

Abstract
The genome of the hyperthermophile Thermotoga maritima contains three isoforms of maltose binding protein (MBP) that are high affinity receptors for di-, tri- and tetra-saccharides. Two of these proteins (tmMBP1 and tmMBP2) share significant sequence identity, approximately 90%, while the third (tmMBP3) shares less than 40% identity. MBP from Escherichia coli (ecMBP) shares 35% sequence identity with the tmMBPs. This subset of MBP isoforms offer an interesting opportunity to investigate the mechanisms underlying the evolution of substrate specificity and affinity profiles in a genome where redundant MBP genes are present. In this study, the X-ray crystal structures of tmMBP1, tmMBP2 and tmMBP3 are reported in absence and presence of oligosaccharides. tmMBP1 and tmMBP2 show larger binding pockets than tmMBP3 enabling them to bind to larger substrates, while tmMBP1 and tmMBP2 also undergo larger substrate induced hinge bending motions (~55°) than tmMBP3 (~35°). Small angle X-ray scattering was used to compare protein behavior in solution and computer simulations provided insights into dynamics of these proteins. Comparing quantitative protein-substrate interactions and dynamical properties of tmMBPs with the promiscuous ecMBP and di-saccharide selective Thermococcus litoralis MBP provide insights into the features that enable selective binding. Collectively, the results provide insights into how the structure and dynamics of tmMBP homologs enable them to differentiate between a myriad of chemical entities whilst maintaining their common fold.
Keywords: Maltose binding protein, ABC transporters, Thermotoga maritima, bacterial membrane transport, binding specificity, protein dynamics, computer simulations, small angle X-ray scattering, X-ray crystallography
Graphical Abstract
INTRODUCTION
Bacteria utilize diverse mechanisms to survive in challenging environmental niches, which include borrowing and duplicating genes.1–3 This provides functional robustness to bacterial genomes by allowing the duplicated components to compensate for any loss of function due to changes in the original gene.4 Operon redundancy can also result in function segregation through specialization of protein isoforms that share significant sequence similarity.5–7 As selection pressure typically removes redundancy, the duplicated genes frequently diverge and vary their roles in bacterial survival, thus escaping evolutionary scrutiny.8, 9 Even when there is reduced sequence identity, such homologs could maintain the same structural fold, allowing the organism to increase functional diversification with minimal structural changes.10–12
Periplasmic binding proteins constitute a large protein super-family commonly involved in substrate binding and translocation of metabolites across the bacterial membrane. In such a large super-family, homologous proteins show conservation in their overall structural fold even with low sequence similarities. The members of this super-family bind a myriad of metabolites; their binding affinities are highly tuned to the environmental niche of the host organism, resulting in differing affinity profiles for identical ligands.13–16 Adjustment of protein structure, especially around the binding pocket, is known to play a role in recognition of ligand(s) in periplasmic binding proteins;17, 18 additionally conformational dynamics can also help fine-tune affinity and specificity by altering the thermodynamics and kinetics of ligand binding in these systems.18 While periplasmic binding proteins are subjected to the constraints of the common structural fold, these proteins manage a wide spectrum of specificity profiles by tailoring the non-bonded interactions in and around their binding sites.13, 19 Moreover, it has also been shown that some periplasmic binding proteins contain inbuilt allosteric mechanisms, which are evolutionarily selected to modulate the affinity profiles of these proteins to their preferred ligands by controlling their conformational switching between the apo and ligand bound states,20–22 this property can also be engineered in these proteins to alter their ligand affinities.23 Therefore, periplasmic binding proteins can provide unique insights into how proteins specialize in their designated activities while being subjected to the constraints of a structural fold. They can also aid in understanding the process of transport associated molecular recognition, which is vital for bacterial physiology and survival.
Members of the periplasmic binding protein super-family operate in conjunction with adenosine tri-phosphate binding cassette (ABC) transport systems, where they bind a diverse array of metabolites in the cellular milieu.24, 25 Periplasmic binding proteins have a two domain architecture with the N- and C-terminal domains linked by a flexible linker region that serves as a hinge.15, 26 The substrate binding site resides at the interface of these two domains, and the hinge is responsible for the characteristic ligand dependent domain motions of this super-family.13, 15 It has been proposed that the location of the substrate binding site within this flexible hinged interface, allows the evolution of flexibility in these systems without affecting the overall fold.27 While it is not clear how the changes at the amino acid level result in entirely different affinity profiles of homologs, there is some evidence to suggest that conformational dynamics impact the overall function of ligand binding and release.18, 28, 29 Based on these findings, it is hypothesized that the modification of the binding site and the changes in the overall dynamics of the system is a potential mechanism to encode functional diversity in this protein fold, beyond what can be attained by simply tuning molecular recognition through structural interactions in the binding pocket.
Maltose binding proteins (MBPs) are ubiquitous periplasmic binding proteins that serve as the primary receptors for alpha-linked glucose based oligosaccharides.19 While many bacteria have only a single copy of a more promiscuous MBP, some organisms contain more than one MBP isoform.30 The hyperthermophile Thermotoga maritima was recently shown to have three isoforms of MBP31 (tmMBP1–3) that are high affinity receptors for di-, tri- and tetra-saccharides respectively.32 tmMBP1 (391 residues) and tMBP2 (393 residues) share significant sequence identity (approximately 90%) but tmMBP3 (411 residues) shares less than 40% identity. The tmMBPs share roughly 35% sequence identity with the Escherichia coli MBP (ecMBP).32 Thus, this cluster of MBP isoforms offer an interesting opportunity to investigate the mechanisms underlying the evolution of specificity and affinity profiles in a genome where MBP gene redundancy is present.
In this study, the X-ray crystal structures of tmMBP1, tmMBP2 and tmMBP3 in absence and presence of substrates have been solved, to understand the structural basis of specificity encoded in this subset of proteins. To account for the contribution of protein dynamics in substrate recognition, which are not very well captured in the static X-ray crystal structures, solution-based small angle X-ray scattering, and microsecond time-scale (500 nanoseconds) computer simulations and theoretical modeling were also performed on these systems. A strategy of comparing structural and dynamical properties of the three tmMBP isoforms to the promiscuous ecMBP and the di-saccharide selective MBP from Thermococcus litoralis (tlMBP) was used to characterize features of promiscuous versus non-promiscuous binding by periplasmic binding proteins. This comparison was further aided by quantitative estimates of MBP-substrate interactions obtained using theoretical calculations. Further the results indicate that the structure and dynamics of these homologs and their substrate interaction profiles enable them to differentiate between myriad chemical entities whilst maintaining their common fold.
MATERIAL AND METHODS
Protein expression and purification:
tmMBP proteins were purified using the published protocol with some minor changes.16 Briefly, the hexahistidine-tagged tmMBP isoforms were cloned in a pET21a vector and expressed in BL21 RIL E.coli cells. The apo form of the proteins was produced using Enfor’s minimal media to avoid binding of endogenous carbohydrates from the medium. The substrate bound form of the proteins was grown in Terrific Broth. Both media were incubated at 37° C and induced using 1 mM IPTG. The induced cells were harvested and lysed using sonication before an affinity purification step (HiTrap Chelating HP column). The column was washed with affinity buffer A (20 nM imidazole, 300 mM NaCl) and subsequently with affinity buffer B (75 mM Imidazole and 150 mM NaCl) and the washes were collected. Finally, elution buffer (300 mM Imidazole, 150 mM NaCl) was used to elute the bound protein from the affinity column. Eluted samples were subsequently loaded onto a S75 26/60 gel filtration column (GE Healthcare Life Sciences). The protein fractions were examined using SDS page and the fractions containing tmMBP protein were pooled and concentrated. The concentrated proteins were dialyzed in 20 mM Tris (pH 7.5) and 40 mM NaCl overnight at room temperature.
