). Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat. Protoc 9, 2045–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, and Kroemer G (2012). Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788. [DOI] [PubMed] [Google Scholar]
- Gustafsson CM, Falkenberg M, and Larsson NG (2016). Maintenance and expression of mammalian mitochondrial DNA. Annu. Rev. Biochem 85, 133–160. [DOI] [PubMed] [Google Scholar]
- Hamilton G (2015). The hidden risks for ‘three-person’ babies. Nature 525, 444–446. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, and Hawley SA (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Schaffer BE, and Brunet A (2016). AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, and Dinkova-Kostova AT (2014). The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci 39, 199–218. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsu Y, Katsuoka F, Funayama R, Nagashima T, Nishida Y, Nakayama K, Engel JD, and Yamamoto M (2012). Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 40, 10228–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodel MR, Corbett AH, and Hodel AE (2001). Dissection of a nuclear localization signal. J. Biol. Chem 276, 1317–1325. [DOI] [PubMed] [Google Scholar]
- Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, and Kim SG (2016). AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at Serine 550. Mol. Cell. Biol 36, 1931–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovaisaite V, and Auwerx J (2015). The mitochondrial unfolded protein response—synchronizing genomes. Curr. Opin. Cell Biol. 33, 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki H, Kadowaki T, Wondisford FE, and Taylor SI (1989). Use of polymerase chain reaction catalyzed by Taq DNA polymerase for site-specific mutagenesis. Gene 76, 161–166. [DOI] [PubMed] [Google Scholar]
- Keskin O, Gursoy A, Ma B, and Nussinov R (2008). Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem. Rev 108, 1225–1244. [DOI] [PubMed] [Google Scholar]
- Khoo NK, Cantu-Medellin N, St Croix C, and Kelley EE (2015). In vivo immuno-spin trapping: imaging the footprints of oxidative stress. Curr. Protoc. Cytom 74, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yang G, Kim Y, Kim J, and Ha J (2016). AMPK activators: mechanisms of action and physiological activities. Exp. Mol. Med 48, e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Xiao J, Wan J, Cohen P, and Yen K (2017). Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol 595, 6613–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre-Pellicer A, Moreno-Loshuertos R, Lechuga-Vieco AV, Sánchez-Cabo F, Torroja C, Acín-Pérez R, Calvo E, Aix E, González-Guerra A, Logan A, et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. [DOI] [PubMed] [Google Scholar]
- Lee C, Yen K, and Cohen P (2013). Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol. Metab 24, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, and Cohen P (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q (2013). Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Jovaisaite V, Durieux J, Matilainen O, Jordan SD, Quirós PM, Steffen KK, Williams EG, Mouchiroud L, Tronnes SU, et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming W, Lu G, Xin S, Huanyu L, Yinghao J, Xiaoying L, Chengming X, Banjun R, Li W, and Zifan L (2016). Mitochondria related peptide MOTS-c suppresses ovariectomy-induced bone loss via AMPK activation. Biochem. Biophys. Res. Commun 476, 412–419. [DOI] [PubMed] [Google Scholar]
- Monaghan RM, and Whitmarsh AJ (2015). Mitochondrial proteins moonlighting in the nucleus. Trends Biochem. Sci 40, 728–735. [DOI] [PubMed] [Google Scholar]
- Murley A, and Nunnari J (2016). The emerging network of mitochondriaorganelle contacts. Mol. Cell 61, 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, and Bomsztyk K (2006). Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc 1, 179–185. [DOI] [PubMed] [Google Scholar]
- Quirós PM, Mottis A, and Auwerx J (2016). Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226. [DOI] [PubMed] [Google Scholar]
- Rabinovitch RC, Samborska B, Faubert B, Ma EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J, and Jones RG (2017). AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 21, 1–9. [DOI] [PubMed] [Google Scholar]
- Rohs R, Jin X, West SM, Joshi R, Honig B, and Mann RS (2010). Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem 79, 233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, and Ristow M (2007). Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293. [DOI] [PubMed] [Google Scholar]
- Stadler C, Rexhepaj E, Singan VR, Murphy RF, Pepperkok R, Uhlén M, Simpson JC, and Lundberg E (2013). Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods 10, 315–323. [DOI] [PubMed] [Google Scholar]
- Stephenne X, Foretz M, Taleux N, van der Zon GC, Sokal E, Hue L, Viollet B, and Guigas B (2011). Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 54, 3101–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Garcia G, Bian Q, Steffen KK, Joe L, Wolff S, Meyer BJ, and Dillin A (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CM, and Bassler BL (2005). Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol 21, 319–346. [DOI] [PubMed] [Google Scholar]
- Willyard C (2017). The drug-resistant bacteria that pose the greatest health threats. Nature 543, 15. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, Related to Figure 1. MOTS-c is a nuclear peptide encoded in the mitochondrial genome. (A) Subcellular distribution pattern of MOTS-c by immunoblotting (equal amounts of total protein loaded for C, N, M) in resting HepG2 cells. (B) Immunofluorescence staining for endogenous MOTS-c in resting HepG2 cells. Nucleus is outlined by white dashed lines. Representative images shown (n=3). Cells were co-stained with DAPI (nucleus) and mitotracker red (mitochondria). W: whole cell lysate, C: cytosol, N: nucleus, M: mitochondria. (C) Immunoblot of endogenous MOTS-c from nuclear extracts of resting HepG2 cells. Antibody specificity to MOTS-c was confirmed by neutralizing peptide competition. Lamin B1 and histone H2B were used as nuclear loading controls. (D) Localization of exogenously treated FITC-MOTS-c peptide (1 ¼M) in C2C12 myoblasts by confocal microscopy. (E) Oxygen consumption rate (OCR), normalized to relative protein concentration, of HEK293 cells that were stably transfected with empty vector (EV) or MOTS-c vectors (WT, YIFY mutant, RKLR mutant) (n=15). WT: wild-type MOTS-c, YIFY: 8YIFY11>8AAAA11 mutant MOTS-c, and RKLR: 13RKLR16>13AAAA16 mutant MOTS-c. Scale bar, 10 ¼m. Error bars represent mean ± s.e.m. ***p<0.001 by Student's t−test.
