

Abstract
Introduction:
The outcome for patients with glioblastoma (GBM) remains poor, and there is an urgent need to develop novel therapeutic approaches. T cells genetically modified with chimeric antigen receptor (CARs) hold the promise to improve outcomes since they recognize and kill cells through different mechanisms than conventional therapeutics.
Areas covered:
This article reviews CAR design, tumor associated antigens expressed by GBMs that can be targeted with CAR T cells, preclinical and clinical studies conducted with CAR T cells, and genetic approaches to enhance their effector function.
Expert commentary:
While preclinical studies have highlighted the potent anti-GBM activity of CAR T cells, the initial foray of CAR T-cell therapies into the clinic resulted only in limited benefits for GBM patients. Additional genetic modification of CAR T cells has resulted in a significant increase in their anti-GBM activity in preclinical models. We are optimistic that clinical testing of these enhanced CAR T cells will be safe and result in improved anti-glioma activity in GBM patients.
Keywords: glioblastoma, immunotherapy, T cells, gene therapy, chimeric antigen receptor, tumor microenvironment
1. Introduction
Glioblastoma (GBM) is the most common type of primary malignant brain tumor.[1] Despite aggressive multimodal therapy including surgery, radiation, and chemotherapy outcomes remain poor with median survival rate of 12 to 15 months.[1] Therefore new therapies are needed to improve outcomes. Immunotherapy has the potential to meet this challenge since it does not rely on the cytotoxic mechanism of conventional therapies.
Adoptive cell therapy with T cells, engineered to express chimeric antigen receptors (CARs) specific for CD19, had remarkable success for B-cell lineage acute lymphoblastic leukemia (ALL) and diffuse large B-cell lymphoma (DLBCL),[2–12] resulting in their FDA approval in 2017. Despite the success of CAR T-cell therapies for CD19-positive hematological malignancies, early phase clinical testing of CAR T cells for solid tumors and brain tumors has been less promising.[13–16] While administration of CAR T cells in general has been safe, only few complete responses have been observed. This lack of efficacy is most likely due to several factors, including (1) low level and/or heterogeneous expression of targeted antigens, (2) limited CAR T-cell expansion and persistence, (3) inefficient homing to tumor sites, and (4) the immunosuppressive tumor microenvironment (TME),[17, 18] a hallmark of solid tumors and brain tumors including GBM. In this article we will review CAR design, GBM targets for CAR T-cell therapy, summarize preclinical and clinical studies conducted so far, and discuss additional genetic modification of CAR T cells to enhance their effector function.
2. CAR Design
CARs are synthetic molecules that are designed to combine the properties of an antibody with T-cell receptor (TCR) signaling upon antigen binding.[19–21] The major components of CARs include an ectodomain and endodomain, which are connected by a spacer region and transmembrane (TM) domain (Figure 1A). The ectodomain or antigen recognition domain typically consists of a single chain variable fragment (scFv) of a monoclonal antibody (MAb) specific for a tumor associated antigen (TAA) that is expressed on the cell surface of cancer cells. Alternatively, it can also contain a ligand, if the TAA is a cell surface receptor. Efforts are also underway to generate universal CARs in which the ectodomain consist of a universal docking protein that can be combined with a range of targeting moieties (for example avidin in combination with biotinylated MAbs).[22] Lastly, CAR ectodomains can also be engineered to target intracellular TAAs using scFvs, which recognize a peptide derived from an intracellular protein in the context of a human leukocyte antigen (HLA) molecule.[23]
Figure 1.
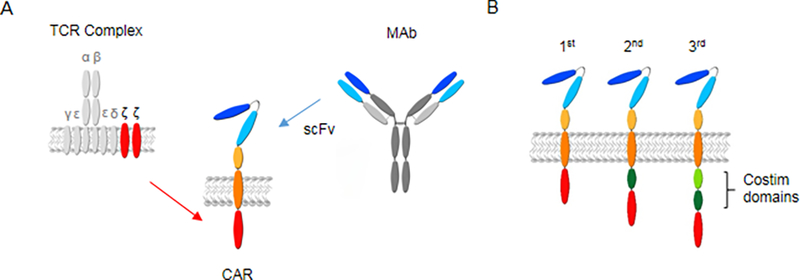
CAR design. (a) CARs consist of an ectodomain, spacer region, transmembrane domain, and endodomain. (b) Depending on the presence of costimulatory domains, CARs are designated as being 1st, 2nd, or 3rd generation. For additional details see text. TCR: T-cell receptor, CAR: chimeric antigen receptor, scFv: single chain variable fragment, MAb: monoclonal antibody, costim: costimulatory.