Crystallization and X-ray data collection:
Dialyzed tmMBP proteins (both apo and holo forms) were concentrated to 15 mg/ml for crystallization. Crystals were grown by hanging drop vapor diffusion with a drop size of 2 μL for each of the protein and the mother liquor and equilibrated against 900 μL of the mother liquor. Maltotetraose (MTT) bound tmMBP1 was crystallized in 25% poly-ethylene glycol (PEG) 4000, 18% isopropanol and 0.1M sodium citrate. MTT bound tmMBP2 was crystallized in 23% PEG 3350, 0.3 M sodium acetate and 0.1 M bis-tris pH 5.5. Crystals of the maltose (MAL) bound tmMBP3 was obtained in 9% PEG 3350 supplemented with 0.4 M sodium thiocynate. Crystals for apo tmMBP2 grew in 26% PEG 3350 and 0.1 M sodium diphosphate. The apo form of the tmMBP3 was crystallized in 20% PEG 4000 and 0.2 M sodium sulfate. tmMBP2 apo and tmMBP2-MTT crystals were transferred to 35% (wt./vol.) PEG 3350 for cryoprotection, mounted in a nylon loop, and flash frozen in liquid nitrogen. X-ray datasets for tmMBP1-MTT, tmMBP3-MAL and tmMBP3 apo were collected at room temperature on a Rigaku 007HFmicromax X-ray generator with a Raxis IV++ detector. Scaling and integration of the X-ray data were carried out using HKL3000.33 The structures of apo tmMBP2 and tmMBP3 were determined to a resolution of 1.90 Å and 2.30 Å, respectively. Structures of tmMBP1 and tmMBP2 bound to maltotetraose were determined at 1.50 Å each. Maltose bound tmMBP3 structure was refined to a resolution of 2.15 Å (Table 1). Atomic coordinates and structure factors for the following systems have been deposited in the Protein Data Bank (PDB): apo tmMBP2 (accession code 6DTT), apo tmMBP3 (6DTR), tmMBP1-maltotetraose complex (6DTU), tmMBP2-maltotetraose complex (6DTS) and tmMBP3-maltose complex (6DTQ).
Table 1:
X-ray data collection and refinement statistics for tmMBPs
| tmMBP1-MTT | apo tmMBP2 | tmMBP2-MTT | apo tmMBP3 | tmMBP3-MAL | |
|---|---|---|---|---|---|
| PDB ID | 6DTU | 6DTT | 6DTS | 6DTR | 6DTQ |
| Data collection | |||||
| Resolution range (Å) | 50.0 – 1.5 (1.55 – 1.50) |
30.0 – 1.9 (1.97 – 1.90) |
50.0 – 1.5 (1.54 – 1.50) |
40.0 – 2.3 (2.38 – 2.30) |
40.0 – 2.1 (2.23 – 2.15) |
| Space group | P 1 | P 21 21 21 | P 1 | C 2 2 21 | P 21 21 21 |
| Unit cell | |||||
| a, b and c (Å) | 34.6, 55.4, 93.5 | 78.9, 91.0, 100.2 | 35.4, 56.1, 88.3 | 65.5, 137.4, 145.0 | 82.3, 124, 174.8 |
| α, β, and γ (°) | 96.8, 93.6, 101.9 | 90.0, 90.0, 90.0 | 89.5, 84.6, 89.6 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Unique reflections | 90224 | 55922 | 98262 | 27256 | 93556 |
| Multiplicity* | 2.1 (2.1) | 3.0 (2.8) | 1.9 (1.9) | 2.4 (2.3) | 3.2 (2.9) |
| Completeness (%) | 83.4 (83.3) | 97.1 (98.3) | 90.3 (85.4) | 92.5 (92.4) | 95.2 (88.5) |
| R-merge | 0.077 (0.464) | 0.056 (0.472) | 0.036 (0.125) | 0.129 (0.641) | 0.086 (0.497) |
| Mean I/σ(I) | 8.3 (3.0) | 16.8 (2.3) | 19.7 (7.1) | 7.6 (2.0) | 11.2 (2.1) |
| Wilson B-factor | 16.99 | 26.62 | 18.53 | 40.09 | 28.14 |
| Refinement | |||||
| Reflections used in refinement | 90177 | 55874 | 98260 | 27237 | 93498 |
| Reflections used for R-free | 1999 | 2000 | 1969 | 1363 | 4624 |
| R-work | 0.205 (0.304) | 0.193 (0.280) | 0.169 (0.203) | 0.164 (0.283) | 0.174 (0.280) |
| R-free | 0.227 (0.318) | 0.234 (0.331) | 0.191 (0.225) | 0.193 (0.319) | 0.205 (0.332) |
| Number of non-hydrogen atoms in refinement | |||||
| macromolecules | 5930 | 5854 | 5891 | 3145 | 12526 |
| ligands | 90 | 0 | 90 | 5 | 96 |
| solvent | 524 | 389 | 730 | 88 | 514 |
| RMS Bond length (Å) | 0.007 | 0.006 | 0.013 | 0.004 | 0.002 |
| RMS Bond angle (°) | 0.9 | 0.8 | 1.2 | 0.7 | 0.6 |
| Ramachandran allowed (%) | 0.8 | 1.7 | 0.7 | 2.8 | 1.5 |
| Ramachandran outliers (%) | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 |
| Ramachandran favored (%) | 99.2 | 98.2 | 99.3 | 97.2 | 98.5 |
| Clashscore | 3.5 | 2.5 | 3.1 | 4.4 | 2.2 |
| Average B-factor | 20 | 28 | 21 | 48 | 31 |
| macromolecules | 20 | 28 | 21 | 48 | 31 |
| ligands | 16 | N/A | 17 | 44 | 19 |
| solvent | 26 | 33 | 29 | 46 | 33 |
Number in parentheses represent values in the highest resolution shell
Small-angle X-ray scattering:
The dialyzed proteins were concentrated to 8 mg/ml. Data were collected at 50° C and in the presence of 1mM ligand for the holo forms of the protein. Circular averaging of the scattering data was used to determine the relative scattering intensity (I) as a function of momentum transfer vector, q (q = 4πsinθ/λ). Data were buffer subtracted prior to analysis. The solution scattering data were analyzed using the ATSAS software package;34 the GNOM software package35 was used for all P(r) and Io analyses. The three-dimensional envelope was generated from the SAXS data using the GASBOR program.36 Each calculation was repeated at least ten times with different random starting points for the simulated annealing algorithm; no predefined shape or symmetry constraints were used. Subsequently, these 10 model structures were averaged using DAMAVER.37 These models were used to generate an average ensemble, which was subsequently used to generate the envelope structure. To superimpose the modeled envelope on to the PDB structure, the SUPCOMB program was used.38 The experimental scattering data was fitted on to the simulated scattering data from the crystal structure using CRYSOL.39
Computational Modeling:
Molecular dynamics (MD) simulations were performed to model MBPs in complex with various oligosaccharides in explicit water solvent. Model preparation and simulations were performed using the AMBER v14 suite of programs for biomolecular simulations.40 AMBER’s ff14SB41 and GLYCAM 06Epb (for sugar units)42 force-fields were used for all simulations. MD simulations were performed using NVIDIA graphical processing units (GPUs) and AMBER’s pmemd.cuda simulation engine using our lab protocols published previously.43, 44
A total of 21 separate simulations were performed, based on the crystal structures determined in this study, and previously solved crystal structures, as well as computationally modeled protein-substrate complexes (see Table S1 in supporting information for details). The alternate substrates, where coordinates were not available from X-ray structures, were modeled based on other crystal structures available for the protein with other substrates. These alternate substrates were modeled by deleting one or more of the sugar units present in the available crystal structure and the missing hydrogen atoms were added by AMBER’s tleap program. After processing the coordinates of the protein and substrate, all systems were neutralized by addition of counter-ions and the resulting system were solvated in a rectangular box of SPC/E water, with a 10 Å minimum distance between the protein and the edge of the periodic box. The prepared systems were equilibrated using a protocol described previously.45 The equilibrated systems were then used to run 0.5 μs of production MD under constant energy conditions (NVE ensemble). The use of NVE ensemble is preferred as it offers better computational stability and performance,46 and as other ensembles (constant temperature or constant pressure) could affect dynamics of the system due to coordinates/velocities scaling. The production simulations were performed at a temperature of 325 K to match the conditions of SAXS experiments. As NVE ensemble was used for production runs, these values correspond to initial temperature at start of simulations. Temperature adjusting thermostat was not used in simulations; over the course of 0.5 μs simulations the temperature fluctuated around 325 K with RMS fluctuations between 2–4 K, which is typical for well equilibrated systems. A total of 10,000 conformational snapshots (stored every 50 ps) collected for each system (apo and substrate bound) was used for analysis.
