

Abstract
The present study tested the hypothesis that maternal smoke exposure results in fetal lung growth retardation due to dysregulation in various signaling pathways, including the Wnt (wingless-related integration site)/β-catenin pathway. Pregnant female C57BL/6J mice were exposed to cigarette smoke (100–150 mg/m3) or room air, and offspring were humanely killed on 12.5, 14.5, 16.5, and 18.5 d post coitum (dpc). We assessed lung stereology with Cavalieri estimation; apoptosis with proliferating cell nuclear antigen, TUNEL, and caspase assays; and gene expression with quantitative PCR (qPCR) and RNA sequencing on lung epithelium and mesenchyme retrieved by laser capture microdissection. Results demonstrated a significant decrease in body weight and lung volume of smoke-exposed embryos. At 16.5 dpc, the reduction in lung volume was due to loss of lung mesenchymal tissue correlating with a decrease in cell proliferation (n = 10; air: 61.65% vs. smoke: 44.21%, P < 0.05). RNA sequence analysis demonstrated an alteration in the Wnt pathway, and qPCR confirmed an increased expression of secreted frizzled-related protein 1 (sFRP-1) [n = 12; relative quantification (RQ) 1 vs. 2.33, P < 0.05] and down-regulation of Cyclin D1 (n = 7; RQ 1 vs. 0.61, P < 0.05) in mesenchymal tissue. Furthermore, genome expression studies revealed a smoke-induced up-regulation of Rho-GTPase-dependent actin cytoskeletal signaling that can lead to loss of tissue integrity.—Unachukwu, U., Trischler, J., Goldklang, M., Xiao, R., D’Armiento, J. Maternal smoke exposure decreases mesenchymal proliferation and modulates Rho-GTPase-dependent actin cytoskeletal signaling in fetal lungs.
Keywords: apoptosis, cigarette smoke, in utero, proliferation, Wnt/β-catenin
In utero exposure to maternal tobacco smoke leads to a higher incidence of preterm birth, intrauterine growth restrictions, decreased body weight at birth, and a decline in lung volume and function (1–7). After smoke exposure, fetal lungs exhibit fibrotic changes and aberrantly remodeled respiratory airways, suppressed pulmonary alveolarization, reduced vascularization, increased smooth muscle thickness, and airway responsiveness (8–12). These phenotypes cause long-lasting respiratory defects in children, such as wheezing, asthma, and nocturnal cough (13), and they increase susceptibility to acute bronchopulmonary disorders later in life (2). These disorders were also found to be independent of postnatal smoke exposure (13) and thus causatively linked to prenatal lung development (2). Despite public awareness efforts regarding the negative effects of maternal smoking during pregnancy, as well as vigorous antitobacco campaigns, 15 to 20% of pregnant women still smoke in most developed countries (5, 14). It is thus important to develop effective strategies to protect the growing fetal lung.
Whereas overall fetal growth impairment due to in utero smoke (IUS) exposure in mammals has been studied in large epidemiologic studies after birth (6, 15), the effect of maternal tobacco smoke on lung development is not as well documented (6). Examining gene expression changes during prenatal development is important for the pathophysiologic understanding of the effect of cigarette smoke on lung function. This is important because some of the effects measured after birth might be compensatory effects after smoke cessation due to delivery of the fetus or additional effects of postnatal smoke exposure or nicotine delivery in some models (16). At the molecular level, evidence suggests that smoke-induced pathologies are caused by the differential regulation of various biochemical pathways involved in lung development and differs by exposures in utero or as adults (17). Most notably, changes in nicotinic and VEGF receptor expression, DNA methylation patterns, and the retinoid acid pathway have been reported (9, 16, 18, 19). Some of these changes hint at their morphologic consequences, where modifications to VEGFR likely leads to vascularization deficits (9), and decreased protein expression of the retinoid acid pathway elements contribute to alveolarization insufficiencies (16). Several animal and human studies report that in utero smoke exposure also stimulates transdifferentiation of airway interstitial cells into myofibroblasts lacking lipid surfactants and increases smooth muscle thickness, all hallmark indices of chronic pulmonary disorders (8, 20).
Of importance to our study is the evidence that prenatal smoke exposure represses the canonical Wnt (wingless-related integration site)/β-catenin signaling pathway (21). Canonical Wnt signaling is active throughout lung development and is involved in the differentiation of lung epithelium, mesenchyme, and specific lung cell types (22, 23). Canonical Wnt functions by regulating the amount of the transcriptional coactivator β-catenin in the cytosol and nucleus to activate downstream target genes including matrix metalloproteinases (MMPs) and cell cycle gene Cyclin D1 (24, 25). This signaling via β-catenin controls key developmental gene expression programs, cell proliferation, and embryonic tissue morphogenesis, and it is central to the formation of peripheral airways of the lungs that conduct gas exchanges (26). Elevated levels of Wnt antagonist–secreted frizzled-related protein 1 (SFRP-1) expression are a marker for tissue destruction and abnormal epithelial cell differentiation, as well as for the generation of an emphysematous phenotype (15, 27). The rescue of Wnt signaling through pharmacologic modification has also been shown to prevent emphysematous changes in the lungs of smoke-exposed mice (28). Because the importance of the Wnt pathway in lung development and its modulation by cigarette smoke are well defined (15, 22, 29), we sought to investigate how smoke-induced modification of the Wnt/β-catenin pathway affects embryonic lung morphogenesis. We postulate a cigarette smoke–induced aberrant signaling of the Wnt/β-catenin pathway in the lungs that is developmentally time dependent.
We studied prenatal lung development in an IUS-exposure mouse model by analyzing fetal body weight, lung volume, and stereology; the proportional development of different embryonic tissues; and differential profiles of developmental genes. By synchronizing time-dependent morphologic changes in the expression profiles of the developing fetal lung tissue, we expect to define the genetic basis for cigarette smoke induction on cell fate progression during normal lung organogenesis and to identify novel signaling targets to modify for therapeutic interventions.
MATERIALS AND METHODS
Animals
All animal procedures were approved by the Columbia University Institutional Animal Care and Use Committee. Eight to 9-wk-old male and female C57BL/6J mice were used for mating purposes, and only female mice were exposed to tobacco smoke (3R4F; University of Kentucky, Lexington, KY, USA).
Smoke exposure
Female C57BL/6J mice were exposed to cigarette smoke 7 d before mating and throughout their pregnancy for 5 h/d, 5 d/wk. This protocol was adopted to ensure smoke exposure throughout the duration of the pregnancy and to allow for an initial adjustment period for the dams, which increases the probability of implantation. The importance of early smoke exposure to the human fetal condition has been published (6) and was recapitulated in this setting to improve the significance of the acquired data. Matutinal vaginal plugs were checked, and on the day of observation, gestational age was considered as 0.5 d post coitum (dpc). Smoke exposure was performed using a specially designed chamber (Teague Enterprises, Woodland, CA, USA) that delivers mainstream and sidestream smoke to the mice as previously described (30, 31). The total particulate matter within the smoking chamber was regulated such that mice received a total particulate matter of 100 to 150 mg/m3, simulating moderate to heavy smoking in humans. Control mice received fresh air. Offspring were humanely killed 24 h after the last smoke exposure on 12.5, 14.5, 16.5, and 18.5 dpc and 1 d postpartum (dpp). After death, lungs were carefully dissected and either fixated in formalin, snap frozen at −80°C, or stored in cryogenic optimal cutting temperature (OCT) compound for preparation of frozen sections.
