

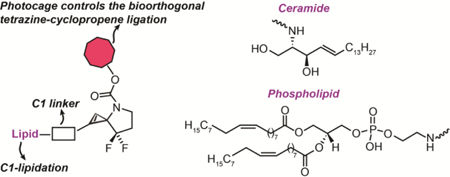
Abstract
Lipidated cyclopropenes serve as useful bioorthogonal reagents for imaging cell membranes due to the cyclopropene’s small size and ability to ligate with pro-fluorescent tetrazines. Previously, the lipidation of cyclopropenes required modification at the C3 position because methods to append lipids at C1/C2 were not available. Herein, we describe C1/C2 lipidation with the biologically active lipid ceramide and a common phospholipid using a cyclopropene scaffold whose reactivity with 1,2,4,5-tetrazines has been caged.
Keywords: Cyclopropene tetrazine, Bioorthogonal chemistry, Click chemistry, Lipid, photocage
Graphical Abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
Cyclopropenes are popular bioorthogonal reagents for exploring native biomolecules’ roles in biology.1 They have been used extensively with sugars,2–4 proteins,5–9 oligonucleotides,10,11 neurotransmitters,12,13 and, to a limited extent, lipids.14,15 The application of cyclopropenes to lipid biology is particularly interesting because lipids are one of the few biomolecules with naturally occurring cyclopropene-containing metabolites, including sterculynic acid,15 sterculic acid,16 and malvalic acid.17 Produced in plants, these naturally occurring cyclopropene-bearing lipids have diverse functions, from promoting liver tumor formation18 to exhibiting anti-parasitic properties, likely through their inhibition of lipid metabolism.19 Similarly fascinating, unnatural cyclopropene-modified lipids have found applications in bioorthogonal chemistry, including, for example, Devaraj and co-workers’ preparation of a C3 lipidated cyclopropene (see Fig. 1 for cyclopropene carbon numbering) and demonstration of its use as a tool for live-cell imaging of mammalian cells.14
Figure 1.
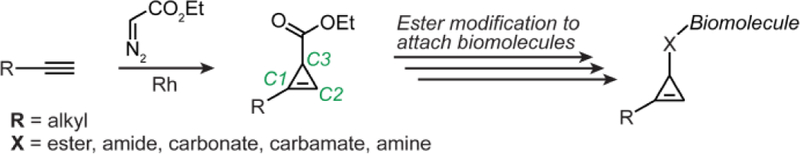
Current strategies to append cyclopropene handles onto biomolecules proceed via modification at the cyclopropene C3 position. Cyclopropenes are first prepared via rhodium catalysed addition of ethyl diazoacetate to alkynes, which are then modified at C3 to append biomolecules such as lipids.
Unfortunately, lipidation of cyclopropenes for biorthogonal chemistry applications is typically restricted to addition of the lipid to the C3 position of the cyclopropene. This is due to the limited number of synthetic procedures that enable direct derivatization of cyclopropenes with biomolecules at C1 or C2. For example, it is possible to generate a cyclopropene that is already modified at C1 with an alkyl alcohol by the rhodium catalyzed addition of ethyl diazoacetate to a protected alcoholic alkyne (R= protected alkyl alcohol, Fig. 1). The protected alcohol could then be deprotected and oxidized to obtain a carboxylic acid handle for further elaboration. However, examples of the oxidation of such cyclopropene C1-alcohols to the corresponding cyclopropene C1-acid are challenging and rare in the literature.13,20 Alternatively, the functionalization handle could be installed using a lipid-modified protected alkyne precursor (R = functional group protected lipid, Fig. 1), but such functionalized alkyne lipids are commercially uncommon, expensive and difficult to synthesize (e.g., Avanti® polar lipids offers four alkyne-modified lipids and the cheapest is ~$150/mg). There are also strategies for cyclopropene modification at C1/C2 on intact cyclopropenes to obtain secondary alcohols21–23 but they require challenging conjugation conditions to attach lipids via an ether or, after a non-trivial oxidation, a ketone.
