

Abstract
Background
An E321G mutation in MYH1 was recently identified in Quarter Horses (QH) with immune‐mediated myositis (IMM) defined by a phenotype of gross muscle atrophy and myofiber lymphocytic infiltrates.
Hypothesis/Objectives
We hypothesized that the MYH1 mutation also was associated with a phenotype of nonexertional rhabdomyolysis. The objective of this study was to determine the prevalence of the MYH1 mutation in QH with exertional (ER) and nonexertional (nonER) rhabdomyolysis.
Animals
Quarter Horses: 72 healthy controls, 85 ER‐no atrophy, 56 ER‐atrophy, 167 nonER horses selected regardless of muscle atrophy.
Methods
Clinical and histopathologic information and DNA was obtained from a database for (1) ER > 2 years of age, with or without atrophy and (2) nonER creatine kinase (CK) ≥ 5000 U/L, <5 years of age. Horses were genotyped for E321G MYH1 by pyrosequencing.
Results
The MYH1 mutation was present in a similar proportion of ER‐no atrophy (1/56; 2%) and in a higher proportion of ER‐atrophy (25/85; 29%) versus controls (4/72; 5%). The MYH1 mutation was present in a significantly higher proportion of nonER (113/165; 68%) than controls either in the presence (39/42; 93%) or in absence (72/123; 59%) of gross atrophy. Lymphocytes were present in <18% of muscle samples with the MYH1 mutation.
Conclusions and Clinical Importance
Although not associated with ER, the MYH1 mutation is associated with atrophy after ER. The MYH1 mutation is highly associated with nonER regardless of whether muscle atrophy or lymphocytic infiltrates are present. Genetic testing will enhance the ability to diagnose MYH1 myopathies (MYHM) in QH.
Keywords: immune‐mediated, myopathy, myosin, myositis, tying‐up
Abbreviations
- AST
aspartate transaminase
- CK
creatine kinase
- ER
exertional rhabdomyolysis
- GYS1
glycogen synthase 1
- IMM
immune‐mediated myositis
- MH
malignant hyperthermia
- MYH1
myosin heavy chain 1
- MYH2
myosin heavy chain 2
- NMDL
Neuromuscular Diagnostic Laboratory, Michigan State University
- nonER
nonexertional rhabdomyolysis
- PSSM1
type 1 polysaccharide storage myopathy
- QH
Quarter Horse
1. INTRODUCTION
Immune‐mediated myositis (IMM) is a well‐recognized cause of rapid onset muscle atrophy in Quarter Horse (QH)‐related breeds.1, 2 Atrophy often follows exposure to environmental stimuli, such as respiratory pathogens or vaccination, but the triggering factor remains unknown in >40% of cases.1 An E321G mutation in MYH1 recently was found to be strongly associated with IMM in QH.3 Horses with IMM were both homozygous (My/My) and heterozygous (My/N) for the MYH1 mutation. The mutation was identified by means of a genome‐wide association study, with the phenotype for IMM defined as gross muscle atrophy and lymphocytic myofiber infiltrates. Subsequent sequencing of genes within the associated locus identified a missense mutation in a highly conserved region of MYH1 that segregated with disease status.3 The MYH1 gene encodes myosin heavy chain 2X, which is found in fast twitch type 2X muscle fibers. In the absence of inflammation, the proportion of type 2X muscle fibers in IMM muscle was within normal limits. However, in IMM horses with gross muscle atrophy, lymphocytes infiltrated type 2X myofibers and fewer type 2X fibers were present.3 The equine MYH1 mutation is the first reported disease‐causing mutation in MYH1 in any species. The biological relevance of the E321G MYH1 mutation is strengthened by the fact that glutamic acid (E) is highly conserved across species (Sus, Bos, Homo, Oryctol, Rat, and Canis) and present in the corresponding position of the myosin heavy chain of type 2A, perinatal, extraocular, embryonic, and cardiac/slow myosin in many species, including the horse.4
The most common muscle disorder in QHs, rhabdomyolysis, shares the characteristic of increased serum creatine kinase (CK) activity with IMM.5 Causes of rhabdomyolysis range from toxins such as hypoglycin A to genetic disorders such as type 1 polysaccharide storage myopathy (PSSM1) and malignant hyperthermia (MH).6 However, in many cases of rhabdomyolysis, the specific etiology is unrecognized.5 The features distinguishing rhabdomyolysis from IMM are the lack of rapid‐onset muscle atrophy and the absence of lymphocytic infiltrates in gluteal or epaxial muscle biopsy samples.1, 5, 7 A diagnosis of IMM in horses could be missed, however, when the primary presenting complaint is rhabdomyolysis because the most common muscle biopsy sample submitted for evaluation of rhabdomyolysis is from the semimembranosus (SM) or semitendinosus (ST) muscles. These muscles often lack lymphocytic infiltrates in IMM horses.1, 2
We hypothesized that the E321G MYH1 mutation would be prevalent in horses presenting with nonexertional rhabdomyolysis (nonER) regardless of whether or not horses presented with or without muscle atrophy. The purpose of our retrospective study was to determine if the E321G MHY1 mutation associated with IMM also was associated with the phenotype of rhabdomyolysis with or without gross muscle atrophy and with or without lymphocytic infiltrates in muscle samples. The objectives were: (1) to determine the prevalence of the MYH1 mutation in QH presenting with ER with or without gross muscle atrophy; (2) to determine the prevalence of the MYH1 mutation in young QH presenting with nonER regardless of gross muscle atrophy; and (3) to characterize the histopathologic findings in muscle biopsy samples of horses with rhabdomyolysis and the MYH1 mutation.
2. MATERIALS AND METHODS
2.1. Horses
2.1.1. Controls
The prevalence of the MYH1 mutation in healthy QH was determined in 72 healthy QH, which were housed on farms across the US, had no known history of muscle disease and had DNA available for testing (Table 1).
Table 1.