RMSF and RMSF10 calculations:
All atoms root mean square fluctuations (RMSF) were computed based on the conformational snapshots collected during the MD simulations. To identify global motions on slower time-scales from MD, for each of the 21 systems the fluctuations associated with the first (slowest) 10 quasi-harmonic modes (RMSF10) were also computed and aggregated. It is well known that slowest 10 modes contribute to the majority of fluctuations in proteins (>80%) and the use of RMSF10, instead of all modes (RMSF), removes the faster stochastic motions of the protein, allowing focus on intrinsic dynamics of proteins.47 Both these calculations were performed using AMBER’s ptraj analysis program. All trajectory conformations were first aligned to a common structure, to remove any translation and overall molecular rotation during the simulations.
Radius of gyration (Rg):
AMBER’s ptraj module was used to calculate the Rg for the protein in its apo and substrate bound forms for each stored conformational snapshot.
Protein-substrate interactions:
The energy for the enzyme-substrate interactions (Epro-subs) were calculated as a sum of electrostatic (Eel) and van der Waals energy (Evdw) between atom pairs, based on an approach developed in our group.48, 49 Details of the computational methodology used these calculations are provided in supporting text. All protein and substrate atom pairs were included in the calculations and resulting interaction energies were summed up per residue pair. The energies were calculated for 10,000 snapshots, every 50 ps, sampled during the full 0.5 μs simulation and were averaged over these 10,000 snapshots.
RESULTS
Overall structure of tmMBPs:
A total of five X-ray crystal structures of the T. maritima MBPs (tmMBPs) were determined in this study: tmMBP1 bound to maltotetraose; tmMBP2 in apo form; tmMBP2 bound to maltotetraose; tmMBP3 in apo form; and tmMBP3 bound to maltose. The structure of the apo form of tmMBP1 has previously been deposited to the Protein Data Bank (PDB ID: 2GHB). An effort to co-crystallize tmMBP1 and tmMBP2 with disaccharide (maltose) resulted in structures with the apo form of these two proteins, while an attempt to crystallize tmMBP3 with maltotetraose resulted in protein co-crystallized with disaccharide. Crystallographic data collection and refinement statistics are presented in Table 1.
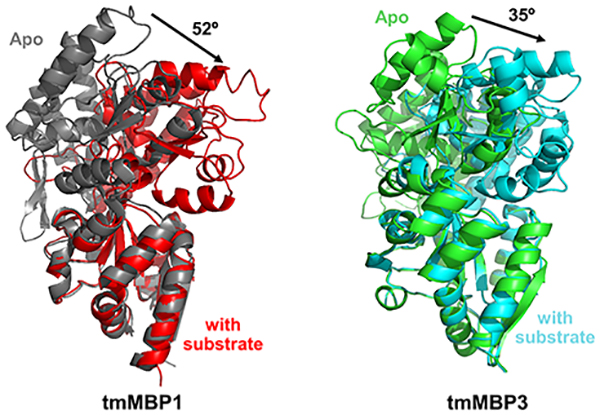
All three tmMBPs possess the signature α/β fold characteristic of MBPs,50 and the three isoforms form several common hydrogen bonds (H-bonds) and hydrophobic interactions with their substrates (Figure 1). tmMBP1 and tmMBP2 have significant structural similarity (RMSD = 0.6 Å) and an essentially identical interaction network with the maltotetraose (Figure 1). These two proteins share high sequence similarity (~90%). tmMBP3 has several amino acid insertions and is larger than the other two isoforms, yet it has a smaller and a more closed off binding pocket. A significant number of water mediated interactions between the protein and the ligands were observed for tmMBP1 and tmMBP2, however, the smaller and closed off binding site of tmMBP3 reduces the number of H-bonded water molecules to three. There is also a considerable difference in the extent of hinge bending motion in the three isoforms upon substrate binding: tmMBP1 (52.3°) and tmMBP2 (51.2°) showing a hinge movement of >15° more than tmMBP3 (35.3°) (Figure 1).
Figure 1: Structural details of the three tmMBP isoforms in complex with maltotetraose and maltose.

(A) Comparison of apo (gray) and maltotetraose bound (red) structures of tmMBP1(left) and the structural details of the substrate interaction in the binding pocket (right). (B) Apo and maltotetraose bound structures of tmMBP2. (C) Apo and maltose bound structures of tmMBP3. In the left panels, substrate is shown as green sticks. In the right panels, the ligands are shown as blue sticks and protein residues as green (hydrophobic contact with substrates) and gray (hydrophilic contact with substrates) stick representation. Direct hydrogen bonds between the protein and the ligands are shown as black dashed lines; magenta spheres indicate oxygen from water molecules observed in crystal structures; and water mediated H-bonds to the ligands as red dashed lines and indirect water mediated H-bonds with binding site residues (cyan sticks) as yellow dashed lines. Some residues making two different types of interactions include: hydrophobic and indirect H-bonds (F41 of tmMBP1); hydrophobic and direct H-bonds (W67 of tmMBP1 and W296 of tmMBP3); and direct and indirect H-bonds (E32 and D133 of tmMBP3).
Structural comparison of tmMBPs with E. coli MBP (ecMBP) and Thermococcus litoralis MBP (tlMBP):
It has been previously reported that the MBP fold contains multiple binding sub-sites in the ligand binding cavity.19, 51 These sites, termed S1, S2, S3 and S4, each accommodate a single sugar ring, with the reducing end of the sugar being placed in the first occupied subsite. The interactions in the subsites are modulated through residue insertions and deletions (indels) which tune substrate specificity and affinity profiles (Figure 2 and Figures S1 and S2). Identified indels include two loops, termed L1 and L2 and three helical insertions H1, H2 and H3 (Figure S1). To characterize the binding of saccharides of different lengths in the conserved sub-sites, the obtained structures of the tmMBPs were compared to the promiscuous E. coli maltose binding protein (ecMBP) and the disaccharide specific T. litoralis trehalose/maltose binding protein (tlMBP).
Figure 2: Differential binding of tetra- and di-saccharides in MBPs.

(A) Maltotetraose bound to tmMP1, tmMBP2, and ecMBP. (B) Maltose bound to tmMBP3, trehalose bound to tlMBP and maltose bound to ecMBP. The bound substrates are shown in blue sticks and protein residues are shown as green (hydrophobic contact with substrates) and gray (hydrophilic contact with substrates) sticks. Sub-sites in the binding pocket (S1, S2, S3 and S4) are separated by gray vertical dashed lines. Loop 1 (L1), and helices H1, H2 and H3 are also marked for each complex.