The CAR endodomain consists of signaling domains derived from CD3ζ and costimulatory molecules such as CD28, 41BB, and/or OX40. CARs are classified as 1st, 2nd, and 3rd generation CARs based on the presence of costimulatory endodomain, being none, one, or two respectively (Figure 1B). Signaling domains derived from CD28 and 41BB are the most commonly used costimulatory domains thus far. While CD28 and 41BB CARs have never been directly compared in individual patients, results of clinical studies with CD19.41BB.ζ or CD19.CD28.ζ CAR T cells indicate that 41BB.ζ CAR T cells persist longer. However, this has not translated in significant differences in clinical outcome.[2–12] A recent preclinical study directly comparing CD19.41BB.ζ and CD19.CD28.ζ CAR T cells has highlighted significant differences between CD28 and 41BB costimulation in CAR T cells.[24] While CD28 signaling induced effector memory differentiation, and glycolytic metabolism, 41BB signaling preserved a central memory phenotype and induced mitochondrial biogenesis, resulting in an increase of oxidative metabolism and spare respiratory capacity. Whether or not the combination of CD28 and 41BB costimulation results in enhanced effector function of CAR T cells remains controversial and model dependent. However, one study suggests that expressing 41BBL on the cell surface of CD28.ζ CAR T cells results in superior effector function in comparison to CD28.ζ or 41BB.ζ CAR T cells by activating the IFN regulator factor 7 (IRF7) pathway.[25] For GBM, preclinical and clinical studies have been conducted with ζ, CD28.ζ and 41BB.ζ CAR T cells. [26–38]
While initially the CAR spacer region and transmembrane domain were only considered a structural component of the CAR, several studies have highlighted their critical contribution to CAR function.[39, 40] The most frequently used spacers are derived from IgG1 hinge (12aa), or the CH2CH3 domain of IgG 1 or 4 (hinge and Fc domain, 229aa).[41, 42] Studies have demonstrated that the Fc domain interacts with cells expressing Fc gamma receptors (FcγR) in vivo, resulting in off-target activation and/or killing of CAR T cells by FcγR-positive innate immune cells.[40, 41, 43] In addition, a CH2CH3-based spacer can cause baseline (aka tonic) CAR signaling without cognate antigen stimulation, resulting in phosphorylation of the CD3ζ chain, IFNγ production, and T-cell exhaustion.[30, 40]
Most commonly used TM domains in CARs are derived from the TM region of CD3ζ, CD4, CD8, or CD28. TM domains have been shown to influence CAR cell surface expression and function. For example, the cell surface expression of CARs containing CD3ζ TM domains is lower than CARs with CD28 TM domains.[39] In addition, TM domains can influence were CARs are located on the plasma membrane. For example, CARs with CD3ζ TM domains are incorporated into TCR complexes, which resulted in improved functionality in one published report.[44]
In summary, at present there is no universal formula for optimal CAR design. Many studies have shown that there is an intricate interplay between epitope location, scFv, signaling domains, and structural components of CARs. Thus, CARs have to be optimized for every targeted antigen. Other factors that have to be considered for CAR design include the gene delivery system that is used to insert CARs into T cells, the promoter that is used to drive CAR expression, and the targeted T-cell populations. For example, retroviral vectors encoding CARs with 41BB.ζ endodomains carry the risk of inducing T-cell apoptosis since they are expressed at higher levels in comparison to CARs delivered by lentiviral vectors.[45] In addition, expressing CARs under the control of physiological promoters, such as the TCRα promoter, has been shown to enhance CAR T-cell function.[46] Lastly, several studies have highlighted that CAR T cells derived from stem cell memory T cells have superior effector function than CAR T cells generated from unselected T-cell populations.[47]
3. GBM targets for CAR T-cell Therapy
An ideal CAR T-cell target is defined by (1) high expression on the tumor cell surface, (2) high degree of homogeneity of expression per tumor, (3) being essential for the malignant phenotype to prevent the development of antigen loss variants, (4) no or limited expression on normal tissues to avoid on-target/off-tumor toxicity, and (5) being ‘not patient specific’, so that one CAR can be used to treat multiple patients. Cluster of differentiation 70 (CD70), epidermal growth factor receptor (EGFR), epidermal growth factor receptor variant 3 (EGFRvIII), ephrin type-A receptor 2 (EphA2), human epidermal growth factor receptor 2 (HER2), and interleukin 13 receptor alpha 2 (IL13Rα2) have been targeted with CAR T cells, and are described in more detail below.