Lung stereology
Formalin-fixed lungs were embedded in paraffin, and serial sections (5 µm) were obtained every 90 µm throughout the whole lung. Slices were flipped 90° in the same direction, and the cut surfaces were overlaid with a point grid with defined distances to calculate lung volume using the Cavalieri estimation stereologic method. An average of 289, 1752, and 4117 grid points were counted for 14.5, 16.5, and 18.5 dpc lungs, respectively. Values represent volume after paraffin embedment and might therefore differ from wet weight. Additionally, lung volume measurements are approximate, as stereology was performed on uninflated lungs as a result of the limitations with lung inflation at earlier embryonic time points (32). Sections were stained with hematoxylin and eosin, and pictures of every lung section were taken at ×20 magnification (OptiScan motorized stage, Prior Scientific and Image-ProPlus; Media Cybernetics, Rockville, MD, USA). Highly standardized processing ensured intergroup comparability. Epithelial and mesenchymal tissues were distinguished morphologically. Statistical analysis for changes in lung volume at multiple time points was performed by 2-way ANOVA and post hoc effect tests.
Proliferation analysis
Slides from serially sectioned lungs were randomly selected, and proliferating cell nuclear antigen (PCNA) staining was performed utilizing the Histomouse Max Mouse-on-Mouse Kit (Thermo Fisher Scientific, Waltham, MA, USA) and PCNA antibody (Sigma-Aldrich, St. Louis, MO, USA). A total of 25 pictures per age group in either smoke or room air conditions were analyzed at random (5 pictures per animal, 5 animals per group), and the ratio of PCNA-reactive cells over total cells was measured at ×20 magnification. The distinction between epithelial and mesenchymal tissues was determined morphologically after counterstaining.
Apoptotic assays
Apoptotic cells in paraffin-embedded lung tissue were detected by TUNEL assay using an in situ cell death detection kit according to the manufacturer’s instructions (Roche Diagnostics, Indianapolis, IN, USA). After removal of paraffin with xylene and rehydration with a graded series of alcohols, tissue sections were digested with proteinase K and stained with TUNEL reaction mix. A converter peroxidase allows light microscopy visualization of the TUNEL reaction after precipitation with a diaminobenzidine substrate. For this assay, a total of 15 pictures per age group per condition were analyzed at random (5 pictures per animal, 3 animals per group), and the ratio of TUNEL-positive cells to total cells was measured at ×20 magnification. Apoptosis was also assessed using the Caspase 3/7 activity assay kit according to the manufacturer’s instructions (Promega, Madison, WI, USA). Luminescence units of caspase activity were measured per milligram of protein using a Tecan Infinite Pro 200 luminometer (Morrisville, NC, USA). Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific).
Laser-capture microdissection
Embryonic lungs were embedded in OCT compound and frozen at −80°C. Frozen sections (10 µm thick) were obtained under RNAse-free conditions. After a quick fixation in 70% ethanol, tissue sections were counterstained with hematoxylin and eosin immediately before laser-capture microdissection. Epithelial and mesenchymal tissues were collected using a wide-field microscope with PALM Microbeam IV in the Confocal and Specialized Microscopy Shared Facility of Columbia University (New York, NY, USA). Areas were selected at ×20 magnification and catapulted into adhesive caps (Carl Zeiss GmbH, Jena, Germany). Tissue dissects were immediately suspended in guanidine thiocyanate buffer and stored on ice for subsequent analysis. Following laser capture microdissection (LCM) protocol, mesenchymal and epithelial tissue origins were verified by quantitative PCR (qPCR).
qPCR analysis
Extracted whole lungs were mechanically homogenized in guanidine thiocyanate buffer. Total RNA from whole lung homogenates and tissue dissects was extracted using RNeasy Mini Kit (Qiagen, Germantown, MD, USA). RNA quality was measured by UV spectroscopy, and DNAse treatment and reverse transcription were performed using the TaqMan Reverse Transcriptase Kit (Thermo Fisher Scientific). qPCR was performed utilizing the following TaqMan probes: Axin2 (Mm00443610_m1), CyclinD1 (Mm00432359_m1), Lef1 (lymphoid enhancer factor; Mm00550265_m1), sFRP-1 (Mm00489161_m1), Bmp4 (bone morphogenetic protein 4; Mm00432087_m1), Wnt2 (Mm00470018_m1), Wnt10b (Mm00442104_m1), Wnt5a (Mm00437347_m1), and Tcf4 (T-cell factor; Mm00443210_m1). Results were analyzed by unpaired t tests. Confirmatory qPCR analysis was also performed on laser microdissected lung tissues using advanced glycosylation end-product specific receptor (AGER; Mm00545815_m1), an epithelial cell marker, to distinguish epithelial tissue digests from the mesenchyme.
RNA isolation and sequencing
RNA was extracted from snap-frozen lung tissue samples of the 16.5 dpc embryonic time point exposed to either room air or cigarette smoke using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA), and RNA quality was assessed electrophoretically (Bioanalyzer 2100; Agilent Technologies, Santa Clara, CA, USA) at the Molecular Pathology Core Facility of Columbia University. Duplicate RNA isolates with verified integrity numbers (RIN = 10) were sequenced at the JP Sulzberger Columbia Genome Center of Columbia University. A poly-A pulldown was used to enrich mRNAs from total RNA samples (200 ng to 1 µg per sample), and library preparation was performed using Illumina TruSeq RNA prep kit (Illumina, San Diego, CA,USA). RNA sequencing was performed on an Illumina HiSeq 2000 platform at 100 bp to a depth of 30 million single-end reads. Real-time analysis (Illumina) was used for base calling and bcl2fastq 1.8.4 for converting BCL to fastq format, coupled with adaptor trimming. RNA-Seq reads were mapped to the mouse reference genome (UCSC/mm9; http://genome.ucsc.edu) using TopHat 2.1.0 (33) with 4 mismatches and 10 maximum multiple hits. Relative abundance of genes and splice isoforms was estimated using Cufflinks 2.0.2 (34) as fragments per kilobase of exon per million fragments mapped. Differentially expressed genes under various conditions were resolved using differential expression analysis for sequence count data (35) based on a negative binomial distribution, and using this approach, 23,385 National Center for Biotechnology Information Reference Sequence Database genes (NCBI RefSeq; https://www.ncbi.nlm.nih.gov/refseq/) were uniquely aligned. The statistical significance of the fold change in expression was determined by paired t tests for difference between expression values obtained under the 2 test conditions in duplicate. The false discovery rate (FDR) was controlled by adjusting the P values using the Benjamini–Hochberg algorithm. Analysis and visualization of differentially expressed genes were conducted by RStudio 0.99.893 (RStudio, Boston, MA, USA). Pathway enrichment analysis and functional annotation was performed for differentially expressed genes with fold change ≥2.0 and FDR ≤ 0.05 using the Ingenuity Pathway Analysis bioinformatics tool version 01-07 (Qiagen, Redwood City, CA, USA).
RESULTS
Maternal smoke–induced reduction in fetal body weight and lung volume occurs at defined developmental time points
IUS embryos exhibited significantly lower whole-body weight beginning at 14.5 dpc and at all prenatal time points up to 1 dpp (Fig. 1A). Measurements using Cavalieri estimation revealed a significant decrease in lung volume only at the 16.5 and 18.5 dpc developmental time points (Fig. 1B). Descriptive statistics for body weight and lung volume measurements are provided in Table 1. Reduction in both body weight and lung volume was observed to increase in severity with prolonged smoke exposure, suggesting that the smoke-induced phenotype accumulated gradually. The changes in fetal lung volume were sufficiently explained by the prediction model of Pearson’s correlation (R2 = 0.9689) in a 2-way ANOVA (Table 2). Exposure to maternal cigarette smoke accounted for a 23.3% difference in lung volume. We additionally observed that the developmental time point accounted for 62.11% of the observed change in fetal lung volume.