Mean ages and genders of horses with exertional (ER) and nonexertional rhabdomyolysis (nonER) grouped according to whether or not they presented with muscle atrophy
| GYS1 status | Mean age years (SD) | Mares | Geldings | Stallions | Not specified | |
|---|---|---|---|---|---|---|
| Control (N = 72) | Both | 8.3 ± 4.9 | 44 | 26 | 1 | 1 |
| ER horses (N = 141) | 8.2 ± 4.2 | 61 | 69 | 4 | 7 | |
| Atrophy (N = 23) | Positive | 7.5 ± 4.6ab | 10 | 13 | 0 | 0 |
| Atrophy (N = 62) | Negative | 7.5 ± 4.0a | 22 | 34 | 3 | 3 |
| No atrophy (N = 56) | Negative | 9.8 ± 3.9b | 29 | 22 | 1 | 4 |
| NonER (N = 165) | 1.5 ± 1.2 | 92 | 34 | 35 | 4 | |
| Atrophy (N = 42) | Negative | 1.3 ± 1.0c | 22 | 10 | 10 | 0 |
| No atrophy (N = 123) | Negative | 1.6 ± 1.3c | 70 | 24 | 25 | 4 |
Horses with ER did not have any restrictions on age for inclusion whereas nonER horses were selected with an upper age‐limit of 4 years of age. Different superscripts indicate significant differences within columns where ER and for nonER analyzed separately. Abbreviation: GYS1, glycogen synthase 1.
2.1.2. Exertional rhabdomyolysis
Records and the sample repository of the Neuromuscular Diagnostic Laboratory at Michigan State University (NMDL) were reviewed between 2012 and 2017 to identify approximately 60 QH > 2 years of age, negative for the GYS1 mutation, with a history of ER and DNA available for testing. Exertional rhabdomyolysis was identified by referring veterinarians based on clinical signs of muscle stiffness after exercise and recorded on submission forms. Serum CK or AST activity were not always reported on submission forms and were not used as selection criteria for ER. Records of 948 ER horses also were searched from 1995 to 2017 to identify QH with hair, buffy coat, or muscle samples available for DNA extraction and that had a history of ER and reported concurrent gross muscle atrophy. If available in the database, the R309H GYS1 genotype for type 1 polysaccharide storage myopathy (PSSM1) was noted and, if unavailable, horses were genotyped for the GYS1 mutation.8 If available in the database, the genotype for the C7360G RYR1 mutation for malignant hyperthermia was noted.9
2.1.3. Nonexertional rhabdomyolysis
Records and the sample repository of the NMDL were reviewed from 1995 to 2017 to identify all QH < 5 years of age that had a history consistent with nonER, documented serum CK activity ≥5000 U/L and hair, buffy coat, or muscle samples available for DNA extraction. If available in the database, the R309H GYS1 genotype for PSSM1 was noted and, if unavailable, horses were genotyped for the GYS1 mutation.8 All horses with the GYS1 mutation were excluded from further analysis to ensure that PSSM1 did not contribute to nonER. If available in the database, the genotype for C7360G RYR1 was noted and, if glycogen depletion had been noted in the histochemical evaluation, analysis was performed for C7360G RYR1. 9 Clinical signs recorded by the referring veterinarian were tabulated and compared between genotypes.
2.2. Histopathology
The sample repository was reviewed to identify those horses that had muscle biopsy samples of the gluteus, epaxial, SM, or ST muscles submitted for evaluation. Archived histologic slides from ER with muscle biopsy samples and all nonER horses were re‐evaluated blinded to genotype. Slides stained with hematoxylin and eosin (HE), periodic acid Schiffs (PAS), and amylase‐PAS were scored for the presence of fiber size variation, anguloid atrophy, internalized myonuclei, acute myofiber degeneration (loss of striations without cellular infiltrate), macrophage infiltrates, myofiber lymphocytic infiltrates, mononuclear cell vascular cuffing, abnormal polysaccharide, and PAS depletion of small or large myofibers. The scoring system used at ×20 magnification to grade the above features was based on: 0 = not present, 1 = present in approximately 10% of fibers in the biopsy sample, 2 = present in approximately 11‐25% of fibers in the sample, and 3 = present in >25% of fibers in the sample. Periodic acid Schiffs stains were scored for the intensity of PAS stain as: absent (0), light (1), moderate (2), or darkly stained (3). Scores were compared across genotypes in different horses within the same muscle group. Scores also were compared between muscle groups within a set of horses that had both gluteal/epaxial and SM/ST muscles sampled.
Myosin ATPase staining was performed in 1 batch of gluteal muscle samples from 10 horses homozygous for the MYH1 mutation and 9 normal/normal (N/N) horses. Samples were preincubated for 5 minutes by an acid pH (4.4), which in our laboratory consistently identifies muscle fiber types 1, 2A, and 2X.10 Muscle fiber type composition was determined by counting the fiber types of at least 250 myofibers. Type 2 fiber types were further evaluated in 2 N/N and 3 MYH1 homozygotes with immunofluorescent (IF) antibody staining for myosin heavy chains as previously described.3, 11 Primary antibodies used included type 2A IgG at 1:6 (A4.74 DSHB) and for both type 2A and 2X IgG at 1:10 (NCL‐MHCf Leica Biosystems, Wetzlar, Germany). Sections were examined by a fluorescence microscope (Olympus, Tokyo, Japan) with filters designed for each of the different emitting wavelengths.
Images were captured and pseudocolored composites generated.
2.3. Genotyping
2.3.1. DNA isolation
The DNA was isolated from hair bulbs, buffy coat, whole blood, or muscle tissue by Gentra Puregene Kits (Qiagen; Germantown, Maryland) and quantified to ensure ≥10 ng/μL of DNA was available with a 260/280 ratio of ≥1.8 and ≤ 2.2. DNA for PCR reactions was diluted to 35 ± 25 ng/μL.