Methods for the addition of bulky biomolecules such as lipids at the C1/2 position of a cyclopropene, instead of C3, would be desirable for several reasons. For example, C1/C2 derivatization strategies will expand the accessible cyclopropene chemical space. Additionally, substituting C1/C2 has been shown to increase the stability of cyclopropenes.10 As a result, lipidation at C1/C2 could serve the dual purpose of stabilizing the cyclopropene while minimizing the size at C3 to limit negative steric effects on the cyclopropene’s reactivity with 1,2,4,5-tetrazines. Indeed, modifications of C1/C2 place the substituent orthogonal to the approach of the tetrazine along the ligation’s reaction coordinate, resulting in only modest reaction rate deceleration relative to unmodified cyclopropenes. For example, Ye and co-workers demonstrated that C1 methylation of a C3-amide-containing cyclopropene produced only a modest decrease of 9% in cyclopropene’s ligation rate.24 Conversely, di-substitutions at C3 cause substantial deceleration or complete inhibition of the cyclopropene-tetrazine ligation.1,7,25
Finally, lipidation at C3 is not directly compatible with our recently described ‘caged cyclopropene’ strategy, which provides external control over the cyclopropene-tetrazine ligation (Fig. 2A).26 These caged cyclopropenes were inspired by the >7500-fold difference in reaction rate between C3 mono-substituted and C3 di-substituted cyclopropenes.25,27 Essentially, a carbamate-caged 3-N spirocyclopropene mimics the unreactive C3 di-substituted cyclopropene, whereas the uncaged, 3-N spirocyclopropene mimics the more reactive C3 di-substituted cyclopropene, with the addition of an electron withdrawing group at C3 critical to prevent the ring opening isomerization to an allene or alkyne.28 The constraints of the spirocyclopropene scaffold complicate efforts to append biomolecules at C3, which contains the required electron withdrawing group. As a result, biomolecules such as lipids must be appended to C1/C2 or elsewhere on the scaffold to create target compounds like CpL1 or CpL2 (Fig. 2B), and both options possess few synthetic precedents.
Figure 2.
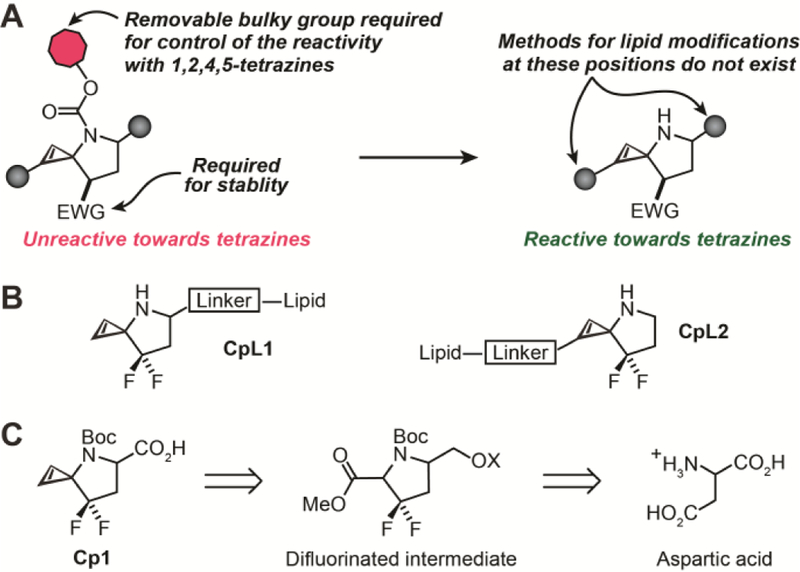
(A) Photocaged 3-N cyclopropenes allow light-mediated control of cyclopropene-tetrazine bioorthogonal ligation. Such 3-N cyclopropene formation require an EWG to prevent rearrangement to an alkyne or allene. Installing a linker on the EWG-stabilized 3-N spirocyclopropenes will enable an alternative to currently available C3 modification for attaching lipids (or other biomolecules) to cyclopropenes. (B) Lipids can be attached to 3-N cyclopropenes either at C1/C2 or on the pyrrolidine ring (C) Retrosynthetic analysis for Cp1 identified aspartic acid as an inexpensive starting point with the ‘difluorinated intermediate’ as a key scaffold.
Herein, we describe the synthesis of a novel class of lipidated cyclopropene. We explore the addition of functionality onto the spirocyclic ring as well as the cyclopropene C1/2 position. Additionally, we describe the application of a reagent that installs an ester group at C1/2 in one step from the corresponding cyclopropene, and we employ this strategy to generate cyclopropenes modified with two important classes of biologically relevant lipids: ceramides and phospholipids.