In the tmMBPs, the reducing end of the saccharide binds to subsite S1 (Figure 2). However, in ecMBP subsite S1 is sterically hindered by two residues found in the L1 loop (Asp14 and Lys15). These residues extend into the S1 subsite and also form H-bonds with the first sugar ring in subsite S2 (Figure 2). In the tmMBPs, subsite S1 has a tryptophan (Trp233 in tmMBP1 and tmMBP2, and Trp230 in ecMBP) residue which forms hydrophobic contacts with the substrate. Further, in the case of tmMBP1 and tmMBP2, the subsite S1 also include a conserved arginine (Arg303) and a serine (Ser12) that interact with both tetra- and tri-saccharide substrates (Figure 2 and Figure S2). Additionally, helix H1 also contains a conserved tyrosine (Tyr213) in tmMBP1 and tmMBP2, which provides for additional hydrophobic interactions with the first sugar ring. In the di-saccharide specific tmMBP3 and tlMBP, multiple H-bonding interactions are formed with sugar ring occupying the S1 subsite. Other common subsite S1 interactions include the conserved Glu32 in tmMBP3 and Glu17 in tlMBP, and Asp133 in tmMBP3 and Asp123 in tlMBP, which form direct H-bonds with sugar ring bound in the S1 subsite (Figure 2). Similarly, a tyrosine that forms hydrophobic interactions is also conserved in both tmMBP3 (Tyr260) and tlMBP (Tyr259). Loop L1 flips in tmMBP3 to allow the opening of subsite S1, in comparison it partially occludes subsite S1 in tlMBP. Furthermore, due to an insertion in helix H1 in tmMBP3 and tlMBP, as compared to the other MBPs, the orientation of pyranose ring is changed in the S1 subsite by almost 90°, where it forms hydrophobic interactions with a conserved tryptophan residue (Trp296 and Trp295 in tmMBP3 and tlMBP, respectively) that protrudes from helix H2 (Figure 2).
Subsite S2 binds to the second glucose ring of the saccharides in tmMBPs, as well as in tlMBP, while it binds the first glucose unit of the saccharides bound to ecMBP (Figure 2 and Figure S2). However, at subsite S2, π-π stacking interactions between aromatic residues (tyrosine, tryptophan or phenylalanine) and the saccharide occurs in all the tmMBPs. Two glutamic acid residues (Glu156 and Glu111) form H-bonds with the second pyranose ring of the saccharide molecule in tmMBP1. However, for trisaccharide bound tmMBP2 the H-bonding occurs only with Glu111; and Asn156 instead forms an interaction with the third glucose ring founds at subsite S3 (Figure S2). Similar to tmMBP1 and tmMBP2, ecMBP shows a significant number of H-bonds between substrate in the S2 subsite and water molecules (Figure 1). Subsite S2 in tmMBP3 and tlMBP binds the second pyranose ring of the maltose and trehalose, respectively, using extensive H-bonding interactions with the side-chains lining the binding cavity (Figure 2), with the interacting amino acids involving charged aspartate and glutamate residues. Moreover, tlMBP has two threonine residues which are on the either ends of the binding pocket and form H-bonds with the substrate at S1 (Thr44) and at S2 (Thr46). Both these residues are present on helix H2 that spans across the entire length of the substrate molecule in tlMBPs (Figure 1B).
Subsite S3 binds the third glucose unit of a triose or a larger sugar in tmMBP1 and tmMBP2 and the second sugar ring in ecMBP. For tmMBP1 the overall H-bond (Asp66, Trp67) and hydrophobic (Trp343) interactions are conserved between maltotriose and maltotetraose for site S3 (Figure 2 and Figure S2). Nonetheless, for tmMBP2 there is a loss of one H-bond with Trp67 at site S3 when tri-saccharide is substituted with a tetra-saccharide in the binding pocket (Figure 2 and Figure S2). In ecMBP, the pyranose ring of the di-, tri- and tetra-saccharides forms H-bonds with an aspartate (Asp65) and a glutamate (Glu153; helix H3) and hydrophobic interactions with tryptophan (Trp340). Additionally, in the disaccharide bound ecMBP it is also observed that there are two additional H-bonds with an arginine residue (Arg66), which is present on the helix adjacent to helix H2 in the N-terminal domain. However, in ecMBP some of the interactions that stabilize the third glucose ring in maltotriose are rewired to support the solvent exposed fourth glucose unit when binding maltotetraose. Both, subsite S3 and S4 are occluded in tmMBP3 and tlMBP due to steric hindrance from helix H2 and H3, which prevents anything larger than a disaccharide from binding.
Subsite S4 is occupied by the third sugar unit of a trisaccharide, or a larger saccharide in ecMBP, and the fourth ring of a tetrasaccharide in tmMBP1 and tmMBP2. Anything larger than a trisaccharide is pushed outside the binding subsite S4 in ecMBP, with only three glucose rings located within the binding pocket. In ecMBP, maltotetraose interactions include an H-bond with Arg66 and Glu44. Additionally, a lysine residue (Lys42), along with an aspartate (Asp65) restricts the movement of the last solvent exposed glucose unit. This reduces the total number of H-bonds associated with third sugar ring at subsite S4 in ecMBP to two (one each with Asp65 and Arg66). In tmMBP1, the fourth glucose ring of maltotetraose is stabilized by a H-bond with backbone of glutamine (Gln42) residue from helix H2. While in tmMBP2, the fourth sugar ring is stabilized by direct a water H-bond, and water mediated H-bonds with residues Ser344, Ser46, and Ala340.
Conformational dynamics in tmMBPs:
Computer simulations of apo and holo (substrate bound) forms of tmMBP and ecMBP, and holo form of tlMBP were performed to obtain insights into the conformational dynamics of the MBPs. Note, all simulations were performed at a temperature of 325 K (~52 °C), to match the limits of the solution characterization conditions of the tmMBPs (discussed below). Although the optimal growth temperature of T. maritima is 80 °C, we expect the results from simulations to be qualitatively similar and indicative of results at higher temperatures. In addition to the solved crystal structure, computer modeling allowed investigation of MBPs in complex with smaller oligomeric sugars for the three isoforms that did not crystallize. The motivation behind generating the models with smaller sugars was to understand the role of protein dynamics and protein-substrate interactions in differential binding of substrates, and further to quantify the preference of each substrate for a specific site in the binding pocket. MD simulations of 0.5 μs duration were performed for each of the four apo systems (tmMBP1, tmMBP2, tmMBP3, and ecMBP) and 17 substrate bound systems (11 simulations for tmMBP, 5 for ecMBP and 1 for tlMBP; see Table S1 for details of the protein-substrate complexes simulated).
Characterization of the computer simulations indicated changes in the apo and substrate bound protein conformations, as well as provided insights into the substrate stability in the binding pocket. Variation in radius of gyration (Rg) provides an indication of changes in protein conformation. On average the Rg of substrate bound systems is smaller by approximately 1.0–1.5 Å (5–7 %) compared to the apo systems (Figure 3). The smaller Rg of substrate bound systems indicate compact conformations, due to increased interactions in the binding pocket located adjacent to the hinge region of the MBPs. The increased interactions in the hinge region are responsible for the observed bending between the N- and C-terminal domains (Figure 2). For the smaller maltose and glucose substrates, the MBPs could have multiple binding positions. These were explored computationally, with two starting positions for the saccharide tested: MAL1 simulations started with maltose in S1+S2 subsites and MAL2 simulations started with maltose in the S2+S3 subsites. The results indicate that MAL2 simulations for tmMBP1 and tmMBP2 are unstable (panel B and F in Figure 3) with protein conformations opening up (increasing Rg values, observed in the blue curves) and returning to the apo conformation, and as the substrate is ejected completely from the binding pocket in case of tmMBP2. However, MAL1 simulations for tmMBP1 and tmMBP2 are stable, indicating that the substrate is stable in S1+S2 subsites, which is consistent with the observed X-ray structures. Similarly, for tmMBP3, glucose can bind in alternate sites S1(GLU1 simulation, panel I in Figure 3) or S2 (GLU2 simulation panel J in Figure 3). The simulation results indicate stable binding in S1 subsite, however, S2 subsite was less preferred as the simulations indicated relative opening up of the protein. Larger RMSD values were also seen in simulations where substrates were ejected (Figure S3).
Figure 3: Radius of gyration (Rg) computed from MD simulations.

Panels A-D show tmMBP1 results for MAL1, MAL2, MTR and MTR; panels E-H show tmMBP2 results for MAL1, MAL2, MTR and MTR; and panels I-K show tmMB1 results for GLU1, GLU2, and MAL. Rg for apo simulations depicted in red and the substrate bound simulations in blue, and average value of Rg computed from all simulation snapshots are shown by horizontal lines. MAL1 and MAL2 indicate simulations with maltose starting in two alternate positions; site S1+S2 for MAL1 and S2+S3 for MAL2. GLU1 simulation started with glucose in S1 binding pocket and GLU2 in the S2 binding pocket.