Figure 1.
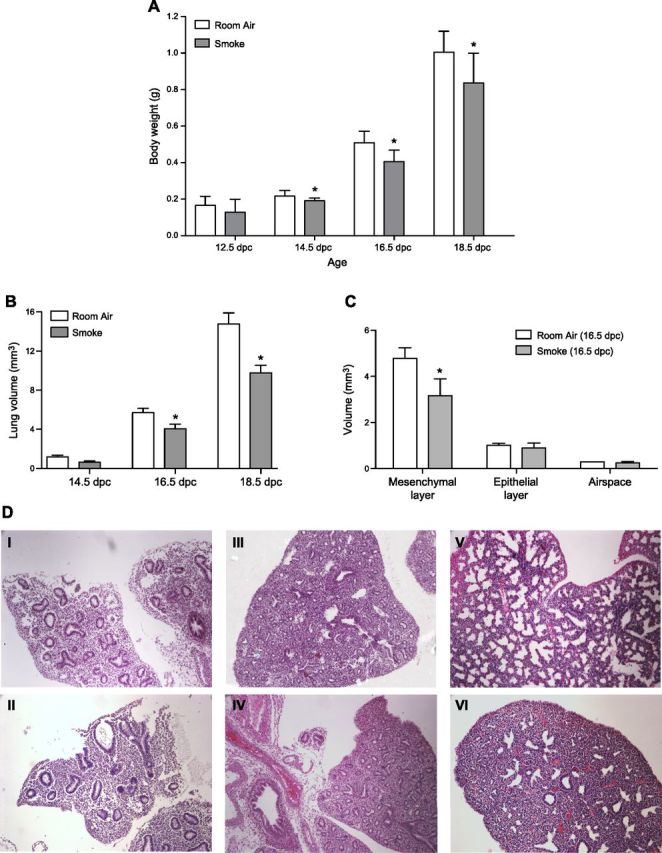
A) Whole body weight measurements of room air– and smoke-exposed fetal lungs. Significant decrease in embryonic body weight due to maternal smoke is detectable beginning at 14.5 dpc. B) Comparative lung volume measurements of room air– and smoke-exposed fetal lungs. Cavalieri estimations reveal significant decrease in fetal lung volume due to maternal smoke beginning at 16.5 and 18.5 dpc. C) Mesenchymal, epithelial, and airspace volumes of room air– and smoke-exposed 16.5 dpc fetal lungs. Smoke-induced reduction in lung volume at 16.5 dpc is attributable to decrease in lung mesenchyme (room air: 4.78 ± 0.47 mm3; smoke: 3.17 ± 0.73 mm3; *P = 0.032). Asterisks denote significant difference in means at P ≤ 0.05 by Student’s t test. D) Representative images of lung tissue slices obtained from room air– and smoke-exposed mice. Serial sections (5 μm) of paraffin-embedded lungs were used to obtain measurements of mesenchymal, epithelial, airspace, and total lung volumes. Lung slices of room air–exposed fetal mice at 14.5 (I), 16.5 (III), and 18.5 (V) dpc are displayed, along with comparative lung slices of in utero smoke-exposed mice at 14.5 (II), 16.5 (IV), and 18.5 (VI) dpc. Original magnification, ×20.
TABLE 1.
Descriptive statistics for body weight and lung volume for mean differences between room air– and smoke-exposed mice
| Age | Condition | n | Body weight (g) | Lung volume (mm3) | ||
|---|---|---|---|---|---|---|
| Mean ± sd | P | Mean ± sd | P | |||
| 12.5 dpc | Room air | 7 | 0.167 ± 0.05 | |||
| Smoke | 21 | 0.128 ± 0.07 | 1.98E-01 | |||
| 14.5 dpc | Room air | 31 | 0.216 ± 0.03 | 1.178 ± 0.31 | ||
| Smoke | 31 | 0.191 ± 0.02 | 3.09E-04* | 0.756 ± 0.09 | 0.09 | |
| 16.5 dpc | Room air | 28 | 0.509 ± 0.06 | 6.087 ± 0.49 | ||
| Smoke | 69 | 0.406 ± 0.06 | 9.14E-11* | 4.311 ± 0.98 | 0.048* | |
| 18.5 dpc | Room air | 49 | 1.005 ± 0.11 | 14.756 ± 1.97 | ||
| Smoke | 30 | 0.837 ± 0.16 | 7.40E-07* | 9.780 ± 1.32 | 0.022* | |
| 1 dpp | Room air | 18 | 1.275 ± 0.10 | |||
| Smoke | 29 | 1.113 ± 0.08 | 9.90E-08* | |||
Statistically significant.
TABLE 2.
Multifactorial analysis of fetal lung volume changes
| ANOVA | Sum of squares (type III) | DF | MS | F(DFn, DFd) | P |
|---|---|---|---|---|---|
| Interaction | 16.92 | 2 | 8.461 | F(2, 15) = 7.622 | 0.0052** |
| Condition | 29.19 | 1 | 29.19 | F(1, 15) = 26.3 | 0.0001*** |
| Time point | 421.7 | 2 | 210.9 | F(2, 15) = 190 | <0.0001**** |
| Residual | 16.65 | 15 | 1.11 |
Two-way ANOVA of effects of developmental time point (14.5, 16.5, 18.5 dpc) and environmental condition (room air or smoke exposure) on changes in fetal lung volume was sufficiently explained by the prediction model of Pearson’s correlation (R2 = 0.9689). Results show that changes in fetal lung volume depends largely on the developmental time point of the fetus (F(2, 15) = 190, P < 0.0001), which accounted for 62.11% of the observed change vs. 23.3% due to the effect of maternal cigarette smoke exposure (F(1, 15) = 26.3, P < 0.0001). **P < 0.01, ***P < 0.001, ****P < 0.0001.
Mesenchymal tissue is overproportionally affected by smoke-induced lung growth retardation
The effects of IUS exposure were further analyzed at 16.5 dpc to determine the tissue type specifying the significant decrease in lung parenchymal volume. This time point represents the developmental period with the largest increase in fetal lung growth accompanied by massive vascularization and angiogenesis (36), and it also marks the initiation of smoke-induced effects in lung volume during murine fetal development (Fig. 1B). For 16.5 dpc lungs, the initial analysis was therefore followed by separate volume estimations for epithelial and mesenchymal tissues in the lung. As shown in Fig. 1C, mesenchymal tissue is overproportionally affected by lung growth retardation due to IUS exposure, exhibiting a significant decrease (4.78 ± 0.47 vs. 3.17 ± 0.73 mm3, P < 0.05), whereas the marginal decrease in epithelial volume did not reach significance. Average mesenchyme-to-epithelium ratios of 3.53 and 4.72 were obtained for IUS and control lungs, respectively. Representative images of tissue slices used for lung volume calculations are provided in Fig. 1D.
Smoke-induced reduction in lung mesenchymal volume results from decreased cellular proliferation and not increased apoptosis
To define the cause of the decrease in mesenchymal volume, PCNA staining confirmed a significant decline in cell proliferation of IUS-exposed lung mesenchyme [61.65% (control) vs. 44.21% (IUS) PCNA-positive staining, P < 0.05], while the observed decrease in epithelial proliferation did not reach significance (28.29% mesenchymal vs. 10.19% epithelial proliferation decrease) (Fig. 2A). Additionally, there was no significant difference in the rate of cellular apoptosis at 14.5, 16.5, and 18.5 dpc using either the Caspase 3/7 activity assay (Fig. 2D) or TUNEL staining (Fig. 2E). Representative images of tissue slices used for analysis of PCNA staining and TUNEL staining are provided in Fig. 2B, C, F, respectively.