To begin, modification of the caged cyclopropene scaffold on the pyrrolidine ring of the spirocyclic framework represents the best possible option: no rate deceleration would be expected from the addition of a bulky group adjacent to the tetrazine approach trajectory, and we would not have to alter the nature of the difluoromethylene substituent at the cyclopropene C3. To explore this possibility, we sought to synthesize difluorinated spirocyclopropene Cp1, which contains a carboxylic acid-bearing linker on the carbon adjacent to the caged nitrogen group (Fig. 2C).
We began the synthesis of Cp1 with a NaBH4-mediated reduction of bis-protected L-aspartic acid 1 to the corresponding alcohol S1 (Scheme S1 in the ESI), which we protected using TBDPSCl to obtain 2 in 92% yield over two steps (Scheme 1). We then subjected 2 to Pd-mediated deprotection of the benzyl ester to give carboxylic acid 3 in 85% yield, activated the resulting acid using CDI, and proceeded directly to the Mg-mediated production of β-keto ester 4 in 80% yield over two steps. The acidity of the methylene sandwiched between the ketone and ester groups allowed us to convert 4 to the corresponding diazo compound 5. Subsequently, compound 5 was cyclized using Rh insertion29 with a non-stereoselective catalyst30 to obtain the five membered, 3-oxopyrrolidine-2-carboxylate based scaffold, 6 in 96% yield as a mixture of diastereomers.
Scheme 1.
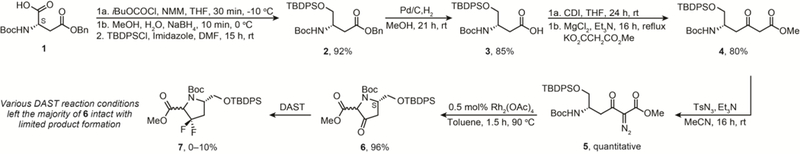
Synthesis of five-membered scaffold 6 with linker for 3-N spirocyclopropene formation.
The ketone intermediate 6 was an ideal candidate for installation of the difluoro group, which is necessary for stability of the cyclopropene in this bicyclic scaffold28. To perform the difluorination, we employed DAST, which has been successfully used to carry out difluorinations of similar proline analogs.31 Our initial attempts using equimolar equivalents of DAST at low or room temperature for varying amounts of time produced no reaction. Doubling the molar equivalents of DAST and adding it under cold conditions afforded 7 in 5% yield and tripling the DAST equivalents improved the yield to 10%. However, extensive efforts to further optimize the reaction through alterations to time, solvents, and temperature, or through the use of a different fluorinating agent Selectfluor, produced no further improvement to the reaction’s yield.
We hypothesized that the bulky TBDPS group limits DAST’s access to the ketone, so we switched to the less bulky MOM protecting group (Scheme S2 in the ESI). For these transformations, diazo 5 was stable to the requisite de- and re-protection conditions, proceeding without issue to give deprotected alcohol S2 in 90% yield. Interestingly, this is only the second report of fluoride-mediated silyl ether deprotection in the presence of diazo functionality and the first report employing TBAF deprotection of TBDPS in these systems.32 Next, we MOM-protected the free alcohol S2 to obtain S3 in 68% yield. Subsequent rhodium catalyzed cyclization afforded S4, the MOM-protected derivative of 7, in 93% yield as a mixture of diastereomers. However, despite the diminutive MOM protecting group, our extensive efforts at difluorinating ketone S4 produced S5 with yields in the 5% range and never fully consumed the starting material. Ultimately, although a maximum yield of 10% for the difluorinated 7 may have been acceptable at late stages in the synthesis, it was not feasible to carry out the next nine linear steps to generate the desired lipidated cyclopropenes. Further, as described earlier, the alkyne containing ceramides and phospholipids are either non-existent or difficult to synthesize, suggesting that rhodium-catalyzed cyclopropenation with an alkyne lipid would not be straightforward; therefore, we explored the possibility of lipidation at C1/C2, after the formation of the cyclopropene scaffold.