2.3.2. Polymerase chain reaction
Thermocycling was carried out by recombinant AmpliTaqs Gold polymerase (Applied Biosystems, Foster City, California) in a PTC‐200 thermocycler (MJ Research Inc., Watertown, Massachusetts). Each 50 μL PCR reaction contained 19 μL Platinum Green PCR 2X MM reagent (Invitrogen, Carlsbad, California), 1 μL of each the forward and reverse primers, 200 ± 25 ng of purified chromosomal DNA, with the remaining volume made up with sterile double‐distilled H2O. Amplification was performed under the following conditions: the initial denaturation temperature was set at 95°C for 10 minutes and the subsequent denaturation at 94°C for 30 seconds, annealing at 46°C for 30 seconds, and a primer extension at 72°C for 30 seconds for 40 cycles with a final extension time of 10 minutes at 72°C. The PCR products were run on a 1.0% agarose gel and stained with ethidium bromide.
2.3.3. Pyrosequencing
Forward, reverse, and sequencing primers were designed by Pyrosequencing Assay Design Software (version 1.0; Biotage, Uppsala, Sweden) and biotin labeled, and regular primers were ordered from Integrated DNA Technologies (IDT), Skokie, Illinois. Forward (5′‐TAAAAAGCTGCATGTGTA‐3′) and reverse (5′‐AAAACACATACCCTGAAT‐3′) primers were used to amplify a 159‐bp region (ECA11: 52993805‐52993963) containing a coding single nucleotide polymorphism (SNP) (T/C, ECA11: 52993877) of the MYH1 mutation. The resulting amplicon was annealed with the sequencing primer (5’‐TGCTGGGGACTGTGA‐3′) and relative expression of each allele was determined by pyrosequencing following manufacturer's protocol (Qiagen; Germantown, Maryland). All sample plates were analyzed with suitable positive controls and template control containing no DNA, with resulting data manually evaluated to determine genotypes.
2.4. Data analysis
Normality was tested via a Shapiro‐Wilk normality test. A 1‐way analysis of variance (ANOVA) was used to compare ages of horses. A chi‐square test was used to compare the genotypes of random QH controls, ER and nonER horses with specific clinical signs as well as to compare the number of horses with lymphocytic infiltrates and large myofiber glycogen depletion. Scores for muscle histopathology were compared by Mann‐Whitney test. Serum CK and AST activities in nonER horses were log transformed to normalize data and compared by one‐way ANOVA. Statistical analyses were performed in GraphPad Prism 7 (GraphPad Prism version 7, GraphPad Software, La Jolla, California, http://www.graphpad.com). Results are presented as mean and standard deviations (SD). Significance was set at P < .05.
3. RESULTS
There were 72 control horses, similar in age to ER horses (Table 1). Fifty‐six ER horses without atrophy and 85 ER horses with atrophy (62 GYS1 negative, 23 GYS1 positive) were included in the study (Table 1). Among GYS1‐animals, horses with muscle atrophy were significantly younger than horses without muscle atrophy (P < .006). There were 165 nonER horses, 42 with atrophy and 123 without atrophy (Table 1). The mean age of nonER horses was only 1 year despite inclusion criteria that specified all nonER horses <5 years of age.
Control genotypes: The MYH1 mutation was present in 5% of controls (4/72).
3.1. Exertional rhabdomyolysis
3.1.1. ER genotypes
The MYH1 mutation was present in 2% (1/56) of ER horses without gross muscle atrophy (Figure 1). The MYH1 mutation was present in 29% (25/85) of ER with atrophy including both those GYS1+ (5/23; 22%) and GYS1‐ (20/62; 32%), which was a significantly higher proportion than controls (P < .001; Figure 1). The mutation for C7360G RYR1 was not found in any of the 30 ER horses tested.
Figure 1.
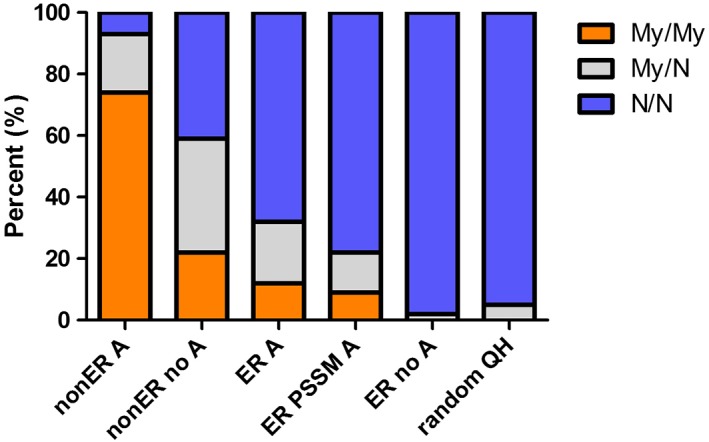
The distribution of MYH1 genotypes in nonER horses with atrophy (nonER A), nonER horses without atrophy (nonER no A), ER horses without atrophy (ER no A), ER horses with atrophy positive for PSSM1 (ER PSSM1 A), ER with atrophy, negative for PSSM1 (ER A) and controls (random QH). Significantly, more nonER horses and ER horses with atrophy possessed the MYH1 mutation (My/My and My/N) compared to random QH
3.1.2. ER histopathology
Slides from 61 muscles were available for evaluation. Only 7 of these horses (8 muscle samples) were homozygous or heterozygous for the MYH1 mutation (2 gluteal/epaxial and 6 SM/ST) and no significant differences were found when histopathology scores for MYH1 homozygotes (My/My) or heterozygotes (My/N) horses combined were compared to N/N horses.
3.2. Nonexertional rhabdomyolysis
3.2.1. NonER genotypes
Significantly, more nonER horses (111/165; 67%) possessed the E321G MYH1 mutation than controls (P < .001). NonER horses were both homozygous (75%) and heterozygous (25%) for the MYH1 mutation (Figure 1). The MYH1 mutation was highly prevalent in nonER horses regardless of atrophy (39/42, 93% with atrophy; 72/123, 59% without atrophy). The mutation for C7360G RYR1 was not present in any of the 81 nonER horses tested.