3.1. CD70
CD70 is a type I glycoprotein and a member of the TNF receptor family. It is expressed by activated B and T cells and plays a role in T-cell activation and generation of cytolytic T cells. While CD70 is mainly expressed by cells of the lymphoid lineage, subsets of hematologic and solid tumors, and gliomas overexpress CD70.[48, 49] In the brain, CD70 is expressed by neoplastic glial cells and CD70 expression is induced by irradiation of glioma cells.[49] A screen of human glioma cell lines demonstrated that about 92% of tested cell lines expressed CD70 at mRNA and protein level.[49] However, an in-depth evaluation of CD70 protein expression in a larger patient cohort needs to be performed. CD70 expression in high-grade gliomas can cause immunosuppression by inducing T-cell dysfunction and apoptosis, [50] and is associated with significant increase in tumor-associated macrophages.[51] Lastly, a recent study suggests that CD70 expression is involved in mediating tumor aggressiveness and immunosuppression through recruitment of tumor-associated macrophages.[52]
3.2. ZEGFR and EGFRvIII
EGFR is a transmembrane protein that is a receptor for members of the epidermal growth factor family (EGF family). It is overexpressed in primary (63%) and secondary (10%) glioblastomas.[53] EGFR influences the migration of neural stem cells during development, and promotes cellular proliferation through activation of MAPK and PI3K-Akt pathways.[54] As for expression in normal tissue, EGFR is expressed in tissues of epithelial, mesenchymal and neuronal origin and plays a major role in normal cellular processes such as proliferation and differentiation. In GBM, EGFR expression frequently involves amplifications or alterations of the EGFR gene which results in expression of various mutations. The most common EGFR mutant variant found in GBM is EGFR variant 3 (EGFRvIII). It arises from mutated/gene amplified EGFR causing deletion of exon 2–7, which results in a functional membrane protein with an extracellular domain mutation. Published data suggest that expression of EGFRvIII on GBMs enhances cell tumorigenicity, invasiveness and therapeutic resistance, [53, 55], however one recent study suggests that EGFRvIII is not a prognostic factor for GBM and its aggressiveness might be related to other proteins rather than EGFRvIII.[56]
3.3. EphA2
EphA2 or epithelial cell receptor protein tyrosine kinase, is strongly overexpressed in 60% of GBMs and expressed at a moderate or strong levels in 90–98% of GBM specimens. [57, 58] Expression is detected at low levels on adult proliferating epithelial cells as well as brain tissue and enriched within sites of cell-cell adhesion in normal epithelial cells.[57] As for its role in the malignant phenotype of GBMs, EphA2 is an important regulator of tumor initiation, neo-vascularization, tumor cell migration, invasion and angiogenesis.
3.4. HER2
HER2 is a transmembrane tyrosine/kinase receptor also known as erbB-2. It is well-characterized tumor antigen which is important for the regulation of cancer growth. For example, it is a prognostic marker in metastatic breast carcinoma, and its overexpression is also associated with a poor prognosis in GBM. HER2 expression level increases with the degree of poor glial cell structural differentiation and other anaplastic related features.[59, 60] In a retrospective study, HER2 expression was detected in 76% of primary GBM cell lines [61]. HER2 expression in pediatric brain tumors was detected in 54% of cases as judged by mRNA/gene profiling analysis.[62]
3.5. IL13Ra2
IL13Rα2 is overexpressed in about 76% of GBMs at a moderate or strong level.[58, 63, 64] A slightly higher percentage (up to 83%) has been reported for pediatric brain tumors, and overexpression is associated with poor prognosis.[62, 65–67] Two studies have also evaluated the expression of IL13Rα2 in diffuse intrinsic pontine gliomas (DIPGs).[62, 66] In the first study 10 out of 15 DIPGs were positive for IL13Rα2, and in the second study 17 of 28 respectively.