Figure S2, Related to Figure 2. MOTS-c translocates to the nucleus in response to metabolic stress. (A) MOTS-c levels in the nucleus with and without tBHP (150 μM) or paraquat (PQ) at the indicated concentrations. (B) Cytosolic MOTS-c levels detected by immunoblotting with and without GR or SD (1 hr). 50 ug of protein was loaded and the membrane was blotted with 2 ng/uL of MOTS-c antibody (final concentration) and exposed for 4 minutes to detect cytosolic MOTS-c. (C-I) Spatial and temporal assessment of MOTS-c localization in HepG2 cells after glucose restriction (GR; 0.5 g/L), serum deprivation (1% FBS), and tert−butyl hydrogen peroxide (tBHP; 100 μM) by (C-E) subcellular fraction immunoblots and (F-I) immunofluorescence microscopy. Subcellular fractions were purified at 0, 3, and 24 hours post-stress, and confocal microscopy images were acquired 3 hours post-stress. Representative images shown (n=3). Cells were co-stained with DAPI (nucleus) and mitotracker red (mitochondria). Scale bar, white: 10 μm, yellow: 2 μm.
Figure S3, Related to Figure 3. AMPK inhibition prevents stress-induced nuclear translocation of MOTS-c. (A) Flow cytometry based on DHE to assess time-dependent reactive oxygen species (ROS) production in response to a 1-hour treatment with tBHP (100 μM), metformin (5mM), and AICAR (2mM) in HEK293 cells (n=4). (B-E) The level of nuclear MOTS-c by immunofluorescence microscopy in (B) resting HEK293 cells and after one hour of (C) GR, (D) SD, (E) tBHP (100 μM) co-treated with compound C (10 μM), an AMPK inhibitor, for 30 minutes prior to stress. Representative images shown (n=3). Cells were co-stained with DAPI (nucleus). Error bars represent mean ± s.e.m. ***p<0.001 by Student's t−test. Scale bar, 10 μm.
Figure S4, Related to Figure 4. MOTS-c interacts with NRF2 and binds to DNA fragments containing consensus ARE sequences. (A) Co-immunoprecipitation of EGFP-MOTS-c (WT and RKLR mutant) and Myc-tagged Nrf2 (Myc-Nrf2) in HEK293 cells determined by immunoblotting. (B) MOTS-c levels in nuclear extracts in response to GR (3 hrs) with and without NRF2 knockdown using siRNA. (C) NRF2 levels in nuclear extracts with and without actinonin treatment (100 μM; 72 hours), which depletes MOTS-c (Lee et al., 2015). (D-F) EMSA analysis of ARE motifs in several Nrf2 target genes by incubation with (D) WT, (E) YIFY mutant MOTS-c, or (F) RKLR mutant MOTS-c peptide at 0, 0.5, 1.5, 3, and 6 μg. WT: wild-type MOTS-c, YIFY: 8YIFY11>8AAAA11 mutant MOTS-c, and RKLR: 13RKLR16>13AAAA16 mutant MOTS-c. Representative images are shown (n=3). (G) ARE luciferase reporter activity on cells transfected with pEV or pMOTS-c in HepG2 cells. Firefly luciferase activity was normalized to Renilla luciferase activity (n=4). (H) qRT-PCR analysis of HO-1 expression in response to the sulforaphane (SFN; 10 μM) and actinonin (150 μM) in HEK293 cells (n=3). (I) Co-immunoprecipitation of MOTS-c and NRF1 from nuclear extracts and chromatin extracts derived thereof of HEK293 cells after GR (3 hrs) determined by immunoblotting. (J-O) RNA-seq-derived Venn diagrams of genes upregulated by MOTS-c and bona fide target genes as indicated (n=6). (P-R) HEK293 cells stably transfected with MOTS-c plasmid (or EV) underwent GR+SD for 4 days and allowed to recover for 7 days in complete media. Survival was assessed by (P) live cell counts using trypan blue exclusion, (Q) phase contrast images, and (R) crystal violet staining of cells (n=6). Error bars represent mean ± s.e.m. **p<0.01, ***p<0.001 by Student's t−test.
Real-time PCR primer sequences
ChIP-qPCR primer sequences
EMSA oligonucleotide sequences
Table. S1, Related to Figure 4. List of primers and oligonucleotides used for real-time PCR, ChIP-qPCR, and EMSA.
Table. S2, Related to Figure 4. Gene ontology (GO) analysis on RNA-seq performed on HEK293 cells that overexpressed MOTS-c (vs EV) under GR (3 hrs).