3.2.2. NonER clinical signs
When specifically evaluating nonER horses with the MYH1 mutation, 65% (72/111) were not reported to have gross muscle atrophy and 35% (39/111) were reported to have atrophy. The MYH1 mutation increased the risk for muscle atrophy 12× in nonER horses (P < .001: OR 12.3). A history of fever was significantly more common in nonER horses with the MYH1 mutation (P < .038; odds ratio [OR], 7.6). Weakness (P < .003; OR, 0.06) was less common in horses with the MYH1 mutation compared to nonER horses without the mutation. The frequencies of recumbency, swollen muscles, stiffness, colic, or history of infection were not significantly different between nonER horses with and without the MYH1 mutation. Serum CK activities were markedly increased in nonER with maximum activities >1 million U/L in all genotypes (Figure 2). Log‐transformed serum CK (P < .006) and AST (P < .001) activities were significantly higher in MYH1 heterozygotes and homozygotes compared to nonER horses without the mutation, with no significant differences between homozygotes and heterozygotes.
Figure 2.
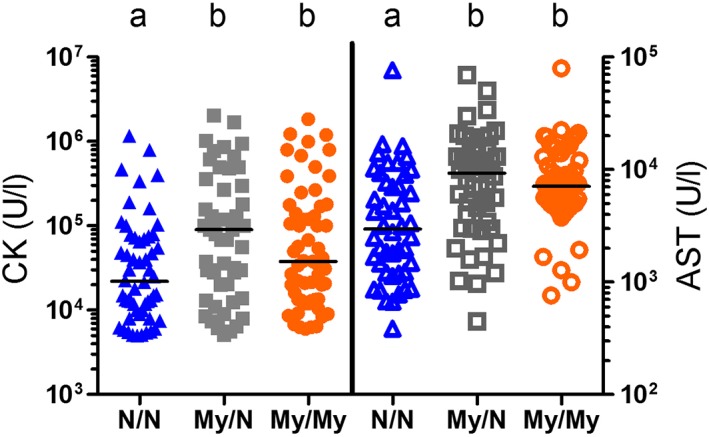
Serum CK and AST activities in individual nonER horses homozygous (My/My), heterozygous (My/N), or unaffected (N/N) for the MYH1 mutation. Different letters indicate significant differences within CK and AST
3.2.3. NonER histopathology
Slides from 153 nonER muscle samples were available for evaluation. The gluteal/epaxial muscles and the SM/ST muscles of My/My horses had significantly higher histopathologic scores for fiber size variation, anguloid atrophy, and centrally displaced myonuclei than N/N nonER horses (Figures 3 and 4, Table 2). Scores for acute myofiber degeneration, lymphocytic infiltrates, and macrophages were significantly higher in gluteal/epaxial muscles of My/My horses than in N/N nonER horses with no significant difference for these features between genotypes in SM/ST muscles (Table 2). This finding agrees with comparisons of the 2 muscle groups within the same MYH1 homozygotes where the gluteal/epaxial muscles had significantly higher scores for acute necrosis, macrophages, and lymphocytes than did the SM/ST muscles (Table 3). Each individual score for heterozygotes was intermediate between My/My and N/N nonER horses but not statistically different from My/My or N/N (Figure 3, Table 2). Fewer than 18% of horses with the MYH1 mutation had lymphocytic infiltrates in myofibers (Table 4) and the frequency of samples with lymphocytic infiltrates did not differ between horses with and without the MYH1 mutation (Table 4). Total myopathic scores were significantly higher in My/My and My/N heterozygotes for gluteal/epaxial and SM/ST (Table 2).
Figure 3.
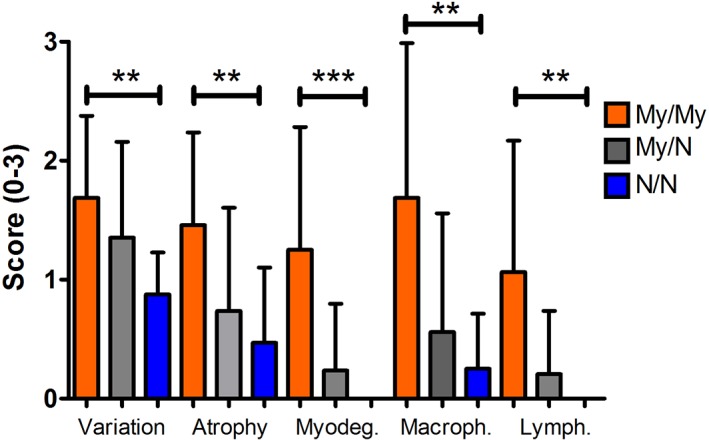
Histopathological scores for skeletal muscle fiber type variation, anguloid atrophy, acute myofiber degeneration (myodegen), macrophages (macroph), and lymphocytes (lymph) in large myofibers in the gluteal muscle of horses homozygous (My/My), heterozygous (My/N), or unaffected (N/N) for the MYH1 mutation. *P < .05, **P < .01, ***P < .001
Figure 4.