GBM CAR targets explored so far, do not meet all the ‘ideal target criteria’ outlined in the beginning of this section. Thus, there is a continued need to discover additional GBM antigens that can be targeted with CAR T cells. Genetic engineering approaches that restrict full CAR T-cell activation to site at which two antigens are expressed could potentially increase the pool of targeted antigens.[68] Lastly, the recent development of CARs that allow the targeting of HLA/peptide complexes, containing peptides derived from intracellular proteins, should also increase the array of potential antigens including BIRC5 (survivin) and/or mutated IDH1.
4. Pre-clinical studies with CAR T cells
The majority of preclinical studies have used xenograft models. Initial studies focused on targeting IL13Ra2-positive glioma with T cells expressing a first-generation CAR that used a mutated IL13 (IL13 mutein) as an antigen-binding domain.[26] IL13Ra2-CAR T cells had potent anti-glioma activity in vitro and in vivo. Subsequently, 2nd generation IL13 mutein-based CARs were developed, which showed improved anti-glioma activity,[27] and a CAR with a 41BB.ζ endomain is currently undergoing clinical evaluation. IL13Rα2-specific CARs have also been developed that take advantage of an IL13Rα2-specific scFv as an antigen-binding domain.[30] Use of a scFv-based IL13Rα2-CAR might be advantageous since these CARs do not recognize IL13Rα1, a broadly expressed molecule that has been reported to be recognized by some of the designed IL13 mutein-based CARs.[31]
Since the pioneering studies with IL13 mutein-based CARs, other CARs have been developed to target GBMs. T cells expressing a 2nd generation HER2-specific CAR with a CD28.ζ endodomain demonstrated potent antiglioma activity not only in xenograft models, but also in patient-derived xenograft (PDX) models in which patient-derived GBMs were treated with autologous HER2-CAR T cells.[28] In addition, this study highlighted that CAR T cells can kill glioma initiating cells, which was subsequently confirmed in studies targeting other GBM-associated antigens.[29, 34, 69] Besides targeting IL13Rα2 and HER2, CARs targeting other glioma-associated antigens such as EphA2, EGFR, EGVRvIII, and CD70 have been developed.[29, 32–38] All CARs were scFv-based except for CARs targeting CD70, which contained the receptor ectodomain of CD70 (CD27) as a binding domain. [37, 38] Since there are on target/off cancer toxicity concerns with targeting EphA2 and EGFR, investigators either used a scFv that recognizes a conformational epitope (EphA2),[29, 70, 71] or a scFv with reduced affinity (EGFR) to mitigate this risk.[32]
Xenograft studies with HER2- or IL13Rα2-CAR T cells have highlighted the risk of an antigen loss variants when a single antigen is targeted.[72–74] Investigators have shown that targeting two antigens, HER2 and IL13Rα2, simultaneously can prevent immune escape.[74] How to best target two antigens remains controversial and several strategies are currently being explored, including infusing (1) two T-cell populations expressing each a single CAR, (2) T cells expressing two CARs, or (3) T cells expressing a single CAR with two antigen-binding domains.[73–75] Lastly, the expression of three CARs in a single T cells has been reported to prevent immune escape.[76]
In an effort to more closely mimic the interactions of the infused CAR T cells with resident immune cells, investigators have also started to explore the use of immune competent animal models to evaluate GBM-targeted CAR T cells. Murine EGFRvIII-CAR T cells were evaluated in an immune competent syngeneic model in which glioma cells were genetically modified to express EGFRvIII.[77] The anti-glioma activity of EGFRvIII-CAR T cells was dependent on lymphodepletion mice prior to T-cell infusion. In addition, glioma-bearing mice that were cured by EGFRvIII-CAR T cells were resistant to re-challenge with parental, EGFRvIII-negative glioma cells, indicating the development of host immunity against other glioma-associated antigens.[77] CD70-CAR T cells have also demonstrated potent anti-tumor activity in an immune competent syngeneic mouse model of glioma.[38]
In summary, preclinical studies with CAR T cells targeting GBMs have shown significant anti-glioma activity, which led to Phase 1 testing of IL13Rα2- (NCT02208362), HER2- (NCT02442297, NCT01109095, NCT02713984), EGFRvIII- (NCT01454596, NCT02209376, NCT02664363, NCT02844062), and EphA2-CAR (NCT02575261) T cells. Studies with IL13Rα2-, HER2-, and EGFRvIII-CAR T cells have been published and will be reviewed in the next section.