For the larger maltotriose and maltotetraose substrates, tmMBP1 and tmMBP2 show stable binding as indicated by the smaller Rg values throughout the simulations (panels C, D, G and H Figure 3). Similarly, for proteins with smaller binding pockets, tmMBP3 and tlMBP, binding to di-saccharide substrates showed stable simulations (panel K in Figure 3 and panel F of Figure S4). The promiscuous ecMBP shows interesting behavior (Figure S4) in simulations, where the smaller glucose substrate was unstable and ejected out of the binding pocket in both S1 and S2 subsites, however, maltose and maltotriose show stable binding in the pocket. Maltotetraose shows mostly stable binding with some minor fluctuations, possibly due to the fact that the last sugar unit does not have room in the binding pocket.
The computational Rg trends in the tmMBPs were confirmed by small-angle X-ray scattering (SAXS) studies of these proteins in the solution state. The fits of the experimental SAXS data to theoretical scattering data generated from the crystal structures (Figure S5) are in agreement (Table 2). The experimental Rg decreases by ~1.5 Å after substrate binding (Table 2), which is comparable to the observed computer simulation results (Figure 3 and Figure S4). It is also interesting to note that the apo proteins in MD simulations for all MBPs show stable Rg values throughout the period of simulation, consistent with a body of literature demonstrating the MBP proteins do not populate the ligand bound conformation in the absence of ligand.28, 29, 52, 53 The hinge angle of the apo proteins shows little deviations over the course of MD simulations (Figure S6), ranging between −10° to +10° from the first frame of the simulations and consistent with previous simulations of ecMBP.28
Table 2:
Radius of gyration (Rg) values of tmMBPs
| Protein | SAXS Conc. (mg/ml) | SAXS Rg (Å) | SAXS Dmax (Å) | χ2* | Averaged Rg (Å), from computations |
|---|---|---|---|---|---|
| apo tmMBP1 | 2.7 | 24.1 | 82.3 | 1.19 | 22.5 |
| tmMBP1-MTT | 2.6 | 22.6 | 78.3 | 1.06 | 21.5 |
| apo tmMBP2 | 2.6 | 23.5 | 79.1 | 1.29 | 22.4 |
| tmMBP2-MTT | 2.4 | 22.2 | 69.9 | 1.21 | 21.2 |
| apo tmMBP3 | 3.6 | 22.4 | 70.3 | 1.18 | 22.1 |
| tmMBP3-MAL | 2.5 | 22.6 | 72.9 | 1.40 | 21.3 |
χ2 generated from the fit of the CRYSOL calculated scattering profile to the experimental data.
The conformational flexibility (or intrinsic dynamics) of the MBP systems were quantified using quasi-harmonic analysis (QHA) of the protein conformations sampled during the MD simulations. The aggregated root means square fluctuation of top 10 slowest QHA modes (RMSF10) provides a good measure of protein’s conformational flexibility. Such an analysis has been successfully used for relating dynamics to function for a number of other proteins.54–56 Both crystallographic B-factors and the RMSF10 showed similar trends of reduction in the overall conformational flexibility of MBPs when they bind to preferred substrates (Figure 4, and Figures S5, S6, S7 and S8). The results indicate that large fluctuations occur in the regions 40–60, 130–150 and 165–195 for apo as well as substrate bound systems for tmMBP1, tmMBP2 and ecMBP. These regions in tmMBP1 and corresponding regions in tmMBP3, are highlighted with ellipses and marked as 1, 2, and 3 in Figure 4 respectively. Region 165–195 shows the largest flexibility, interestingly this region corresponds to region 194–218 in tmMBP3 (see Figure S1); and as seen in Figure 4, part of this region in tmMBP3 (residue 197–211) also shows increased flexibility. This region is located in the C-terminal domain and interacts with the N-terminal domain in the apo form of tmMBP and ecMBP structures. Previous experimental studies have shown altering this loop enhances the substrate binding affinities in ecMBPs,57 and plays a role in the energetics of the conformational dynamics in the apo state of the protein.28 It is therefore likely that this region plays a role in substrate selectivity. Additionally, subtle variations in the dynamics were also observed when the same protein was interacting with different substrates. The lower panels of Figure 4 provide quantitative comparison of the flexibility of apo and different substrate bound cases (dashed red line shows the average over all residues of apo protein for comparison). For ecMBP, tmMBP1 and tmMBP2, it is observed that with an addition of each pyranose ring in the substrate, the dynamics of only certain regions in the protein changes (see Figures S7–S11). These variations in dynamics were not just observed around the binding site, rather the changes occurred at sites distally located from the substrate binding cavity (Figure 4 and Figures S7, S8, S9 and S10), which supports the potential role of these regions in substrate binding or specificity. Unfortunately, the current simulations cannot differentiate whether dynamical variations are caused due to different substrate binding or these motions playing an active role in tuning binding preference.
Figure 4: Comparison of tmMBP1 and tmMBP3 conformational flexibility based on RMSF10.

(A) tmMBP1 and (B) tmMBP3. The tube thickness corresponds to degree of flexibility, with thicker tubes (green/yellow) indicating more flexible regions than the rest of rigid protein (dark blue). Surface exposed regions displaying higher than average conformational flexibility are marked by black ellipses (40–60, 130–150, and 165–195 for tmMBP1 and 50–85, 155–175, and 197–211 for tmMBP3), and the plots below compare the observed values for apo and protein in complex with various substrates. The dashed red line in the plots indicate the average value of RMSF10 observed in the apo proteins for comparison.
Interaction energy of substrates with tmMBPs and ecMBP:
The interaction energies for substrate bound systems (Table 3), calculated from computer simulations, also supports the differential binding in MBPs. These computational estimates only provide enthalpic contributions, as accurate estimates of entropic contributions are difficult to obtain. Larger negative values in Table 3 are considered to be more favorable interactions than smaller negative values. The magnitude of interaction energies, averaged per residue (Table 3), were stronger for the known natural binders (which are known to be maltotriose and maltotetraose for tmMBP1/tmMBP2; maltose for tmMBP3; maltose, maltotriose, and maltotetraose for ecMBP; and trehalose for tlMBP). In the case of the promiscuous ecMBP, maltotriose has the most favorable interaction energy, followed by maltotetraose and maltose, in a trend identical to the previously experimentally measured binding constants for this system,19 which suggests the simulations capture the elements that lead to ligand discrimination accurately. Disaccharide binding to the specific tmMBP3 and tlMBP proteins produced significantly stronger interaction energies when compared to the promiscuous ecMBP. tmMBP1 and tmMBP2 show a similar interaction energy in the case of maltotriose, however, of these two proteins tmMBP2 shows significantly stronger interactions with maltotetraose. The substrate-protein complex co-crystallized in this study exhibited more favorable interaction energy profiles when compared to those substrate-protein interactions that failed to co-crystallize. The cases where the ligand showed relative unstable binding during the computer simulation, indicated weaker interaction energies (marked with a,b in Table 3), compared to the cases of stable substrate binding. Previous studies of oligosaccharide binding to ecMBP, based on isothermal titration calorimetry (ITC), indicated dissociation constant (Kd) for maltose and maltotriose is tighter when compared to maltotetraose.19 The computational results obtained in this study are in agreement with these results as maltotriose shows more favorable binding than maltose and maltotetraose (Table 3).