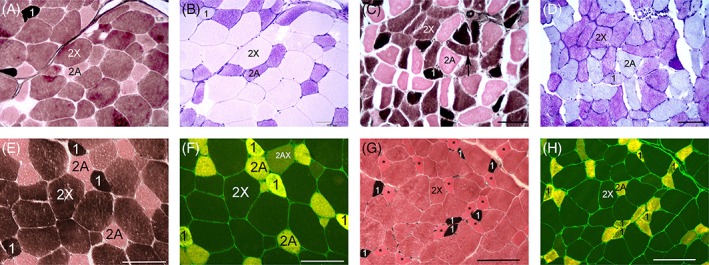
Glycogen depletion patterns (A‐D) and muscle fiber types identified by either myosin ATPase (E,G) or IF myosin heavy chain antibody staining (F,H). Myosin ATPase staining of N/N and My/My horses was performed at the same time. A, Three distinct fiber type (1, 2A, and 2X) with the largest fibers being type 2X in an N/N horse. Myosin ATPase stain (pH 4.4) 20X. B, Complete depletion of muscle glycogen in type 1 and 2X myofibers of the N/N horse with nonexertional rhabdomyolysis. PAS 20X. C, Three distinct fiber types with anguloid atrophy of type 2X myofibers in an My/My horse. Type 2X fibers show anguloid atrophy (arrow) and the largest sized fibers are type 2A. Myosin ATPase stain (pH 4.4) 20X. D, Glycogen depletion is present in type 1 and 2A myofibers of the MY/My horse. PAS 20X. E, Type 2A and 2X fiber types are readily distinguished in myosin ATPase stains of an N/N horses with nonER. Myosin ATPase (pH 4.4) 20X. F, Type 2A, 2X and a few 2AX fiber types are apparent in the N/N non ER horse. Type 1 fibers were distinguished by cross‐matching with black stained type 1 fibers in the ATPase stain. IF stain 20X. G, This My/My nonER horse only had two distinct fiber types (1 and 2) in the myosin ATPase stain. Asterisks represent the type 2A fibers that were identified by serial IF antibody stain. A type 2X fiber identified by IF stain is labeled 2X. Note the generally smaller fiber sizes compared to the control. myosin ATPase (pH 4.4) 20X. H. Type 2A and 2X fibers are distinguished in IF stain of the My/My horse in G. Type 1 fibers were identified by cross‐matching black stained type 1 fibers in the ATPase stain with corresponding fibers in the serial IF stain
Table 2.
Mean (±SD) histopathologic scores in gluteal/epaxial muscles and semimembranosus (SM)/semitendinosus (ST) muscles in horses with nonexertional rhabdomyolysis (nonER) and exertional rhabdomyolysis (ER)
| Muscle | N | Genotype | Fiber size variation | Anguloid atrophy | Internalized myonuclei | Acute myodegen. | Macroph. | Lymph. | Large fiber glycogen depletion | Total score |
|---|---|---|---|---|---|---|---|---|---|---|
| NonER | ||||||||||
| Glut/Epaxial | 24 | My/My | 1.7 ± 0.7a | 1.5 ± 0.8a | 0.6 ± 0.8a | 1.3 ± 1.0a | 1.7 ± 1.3a | 1.1 ± 1.1a | 1.4 ±1.2a | 11.9 ± 4.1a |
| Glut/Epaxial | 17 | My/N | 1.4 ±0.8ab | 0.7 ± 0.9ab | 0.1 ± 0.5ab | 0.2 ± 0.6ab | 0.6 ± 1.0ab | 0.2 ± 0.5ab | 1.1 ± 1.4ab | 6.4 ± 2.8b |
| Glut/Epaxial | 8 | N/N | 0.9 ± 0.4b | 0.5 ± 0.6b | 0.0 ± 0.0b | 0.0 ± 0.0b | 0.3 ± 0.5b | 0.0 ± 0.0b | 0.4 ± 1.1b | 5.0 ± 2.8b |
| SM/ST | 41 | My/My | 1.5 ± 0.7a | 1.2 ± 0.8a | 0.6 ± 0.8a | 0.2 ± 0.7 | 0.4 ± 0.9 | 0.2 ± 0.7 | 1.1 ± 1.3 | 8.0 ± 4.8a |
| SM/ST | 33 | My/N | 1.3 ± 0.5ab | 0.7 ± 0.8ab | 0.1 ± 0.2ab | 0.2 ± 0.6 | 0.2 ± 0.7 | 0.0 ±0.0 | 0.8 ± 1.1 | 5.5 ± 2.8b |
| SM/ST | 30 | N/N | 1.1 ± 0.5b | 0.5 ± 0.7b | 0.1 ± 0.4b | 0.2 ± 0.5 | 0.2 ± 0.6 | 0.1 ±0.2 | 0.8 ± 1.0 | 5.6 ± 2.5b |
| ER | ||||||||||
| SM/ST | 3 | My/My | 1.3 ± 0.3 | 0.8 ± 0.8 | 1.0 ± 0.9 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 5.5 ± 2.7 |
| SM/ST | 4 | My/N | 1.5 ± 1.3 | 0.9 ± 1.0 | 0.5 ± 0.6 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 6.4 ± 4.2 |
| SM/ST | 46 | N/N | 1.4 ± 0.6 | 0.9 ± 0.9 | 0.4 ± 0.7 | 0.0 ± 0.1 | 0.1 ± 0.3 | 0.0 ± 0.2 | 0.1 ± 0.3 | 6.5 ± 3.4 |
Scores ranged from 0 to 3. Different letters indicate significant differences (P < .05) in scores across genotypes within the same muscle. Abbreviations: nonER, nonexertional rhabdomyolysis; ER, exertional rhabdomyolysis; Myodegen, myodegeneration; macroph., macrophage; lymph, lymphocytes.
Table 3.
Mean (±SD) histopathologic scores in paired gluteal/epaxial muscles and semimembranosus/semitendinosus muscles obtained from the same horses with nonexertional rhabdomyolysis (nonER)
| Muscle | N | Genotype | Fiber size variation | Anguloid atrophy | Internalized myonuclei | Acute mydegen. | Macroph. | Lymph. | Large fiberglycogen depletion |
|---|---|---|---|---|---|---|---|---|---|
| NonER | |||||||||
| Glut/Epaxial | 19 | My/My | 1.6 ± 0.7 | 1.5 ± 0.7 | 0.7 ± 0.7 | 1.2 ± 0.9a | 1.4 ± 1.3a | 0.9 ± 0.9a | 1.3 ± 1.1a |
| SM/ST | 19 | My/My | 1.5 ± 0.5 | 1.3 ± 0.6 | 0.8 ± 0.8 | 0.3 ±0.8 b | 0.3 ± 0.8b | 0.2 ± 0.7b | 1.4 ± 1.4b |
| Glut/Epaxial | 7 | My/N | 1.6 ± 0.8 | 1.0 ± 0.9 | 0.2 ± 0.6 | 0.4 ±0.7 | 1.0 ± 1.2 | 0.3 ± 0.7 | 1.3 ± 1.4 |
| SM/ST | 7 | My/N | 1.3 ± 0.5 | 0.6 ± 1.0 | 0.3 ± 0.5 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.2 |
| Glut/Epaxial | 5 | N/N | 1.0 ± 0.0 | 0.3 ± 0.4 | 0.0 ± 0.0a | 0.0 ± 0.0 | 0.2 ± 0.4 | 0.0 ± 0.0a | 0.0 ± 0.0 |
| SM/ST | 5 | N/N | 0.9 ± 0.5 | 0.8 ± 0.4 | 0.4 ± 0.3b | 0.4 ± 0.4 | 0.6 ± 0.5 | 0.4 ± 0.3b | 0.8 ± 0.6 |
Scores ranged from 0 to 3. Different letters indicate significant differences (P < .05) in scores between muscles within the same genotype
Table 4.
