

Abstract
Protein structural analysis by mass spectrometry has gained significant popularity in recent years, including high-resolution protein topographical mapping by fast photochemical oxidation of proteins (FPOP). The ability to provide protein topographical information at moderate spatial resolution makes FPOP an attractive technology for the protein pharmaceutical discovery and development processes. However, current technology limits the throughput and requires significant manual sample manipulation. Similarly, as FPOP is being used on larger samples, sample flow through the capillary becomes challenging. No systematic comparison of the performance of static flash photolysis with traditional flow FPOP has been reported. Here, we evaluate a 96-well microtiter-based laser flash photolysis method for the topographical probing of proteins, which subsequently could be used to analyze higher order structure of the protein in a high-throughput fashion with minimal manual sample manipulation. We used multiple metrics to compare microtiter FPOP performance with that of traditional flow FPOP: adenine-based hydroxyl radical dosimetry, oxidation efficiency of a model peptide, and hydroxyl radical protein footprint of myoglobin. In all cases, microtiter plate FPOP performed comparably with traditional flow FPOP, requiring a small fraction of the time for exposure. This greatly reduced sample exposure time, coupled with automated sample handling in 96-well microtiter plates, makes microtiter-based FPOP an important step in achieving the throughput required to adapt hydroxyl radical protein footprinting for screening purposes.
Keywords: Mass spectrometry, fast photochemical oxidation of proteins (FPOP), covalent labeling, hydroxyl radical protein footprinting (HRPF), myoglobin
1. Introduction
Structural characterization of biomolecules using mass spectrometry (MS)-based techniques is being extensively employed over the last few decades [1]. Hydroxyl radical protein footprinting (HRPF) is a mass spectrometry based structural characterization technique that provides a lower level of structural detail than traditional high-resolution structural biology techniques such as X-ray crystallography or multidimensional nuclear magnetic resonance (NMR) spectroscopy [2,3]. HRPF is complementary to other MS-based methods such as hydrogen-deuterium exchange and ion mobility spectroscopy, as it measures changes in the protein topography (i.e. solvent-accessible surface) using a stable and broadly reactive labeling group. Many methods for the generation of hydroxyl radicals for HRPF have been introduced, including the use of UV photolysis of hydrogen peroxide [3]. Afterward, two groups independently introduced two laser-induced methods for pulsed UV photolysis of hydrogen peroxide: Fast Photochemical Oxidation of Proteins (FPOP) and batch photolysis.
Originally developed by Hambly and Gross in 2005[4], FPOP is a technique commonly used today to produce hydroxyl radicals through photolysis of hydrogen peroxide. In FPOP, a mixture of the analyte protein, hydrogen peroxide, and a hydroxyl radical scavenger is pushed through a capillary past a focused KrF excimer laser (248 nm wavelength). The laser is pulsed to flash photolyze hydrogen peroxide, forming a sharp and short-lived burst of hydroxyl radicals that are consumed in under a microsecond [5–8]. Theoretically, with addition of exclusion volume of unirradiated sample between each irradiated volume, each volume of sample is exposed to only a single laser pulse. Under such conditions, the initial and irreversible hydroxyl radical reaction with the protein occurs on approximately a microsecond timescale, and this rapid time frame allows for proteins to be thoroughly surface labeled faster than the protein will undergo large-scale structural changes due to the labeling process [9,10]. In reality, due to the characteristic parabolic flow of the solution within the capillary system under laminar flow conditions, a small volume of sample may be double-exposed. However, the fraction of such double-exposed sample is limited to an undetectable amount when using an appropriately large volume of unirradiated sample between each laser shot, as determined by an analysis of the labeling kinetics of intact proteins by FPOP [8].
One limitation of FPOP is that it is time consuming (especially when dealing with a large number of samples), and FPOP requires significant manual handling of samples for exposure. A conventional flow FPOP method takes nearly 13 seconds of mid-flow time to irradiate 4ul of sample, not including time for sample loading, capillary-resident sample ejection, and inter-sample capillary washing [11]. Additionally, recent developments using FPOP technology for larger systems, including living cells, have presented challenges in maintaining appropriate flow through the system [12,13]. Workflows that eliminate the need for flow path cleaning and sample reloading could substantially reduce the time per sample requirements and eliminate the possibilities of capillary clogging or laser-induced capillary breakage.