Methods for attaching an amine or acid handle at the C1/C2 position of an intact cyclopropene were, with one exception, unprecedented in the literature. The one exception is our recently reported orthoformate strategy that installs an ester in one step from the corresponding cyclopropene.26 Here, we explored a variety of electrophiles in an effort to expand the scope for installation of carboxylic acid functionality on 15 (Scheme S3 in the ESI). For example, we attempted to install an alcohol using oxirane or paraformaldehyde after treatment of the cyclopropene with n-BuLi, but this produced only unreacted starting material. We then explored reaction with a tosyl-activated alcohol linked to dihydrooxazole-protected carboxylic acid. This reagent should participate in base-mediated substitution of the tosylate at the cyclopropene C1/C2, and the dihydrooxazole could be subsequently hydrolysed to the carboxylic acid. However, our attempts at nucleophilic substitution using n-BuLi produced a mixture of unreacted starting material and oxazole decomposition. Ultimately, of all electrophiles tested, the iodo-orthoformates were the only electrophile that efficiently modified C1/C2 on the cyclopropene, producing the orthoformate-modified cyclopropene in high yield that quantitatively hydrolysed to the corresponding ester upon silica gel chromatography, and, after saponification, produced the spirocyclopropene acid 16 (Scheme 2).
Scheme 2.
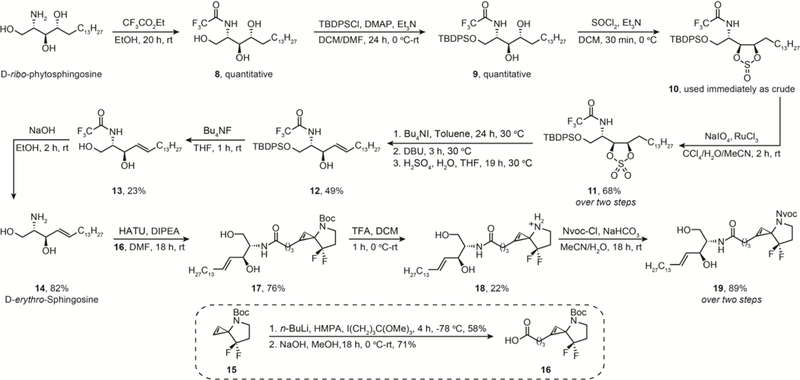
Synthesis of C1-ceramide, photocaged 3-N spirocyclopropene after installation of a C1-acid.
Finally, we explored lipidation of the carboxylic-acid-modified cyclopropene C1 with two different classes of bioactive lipids: ceramide and phospholipid. Ceramides are important components of a eukaryotic cell lipidome. They make up sphingomyelin, a major bilayer lipid, and control a variety of cellular signaling processes.33,34 Consequently, functionalized ceramides, such as a fluorophore-modified BODIPY-ceramide, are employed for tracking the biological functions of ceramides in the cell. Bioorthogonal chemistry-based applications include a recently described trans-cyclooctene-containing ceramide analog that Schepartz and co-workers used for super-resolution imaging of the Golgi in living cells.35 The availability of a cyclopropene analog of ceramide will further increase the available options for bioorthogonal reagents that can be used to study ceramide dynamics.
To obtain a C1-ceramide containing spirocyclopropene we started from d-ribo-phytosphingosine, a commercially available reagent (Scheme 3, and Scheme S4 in the ESI). Ceramides are sphingosines where the amine on the sphingosine contains hydrophobic residues via an amide linkage. We converted d-ribo-phytosphingosine to d-erythro-sphingosine using slight modifications to the synthetic strategy reported by Kim and co-workers36. Briefly, the dual protection of the amine group on d-ribo-phytosphingosine using ethyl trifluoroacetate to obtain 8, and the primary alcohol with TBDPSCl to obtain 9 proceeded in quantitative yield. Next, we converted 9 to cyclic sulfite 10 with thionyl chloride. We found this cyclic sulfite to be unstable to silica gel and overnight storage. Therefore, we subjected it immediately to ruthenium-periodate oxidation to obtain the cyclic sulfate 11 in 68% yield over two steps. Additionally, in our hands, the trityl protected analogues were unstable to the required thionyl chloride/ruthenium-periodate conditions, so we proceeded with ethyl trifluoroacetate protected analogs instead. We performed the 11–12 reaction sequence in one pot because our attempts to purify the intermediates resulted in substantially lower overall yields due to their decomposition on silica gel. Essentially, we converted cyclic sulfate 11 to the sulfate ester-iodide using Bu4NI, dehydrohalogentated the intermediate using DBU to produce the sulfate alkene, and hydrolysed the sulfate under acidic conditions to obtain 12 in 49% yield over three steps. Then, this N,O-bis protected sphingosine 12 was TBDPS-deprotected using the fluoride source Bu4NF to obtain a 23% yield of 13, which was converted to the d-erythro-sphingosine 14 by base-mediated hydrolysis in 82% yield.