Table 3:
Averaged protein-substrate interaction energy.
| Glucose | maltose | maltotriose | maltotetraose | |||
|---|---|---|---|---|---|---|
| GLU1 | GLU2 | MAL1 | MAL2 | |||
| tmMBP1 | −55.5 ± 0.35 (−27.8) | −42.6 ± 0.18 (−21.3)a | −79.4 ± 0.09 (−26.5) | −90.0 ± 0.44 (−22.5)c | ||
| tmMBP2 | −55.7 ± 0.10 (−27.8) | −24.8 ± 0.27(−12.4)b | −79.4 ± 0.13 (−26.5) | −92.0 ± 0.36 (−23.0)c | ||
| tmMBP3 | −40.0 ± 0.18 | −33.6 ± 0.09a | −72.6 ± 0.14 (−36.3)c | |||
| ecMBP | −26.6 ± 0.15a | −33.9 ± 0.20a | −62.2 ± (−31.1) | −81.1 ± (−27.0) | −78.5 ± (−19.6) | |
| tlMBP | −68.3 ± (−34.2) | |||||
All values are shown in kcal/mol. The values in parenthesis are per sugar unit.
indicates that substrate changed significantly from the original orientation as a number of protein-substrate contacts are broken
indicates that substrate was completely ejected out of the binding pocket. GLU1/GLU2 and MAL1/MAL2 indicate two different simulations with substrate alternate sites (see Figure 4 and its legend for details).
indicates the substrate in complex with the protein for X-ray solved in this study. The errors associated are provided; see SI text for details on how the errors were calculated.
Figure 5 provides more details of the interaction energy at protein residue and substrate unit pair level. The protein residues showing most favorable contacts are highlighted. For each protein, there were some common residues that showed favorable interactions across substrates of different lengths (Figure 5). These include: Glu13, Phe41, Glu111, Tyr158, Trp233 and Arg303 for tmMBP1 and tmMBP2; Glu32, Asp85, and Trp296 for tmMBP3; Trp62, Asp65, Arg66, Tyr155 and Trp230 for ecMBP. A full list of residues is available in Table S2. Not surprisingly these residues are also the ones which make hydrophilic and hydrophobic contacts in the obtained tmMBP X-ray structures (see Figures 1 and 3; Figure S2). These interactions do not show up in the case of weak binding substrates.
Figure 5: Protein-substrate interaction energy.

The interaction energy was computed as a sum of electrostatics and van der Waals contributions between each protein residue and substrate unit pair. The yellow-green-blue areas with more favorable contacts are highlighted, and the corresponding protein residue is marked. See Figure 3 legend in main manuscript for the substrate key.
DISCUSSION
The periplasmic binding protein super-family is a collection of proteins with a conserved structural fold; however, these proteins show considerable diversity in the size of substrates that they bind.58 One subset of the periplasmic binding protein super-family, oligosaccharide binding MBPs, is of interest with regards to understanding the binding specificity in these proteins, as members of this family share a common protein fold and bind a wide diversity of substrates.59 Intriguingly three different MBP genes are present in T. maritima, which encode for three different isoforms with different substrate binding preferences. Two of these isoforms, tmMBP1 and tmMBP2, show 90% sequence similarity while the third, tmMBP3, only shares 40%; however, all three share the same structural fold. In this study, we have used structural and computational studies to characterize the substrate binding preferences of these three isoforms, which provide insights into presence of multiple genes in T. maritima, with related function. The obtained X-ray structures of tmMBP1, tmMBP2 and tmMBP3, in substrate free and substrate bound forms, provide detailed information about the interaction of native oligosaccharide substrates in the binding pocket. The binding pockets of tmMBP1 and tmMBP2 are more open, and suitable to accommodate larger substrates, however, in the case of tmMBP3 the binding pocket is sterically hindered by helices H2 and H3. This observation is similar to the binding characteristics of T. litoralis trehalose/maltose binding protein reported previously.60
One of the largest structural change observed between the three isoforms, upon substrate binding, is the extent of the hinge bending motion between the N- and C-terminal domains. The binding pocket of periplasmic binding proteins is partially constituted by the residues around the hinge region. In the case of tmMBP1 and tmMBP2 the substrate binding induces the relative orientation of the two domains to change by ~50° while in the case of tmMBP3 the change is only ~35° (see Figure 2). Previous reports have indicated that the substrate binding affinities are affected by the residues (and mutations) in the hinge region,18 which interestingly also manifests through observed differences in the magnitude of the hinge bending motion.52
The larger binding pockets of tmMBP1 and tmMBP2 also show the presence of water molecules in and around the binding pocket, which have previously been shown to contribute towards ligand specificity in periplasmic binding proteins.61 In both tmMBP1 and tmMBP2, the majority of water involving interactions are found near subsite S1. Similarly, in tmMBP3 a number of H-bond and hydrophobic interactions stabilize the first pyranose ring of the bound maltose. It was observed that in all the stable computer simulations (where the substrate stays in the binding cavity for the entire simulation) subsite S1 was occupied consistently. Interestingly, simulations that started with a vacant S1 subsite (substrate present in other subsites) showed unstable substrate binding in other subsites as well (see Figure 4). These results suggest that there is possibly a subsite preference for a tighter binding of malto-oligosaccharides in MBPs, which is mediated by bulk water along with the protein-substrate H-bonds. Further, it is possible that binding at the subsite S1 may help in aligning larger oligosaccharides in a suitable orientation within the binding pocket.
For tmMBP1 and tmMBP2, which show similar binding to maltotriose and maltotetraose, there are subtle differences in the interactions with residues present in the binding pocket, and a similar magnitude of conformational changes upon encountering ligand. In the case of tmMBP3, however, the binding of substrate did not induce a similar magnitude of conformational changes in solution or in simulations. The computer simulations indicate that the conformational flexibility of these 3 isoforms significantly reduced after binding to the substrates, which has been previously shown for other sugar binding proteins.62 The degree of conformational flexibility of the protein plays a functional role in the conformational selection associated with binding of the native substrate. An important observation in the current study is that the conformational changes were not limited to the canonical hinge bending motion, but distally located solvent exposed regions were modulated as well in response to ligand binding (Figure 4). In simulations, this behavior varied between the apo/substrate bound forms as well as the three isoforms investigated. It is interesting to note that previous reports20, 21 have suggested the presence of allosteric modulation in the broader periplasmic binding protein super-family.23, 57 An increasing number of proteins/enzymes are showing that protein dynamics is an important contributor to allosteric regulation.63–65 Further studies are needed to characterize the relation, if any, between dynamics and allosteric modulation in MBPs. Such insights will have important implications towards an understanding of the regulation of periplasmic binding protein mediated transmembrane metabolite transport.
Computer simulations provide unique insights into substrates binding by different tmMBP isoforms. The computed interaction energies (based on electrostatics and van der Waals terms) between the protein residues and substrate provide quantitative information about the preferred interactions in the binding pocket. A number of alternate simulations were performed with the substrate in the different sub-sites within the binding pocket. The native substrates which crystallized showed the most stable interactions confirming that the observed structures are the preferred binding sites. Interestingly, the simulations with ligand positions different from the crystallized structures demonstrated non-stable interactions, supporting the combined requirements of correct ligand binding site, dynamics and conformational energetics.
In the broader context, the present study touches upon the interesting interplay among gene duplication and gene specialization in ABC transporters. Yet the more omnipresent question that still remains is, why E. coli uses a promiscuous MBP, while T. maritima uses redundant multiple copies of similar proteins? One possible reason for presence of multiple genes is that binding of longer, or shorter saccharides is optimized, subtlety, in the particular fold (e.g. tmMBP1/2 versus tmMBP3), which is not as efficiently achieved in a single protein such as in ecMBP. Indeed, T. martima utilizes promiscuous periplasmic carbohydrate binding proteins, yet the oligopeptide binding PBP fold is used rather than MBP.16, 30, 51, 66 From an evolutionary prospective the encoding of promiscuity is tied to the evolutionary drive to minimize genome size and gene redundancy. Yet, E. coli and T. maritima live in significantly different environmental niches, one that fluxes between high and low metabolite availability and the other which is vastly carbohydrate deficient. This in-turn has led to different carbohydrate ABC transport systems in these two organisms, with T. maritima encoding transport operons for a variety of beta linked carbohydrates that are lacking from E. coli.30 It is thus possible that due to the carbohydrate limiting environment of T. maritima, the energy expenditure of operon redundancy in carbohydrate acquisition outweighs the metabolic benefit of the ability to scavenge saccharides. Collectively the results presented here, and the future studies could provide vital clues to understanding of how the limitations imposed on members of a protein super-family due to a structural fold can be compensated by gene duplication and neofunctionalization.