Sze and coworkers demonstrated protein oxidation in a microfuge tube on a nanosecond time scale with a single pulse irradiation in 2005 [9]. Later the same group demonstrated the protein footprinting of EGFR in 12 well plates [14]. Recently, Schriemer and co-workers performed protein footprinting of calmodulin in static set-up using photoleucine as a diazarine labelling agent [15–16]. Subsequently, Gross and co-workers used photoleucine to footprint calmodulin using a capillary flow system [17]; while the two results were grossly similar, there were significant differences in the peptide-level footprint. As these experiments were not carried out with the intention of a direct comparison between the two techniques, it is quite possible that the differences were due to variables other than flow exposure versus static exposure, but the role of the different exposure techniques in the observed different footprints has not been explored. Similarly, while the correlation between labeling by FPOP and amino acid side chain solvent accessibility has been established [18–20], we are not aware of any published comparison of single pulse batch photolysis and FPOP results that establish the equivalency of the two technologies.
Inspired by work pioneered by Sze and co-workers, we report our first investigations into the technologies required to develop FPOP and related technologies into a high throughput format: the development and evaluation of a 96-well microtiter plate-based FPOP exposure system. We describe efforts involved in ensuring reliability and reproducibility in microtiter FPOP and evaluate microtiter FPOP against traditional flow FPOP using three systems: adenine-based hydroxyl radical dosimetry; oxidation of the model peptide [Glu]1-Fibrinopeptide B (GluB); and HRPF analysis of the model protein myoglobin. With low sample requirements (4 µL or less), very fast exposure times (<5 seconds per sample, which could be automated to <1 second per sample), no sample dilution from unirradiated volumes, and compatibility with common 96-well microtiter-based sample handling workflows already in place in biochemistry and proteomics, our results demonstrate the considerable promise of microtiter-based single pulse batch photolysis for increasing the throughput of HRPF, an emerging technique for the analysis of protein pharmaceuticals [21,22].
2. Materials and methods
2.1. Chemicals and reagents
Myoglobin, GluB, and catalase were purchased from Sigma Aldrich Corp (St. Louis, MO). Sequencing grade modified trypsin was purchased from Promega (Madison, WI). Adenine and glutamine were purchased from Acros Organics (Fair Lawn, NJ) and methionine amide was from Bachem (Torrance, CA). Hydrogen peroxide (30%), LC-MS grade water, acetonitrile, formic acid were purchased from Fisher Scientific (Fair Lawn, NJ). Corning™ Costar™ flat bottom 96 well plates were from Fisher Scientific (Fair Lawn, NJ) and V-shaped 96 well microtiter plates were purchased from Greiner Bio-One (Monroe, NC).
2.2. Sample oxidation
Fast photochemical oxidation of protein (FPOP) was performed with a Compex Pro 102 excimer laser (Coherent, Germany) at 248 nm wavelength as described previously [23]. Adenine dosimetry was used to ensure the consistency of free radicals generated during FPOP and microtiter plate single pulse photolysis among different samples illuminated with different laser fluences [24]. For microtiter plate single pulse photolysis, a TECHSPEC® Excimer laser line mirror from Edmund Optics (Barrington, NJ) was placed at 45° of the laser beam at ~15 cm distance of the source as shown in Figure 1. The laser beam was deflected by the mirror and subsequently focused by an FL uncoated, UV plano-convex lens (Edmund Optics). The focused beam was allowed to score an index card target mounted on the optical bench to assist with alignment of the plate with the beam. Another card positioned at the height of the surface of the sample was used to position the focusing lens to ensure the laser spot covered the entire surface area of the sample. A clear V-shaped well polystyrene microtiter plate was placed beneath the lens that contained the sample to be irradiated with the laser.
Figure 1: Schematic cartoon for microtiter FPOP.