Scheme 3.
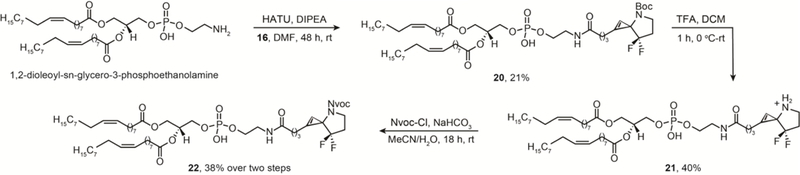
Synthesis of C1-phospholipid, photocaged 3-N spirocyclopropene via a C1-acid containing cyclopropene.
The free amine sphingosine 14 was coupled to the 3-N spirocyclopropene acid 16 using HATU to obtain the C1-ceramide Boc-cyclopropene 17 in 76% yield. Finally, the lipidated-cyclopropene was Boc-deprotected to obtain free amine C1-ceramidated 3-N spirocyclopropene 18 in 22% yield. Importantly, the low yields of 18 are due to difficulties inherent to the purification of this charged lipid, not decomposition, because similar cyclopropene scaffolds, including the Boc- and Nvoc-protected variants of 18, have survived acidic HPLC and deprotection conditions unscathed26. Separately, the Nvoc photocaged version was prepared directly without purifying the Boc-deprotected intermediate 18 to obtain 19 in 89% yield over two steps.
Like ceramides, phospholipids represent another important class of biologically relevant lipids. The amphiphilic character of phospholipids makes them a major component of the lipid bilayer in the cell plasma membrane. The availability of a photocaged, cyclopropene-containing phospholipid will permit its applications as a bioorthogonal probe to study membrane biology. We installed a commercially available phospholipid, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (18:1 (Δ9-cis) PE), at the C1 position of cyclopropene 16 to obtain C1 phospholipidated 3-N spirocyclopropene 20 in 21% yield, which, upon Boc deprotection, afforded 21 in 40% yield. Similar to the photocaged ceramide cyclopropene lipid, the Nvoc photocaged version 22 was prepared without purifying the Boc-deprotected intermediate 21 to obtain 22 in 38% yield over two steps (Scheme 3). However, the amounts obtained complicated our efforts at full spectroscopic characterization other than HRMS due to the complexity of the molecule, modest reaction yields, and high cost of the phospholipid starting material.
In summary, we describe a method for the C1-lipidation of cyclopropenes. We have generated two biologically relevant, C1-lipidated cyclopropenes using an acid functionalized 3-N spirocyclopropene scaffold. Both the C1-ceramide- and phospholipid-linked spirocyclopropene represent the first examples of cyclopropene modifications with biomolecules on the alkenic C1/C2 position.
Supplementary Material
Acknowledgments
The authors thank B. O’Neill for critical reading of the manuscript. This work was supported by a grant to S.T.L. from the National Science Foundation (CHE1451366).