Figure 2.

Proliferation and apoptosis assays. A) Cell proliferation assessed using PCNA staining decreased significantly by 28.29% in embryonic lung mesenchyme compared with nonsignificant 10.19% decrease in fetal lung epithelium. *P ≤ 0.05. B, C) Representative images of room air–exposed (B) and cigarette smoke–exposed (C) lung tissue slices stained with PCNA antigen are also provided. Brown nuclear staining depicts positive result. Original magnification, ×20. D) Apoptosis in room air– and smoke-exposed fetal lung tissue was assessed by luminescence of cleavage product of caspase 3/7 enzymes measured as function of cell death per milligram protein. E) DNA double strand breaks produced after apoptosis were also labeled using terminal deoxynucleotidyl transferase (Tdt) in TUNEL assay. These results show that fetal lung exposure to maternal smoke at all embryonic time points measured does not result in significant fetal lung tissue death compared to fetal lungs exposed to room air conditions. F) Representative lung tissue slices depicting TUNEL staining results. I, II) Negative control slides stained without TUNEL reaction mixture (I) or with reaction mixture for positive control (II) to induce double-stranded DNA breaks were used to compare TUNEL staining in experimental slides as measure of apoptosis after in utero exposure to maternal cigarette smoke. III–VIII) Room air–exposed lung slices (III, V, VII) and comparative smoke-exposed lung slices (IV, VI, VIII) obtained from 14.5 (III, IV), 16.5 (V, VI), and 18.5 (VII, VIII) dpc mouse fetuses. Original magnification, ×20.
In utero smoke exposure alters expression of cell cycle genes and Wnt signaling pathway
To understand the genetic basis for the maternal smoke-induced decrease in fetal lung mesenchymal volume, we first analyzed the effect IUS exposure had on the Wnt signaling pathway and Wnt target genes, given the established role of Wnt signaling in regulating aspects of lung organogenesis, specifically epithelial/mesenchymal regional specialization (36–38). Laser microdissected epithelial tissues were distinguished from mesenchyme by confirmatory qPCR analysis of AGER expression after correction for GAPDH levels (Supplemental Fig. 1). We performed RNA sequencing of room air– and smoke-exposed fetal whole lung homogenates at 16.5 dpc and determined comparative expression levels specific to Wnt pathway genes. Results visualized using IPA bioinformatics in Fig. 3A reveal that Wnt antagonists sFRPs are up-regulated while the frizzled receptor and coreceptor, LDL receptor (LRP5/6), are down-regulated as a result of fetal lung exposure to maternal smoke. Given the consequent absence of Wnt signaling, cytosolic β-catenin ubiquitination is expected, and its translocation to the nucleus is not predicted to occur, leading to LEF/TCF-mediated repression of the downstream transcription of select nuclear Wnt target genes Axin2, Lef1, Mmp7, and Cyclin D1 (Fig. 3A). At these expression states, cell proliferation will thus be inhibited (26, 29, 39), stymying embryonic lung growth, as has been phenotypically observed in our studies.
Figure 3.
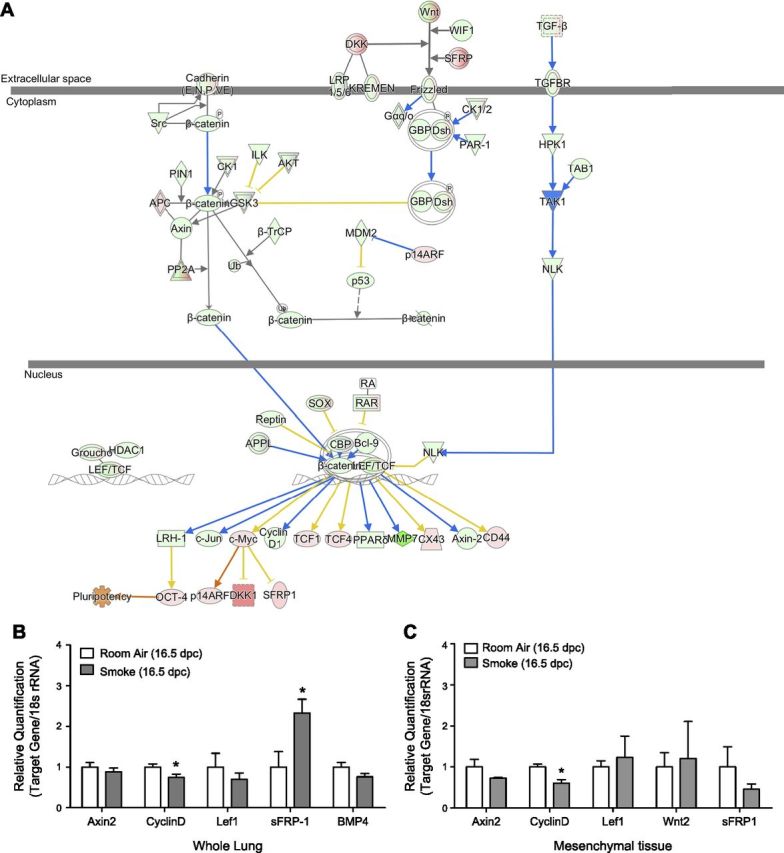
A) Overlay of Wnt/β-catenin signaling pathway with 16.5 dpc transcriptomic data. Gene expression states of Wnt signaling genes after maternal smoke exposure predict that up-regulation of SFRPs and down-regulation of frizzled receptor will lead to phosphorylation and ubiquitination of cytosolic β-catenin, preventing its translocation to nucleus. At these expression states, LEF/TCF-mediated repression of transcription of Wnt downstream targets CyclinD1, Axin2, and Mmp7 is predicted, as shown, causing decreased cell proliferation phenotypically observed in our studies. B, C) Comparative Wnt signaling in room air– and smoke-exposed fetal mouse lungs. B) qPCR analysis resolves smoke-induced up-regulation of sFRP-1 in 16.5 dpc fetal mouse lungs and down-regulation of Cyclin D1, validating RNA sequencing results presented in A. These expression states account for decreased cell proliferation rates in whole lung homogenates. C) Correlative results obtained from LCM mesenchymal tissue show Cyclin D1 is similarly down-regulated.
We further validated our RNA sequencing results using qPCR analysis to quantify the expression of select Wnt target genes in whole lung and laser-capture microdissected epithelial and mesenchymal tissue homogenates. Of the target gene amplification products quantified, sFRP-1 expression was significantly up-regulated in whole lung homogenates of 16.5 dpc IUS-exposed offspring [relative quantification (RQ) 1 vs. 2.33, P < 0.05, n = 6 lungs per group], whereas CyclinD1 was down-regulated (RQ 1 vs. 0.74, P = 0.05, n = 3–4 lungs per group) (Fig. 3B) in concordance with our RNA sequencing results. Up-regulated SFRP-1 expression levels in the placenta of smoke-exposed mouse dams have previously been associated with restricted fetal growth (40). Because the tested genes are differentially expressed in epithelial and mesenchymal tissues (38, 41), tissue-specific expression profiles for the genes were assessed. In mesenchymal tissues, CyclinD1 was down-regulated (RQ 1 vs. 0.61, P < 0.05), matching the proliferation decrease observed in mesenchymal tissue (Fig. 3C). Descriptive statistics of qPCR analysis of whole lung and tissue-specific RNA digests are provided in Table 3. No correlative change in expression was detected in epithelial tissues (data not shown). Significant changes in sFRP-1 expression could not be detected in either lung epithelium or mesenchyme (Fig. 3C)
TABLE 3.