Frequency of large fiber glycogen depletion and lymphocytic infiltrates in gluteal/epaxial (Glut/epax) muscles and semimembranosus/ semitendinosus (SM/ST) muscles of horses genotyped for the MYH1 mutation
| Muscle | N total | Genotype | Lymphocytic infiltrates N (%) | Large fiber glycogendepletion N (%) |
|---|---|---|---|---|
| Glut/Epaxial | 24 | My/My | 3 (12.5) | 7 (29.2) |
| Glut/Epaxial | 17 | My/N | 3 (17.6) | 5 (29.4) |
| Glut/Epaxial | 8 | N/N | 0 (0) | 1 (12.5) |
| SM/ST | 41 | My/My | 4 (9.8) | 11 (26.8)a |
| SM/ST | 33 | My/N | 0 (0) | 2 (6.1)b |
| SM/ST | 30 | N/N | 3 (10.0) | 2 (6.7)b |
Letters indicate significant differences in glycogen depletion within the same muscle. No differences were found in the frequency of lymphocytes across genotypes. Different letters indicate significant differences (P < .05) in scores across genotypes within the same muscle
No significant differences were found in scores for PAS stain intensity, glycogen depletion in small myofibers, or abnormal cytoplasmic glycogen between My/My, My/N, or N/N nonER horses. However, significant differences were identified in scores for glycogen depletion in large myofibers of the gluteal/epaxial muscles across genotypes, with My/My horses having significantly higher scores than N/N nonER horses (Figure 4, Table 2). The number of horses with total glycogen depletion in large fibers of SM/ST also was higher in My/My versus N/N nonER horses (Table 4). Subjectively, the largest fibers were typical type 2X fibers in N/N nonER horses, whereas in My/My horses, the largest fibers were typically type 2A fibers (Figure 4).
In myosin, ATPase stains of N/N horses, 3 distinct fiber types were readily distinguished (Figure 4), with the mean fiber type composition being type 1:8 ± 9%, type 2A: 24 ± 11% and type 2X: 67 ± 12%. Three distinct fiber types were apparent in 4/10 My/My horses, with highly variable fiber type compositions of type 1:13 ± 17%, type 2A: 49 ± 26%, and type 2X: 43 ± 21%. In 6/10 nonER MYH1 homozygotes, it was difficult to discern type 2A and type 2X fibers ATPase stains (Figure 4). By IF staining for type 2 myosin heavy chains, type 2 fiber subtypes, 2A, 2AX, and 2X were readily apparent in My/My and N/N horse muscle.
4. DISCUSSION
The major finding in our study was the strong association between the E321G MYH1 mutation and nonER in QH‐related breeds. In total, 67% of nonER QH possessed the MYH1 mutation compared to 5% of randomly selected healthy QH. Rhabdomyolysis in the nonER horses was severe with median serum CK activities above 30 000 U/L and maximal activities of over 1 million U/L, much higher than median activities previously reported for IMM.1 Neither PSSM1, which affects 11% of QH, nor MH which affects <1% of QH (16/7334 tested 2008‐2014; personal observation), were likely causes of nonER in our study because PSSM1 horses were excluded from the analysis and genetic testing was negative for MH in all horses tested.12
The E321G MYH1 mutation appeared to be dominant with respect to rhabdomyolysis because both heterozygotes and homozygotes had serum muscle enzyme activities that were significantly higher than those reported for horses with rhabdomyolysis because of other causes. The MYH1 mutation appeared to have a codominant effect on muscle histopathology in that nonER homozygotes had higher total histopathologic scores than heterozygotes in both gluteal/epaxial and SM/ST muscles and scores for each histopathologic characteristic in heterozygotes were intermediate between homozygotes and N/N horses. Unlike PSSM1 and MH, exercise was not a trigger for rhabdomyolysis in horses with the MYH1 mutation, with only 2% of horses with ER and normal muscle mass possessing the MYH1 mutation.9, 13
Quarter Horses with gross muscle atrophy were highly likely to possess the MYH1 mutation with 93% of nonER QH with atrophy being My/My or My/N and 29% of atrophied ER horses also were My/N. Quite unexpectedly, however, nearly 60% of nonER horses with the MYH1 mutation did not have gross muscle atrophy, the most common clinical sign reported for IMM.1, 14 It is possible that atrophy developed later after muscle biopsy in some horses or that horses died before developing atrophy. Alternatively, moderate to severe rhabdomyolysis may be an early if not sole myopathic feature of nonER in some QH with the MYH1 mutation. Our results therefore suggest that testing for the MYH1 mutation is indicated in QH with nonER whether or not atrophy is present and in ER horses that develop muscle atrophy.