Supplementary Material
Acknowledgements
The authors would like to thank Prof. Elizabeth Howell, Dr. Hugh O’Neill and Dr. Jerry Parks for their valuable inputs.
Funding Sources
This work was supported in part by a multi-PI grant from NIH to PKA (GM105978).
Abbreviations:
- ecMBP
Escherichia coli MBP
- GLU
glucose
- MAL
maltose
- MBP
maltose binding protein
- MTR
maltotriose
- MTT
maltotetraose
- tlMBP
Thermococcus litoralis MBP
- tmMBP
Thermotoga maritima MBP
Footnotes
The authors declare no competing financial interest.
Supporting information
A PDF file containing additional details about structures used computational modeling, results from sequence alignment for MBPs, differential binding of trisaccharide substrates, Rg for ecMBP and tlMBP MD simulations, SAXS profiles, comparison of B-factors and RMSF, full results for the RMSF10, RMSF10 for tlMBP and list of residues showing significant interaction energy with the substrates.
REFERENCES
- [1].Lawrence JG (1999) Gene transfer, speciation, and the evolution of bacterial genomes, Current Opinion in Microbiology 2, 519–523. [DOI] [PubMed] [Google Scholar]
- [2].Zhang J (2003) Evolution by gene duplication: an update, Trends in Ecology & Evolution 18, 292–298. [Google Scholar]
- [3].Ochman H, Lawrence JG, and Groisman EA (2000) Lateral gene transfer and the nature of bacterial innovation, Nature 405, 299. [DOI] [PubMed] [Google Scholar]
- [4].Mendonça AG, Alves RJ, and Pereira-Leal JB (2011) Loss of genetic redundancy in reductive genome evolution, PLoS Computational Biology 7, e1001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shukla S, Bafna K, Sundar D, and Thorat SS (2014) The bitter barricading of prostaglandin biosynthesis pathway: understanding the molecular mechanism of selective cyclooxygenase-2 inhibition by amarogentin, a secoiridoid glycoside from Swertia chirayita, PLoS ONE 9, e90637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Recinos DA, Sekedat MD, Hernandez A, Cohen TS, Sakhtah H, Prince AS, Price-Whelan A, and Dietrich LE (2012) Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity, Proceedings of the National Academy of Sciences USA 109, 19420–19425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cole J (1996) Nitrate reduction to ammonia by enteric bacteria: redundancy, or a strategy for survival during oxygen starvation?, FEMS Microbiology Letters 136, 1–11. [DOI] [PubMed] [Google Scholar]
- [8].Hughes AL (1994) The evolution of functionally novel proteins after gene duplication, Proc. R. Soc. Lond. B 256, 119–124. [DOI] [PubMed] [Google Scholar]
- [9].Qian W, and Zhang J (2014) Genomic evidence for adaptation by gene duplication, Genome Research 24, 1356–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Russell RB, Saqi MA, Sayle RA, Bates PA, and Sternberg MJ (1997) Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation1, Journal of Molecular Biology 269, 423–439. [DOI] [PubMed] [Google Scholar]
- [11].Cygler M, Schrag JD, Sussman JL, Harel M, Silman I, Gentry MK, and Doctor BP (1993) Relationship between sequence conservation and three‐dimensional structure in a large family of esterases, lipases, and related proteins, Protein Science 2, 366–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Friedberg I, and Margalit H (2002) Persistently conserved positions in structurally similar, sequence dissimilar proteins: roles in preserving protein fold and function, Protein Science 11, 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Quiocho FA, and Ledvina PS (1996) Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes, Molecular Microbiology 20, 17–25. [DOI] [PubMed] [Google Scholar]
- [14].Dwyer MA, and Hellinga HW (2004) Periplasmic binding proteins: a versatile superfamily for protein engineering, Current Opinion in Structural Biology 14, 495–504. [DOI] [PubMed] [Google Scholar]
- [15].Quiocho FA (1990) Atomic structures of periplasmic binding proteins and the high-affinity active transport systems in bacteria, Phil. Trans. R. Soc. Lond. B 326, 341–352. [DOI] [PubMed] [Google Scholar]
- [16].Ghimire-Rijal S, Lu X, Myles DA, and Cuneo MJ (2014) Duplication of genes in an ATP-binding cassette transport system increases dynamic range while maintaining ligand specificity, Journal of Biological Chemistry 289, 30090–30100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vermersch PS, Lemon DD, Tesmer JJ, and Quiocho FA (1991) Sugar-binding and crystallographic studies of an arabinose-binding protein mutant (Met108Leu) that exhibits enhanced affinity and altered specificity, Biochemistry 30, 6861–6866. [PubMed] [Google Scholar]
- [18].Seo M-H, Park J, Kim E, Hohng S, and Kim H-S (2014) Protein conformational dynamics dictate the binding affinity for a ligand, Nature Communications 5, 3724. [DOI] [PubMed] [Google Scholar]
- [19].Quiocho FA, Spurlino JC, and Rodseth LE (1997) Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport/chemosensory receptor, Structure 5, 997–1015. [DOI] [PubMed] [Google Scholar]
- [20].Kadaba NS, Kaiser JT, Johnson E, Lee A, and Rees DC (2008) The high-affinity E. coli methionine ABC transporter: structure and allosteric regulation, Science 321, 250–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li L, Ghimire-Rijal S, Lucas SL, Stanley CB, Wright E, Agarwal PK, Myles DA, and Cuneo MJ (2017) Periplasmic Binding Protein Dimer Has a Second Allosteric Event Tied to Ligand Binding, Biochemistry 56, 5328–5337. [DOI] [PubMed] [Google Scholar]
- [22].Marinelli F, Kuhlmann SI, Grell E, Kunte H-J, Ziegler C, and Faraldo-Gómez JD (2011) Evidence for an allosteric mechanism of substrate release from membrane-transporter accessory binding proteins, Proceedings of the National Academy of Sciences USA 108, E1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rizk SS, Paduch M, Heithaus JH, Duguid EM, Sandstrom A, and Kossiakoff AA (2011) Allosteric control of ligand-binding affinity using engineered conformation-specific effector proteins, Nature Structural and Molecular Biology 18, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rees DC, Johnson E, and Lewinson O (2009) ABC transporters: the power to change, Nature reviews Molecular Cell Biology 10, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Locher KP (2016) Mechanistic diversity in ATP-binding cassette (ABC) transporters, Nature Structural and Molecular Biology 23, 487. [DOI] [PubMed] [Google Scholar]
- [26].Orelle C, Oldham ML, and Davidson AL (2014) The maltose ABC transporter: where structure meets function, In Membrane Transport Mechanism, pp 181–205, Springer. [Google Scholar]
- [27].Marvin JS, and Hellinga HW (2001) Conversion of a maltose receptor into a zinc biosensor by computational design, Proceedings of the National Academy of Sciences USA 98, 4955–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bucher D, Grant BJ, Markwick PR, and McCammon JA (2011) Accessing a hidden conformation of the maltose binding protein using accelerated molecular dynamics, PLoS Computational Biology 7, e1002034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tang C, Schwieters CD, and Clore GM (2007) Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR, Nature 449, 1078. [DOI] [PubMed] [Google Scholar]
- [30].Nanavati DM, Nguyen TN, and Noll KM (2005) Substrate specificities and expression patterns reflect the evolutionary divergence of maltose ABC transporters in Thermotoga maritima, Journal of Bacteriology 187, 2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Boucher N, and Noll KM (2011) Ligands of thermophilic ABC transporters encoded in a newly sequenced genomic region of Thermotoga maritima MSB8 screened by differential scanning fluorimetry, Applied and Environmental Microbiology 77, 6395–6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wassenberg D, Liebl W, and Jaenicke R (2000) Maltose-binding protein from the hyperthermophilic bacterium Thermotoga maritima: stability and binding properties1, Journal of Molecular Biology 295, 279–288. [DOI] [PubMed] [Google Scholar]