Descriptive statistics of whole lung and mesenchymal tissue qPCR analysis
| Gene | Whole lung | Mesenchyme | ||||
|---|---|---|---|---|---|---|
| Room air | Smoke | P | Room air | Smoke | P | |
| Axin2 | 1.00 ± 0.11 | 0.89 ± 0.09 | 0.43 | 1.00 ± 0.18 | 0.73 ± 0.02 | 0.28 |
| CyclinD1 | 1.00 ± 0.08 | 0.74 ± 0.08 | 0.05* | 1.00 ± 0.07 | 0.61 ± 0.09 | 0.02* |
| sFRP-1 | 1.00 ± 0.38 | 2.33 ± 0.34 | 0.03* | 1.00 ± 0.49 | 0.46 ± 0.13 | 0.27 |
| Lef1 | 1.00 ± 0.29 | 0.70 ± 0.16 | 0.41 | 1.00 ± 0.15 | 1.23 ± 0.52 | 0.69 |
| Bmp4 | 1.00 ± 0.11 | 0.76 ± 0.08 | 0.10 | |||
| Wnt2 | 1.00 ± 0.35 | 1.21 ± 0.90 | 0.86 | |||
Tissue-specific digests were obtained by laser capture microdissection of 16.5 dpc lungs. All qPCR analyses were performed in triplicate each for 3 to 6 lungs per group. Data are presented as means ± sem. P values were determined by Student’s t test. *P < 0.05.
Maternal smoke exposure modulates Rho-GTPase-dependent actin cytoskeleton signaling and morphogenesis of 16.5 dpc fetal lungs
For genomewide assessment of other novel signaling pathways induced by maternal tobacco smoke in 16.5 dpc mouse fetal lungs, we further utilized the RNA sequencing analysis pipeline depicted in Fig. 4A and resolved changes in 43 genes that express at least a 2-fold change and a FDR of q ≤ 0.05, as shown in the volcano plot displayed in Fig. 4B. These expressed genes are listed in Supplemental Table 1. Core analysis and functional annotation using IPA bioinformatics mapped the resolved genes to 6 canonical signaling pathways, with Z scores −2.0 ≤ x ≥ 2.0 denoting significant biochemical inhibition or activation of the canonical pathways by the expression of the candidate genes (Fig. 4C). Smoke-induced activation of actin cytoskeletal signaling (Z = 2.45; P = 1.36 × 10−9) and inhibition of RhoGDI signaling (Z = −2.0; P = 6.73 × 10−6) were among the top canonical signaling pathways resolved (Table 4). Disease and function annotation of the candidate 43 genes to predefined gene sets in the IPA knowledge base by right-tailed Fisher’s exact test identified cardiovascular function (P = 1.02 × 10−12), fibrosis (P = 1.28 × 10−7), abnormal cell morphology (P = 2.34 × 10−5), and disruption of the cytoskeleton (P = 3.37 × 10−3) as aspects of the fetal lung physiology that could be affected by exposure to maternal cigarette smoke (Table 5). The canonical signaling pathways resolved suggest that fetal lung exposure to maternal smoke activates Rho-GTPase-regulated cytoskeletal reorganization, a phenomenon typically associated with permeability of tight junctions and disruption of lung endothelial and epithelial tissue barriers (42–44).
Figure 4.
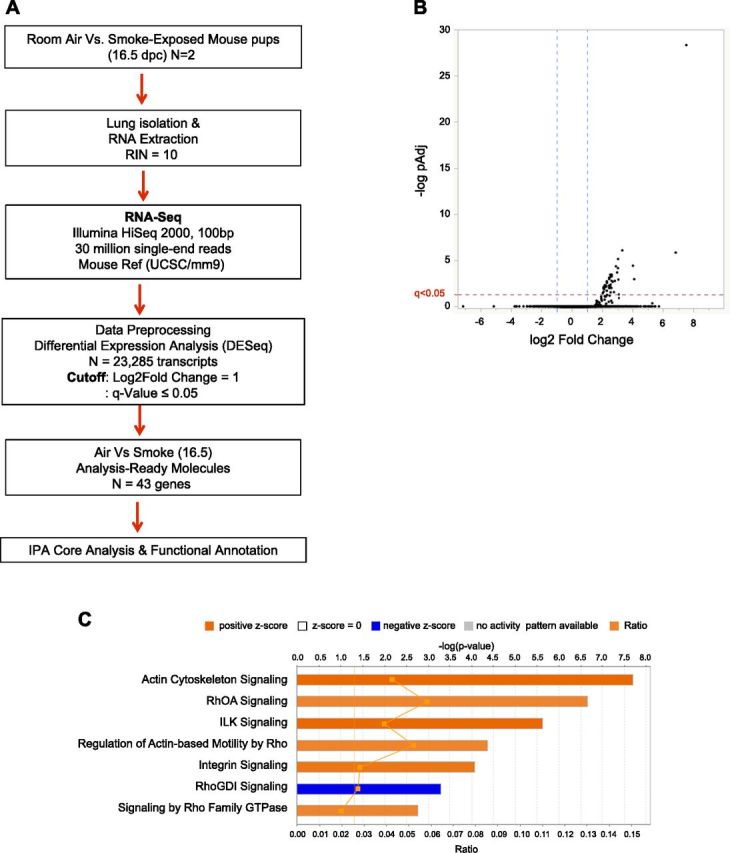
A) Analysis pipeline for RNA sequencing. B) Volcano plot demarcating analysis-ready genes (n = 43) with 2-fold change in expression (blue dotted lines) and P value with FDR correction (q ≤ 0.05), which ensures that observed change in gene expression occurs by chance alone (red dotted line). Total number of transcripts is 23,285. Analysis-ready genes are all up-regulated and located in right quadrant of diagram. C) Canonical pathways induced by fetal lung exposure to maternal smoke. Top signaling pathways with significant association to differential gene expression data set for 16.5 dpc whole-lung RNA lysates (FDR ≤ 0.05) were calculated by right-tailed Fisher’s exact test. Z scores predict activation (positive, orange) or inhibition (negative, blue) of biochemical signaling. Summary results reveal activation of Rho-GTPase-regulated cytoskeletal reorganization that typifies barrier disruption and vascular permeability in endothelial cells and subsequently epithelial tissues.
TABLE 4.
Canonical signaling associated with 43 analysis-ready genes resolved from RNA sequence analysis
| Ingenuity canonical pathways | P | Ratio | Z |
|---|---|---|---|
| Actin cytoskeleton signaling | 1.37 × 10−9 | 4.26 × 10−2 | 2.449 |
| RhoA signaling | 5.83 × 10−9 | 5.83 × 10−2 | 2.000 |
| ILK signaling | 3.13 × 10−8 | 3.9 × 10−2 | 2.449 |
| Regulation of actin-based motility by Rho | 3.94 × 10−7 | 5.19 × 10−2 | 2.000 |
| Integrin signaling | 8.22 × 10−7 | 2.79 × 10−2 | 2.236 |
| RhoGDI signaling | 6.73 × 10−6 | 2.72 × 10−2 | -2.000 |
| Signaling by Rho family GTPases | 3.90 × 10−5 | 1.97 × 10−2 | 2.000 |
Selected genes annotate to predefined gene sets in IPA knowledge base by right-tailed Fisher’s exact test. P < 0.05. Z scores predict biochemical activation or inhibition, and ratios define proportion of our analysis-ready genes that overlap with canonical pathway genes.