One of the previously reported features of muscle from IMM horses with the MYH1 mutation is immune‐mediated lymphocytic destruction of type 2X muscle fibers, which comprise at least 60% of fibers in healthy gluteal muscle.3 Both innate and adaptive immunity could trigger IMM. The innate immune response could drive reactivity to self and affect the type of adaptive immune response that occurs when the mutant form of type 2X myosin heavy chain is released from myofibers after muscle damage (ER, trauma, and vaccination).14, 15 Shared epitopes among bacteria, such as the M protein of group A Streptococcus spp. and the altered myosin in horses with the MYH1 mutation are potential instigators of an adaptive immune response.3, 16 Of note, 2 young horses in our study that were homozygous and heterozygous for MYH1 were previously reported to have fatal S. equi‐associated rhabdomyolysis.17 This observation suggests that the MYH1 mutation may be the cause of previously reported S. equi‐associated rhabdomyolysis in horses. Agents such as S. equi zooepidemicus, C. pseudotuberculosis, equine herpes virus, and equine influenza virus previously have been associated with triggering IMM and therefore potentially could be triggers for rhabdomyolysis in QH with the MYH1 mutation through the innate response either to primary muscle damage or by adaptive immunity.14 Atrophy, fever, and infection in the presence of rhabdomyolysis appear to be strong indicators to perform MYH1 genetic testing in QH.
A confounding factor in connecting rhabdomyolysis to cell‐mediated destruction of type 2X fibers in our study was the fact that <18% of horses with the MYH1 mutation had lymphocytic infiltrates in muscle biopsies. The lack of lymphocytic infiltrates in many muscle samples suggests that either the cell‐mediated immune reaction in skeletal muscle was not captured in the muscle biopsies evaluated or that an immune‐mediated mechanism is not entirely responsible for rhabdomyolysis. The SM muscle alone was sampled in 48% of nonER horses and inflammatory infiltrates are more likely to be present in epaxial and gluteal muscles in IMM horses.2 Therefore, the selection of the SM muscle for biopsy could have decreased the ability to identify lymphocytic infiltrates. In 59% percent of horses with the MYH1 mutation, however, gluteal/epaxial biopsy samples did not contain lymphocytic infiltrates. Timing of biopsies could have affected inflammatory infiltrates such that biopsies taken several weeks after the onset of atrophy potentially lacked inflammatory cells. Smaller needle biopsies of epaxial and gluteal muscles also could have resulted in missed areas of inflammation. Alternatively, other mechanisms may exist for rhabdomyolysis in horses with the MYH1 mutation.
Some myopathies associated with mutations in MYH2, encoding type 2A myosin heavy chain, exist in humans and are not believed to have an immune‐mediated basis.18, 19, 20 Weakness rather than rhabdomyolysis are primary presenting complaints in these cases. Histopathologic findings in late onset dominant MYH2 mutations overlap features of equine MYH1 myopathy in our study and include increased variation in muscle fiber sizes, anguloid myofiber atrophy and centrally located nuclei. Type 2A fiber are either atrophic with dominant MYH2 mutations or absent in children with recessive deletions.18, 21 Atrophic and atrophied type 2X fibers were present in some horses with MYH1 myopathy, suggesting the mutation itself may be a cause of atrophy. Mutations in MYH2 in humans, however, have not been associated with rhabdomyolysis.
Our study provided an indication the MYH1 mutation affect the activity of the myosin ATPase enzyme. The ATPase enzyme is located in the globular head of type 2X myosin near the site of the MYH1 mutation.3 Modeling suggests that the E321G MYH1 mutation decreases contact between the SWITCH1 and helix 1 domains of the myosin 2X globular head and decreases stability of the protein.3 This altered protein interaction could affect the myosin ATPase enzyme, which is commonly used in histochemical assays to detect fiber types. In our study, IF staining and myosin ATPase assays both differentiated 2A and 2X fiber types consistently in N/N horses. In 60% of horses, homozygous for the MYH1 mutation however, myosin ATPase staining failed to clearly distinguish 2A and 2X, even though the 2A and 2X fibers were identified by IF antibody staining. All samples stained with myosin ATPase were handled in the same fashion in that they were shipped overnight to the laboratory before freezing. The difference in staining of fast twitch fibers by ATPase activity in N/N and My/My horses suggests that the functional effect of the MYH1 mutation on the speed of contraction and myosin ATPase enzyme should be further investigated.
Complete depletion of glycogen is an unusual feature of equine muscle biopsy samples. Approximately 30% of nonER MYH1 homozygotes showed marked glycogen depletion in large myofibers of both epaxial/gluteal and SM/ST samples. Glycogen depletion also was present in 7‐13% of N/N horses with rhabdomyolysis. Glycogen depletion is a feature of episodes of MH in which increased myoplasmic calcium increases actin/myosin interaction, markedly increases ATP consumption and leads to marked anaerobic glycolysis and glycogen degradation.9, 22 Malignant hyperthermia was unlikely in the horses in our study because none of the horses tested, including those with glycogen depletion scores of 3, had the equine mutation for MH. Based on the location of the MYH1 mutation in the myosin 2X globular head near the ATP binding pocket, and the inconsistent demonstration of myosin ATPase activity in type 2X fibers, one could postulate a potential role for abnormal actin/myosin interaction in a fashion similar to MH, resulting in excessive glycogen metabolism and rhabdomyolysis in the non ER horses.3 Determining whether there is a potential role of altered myosin ATPase enzyme activity and excessive glycogen metabolism in the development of nonER in horses with MYH1 is an intriguing possibility that requires further exploration.
In conclusion, the E321G MYH1 mutation is strongly associated with nonER in QH‐related breeds whether or not generalized muscle atrophy is present. Because most nonER horses with the MYH1 mutation did not present with muscle atrophy or have lymphocytic infiltrates in muscle biopsy samples, we suggest that the term MYH1 myopathy (MYHM) be applied specifically to horses with atrophy or rhabdomyolysis that test positive for the MYH1 mutation. This term rather than IMM for horses with this genetic mutation eliminates the confusion about whether the underlying mechanism for rhabdomyolysis and atrophy is immune‐mediated and distinguishes those inflammatory or immune‐mediated myopathies that are not caused by the MYH1 mutation.
CONFLICT OF INTEREST DECLARATION
Dr Valberg is one of the owners of the patent for the PSSM genetic test and receives sales income from its use. Her financial interest has been reviewed and managed by the University in accordance with its conflict of interest policies. Michigan State University and the University of California, Davis have applied for a patent for the E321G MYH1 genetic mutation test.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Authors declare no IACUC or other approval was needed.