- [33].Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes, Acta Crystallographica Section D: Biological Crystallography 62, 859–866. [DOI] [PubMed] [Google Scholar]
- [34].Franke D, Petoukhov M, Konarev P, Panjkovich A, Tuukkanen A, Mertens H, Kikhney A, Hajizadeh N, Franklin J, and Jeffries C (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions, Journal of Applied Crystallography 50, 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Svergun D (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria, Journal of Applied Crystallography 25, 495–503. [Google Scholar]
- [36].Svergun DI, Petoukhov MV, and Koch MH (2001) Determination of domain structure of proteins from X-ray solution scattering, Biophysical Journal 80, 2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Volkov VV, and Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering, Journal of Applied Crystallography 36, 860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kozin MB, and Svergun DI (2001) Automated matching of high-and low-resolution structural models, Journal of Applied Crystallography 34, 33–41. [Google Scholar]
- [39].Svergun D, Barberato C, and Koch MH (1995) CRYSOL–a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates, Journal of Applied Crystallography 28, 768–773. [Google Scholar]
- [40].Case DA, Berryman JT, Betz RM, Cerutti DS, Cheatham III TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Götz AW, Homeyer N, Janowski S,I, Kaus P, Kovalenko J, Lee A, LeGrand TS, Li S, Luchko P, Luo T, Madej R, Merz B, Monard KM, Needham G, Nguyen P,H,N, Omelyan HT, Onufriev I, Roe A, Roitberg DR, Salomon-Ferrer A, Simmerling R, Smith CL, Swails J, Walker J, Wang RC, Wolf J, Wu RM, York X, M. D, and Kollman PA (2015) AMBER 15, University of California, San Francisco. [Google Scholar]
- [41].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, Journal of Chemical Theory and Computation 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kirschner KN, Yongye AB, Tschampel SM, González‐Outeiriño J, Daniels CR, Foley BL, and Woods RJ (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates, Journal of Computational Chemistry 29, 622–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ramanathan A, Agarwal PK, Kurnikova M, and Langmead CJ (2010) An online approach for mining collective behaviors from molecular dynamics simulations, Journal of Computational Biology 17, 309–324. [DOI] [PubMed] [Google Scholar]
- [44].Ramanathan A, Savol AJ, Agarwal PK, and Chennubhotla CS (2012) Event detection and sub-state discovery from biomolecular simulations using higher-order statistics: application to enzyme adenylate kinase, Proteins 80, 2536–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ramanathan A, and Agarwal PK (2011) Evolutionarily Conserved Linkage between Enzyme Fold, Flexibility, and Catalysis, PLoS Biology 9, e1001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Beck DA, and Daggett V (2004) Methods for molecular dynamics simulations of protein folding/unfolding in solution, Methods 34, 112–120. [DOI] [PubMed] [Google Scholar]
- [47].Ramanathan A, and Agarwal PK (2009) Computational identification of slow conformational fluctuations in proteins, The Journal of Physical Chemistry B 113, 16669–16680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Agarwal PK (2004) Cis/trans isomerization in HIV-1 capsid protein catalyzed by cyclophilin A: insights from computational and theoretical studies, Proteins 56, 449–463. [DOI] [PubMed] [Google Scholar]
- [49].Gagne D, Narayanan C, Nguyen-Thi N, Roux LD, Bernard DN, Brunzelle JS, Couture JF, Agarwal PK, and Doucet N (2016) Ligand Binding Enhances Millisecond Conformational Exchange in Xylanase B2 from Streptomyces lividans, Biochemistry 55, 4184–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cuneo MJ, Changela A, Warren JJ, Beese LS, and Hellinga HW (2006) The crystal structure of a thermophilic glucose binding protein reveals adaptations that interconvert mono and di-saccharide binding sites, Journal of Molecular Biology 362, 259–270. [DOI] [PubMed] [Google Scholar]
- [51].Cuneo MJ, Changela A, Beese LS, and Hellinga HW (2009) Structural adaptations that modulate monosaccharide, disaccharide, and trisaccharide specificities in periplasmic maltose-binding proteins, Journal of Molecular Biology 389, 157–166. [DOI] [PubMed] [Google Scholar]
- [52].Millet O, Hudson RP, and Kay LE (2003) The energetic cost of domain reorientation in maltose-binding protein as studied by NMR and fluorescence spectroscopy, Proceedings of the National Academy of Sciences USA 100, 12700–12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bucher D, Grant BJ, and McCammon JA (2011) Induced fit or conformational selection? The role of the semi-closed state in the maltose binding protein, Biochemistry 50, 10530–10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Narayanan C, Bernard DN, Bafna K, Gagné D, Chennubhotla CS, Doucet N, and Agarwal PK (2018) Conservation of dynamics associated with biological function in an enzyme superfamily, Structure 26, 426–436. e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gagné D, Narayanan C, Bafna K, Charest L-A, Agarwal PK, and Doucet N (2017) Sequence-specific backbone resonance assignments and microsecond timescale molecular dynamics simulation of human eosinophil-derived neurotoxin, Biomolecular NMR Assignments 11, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Agarwal PK, Schultz C, Kalivretenos A, Ghosh B, and Broedel SE Jr (2012) Engineering a Hyper-catalytic Enzyme by Photoactivated Conformation Modulation, The Journal of Physical Chemistry Letters 3, 1142–1146. [Google Scholar]
- [57].Telmer PG, and Shilton BH (2003) Insights into the conformational equilibria of maltose-binding protein by analysis of high affinity mutants, Journal of Biological Chemistry 278, 34555–34567. [DOI] [PubMed] [Google Scholar]
- [58].Lawson DM, Williams CE, Mitchenall LA, and Pau RN (1998) Ligand size is a major determinant of specificity in periplasmic oxyanion-binding proteins: the 1.2 Å resolution crystal structure of Azotobacter vinelandii ModA, Structure 6, 1529–1539. [DOI] [PubMed] [Google Scholar]
- [59].Locher KP (2004) Structure and mechanism of ABC transporters, Current Opinion in Structural Biology 14, 426–431. [DOI] [PubMed] [Google Scholar]
- [60].Diez J, Diederichs K, Greller G, Horlacher R, Boos W, and Welte W (2001) The crystal structure of a liganded trehalose/maltose-binding protein from the hyperthermophilic archaeon Thermococcus litoralis at 1.85 Å1, Journal of Molecular Biology 305, 905–915. [DOI] [PubMed] [Google Scholar]
- [61].Quiocho F, Wilson DK, and Vyas N (1989) Substrate specificity and affinity of a protein modulated by bound water molecules, Nature 340, 404. [DOI] [PubMed] [Google Scholar]
- [62].Gagné D, Narayanan C, Nguyen-Thi N, Roux LD, Bernard DN, Brunzelle JS, Couture J-F, Agarwal PK, and Doucet N (2016) Ligand binding enhances millisecond conformational exchange in xylanase B2 from Streptomyces lividans, Biochemistry 55, 4184–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bouskila M, Hunter RW, Ibrahim AF, Delattre L, Peggie M, Van Diepen JA, Voshol PJ, Jensen J, and Sakamoto K (2010) Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle, Cell Metabolism 12, 456–466. [DOI] [PubMed] [Google Scholar]
- [64].Freiburger LA, Baettig OM, Sprules T, Berghuis AM, Auclair K, and Mittermaier AK (2011) Competing allosteric mechanisms modulate substrate binding in a dimeric enzyme, Nature Structural and Molecular Biology 18, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dagliyan O, Tarnawski M, Chu P-H, Shirvanyants D, Schlichting I, Dokholyan NV, and Hahn KM (2016) Engineering extrinsic disorder to control protein activity in living cells, Science 354, 1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Munshi P, Stanley CB, Ghimire-Rijal S, Lu X, Myles DA, and Cuneo MJ (2013) Molecular details of ligand selectivity determinants in a promiscuous beta-glucan periplasmic binding protein, BMC Struct Biol 13, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.