5. Clinical studies with CAR T cells
The first clinical study used autologous CD8-positive T-cell clones that were genetically modified to express a first generation IL13Rα2-CAR with an IL13 mutein as an antigen binding domain, the hygromycin gene for selection, and the Herpes Simplex Virus thymidine kinase (HSV-tk) gene as a suicide switch and to allow positron emission tomography (PET)-based imaging.[15, 78] Three patients received up to 12 local infusions of CAR T cells into the GBM resection cavity. Infusions were well tolerated at a cell dose of 1×107 and 5×107, however at the highest cell dose tested (1×108 CAR T cells), 2/3 developed Grade 3 headaches. MRI imaging revealed inflammation, and MR spectroscopy in one patient was consistent with tumor necrosis. Lastly, one tumor showed decreased IL13Rα2 expression in comparison to a pre-infusion sample, suggestive of in vivo killing of IL13Rα2-positive glioma cells.
Two additional patients that received autologous CD8-positive IL13Rα2-CAR T-cell clones, and four that received allogeneic CD8-positive IL13Rα2-CAR T-cell clones were subsequently reported.[79] This publication focused on the utility of using the HSV-tk gene for non-invasive PET imaging of infused CAR T cells. Investigators could demonstrate that 9-[4-[18F]fluoro-3-(hydroxymethyl)butyl]guanine ([18F]FHBG) imaging of infused CAR T cells is feasible, safe, and allows for the longitudinal in vivo imaging of CAR T cells expressing HSV-tk.
Subsequently, the same group of investigators developed a 2nd generation mutein-based IL13Rα2-CAR with a 41BB.ζ endodomain.[27] A study evaluating the safety and efficacy of intracranially infused autologous, polyclonal, lentiviral transduced IL13Rα2-CAR T cells is in process. One patient had a remarkable clinical response, with regression of intracranial and spinal tumors, which continued for 7.5 months,[80] demonstrating that CAR T cells can have significant antitumor activity against highly aggressive tumors such as GBM.
The other two clinical studies infused CAR T cells intravenously. One study utilized a HER2-CAR.CD28.ζ T-cell product in which genetically modified T cells were enriched for T cells that recognized Cytomegalovirus (CMV), Epstein Barr Virus (EBV) or Adenovirus (Adv).[14] These CAR T cells not only provide anti-tumor activity through their CAR, but may also receive stimulation following their endogenous T-cell receptor by cells that present viral antigens. In addition, they might have anti-glioma activity through endogenous T-cell receptor since several studies have suggested that GBMs are CMV positive.[81] Seventeen patients with recurrent/refractory GBM received up to 1×108/m2 autologous HER2-CAR T cells. Infusions were well tolerated, and of 16 evaluable patients one patient had a partial response, seven had stable disease for 8 weeks to 29 months, and eight progressed after T-cell infusion. Correlative studies were limited to tracking CAR T cells in the peripheral blood, and while CAR T cells could be detected six weeks post infusion, no significant in vivo expansion of infused CAR T cells was observed.
The last published study, infused up to 5×108 EGFRvIII-CAR.41BB.ζ CAR T cells into 10 patients with recurrent/refractory GBM.[13] Seven of 10 patients had their tumor resected post infusion, allowing the investigators to perform comprehensive correlative studies. CAR T cells were detected in resected GBMs, and 5/7 GBMs expressed decreased levels of EGFRvIII in comparison to pre-infusion samples, suggestive for on-target CAR T-cell activity. In addition, there was an increase in inhibitory molecules such as indoleamine 2,3 dioxygenase (IDO) and IL-10, and influx of regulatory T cells in post-infusion GBMs, highlighting the ability of GBMs to actively suppress effector T cells at tumor sites. Although progression-free survival could not be evaluated since the majority of patients had their GBM removed post CAR T-cell therapy, the median overall survival was 251 days. At the time of the study’s publication, one patient had achieved a lasting response and was alive and well 18 months post infusion.