TABLE 5.
Diseases and biological functions associated with 43 analysis-ready genes resolved from RNA sequence analysis
| Disease/biological function | P | Molecules (n) |
|---|---|---|
| Functioning of cardiovascular system | 1.02 × 10−12 | 16 |
| Fibrosis | 1.28 × 10−7 | 12 |
| Abnormal cell morphology | 2.34 × 10−5 | 16 |
| Cell morphology | 5.04 × 10−5 | 19 |
| Apoptosis of endodermal cells | 7.63 × 10−3 | 2 |
P values quantify statistically significant mapping of analysis-ready genes to the diseases and functions categories and the number of the genes in each category are presented.
DISCUSSION
Our findings characterize the biochemical events yielding deleterious phenotypes associated with IUS exposure. We demonstrate in the active smoke model that the body weight of smoke-exposed mouse fetuses begins to lag behind at 14.5 dpc. A marked fetal lung volume decrease at the next measured embryonic time point of 16.5 dpc coincides with the canalicular mouse developmental stage. This period in lung development is characterized by a massive increase in vascularization and capillary supply (36), possibly allowing smoke toxins to be transported via the maternal placenta to vital organs of the fetus such as the lungs. An even greater decrease in fetal lung volume is observed at 18.5 dpc, which might indicate an accumulation of cigarette smoke–induced effects over time on fetal lung volume. Indeed, smoke-induced fetal growth restriction has been associated with significant up-regulation of SFRP-1 mRNA and proteins in the placenta of smoke-exposed dams compared to non-smoke-exposed dams (40), supporting this deduction. It is noteworthy that lung volume measurements are approximate values because measurement was performed on uninflated lungs.
The data from this study suggest that the decrease in lung volume can be attributed to a reduction in mesenchymal tissue. At 16.5 dpc, in utero smoke exposure causes a significant decrease in the proliferation of the lung mesenchyme of the developing fetus, thus explaining the loss of lung volume. The epithelium at its nascent stage of formation and differentiation was not affected. Even though changes in the cellular structure of the lung due to smoke exposure in adults seems to be at least partly driven by increased apoptosis (45), we did not resolve a significant increase in apoptosis in the in utero smoke-exposed lung. This is especially remarkable because apoptosis is a regular feature of structural changes in the developing lung (46), but it is similar to previous reports of Wnt-mediated decrease in epithelial and mesenchymal proliferation in the developing distal mouse lung epithelial tip without significant increases in cell death (29). After the presented results, growth retardation is mainly caused by decreased proliferation of the developing lung mesenchyme. The observed decrease in proliferation rates was confirmed at the molecular level to be due to the smoke-induced decrease in expression of Wnt/β-catenin pathway targets Cyclin D1 in mesenchymal lung tissue by qPCR analysis. However, up-regulated sFRP-1 expression in whole lung homogenates by qPCR analysis was not recapitulated in the mesenchymal or epithelial tissue extracted by LCM. Because the Wnt/β-catenin pathway is not the only pathway influencing downstream CyclinD1 expression, it is possible that other pathways, such as the Ras-mediated pathways, play a role in the decreased CyclinD1 expression. Additionally, given that sFRP-1 is secreted into the extracellular space, its spatial expression analysis is challenging. Because the gene expression differences among other Wnt ligands and targets like Lef1, Axin2, Tcf4, Wnt2, Wnt5a, and Wnt10b also did not reach levels of statistical significance, it is possible that the RNA degradation typically accompanying LCM tissue processing, and the quantity of recoverable RNA from the limited amount of laser-captured epithelial and mesenchymal tissues compared to whole lung specimens, might account for the insignificant gene expression levels obtained.
To further identify other crucial pathways and new, relevant genes that are affected by in utero smoke exposure, the transcriptomic profiles of smoke-exposed fetal lungs were compared to room air controls. To our knowledge, there are no published reports of RNA sequence analysis of in vivo smoke-exposed murine fetal lungs. Thus far, in lung developmental studies, this analysis technique has been used to characterize stem or progenitor cell populations (47, 48) and to identify active molecular pathways in hyperoxia models (49). Our results demonstrate that in utero smoke exposure activated Rho-GTPase-dependent signaling for actin polymerization and cytoskeletal reorganization. This signal activation has previously been shown to modulate the integrity of lung epithelial and endothelial tissue, increase permeability of the vascular endothelium, and modify branching morphogenesis (42, 44, 50, 51). These phenotypes have also been reportedly caused by exposure to cigarette smoke in adult lungs (52, 53). Similar results were obtained even without Benjamini–Hochberg FDR corrections for multiple-test P values (data not shown). Because the morphologic results in this study confirm an overproportional effect of in utero smoke exposure on the mesenchymal compartment, it is not surprising that pathways in the endothelium, which derives from mesenchymal tissues, are affected. Rho-GTPase-induced barrier disruption can occur either by weakened intercellular junctions or cell–matrix tethering structures, or by increased contractility of the actin–myosin cytoskeleton via myosin light chain (MLC) phosphorylation/activation and dynamic actin polymerization (51, 54). Given the up-regulated state of MLC genes in our analysis-ready gene data set (Supplemental Table 1), we believe that smoke-induced barrier disruption of the 16.5 dpc lung endothelium and epithelium may occur via MLC activation, which would lead to delayed lung morphogenesis and compromised lung integrity.
On the basis of the findings of this study, we postulate that maternal tobacco smoke acts as an edemagenic agent to the vascular endothelium via activation of RhoA-mediated phosphorylation of the MLC. Given the intimate association between the lung epithelium and endothelium, where they form a tight blood-gas barrier, it is possible that endothelial barrier dysfunction will potentially affect the maturation and integrity of epithelial tissue, as has been observed in the air spaces of adult smokers (55). Future experiments will validate the postulated smoke-induced loss of barrier integrity by investigating signaling regulating endothelial barrier function (56) and performing transepithelial electrical resistance studies on smoke-exposed fetal lung endothelium (57). These findings also highlight the possibility that administration of therapeutic agents that promote barrier protective responses in the developing fetal lung may help alleviate the effects of maternal smoke exposure. Additionally, resident progenitor cell populations may play a role in reestablishing barrier integrity in fetal lungs after injury by tobacco smoke (58).
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank T. Zelonina for assistance with animal studies and histology, and K. Stearns (both from Columbia University, New York, NY, USA) for smoke experiments and apoptotic assays. U.U. and J. T. contributed equally to this work as co–first authors.
Glossary
- AGER
advanced glycosylation end-product specific receptor
- BMP
bone morphogenetic protein
- dpc
days post coitum
- dpp
days postpartum
- FDR
false discovery rate
- IPA
ingenuity pathway analysis
- IUS
in utero smoke
- LCM
laser capture microdissection
- LEF
lymphoid enhancer factor
- MLC
myosin light chain
- MMP
matrix metalloproteinase
- PCNA
proliferating cell nuclear antigen
- qPCR
quantitative PCR
- RQ
relative quantification
- sFRP
secreted frizzled-related protein
- TCF
T-cell factor
- Wnt
wingless-related integration site
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
J. Trischler, U. Unachukwu, and J. D’Armiento conceived and designed the research study; J. Trischler, U. Unachukwu, and M. Goldklang performed smoke exposure studies and experiments; J. Trischler, U. Unachukwu, and R. Xiao analyzed and interpreted the data; and J. Trischler, U. Unachukwu, M. Goldklang, R. Xiao, and J. D’Armiento wrote and critically revised the paper.