References
- 1.Ravasco JMJM, Monteiro CM, Trindade AF. Org Chem Front 2017; 4: 1167–1198. [Google Scholar]
- 2.Patterson DM, Jones K a, Prescher J a. Mol Biosyst 2014: 1693–1697. [DOI] [PubMed] [Google Scholar]
- 3.Cole CM, Yang J, Šečkutė J, Devaraj NK. ChemBioChem 2013; 14: 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Späte A-K, Bußkamp H, Niederwieser A, Schart VF, Marx A, Wittmann V. Bioconjug Chem 2014; 25: 147–154. [DOI] [PubMed] [Google Scholar]
- 5.Smith NJ, Rohlfing K, Sawicki LA, Kharkar PM, Boyd SJ, Kloxin AM, Fox JM. Org Biomol Chem 2018; 16: 2164–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Z, Wang D, Li L, Pan S, Na Z, Tan CYJ, Yao SQ. J Am Chem Soc 2014; 136: 9990–9998. [DOI] [PubMed] [Google Scholar]
- 7.Kamber DN, Nazarova LA, Liang Y, Lopez SA, Patterson DM, Shih H-W, Houk KN, Prescher JA. J Am Chem Soc 2013; 135: 13680–13683. [DOI] [PubMed] [Google Scholar]
- 8.Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA. J Am Chem Soc 2012; 134: 18638–43. [DOI] [PubMed] [Google Scholar]
- 9.Yu Z, Pan Y, Wang Z, Wang J, Lin Q. Angew Chem Int Ed Engl 2012; 51: 10600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Liang Y, Šečkutė J, Houk KN, Devaraj NK. Chemistry 2014; 20: 3365–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eggert F, Kath-Schorr S. Chem Commun 2016; 52: 7284–7287. [DOI] [PubMed] [Google Scholar]
- 12.Paulini K, Reissig H-U. Synlett 1992; 1992: 505–506. [Google Scholar]
- 13.Kumar P, Shukhman D, Laughlin ST. Tetrahedron Lett 2016; 57: 5750–5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Šečkutė J, Cole CM, Devaraj NK. Angew Chemie Int Ed 2012; 51: 7476–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baird MS, Dale CM, Lytollis W, Simpson MJ. Tetrahedron Lett 1992; 33: 1521–1522. [Google Scholar]
- 16.Nunn JR. J Chem Soc 1952; 0: 313–318. [Google Scholar]
- 17.Macfarlane JJ, Shenstone FS, Vickery JR. Nature 1957; 179: 830–831. [DOI] [PubMed] [Google Scholar]
- 18.Lee DJ, Wales JH, Ayres JL. Cancer Res 1968; 28: 2312–2318. [PubMed] [Google Scholar]
- 19.Hao P, Alaraj IQM, Dulayymi JR Al, Baird MS, Liu J, Liu Q. Korean J Parasitol 2016; 54: 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kogen H, Kiho T, Tago K, Miyamoto S, Fujioka T, Otsuka N, Suzuki-Konagai K, Ogita T. J Am Chem Soc 2000; 122: 1842–1843. [DOI] [PubMed] [Google Scholar]
- 21.Kim R, Sherrill WM, Rubin M. Tetrahedron 2010; 66: 4947–4953. [Google Scholar]
- 22.Edwards A, Rubina M, Rubin M. Curr Org Chem 2016; 20: 1862–1877. [Google Scholar]
- 23.Liao L, Yan N, Fox JM. Org Lett 2004; 6: 4937–9. [DOI] [PubMed] [Google Scholar]
- 24.Xiong D-C, Zhu J, Han M-J, Luo H-X, Wang C, Yu Y, Ye Y, Tai G, Ye X-S. Org Biomol Chem 2015; 13: 3911–7. [DOI] [PubMed] [Google Scholar]
- 25.Thalhammer F, Wallfahrer U, Sauer J. Tetrahedron Lett 1990; 31: 6851–6854. [Google Scholar]
- 26.Kumar P, Jiang T, Li S, Zainul O, Laughlin ST. Org Biomol Chem 2018; 16: 4081–4085. [DOI] [PubMed] [Google Scholar]
- 27.Sauer J, Heldmann DK, Hetzenegger J, Krauthan J, Sichert H, Schuster J. European J Org Chem 1998: 2885–2896. [Google Scholar]
- 28.Kumar P, Zainul O, Laughlin ST. Org Biomol Chem 2018; 16: 652–656. [DOI] [PubMed] [Google Scholar]
- 29.Moreau RJ, Sorensen EJ. Tetrahedron 2007; 63: 6446–6453. [Google Scholar]
- 30.Adams J, Spero DM. Tetrahedron 1991; 47: 1765–1808. [Google Scholar]
- 31.Doebelin C, He Y, Kamenecka TM. Tetrahedron Lett 2016; 57: 5658–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abid I, Gosselin P, Mathé-Allainmat M, Abid S, Dujardin G, Gaulon-Nourry C. J Org Chem 2015; 80: 9980–9988. [DOI] [PubMed] [Google Scholar]
- 33.Bieberich E Future Lipidol 2008; 3: 273–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morad SAF, Cabot MC. Nat. Rev. Cancer, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Erdmann RS, Takakura H, Thompson AD, Rivera-Molina F, Allgeyer ES, Bewersdorf J, Toomre D, Schepartz A. Angew Chemie - Int Ed 2014; 53: 10242–10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee Y, Lee S, Jeon H, Baek D, Seo J, Kim D, Kim S. Synthesis (Stuttg) 2011; 2011: 867–872. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.