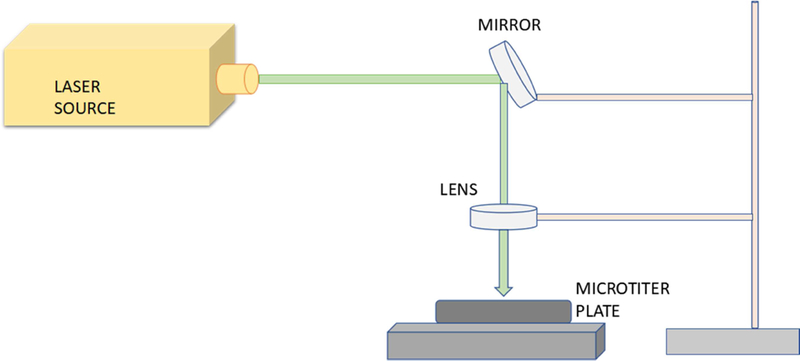
A mirror placed at 450 angles of laser path deflects the laser beam onto a lens, which then focuses the light onto the well of the microtiter plate.
The sample was mixed to contain 5 µM of GluB peptide, 5 µM myoglobin, 1mM adenine, 17 mM glutamine, 100 mM hydrogen peroxide, and 50 mM sodium phosphate buffer (pH 7.4). Immediately after mixing, 4 µl of sample was placed in each microtiter well. The samples were oxidized with a single shot laser pulse and immediately quenched by addition of 5 µl of quenching solution consisting for a final concentration of catalase (0.5 µg/µl) and methionine amide (0.5 µg/µl) to remove the residual hydrogen peroxide and other secondary oxidants. Samples were illuminated in triplicate at each laser fluence tested. The laser fluence was calculated from the laser energy and laser beam. The estimated fluence was calculated based on the spot size of the beam at the approximate site of contact with the sample, assuming no loss of light from the mirror and focusing. After quenching for one hour, the absorbance of adenine was measured by Nanodrop UV-Vis spectrophotometer from Thermo Fisher Scientific (Waltham, MA).
Traditional capillary flow FPOP was performed as previously described [18]. Briefly, 20µl of the sample containing 5 µM of GluB peptide, 5 µM myoglobin, 1mM adenine, 17 mM glutamine, 100 mM hydrogen peroxide, and 50 mM sodium phosphate buffer (pH 7.4) for the dosimetry measurement, were flowed through a 100 µm ID capillary through the path of a focused laser. The laser spot width and laser pulse rate were calculated to give an exclusion volume of 15%. Samples were immediately collected in a 1.5 mL microfuge tube containing 25µl quenching solution (final concentration of (0.5 µg/µl) catalase and (0.5 µg/µl) methionine amide) to remove extra hydrogen peroxide and secondary oxidants, and adenine dosimetry was performed as described above.
2.3. Tryptic digestion
50 mM Tris-HCl and 5 mM DTT were added to the sample and the final volume of the samples was adjusted to 40µl. In order denature the myoglobin, samples were heated in an oven at 90° C for 15 min. Samples were cooled down to room temperature and sequencing grade trypsin was added to a 1:5 weight ratio of trypsin/protein to the protein sample and incubated it at 37° C overnight. The digested samples were stored at −20° C before analysis on for LC-MS system.
2.4. LC-MS/MS analysis
The protein samples were analyzed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) coupled to a Dionex Ultimate nano 3000 (Dionex, Sunnyvale, CA). Samples were loaded onto an Acclaim PepMap 100 C18 nanocolumn (0.75 mm × 150 mm, 2 µm, Thermo Fisher Scientific). The peptides were eluted with a gradient consisting of 2% to 35% solvent B (0.1% formic acid in acetonitrile) with a balance of solvent A (0.1% formic acid in water) over 28 min, ramped to 95% B over 5 min, held for 2 min, and then returned to 2% B over 1 min and held for 9 min. Peptides were eluted directly into the nanospray source using a conductive nanospray emitter. All data were acquired in positive ion mode at a spray voltage of 2500 V and a heated capillary temperature of 300° C. Both collision-induced dissociation (CID) and electron transfer dissociation (ETD) were used to fragment peptides, with an isolation width of 3 m/z units. In CID mode, full MS scans were acquired from m/z 350 to 2000 followed by eight subsequent MS2 scans on the top eight most abundant peptide ions. In ETD mode of fragmentation, calibrated charge dependent ETD parameters were used with 5% supplementation activation of collision energy.