REFERENCES
- 1.Gilliland F. D., Berhane K., McConnell R., Gauderman W. J., Vora H., Rappaport E. B., Avol E., Peters J. M. (2000) Maternal smoking during pregnancy, environmental tobacco smoke exposure and childhood lung function. Thorax , 271–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moshammer H., Hoek G., Luttmann-Gibson H., Neuberger M. A., Antova T., Gehring U., Hruba F., Pattenden S., Rudnai P., Slachtova H., Zlotkowska R., Fletcher T. (2006) Parental smoking and lung function in children: an international study. Am. J. Respir. Crit. Care Med. , 1255–1263 [DOI] [PubMed] [Google Scholar]
- 3.Esposito E. R., Horn K. H., Greene R. M., Pisano M. M. (2008) An animal model of cigarette smoke–induced in utero growth retardation. Toxicology , 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larcombe A. N., Foong R. E., Berry L. J., Zosky G. R., Sly P. D. (2011) In utero cigarette smoke exposure impairs somatic and lung growth in BALB/c mice. Eur. Respir. J. , 932–938 [DOI] [PubMed] [Google Scholar]
- 5.Rogers J. M. (2009) Tobacco and pregnancy. Reprod. Toxicol. , 152–160 [DOI] [PubMed] [Google Scholar]
- 6.Iñiguez C., Ballester F., Costa O., Murcia M., Souto A., Santa-Marina L., Aurrekoetxea J. J., Espada M., Vrijheid M., Alvarez-Avellón S. M., Alvarez-Pedrerol M., Rebagliato M.; INMA Study Investigators (2013) Maternal smoking during pregnancy and fetal biometry: the INMA Mother and Child Cohort Study. Am. J. Epidemiol. , 1067–1075 [DOI] [PubMed] [Google Scholar]
- 7.Anblagan D., Jones N. W., Costigan C., Parker A. J., Allcock K., Aleong R., Coyne L. H., Deshpande R., Raine-Fenning N., Bugg G., Roberts N., Pausova Z., Paus T., Gowland P. A. (2013) Maternal smoking during pregnancy and fetal organ growth: a magnetic resonance imaging study. PLoS One , e67223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blacquière M. J., Timens W., Melgert B. N., Geerlings M., Postma D. S., Hylkema M. N. (2009) Maternal smoking during pregnancy induces airway remodelling in mice offspring. Eur. Respir. J. , 1133–1140 [DOI] [PubMed] [Google Scholar]
- 9.Petre M. A., Petrik J., Ellis R., Inman M. D., Holloway A. C., Labiris N. R. (2011) Fetal and neonatal exposure to nicotine disrupts postnatal lung development in rats: role of VEGF and its receptors. Int. J. Toxicol. , 244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliot J., Carroll N., Bosco M., McCrohan M., Robinson P. (2001) Increased airway responsiveness and decreased alveolar attachment points following in utero smoke exposure in the guinea pig. Am. J. Respir. Crit. Care Med. , 140–144 [DOI] [PubMed] [Google Scholar]
- 11.Elliot J., Vullermin P., Robinson P. (1998) Maternal cigarette smoking is associated with increased inner airway wall thickness in children who die from sudden infant death syndrome. Am. J. Respir. Crit. Care Med. , 802–806 [DOI] [PubMed] [Google Scholar]
- 12.Collins M. H., Moessinger A. C., Kleinerman J., Bassi J., Rosso P., Collins A. M., James L. S., Blanc W. A. (1985) Fetal lung hypoplasia associated with maternal smoking: a morphometric analysis. Pediatr. Res. , 408–412 [DOI] [PubMed] [Google Scholar]
- 13.Pattenden S., Antova T., Neuberger M., Nikiforov B., De Sario M., Grize L., Heinrich J., Hruba F., Janssen N., Luttmann-Gibson H., Privalova L., Rudnai P., Splichalova A., Zlotkowska R., Fletcher T. (2006) Parental smoking and children’s respiratory health: independent effects of prenatal and postnatal exposure. Tob. Control , 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caleyachetty R., Tait C. A., Kengne A. P., Corvalan C., Uauy R., Echouffo-Tcheugui J. B. (2014) Tobacco use in pregnant women: analysis of data from demographic and health surveys from 54 low-income and middle-income countries. Lancet Glob. Health , e513–e520 [DOI] [PubMed] [Google Scholar]
- 15.Wang R., Ahmed J., Wang G., Hassan I., Strulovici-Barel Y., Hackett N. R., Crystal R. G. (2011) Down-regulation of the canonical Wnt β-catenin pathway in the airway epithelium of healthy smokers and smokers with COPD. PLoS One , e14793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manoli S. E., Smith L. A., Vyhlidal C. A., An C. H., Porrata Y., Cardoso W. V., Baron R. M., Haley K. J. (2012) Maternal smoking and the retinoid pathway in the developing lung. Respir. Res. , 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao R., Perveen Z., Paulsen D., Rouse R., Ambalavanan N., Kearney M., Penn A. L. (2012) In utero exposure to second-hand smoke aggravates adult responses to irritants: adult second-hand smoke. Am. J. Respir. Cell Mol. Biol. , 843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breton C. V., Byun H. M., Wenten M., Pan F., Yang A., Gilliland F. D. (2009) Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. , 462–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sekhon H. S., Jia Y., Raab R., Kuryatov A., Pankow J. F., Whitsett J. A., Lindstrom J., Spindel E. R. (1999) Prenatal nicotine increases pulmonary alpha7 nicotinic receptor expression and alters fetal lung development in monkeys. J. Clin. Invest. , 637–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehan V. K., Asotra K., Torday J. S. (2009) The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis, and repair. Lung , 281–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blacquière M. J., Timens W., van den Berg A., Geerlings M., Postma D. S., Hylkema M. N. (2010) Maternal smoking during pregnancy decreases Wnt signalling in neonatal mice. Thorax , 553–554 [DOI] [PubMed] [Google Scholar]
- 22.Li C., Xiao J., Hormi K., Borok Z., Minoo P. (2002) Wnt5a participates in distal lung morphogenesis. Dev. Biol. , 68–81 [DOI] [PubMed] [Google Scholar]
- 23.Minoo P., Hamdan H., Bu D., Warburton D., Stepanik P., deLemos R. (1995) TTF-1 regulates lung epithelial morphogenesis. Dev. Biol. , 694–698 [DOI] [PubMed] [Google Scholar]
- 24.Shtutman M., Zhurinsky J., Simcha I., Albanese C., D’Amico M., Pestell R., Ben-Ze’ev A. (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA , 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamamura Y., Otani T., Kanatani N., Koyama E., Kitagaki J., Komori T., Yamada Y., Costantini F., Wakisaka S., Pacifici M., Iwamoto M., Enomoto-Iwamoto M. (2005) Developmental regulation of Wnt/beta-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J. Biol. Chem. , 19185–19195 [DOI] [PubMed] [Google Scholar]
- 26.MacDonald B. T., Tamai K., He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell , 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foronjy R., Imai K., Shiomi T., Mercer B., Sklepkiewicz P., Thankachen J., Bodine P., D’Armiento J. (2010) The divergent roles of secreted frizzled related protein-1 (SFRP1) in lung morphogenesis and emphysema. Am. J. Pathol. , 598–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kneidinger N., Yildirim A. O., Callegari J., Takenaka S., Stein M. M., Dumitrascu R., Bohla A., Bracke K. R., Morty R. E., Brusselle G. G., Schermuly R. T., Eickelberg O., Königshoff M. (2011) Activation of the WNT/β-catenin pathway attenuates experimental emphysema. Am. J. Respir. Crit. Care Med. , 723–733 [DOI] [PubMed] [Google Scholar]