In conclusion, initial clinical experience with locally or systemically injected CAR T cells targeting IL13Rα2, HER2, and EGFRvIII has demonstrated their safety and antitumor activity in subsets of patients; however, only few durable responses were observed. Lack of efficacy is most likely multifactorial, including heterogeneous antigen expression, limited T-cell homing to GBM sites, and the immunosuppressive TME. Among these, the immunosuppressive TME is considered a major impediment,[17, 18] and we will therefore focus on it in the next section.
6. Improving CAR T cell therapy for GBM
A major hurdle to the efficacy of CAR T-cell therapy for GBM is its immunosuppressive TME as the tumor and surrounding cells foster a toxic environment not conducive to T-cell survival or effector function.[17, 18] Within the TME, T cells encounter ligands for inhibitory T-cell receptors such as programmed cell death protein 1 (PD-1), engagement of which negatively regulates T-cell activation in the presence of antigen and can promote anergy or apoptosis.[82–84] Immunosuppressive cytokines such as TGFβ, IL-4, and IL-10 also inhibit anti-tumor activity by suppressing T-cell proliferation and cytolytic function.[85] Additionally, the high metabolic activity of the tumor cells depletes the TME of glucose and oxygen and produces high amounts of lactic acid.[86] This combined with the production of enzymes such as IDO, which breaks down tryptophan, hampers T-cell metabolism and results in nutrient deprivation.[86] Improving the therapeutic benefit of CAR T cells for brain tumors will most likely require additional modifications to strengthen them against this hazardous environment (Figure 2). Several genetic modifications have been explored to make CAR T cells less susceptible to TME-mediated immunosuppression, which we describe in detail below. Specifically, we will focus on (1) transgenic expression of cytokines and cytokine receptors, (2) dominant negative and chimeric switch receptors, and (3) genetic approaches to overcome checkpoint blockade.
Figure 2.
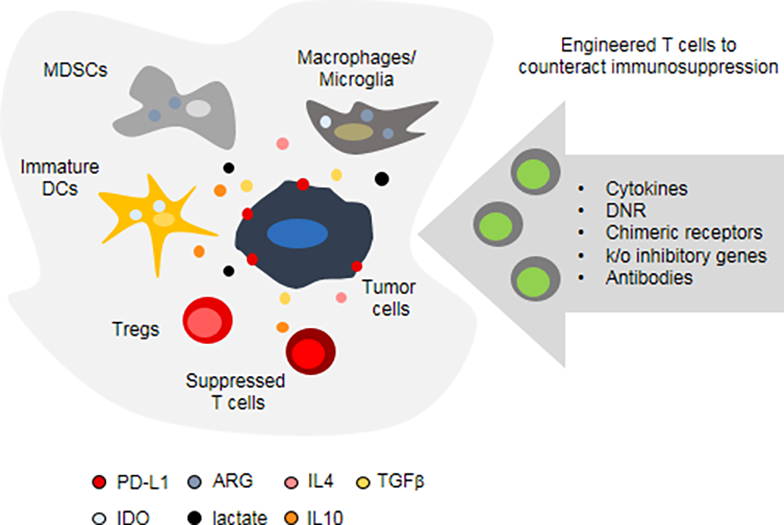
Engineered T cells to counteract the immunosuppressive GBM microenvironment. Major immunosuppressive cell populations and molecules are depicted. For additional details see text. MDSCs: myeloid derived suppressor cells, DCs: dendritic cells, Tregs: regulatory T cells, PD-L1: programmed cell death protein ligand 1,
IDO: indoleamine 2,3 dioxygenase, ARG: arginase, TGFβ: transforming growth factor β. DNR: dominant negative receptor, k/o: knockout.