2.5. Data analysis
Initial data analysis was performed using ByOnic (Protein Metrics, San Carlos CA) with major oxidation products inserted as variable modification. Data analysis was completed manually, using Xcalibur version 4.1.31.9. Peak intensities of the unoxidized peptides and their corresponding oxidation products observed in LC-MS were used to calculate the average oxidation events per peptide in the sample with maximum 7 ppm mass error. Peptide level oxidation was calculated by summing the ion intensities of all the oxidized peptides multiplied by the number of oxidation events required for the mass shift (e.g., one event for +16, two events for +32) and then divided by the sum of the ion intensities of all unoxidized and oxidized peptide masses as represented by equation 1.
| (1). |
where P is the average oxidation events per peptide, and I values are the peak intensities of corresponding oxidized and unoxidized peptides.
3. Results and Discussion
3.1. Adenine radical dosimetry
Initial measurements were made to determine the efficiency with which we generate hydroxyl radicals in microtiter FPOP. To test this, we measured the loss of absorbance at 260 nm of 1 mM adenine after exposure to hydroxyl radicals. As we previously demonstrated, this loss of adenine absorbance is linear with effective radical concentration [24]. Our first efforts were performed using traditional flat-bottomed microtiter plates. However, the resulting data were highly irreproducible and showed very poor correlation, as shown in a representative data set in Figure 2A. Based on the success with batch laser photolysis reported both by our group and Aye and coworkers [9, 14, 25], we hypothesized that irreproducibility in sample illumination was the cause. Polystyrene is largely opaque at 248 nm; we hypothesized that UV shadowing of the sample near the wall of the well resulting from an imperfectly perpendicular incident laser beam and/or an imperfectly aligned well may be leading to the poor correlation and reproducibility observed. To test this hypothesis, we performed similar adenine dosimetry experiments by placing the sample at the center of the well in a V-shaped well plate, where the edges of the sample are far from the sides of the well. The results are shown in Figure 2B. Adenine dosimetry indicated an effective hydroxyl radical dose comparable to that of traditional flow FPOP, and the radical dose increased linearly with estimated laser fluence. All further experiments described were carried out in a V-shaped 96-well microtiter plate, with the sample centered in the well away from the walls to prevent shadowing.
Figure 2. Adenine dosimetry of microtiter FPOP.
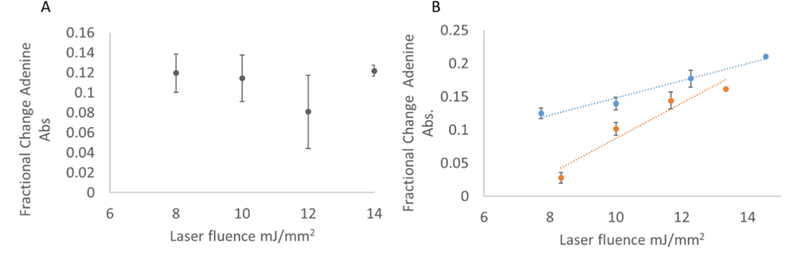
(A): Representative data set of microtiter single pulse photolysis carried out in a flat-bottomed 96-well microtiter plate. (B) Microtiter single pulse photolysis carried out in a (orange) V-shaped 96-well microtiter plate or (blue) traditional flow FPOP in a fused silica capillary. Error bars represent one standard deviation from a triplicate data set.