ACKNOWLEDGMENTS
The assistance of Zoe Williams and Megan Bertels is gratefully acknowledged. The project was performed at Michigan State University. Funding by the American Quarter Horse Foundation and the endowment of the Mary Anne McPhail Equine Dressage Chair in Equine Sports Medicine. Support for CJF was provided by the National Institutes of Health (NIH; 1K01OD015134 and L40TR001136). The funding sources did not contribute to study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Valberg SJ, Henry ML, Perumbakkam S, Gardner KL, Finno CJ. An E321G MYH1 mutation is strongly associated with nonexertional rhabdomyolysis in Quarter Horses. J Vet Intern Med. 2018;32:1718–1725. 10.1111/jvim.15299
Funding information American Quarter Horse Foundation; Foundation for the National Institutes of Health, Grant/Award Number: 1K01OD015134L40 TR001136
[Corrections added after publication 06 August 2018: Pages 2, 4, 6, and 8 have been updated to remove redundant text, to fix grammatical items, and update for clarity.]
REFERENCES
- 1. Lewis SS, Valberg SJ, Nielsen IL. Suspected immune‐mediated myositis in horses. J Vet Intern Med. 2007;21:495‐503. [DOI] [PubMed] [Google Scholar]
- 2. Durward‐Akhurst SA, Valberg SJ. Immune‐mediated muscle diseases of the horse. Vet Pathol. 2017;55:68‐75. [DOI] [PubMed] [Google Scholar]
- 3. Finno CJG,G, Perumbakkam S, Williams ZJ, et al. A missense mutation in MYH1 is associated with susceptibility to immune‐mediated myositis in quarter horses. Skelet Muscle. 2018;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weiss A, Schiaffino S, Leinwand LA. Comparative sequence analysis of the complete human sarcomeric myosin heavy chain family: implications for functional diversity. J Mol Biol. 1999;290:61‐75. [DOI] [PubMed] [Google Scholar]
- 5. Valberg SJ. Diseases of muscle In: Smith BP, ed. Large Animal Internal Medicine. St. Louis, MO: Mosby; 2014:1276‐1308. [Google Scholar]
- 6. Valberg SJ. Disorders of the musculoskeletal system In: Reed SB,W, Sellon D, eds. Equine Internal Medicine. St. Louis MO: Elsevier; 2018:542‐549. [Google Scholar]
- 7. Durward‐Akhurst SA, Finno CJ, Barnes N, et al. Major histocompatibility complex I and II expression and lymphocytic subtypes in muscle of horses with immune‐mediated myositis. J Vet Intern Med. 2016;30:1313‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McCue ME, Valberg SJ, Miller MB, et al. Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 2008;91:458‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aleman M, Nieto JE, Magdesian KG. Malignant hyperthermia associated with ryanodine receptor 1 (C7360G) mutation in quarter horses. J Vet Intern Med. 2009;23:329‐334. [DOI] [PubMed] [Google Scholar]
- 10. Brooke MH, Kaiser KK. Muscle fiber types: how many and what kind? Arch Neurol. 1970;23:369‐379. [DOI] [PubMed] [Google Scholar]
- 11. Tulloch LK, Perkins JD, Piercy RJ. Multiple immunofluorescence labelling enables simultaneous identification of all mature fibre types in a single equine skeletal muscle cryosection. Equine Vet J. 2011;43:500‐503. [DOI] [PubMed] [Google Scholar]
- 12. Tryon RC, Penedo MC, McCue ME, et al. Evaluation of allele frequencies of inherited disease genes in subgroups of American quarter horses. J Am Vet Med Assoc. 2009;234:120‐125. [DOI] [PubMed] [Google Scholar]
- 13. Valberg SJ, Cardinet GH 3rd, Carlson GP, et al. Polysaccharide storage myopathy associated with recurrent exertional rhabdomyolysis in horses. Neuromuscul Disord. 1992;2:351‐359. [DOI] [PubMed] [Google Scholar]
- 14. Hunyadi L, Sundman EA, Kass PH, Williams DC, Aleman M. Clinical implications and hospital outcome of immune‐mediated myositis in horses. J Vet Intern Med. 2017;31:170‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Root‐Bernstein R, Fairweather D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J Theor Biol. 2015;375:101‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pahlman LI, Morgelin M, Eckert J, et al. Streptococcal M protein: a multipotent and powerful inducer of inflammation. J Immunol. 2006;177:1221‐1228. [DOI] [PubMed] [Google Scholar]
- 17. Sponseller BT, Valberg SJ, Tennent‐Brown BS, Foreman JH, Kumar P, Timoney JF. Severe acute rhabdomyolysis associated with Streptococcus equi infection in four horses. J Am Vet Med Assoc. 2005;227:1800‐1807. 1753‐1754. [DOI] [PubMed] [Google Scholar]
- 18. Martinsson T, Oldfors A, Darin N, et al. Autosomal dominant myopathy: missense mutation (Glu‐706 ‐‐> Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci USA. 2000;97:14614‐14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tajsharghi H, Hilton‐Jones D, Raheem O, Saukkonen AM, Oldfors A, Udd B. Human disease caused by loss of fast IIa myosin heavy chain due to recessive MYH2 mutations. Brain. 2010;133:1451‐1459. [DOI] [PubMed] [Google Scholar]
- 20. Tajsharghi H, Darin N, Rekabdar E, et al. Mutations and sequence variation in the human myosin heavy chain IIa gene (MYH2). Eur J Hum Genet. 2005;13:617‐622. [DOI] [PubMed] [Google Scholar]
- 21. D'Amico A, Fattori F, Bellacchio E, et al. A new de novo missense mutation in MYH2 expands clinical and genetic findings in hereditary myosin myopathies. Neuromuscul Disord. 2013;23:437‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hall GM, Lucke JN. Porcine malignant hyperthermia. IX: changes in the concentrations of intramuscular high‐energy phosphates, glycogen and glycolytic intermediates. Br J Anaesth. 1983;55:635‐640. [DOI] [PubMed] [Google Scholar]