- 29.Rajagopal J., Carroll T. J., Guseh J. S., Bores S. A., Blank L. J., Anderson W. J., Yu J., Zhou Q., McMahon A. P., Melton D. A. (2008) Wnt7b stimulates embryonic lung growth by coordinately increasing the replication of epithelium and mesenchyme. Development , 1625–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cloonan S. M., Glass K., Laucho-Contreras M. E., Bhashyam A. R., Cervo M., Pabón M. A., Konrad C., Polverino F., Siempos I. I., Perez E., Mizumura K., Ghosh M. C., Parameswaran H., Williams N. C., Rooney K. T., Chen Z.-H., Goldklang M. P., Yuan G.-C., Moore S. C., Demeo D. L., Rouault T. A., D’Armiento J. M., Schon E. A., Manfredi G., Quackenbush J., Mahmood A., Silverman E. K., Owen C. A., Choi A. M. K. (2016) Mitochondrial iron chelation ameliorates cigarette smoke–induced bronchitis and emphysema in mice. Nat. Med. , 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollack D., Xiao Y., Shrivasatava V., Levy A., Andrusier M., D’Armiento J., Holz M. K., Vigodner M. (2015) CDK14 expression is down-regulated by cigarette smoke in vivo and in vitro. Toxicol. Lett. , 120–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsia C. C., Hyde D. M., Ochs M., Weibel E. R.; ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure (2010) An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am. J. Respir. Crit. Care Med. , 394–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trapnell C., Pachter L., Salzberg S. L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics , 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., Pachter L. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. , 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anders S., Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol. , R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warburton D., El-Hashash A., Carraro G., Tiozzo C., Sala F., Rogers O., De Langhe S., Kemp P. J., Riccardi D., Torday J., Bellusci S., Shi W., Lubkin S. R., Jesudason E. (2010) Lung organogenesis. Curr. Top. Dev. Biol. , 73–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Langhe S. P., Reynolds S. D. (2008) Wnt signaling in lung organogenesis. Organogenesis , 100–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pongracz J. E., Stockley R. A. (2006) Wnt signalling in lung development and diseases. Respir. Res. , 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ornitz D. M., Yin Y. (2012) Signaling networks regulating development of the lower respiratory tract. Cold Spring Harb. Perspect. Biol. , a008318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang A., Zsengellér Z. K., Hecht J. L., Buccafusca R., Burke S. D., Rajakumar A., Weingart E., Yu P. B., Salahuddin S., Karumanchi S. A. (2015) Excess placental secreted frizzled-related protein 1 in maternal smokers impairs fetal growth. J. Clin. Invest. , 4021–4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim B.-M., Buchner G., Miletich I., Sharpe P. T., Shivdasani R. A. (2005) The stomach mesenchymal transcription factor Barx1 specifies gastric epithelial identity through inhibition of transient Wnt signaling. Dev. Cell , 611–622 [DOI] [PubMed] [Google Scholar]
- 42.Schweitzer K. S., Hatoum H., Brown M. B., Gupta M., Justice M. J., Beteck B., Van Demark M., Gu Y., Presson R. G. Jr., Hubbard W. C., Petrache I. (2011) Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides. Am. J. Physiol. Lung Cell. Mol. Physiol. , L836–L846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olivera D. S., Boggs S. E., Beenhouwer C., Aden J., Knall C. (2007) Cellular mechanisms of mainstream cigarette smoke–induced lung epithelial tight junction permeability changes in vitro. Inhal. Toxicol. , 13–22 [DOI] [PubMed] [Google Scholar]
- 44.Kawkitinarong K., Linz-McGillem L., Birukov K. G., Garcia J. G. (2004) Differential regulation of human lung epithelial and endothelial barrier function by thrombin. Am. J. Respir. Cell Mol. Biol. , 517–527 [DOI] [PubMed] [Google Scholar]
- 45.Kasahara Y., Tuder R. M., Taraseviciene-Stewart L., Le Cras T. D., Abman S., Hirth P. K., Waltenberger J., Voelkel N. F. (2000) Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Invest. , 1311–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scavo L. M., Ertsey R., Chapin C. J., Allen L., Kitterman J. A. (1998) Apoptosis in the development of rat and human fetal lungs. Am. J. Respir. Cell Mol. Biol. , 21–31 [DOI] [PubMed] [Google Scholar]
- 47.Mukhametshina R. T., Ruhs A., Singh I., Hasan D., Contreras A., Mehta A., Nikam V. S., Ahlbrecht K., Carraro G., Cabrera-Fuentes H. A., Jiang D., Voswinckel R., Seeger W., Bellusci S., Scharffetter-Kochanek K., Bagaeva T. V., Preissner K. T., Boettger T., Braun T., Krüger M., Barreto G. (2013) Quantitative proteome analysis of alveolar type-II cells reveals a connection of integrin receptor subunits beta 2/6 and WNT signaling. J. Proteome Res. , 5598–5608 [DOI] [PubMed] [Google Scholar]
- 48.Li C., Li M., Li S., Xing Y., Yang C. Y., Li A., Borok Z., De Langhe S., Minoo P. (2015) Progenitors of secondary crest myofibroblasts are developmentally committed in early lung mesoderm. Stem Cells , 999–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salaets T., Richter J., Brady P., Jimenez J., Nagatomo T., Deprest J., Toelen J. (2015) Transcriptome analysis of the preterm rabbit lung after seven days of hyperoxic exposure. PLoS One , e0136569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim H. Y., Nelson C. M. (2012) Extracellular matrix and cytoskeletal dynamics during branching morphogenesis. Organogenesis , 56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bogatcheva N. V., Verin A. D. (2009) Reprint of “The role of cytoskeleton in the regulation of vascular endothelial barrier function” [Microvascular Research 76 (2008) 202–207]. Microvasc. Res. , 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X. Y., Rahman I., Donaldson K., MacNee W. (1996) Mechanisms of cigarette smoke induced increased airspace permeability. Thorax , 465–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schamberger A. C., Mise N., Jia J., Genoyer E., Yildirim A. O., Meiners S., Eickelberg O. (2014) Cigarette smoke–induced disruption of bronchial epithelial tight junctions is prevented by transforming growth factor-β. Am. J. Respir. Cell Mol. Biol. , 1040–1052 [DOI] [PubMed] [Google Scholar]
- 54.Garcia J. G. N. (2009) Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc. Res. , 1–3 [DOI] [PubMed] [Google Scholar]
- 55.Morrison D., Rahman I., Lannan S., MacNee W. (1999) Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokers. Am. J. Respir. Crit. Care Med. , 473–479 [DOI] [PubMed] [Google Scholar]
- 56.Komarova Y. A., Mehta D., Malik A. B. (2007) Dual regulation of endothelial junctional permeability. Sci. STKE , re8 [DOI] [PubMed] [Google Scholar]
- 57.Wang T., Chiang E. T., Moreno-Vinasco L., Lang G. D., Pendyala S., Samet J. M., Geyh A. S., Breysse P. N., Chillrud S. N., Natarajan V., Garcia J. G. N. (2010) Particulate matter disrupts human lung endothelial barrier integrity via ROS- and p38 MAPK-dependent pathways. Am. J. Respir. Cell Mol. Biol. , 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yanagi S., Tsubouchi H., Miura A., Matsumoto N., Nakazato M. (2015) Breakdown of epithelial barrier integrity and overdrive activation of alveolar epithelial cells in the pathogenesis of acute respiratory distress syndrome and lung fibrosis. BioMed Res. Int. , 573210 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.