3.2. Model peptide oxidation
In order to ensure that the microtiter format was not causing spurious readings in our adenine dosimetry, we also tested the efficiency at which we could oxidize a model peptide, GluB, by microtiter FPOP. We examined the average oxidation events per molecule for 5 µM GluB in both microtiter FPOP and traditional flow FPOP at four different laser fluences. The results are shown in Figure 3. For GluB, the results of microtiter FPOP and traditional flow FPOP were indistinguishable. Differences in the optical interface and the lack of an exclusion volume in FPOP could contribute to measured differences between capillary and microtiter FPOP oxidation efficiency, but both GluB and adenine dosimetry show that oxidation is comparable between the two methods. We are uncertain why adenine showed a small difference in exposure efficiency for microtiter FPOP compared to flow FPOP, while GluB did not. However, in both cases, the change in oxidation was linearly related to estimated laser fluence with strong reproducibility. This suggests that perhaps insufficient oxidized adenine recovery, especially at lower hydroxyl radical doses, may contribute to oberved differences in adenine dosimetry. A separate set of adenine dosimetry triplicate analyses in microtiter plates gave identical results as the microtiter dosimetry results shown in Figure 2B (data not shown) indicating that, whatever the cause of differential slopes of adenine dosimetry response between FPOP and microtiter plate, the dosimetry is reproducible.
Figure 3. Comparison of GluB peptide oxidation by (orange) microtiter single pulse photolysis versus (blue) traditional capillary FPOP.
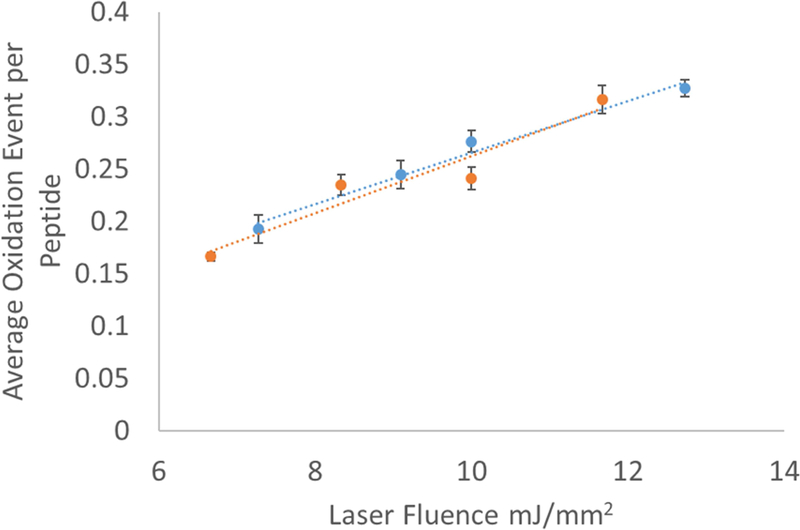
Error bars represent one standard deviation from a triplicate data set.
3.3. HRPF of a model protein
As the primary use of FPOP lies in measuring changes in protein topography, we tested whether performing the oxidation in the microtiter plate environment gave similar topography results as samples tested by traditional flow FPOP. We used myoglobin as our model protein due to its long history of HRPF studies as a model system [3,26]. After the exposure of myoglobin at four different laser fluences, the protein was digested, run on LC-MS and the oxidation level of the peptides was calculated. The sequence coverage of myoglobin was 88.3% on the ByOnic search for microtiter setup. On peptide level analysis of oxidation in microtiter experiment we found that out of ten peptides observed, seven peptides showed oxidation in a linear fashion while the other three peptides did not show any oxidation. The data analysis of oxidation event of seven peptides showed a linear response to increasing laser fluence for all peptides as shown in Figure 4. With the exception of peptide 17–31, all peptides also showed the good precision of measurement.
Fig 4. Correlation among oxidation event for seven peptides of myoglobin.
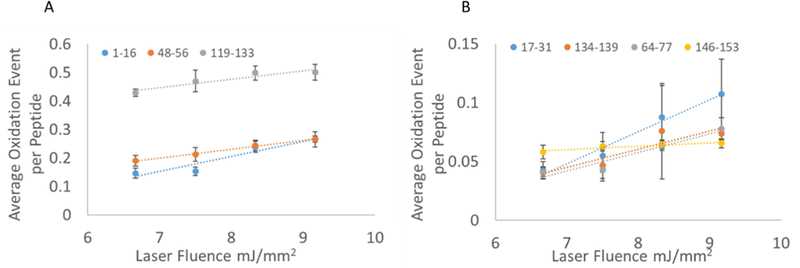
5 µM myoglobin exposed to four different laser fluences keeping reagent concentration constant. Error bars represent one standard deviation from a triplicate data set. Peptides are grouped according to level of oxidation for improved clarity of data; both left and right panels represent peptides from the same set of experiments.
Finally, we performed the FPOP experiment for myoglobin in a capillary setting to compare the results obtained with the microtiter plate. Two relatively similar laser fluences (8.5 mJ/mm2 in capillary compared to 8.3 mJ/mm2 in the plate) were used to compare the peptide level oxidation between two methods. After tryptic digestion and LC-MS run of the capillary set up experiment, we found the sequence coverage to be 99.35%. We found eleven peptides in capillary set up and eight of them showed detectable oxidation. Upon comparing the oxidation of peptides between two methods, no peptide showed statistically significant differences providing the evidence that microtiter plate oxidation of protein produces comparable oxidation to the capillary method as presented in Figure 5.
Fig 5. Comparison of myoglobin peptide oxidation between (blue) capillary FPOP and (orange) microtiter single pulse photolysis.
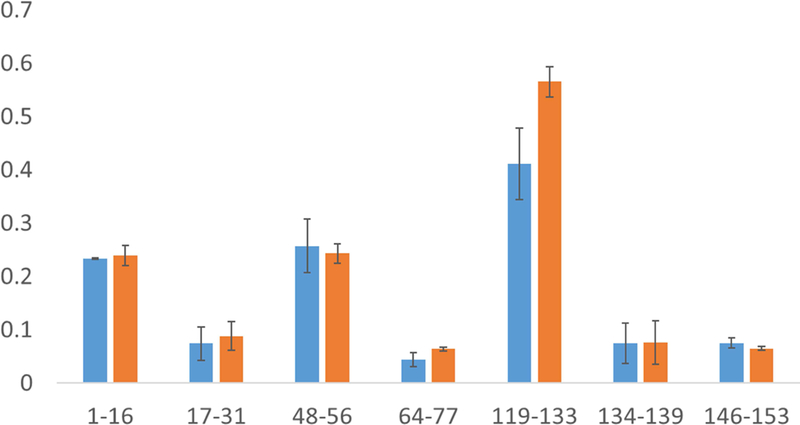
Error bars represent one standard deviation from a triplicate data set. No statistically significant differences in oxidation were detected (α = 0.05).
Myoglobin oxidized by both capillary FPOP and microtiter single pulse photolysis methods yielded equivalent results, indicating that the oxidation methods are equivalent if exposure is appropriately controlled. We observed that the plate set up provided us similar oxidation although it had experienced slightly less laser fluence (8.5mJ/mm2 in capillary compared to 8.3mJ/mm2 in the plate). In FPOP, a 15% exclusion volume was used to prevent illumination of any significant volume of sample with two laser pulses; therefore, approximately 15% of the sample remains unoxidized, lowering the average oxidation measured. However, in microtiter single pulse photolysis, no exclusion volume is necessary; the entire sample volume is illuminated by a single laser pulse. Therefore, assuming equivalent optical properties (e.g. light scatter from the liquid-air interface vs. light scatter from the capillary), microtiter single pulse photolysis should yield more efficient oxidation than FPOP for equivalent laser fluence. The relatively small change in oxidation suggests that differential optical properties of the capillary versus the open microtiter well compensates largely for the exclusion volume.
4. Conclusion
Here, we demonstrate that microtiter single pulse photolysis can achieve comparable oxidation as traditional FPOP, yielding identical topographical measurements of proteins. With the simple and low-cost design of experimental setup, the newly developed technique reduces the experiment time substantially and provides the opportunity to conduct the experiment with low sample volume. Current robotic options for microtiter plate-based assays can be leveraged to minimize human error during the sample handling, especially when dealing with a large number of samples. Automation of the technique will bring a drastic change in experimental time and reduce sample variation due to human errors. This is a key step in the adaptation of HRPF for sample screening based on topographical changes (e.g. screening for allosteric binding). Similarly, the lack of a flow system adds flexibility to HRPF, allowing for the analysis of viscous samples such as some poorly soluble membrane protein preparations or other samples that present problems in a standard flow system. Additionally, as the whole sample is exposed to the laser pulse, the exclusion volume of the capillary set up can be eliminated, alleviating concerns with laminar flow and improving oxidation efficiency.
Microtiter single pulse photolysis has several drawbacks compared to traditional FPOP that should be clearly acknowledged, as well. The area of incidence between the laser light and the sample is considerably larger for microtiter single pulse photolysis. The use of cylindrical lenses to asymmetrically compress the beam to the shape of the capillary allows traditional FPOP to reach higher laser fluences at more modest pulse energies [27]. Since the area illuminated is much larger in the microtiter plate, the laser energy per pulse required is considerably higher, potentially requiring a higher power laser. Online FPOP offers online mixing of reagents, allowing for interesting technologies including fluidic focusing of living cells for footprinting [12] and rapid mixing for time-resolved footprinting experiments [28]. Additionally, traditional FPOP offers flexibility by examining theoretically unlimited volumes of sample without reloading, limited only by the amount of time the investigator wishes to spend on the experiment and the volume of the sample reservoir used. Additionally, if each laser illumination event is viewed as a separate experiment on the same sample, it becomes apparent that FPOP data actually represents an average of many technical replicates of the photochemical event. While our data showed no systematic advantage in oxidation precision to FPOP over microtiter photolysis, one can imagine such a reproducibility advantage when dealing with light sources with high pulse-to-pulse variability.
With UV-opaque microtiter plates, the volume must be kept from the edges of the well unless the researcher is willing to perform highly precise laser/sample alignment to ensure complete and reproducible illumination of all samples. This limits the volume of sample that can be illuminated in a V-shaped 96-well microtiter plate. The use of UV-transparent quartz microtiter plates and automated plate alignment may alleviate this problem, but the sample volume will still be limited by the well volume and the desired path length for the incident UV light. Microtiter single pulse photolysis has a potential role to play as an extension of current HRPF technologies towards particular high throughput needs or specialty sample analysis, rather than a wholesale replacement of FPOP.
Highlights.
HRPF in 96-well microtiter plates can be achieved yielding comparable results to traditional FPOP
V-shaped microtiter plate wells allows for centering of sample, preventing UV shadowing from the well walls
Both adenine and internal reference standard peptide oxidation provide linear dosimetry profiles in 96-well microtiter plate HRPF comparable to traditional flow FPOP
Identical peptide and protein oxidation is obtained from 96-well microtiter plate HRPF as for traditional flow FPOP with a 15% exclusion volume at similar laser fluence
Acknowledgments
This research was funded by the National Institute of General Medical Sciences (R01GM127267) and the NIH Center for Research Excellence in Natural Products Neuroscience (P20GM104932).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure
J.S.S. discloses a significant financial interest in GenNext Technologies, Inc., a small company seeking to commercialize technologies for protein higher order structure analysis.
References
- 1.Konermann L, Vahidi S, Sowole MA, Mass spectrometry methods for studying structure and dynamics of biological macromolecules, Anal. Chem, 86 (2013) 213–232. [DOI] [PubMed] [Google Scholar]
- 2.Kiselar JG, Chance MR, Future directions of structural mass spectrometry using hydroxyl radical footprinting, J. Mass Spectrom, 45 (2010) 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharp JS, Becker JM, Hettich RL, Protein surface mapping by chemical oxidation: Structural analysis by mass spectrometry, Anal. Biochem, 313 (2003) 216–225. [DOI] [PubMed] [Google Scholar]
- 4.Hambly DM, Gross ML, Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale, J. Am. Soc. Mass Spectrom, 16 (2005) 2057–2063. [DOI] [PubMed] [Google Scholar]
- 5.Gau BC, Chen J, Gross ML, Fast photochemical oxidation of proteins for comparing solvent-accessibility changes accompanying protein folding: Data processing and application to barstar, Biochim. Biophys. Acta, 1834 (2013) 1230– 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Rempel DL, Gau BC, Gross ML, Fast photochemical oxidation of proteins and mass spectrometry follow submillisecond protein folding at the amino-acid level, J. Am. Chem. Soc, 134 (2012):18724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gau B, Garai K, Frieden C, Gross ML, Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4, Biochem, 50 (2011) 8117–8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gau BC, Sharp JS, Rempel DL, Gross ML, Fast photochemical oxidation of protein footprints faster than protein unfolding, Anal. Chem, 81 (2009) 6563–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aye TT, Low TY, Sze SK, Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry, Anal. Chem, 77 (2005) 5814–5822. [DOI] [PubMed] [Google Scholar]
- 10.Watson C, Janik I, Zhuang T, Charvátová Olga. Woods RJ, Sharp JS, Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting, Anal. Chem 81 (2009) 2496–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watkinson TG, Calabrese AN, Ault JR, Radford SE, Ashcroft AE, FPOP-LC-MS/MS suggests differences in interaction sites of amphipols and detergents with outer membrane proteins, J. Am. Soc. Mass Spectrom 28 (2016) 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rinas A, Mali VS, Espino JA, Jones LM, Development of a microflow system for in-cell footprinting coupled with mass spectrometry, Anal. Chem, 88 (2016) 10052–10058. [DOI] [PubMed] [Google Scholar]
- 13.Chea EE, Jones LM, Modifications generated by fast photochemical oxidation of proteins reflect the native conformations of proteins, Protein Sci, 27 (2018) 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y, Serra A, Guo T, Park JE, Zhong Q, Sze SK, Application of nanosecond laser photolysis protein footprinting to study EGFR activation by EGF in cells, J. of Prot. Res, 16 (2017) 2282–2293 [DOI] [PubMed] [Google Scholar]
- 15.Jumper CC, Schriemer DC, Mass spectrometry of laser-initiated carbene reactions for protein topographic analysis, Anal. Chem 83 (2011) 2913–2920. [DOI] [PubMed] [Google Scholar]
- 16.Jumper CC, Bomgarden R, Rogers J, Etienne C, Schriemer DC, High-resolution mapping of carbene-based protein footprints, Anal. Chem 84 (2012) 4411–4418. [DOI] [PubMed] [Google Scholar]
- 17.Zhang B, Rempel DL, Gross ML, Protein footprinting by carbenes on a fast photochemical oxidation of proteins (FPOP) platform, J. Am. Soc. Mass Spectrom, 27 (2015) 552–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie B, Sood A, Woods RJ, Sharp JS, Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection, Scientific Rep, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang W, Ravikumar KM, Chance MR, Yang S, Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis, Biophys. J, 108 (2015) 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaur P, Kiselar J, Yang S, Chance MR, Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry (MS), Mol. & Cell. Prot, 14 (2015) 1159–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson C, Sharp JS, Conformational analysis of therapeutic proteins by hydroxyl radical protein footprinting, The AAPS J, 14 (2012) 206–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Storek Kelly M., et al. “Monoclonal antibody targe ting the β-barrel assembly machine of Escherichia Coli is bactericidal.” Proc. Natl. Acad. Sci. U.S.A, 115 (2018) 3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Moniz H, Wang S, Ramiah A, Zhang F, Moremen KW, et al. , High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1-Heparin binding interface, J. Biol. Chem, 290 (2015) 10729–10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie B, Sharp JS, Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method, Anal. Chem, 87 (2015) 10719–10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charvátová O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ, Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: Application to galectin-1, J. Am. Soc. Mass Spectrom, 19 (2008) 1692–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chance MR, Unfolding of apomyoglobin examined by synchrotron footprinting, Biochem. Biophys. Res. Commun, 287 (2001) 614–621 [DOI] [PubMed] [Google Scholar]
- 27.Zhang B, Cheng M, Rempel D, Gross ML, Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology, Methods, 144 (2018) 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vahidi S, Stocks BB, Liaghati-Mobarhan Y, Konermann L, Submillisecond protein folding events monitored by rapid mixing and mass spectrometry-based oxidative labeling, Anal. Chem 85 (2013) 8618–8625 [DOI] [PubMed] [Google Scholar]