6.1. Transgenic expression of cytokines and/or cytokine receptors
Providing CAR T cells with pro-survival cytokines can fortify them against the immunosuppressive microenvironment. Transgenic expression of IL-7, IL-12, IL-15, and IL18 have been explored with promising results. CAR T cells can be engineered to express cytokines that are either tethered to the cell membrane or secreted.[87, 88] Both approaches have shown to be effective for IL-15, with membrane-bound IL-15 promoting a stem-cell memory subset and improving the antitumor activity of CD19-CAR T cells,[87] while secreted IL-15 provided improved persistence and anti-tumor activity of IL13Rα2-CAR T cells in an orthotopic model of glioma.[72] Transgenic expression of IL-12 by CAR T cells has also yielded improvements in persistence and antitumor activity.[89, 90] IL-12 secreting T cells influenced nearby cells as well, with one group reporting the induction of anti-tumor activity by innate macrophages,[91] and another group reporting depletion of immunosuppressive M2 tumor-associated macrophages.[90] Secretion of IL-18 has been shown to enhance the proliferation and anti-tumor of CD19-CAR T cells in both xenograft and immune-competent mouse models.[92] Given the safety concerns associated with systemic administration of IL-12 and IL-18, both cytokines have been evaluated as part of an NFAT-inducible system to selectively produce their respective cytokine at the tumor site.[92, 93]
Constitutively active cytokine receptors can also enhance T cell survival. The expression of IL-7 tethered to IL-7Rα produced constitutive STAT5 signaling and enhances T-cell viability and proliferation.[94] Another group completely circumnavigated the necessity of providing a pro-survival cytokine and expressed a constitutively active IL-7 receptor (C7R) that homodimerizes without its ligand IL-7, once again producing constitutive STAT5 activation.[95, 96] Combining C7R with EphA2-CAR T cells in an orthotopic model of glioma resulted in complete responses in all mice with no recurrences at a T-cell dose at which unmodified EphA2-CAR T cells were ineffective.[96]
6.2. Dominant negative and chimeric switch receptors
In addition to providing pro-survival cytokines, removing immunosuppressive cytokine signaling can also improve T-cell effector function. Glioma cells produce several cytokines that directly inhibit T-cell function, one of the most well studied being transforming growth factor β (TGFβ). Not normally expressed in healthy brain tissue, TGFβ expression is associated with worse outcomes in high grade glioma and directly suppresses T-cell cytolytic function by inhibiting the expression of perforin, granzyme A, granzyme B, Fas ligand, and interferon gamma.[85, 97] Expressing a dominant negative TGFβ receptor (DNR) that binds the cytokine but does not signal has been shown to enhance the antitumor activity of adoptive T cell therapy.[98, 99]
While dominant negative receptors, can only block inhibitory cytokines, receptors have also been developed that convert/invert an inhibitory into a positive signal. These receptors have been called chimeric switch receptors, chimeric cytokine receptors, inverted cytokine receptors, or switch receptors. For example, fusing the ectodomain of the IL-4 receptor to the transmembrane domain and endodomain of the IL-2 or IL-7 receptors resulted in chimeric receptors that converted the immunosuppressive effects of the TH2 cytokine IL-4 into the pro-survival signaling pathways associated with IL-2 or IL-7.[100, 101] In addition, the above described DNR was converted into a chimeric switch receptor by adding the signaling domain of TLR4 as a cytoplasmic endodomain.[102] Clearly, this approach could be extended to other inhibitory cytokines present in the TME of GBMs such as IL-10.
6.3. Genetic approaches to overcome checkpoint blockade
The immune check point programmed cell death protein ligand 1 (PD-L1) is expressed in the majority of GBMs, with higher expression correlating with glioma grade.[103–105] Checkpoint antibodies conferred a T cell-dependent survival advantage in an orthotopic mouse model of glioma.[106] However, the clinical experience with checkpoint blockade for GBM has been disappointing unless the GBM is deficient in mismatched mismatch repair (MMR) genes.[107, 108] While lack of efficacy of checkpoint blockade monotherapy is multifactorial, the blood brain barrier (BBB) is most likely a contributing factor based on the experience with other GBM-targeted MAb therapies. Genetic modification of T cells to render them resistant to checkpoint inhibition might be one strategy to address this problem.
Several T-cell intrinsic methods of disrupting the PD-1/PD-L1 axis have been shown to enhance CAR T-cell efficacy, including genetic disruption of PD-1 using CRISPR/Cas9,[109] secretion of a PD-1 antibody,[110] expression of a dominant negative PD-1 receptor,[111] or expression of a chimeric switch receptor that combines the extracellular domain of PD-1 with the intracellular signaling domain of CD28 in order to convert an inhibitory signal into a co-stimulatory one.[112, 113] Each of these approaches yielded increased survival times compared with adoptive T-cell transfer where PD-1-mediated immunosuppression was not inhibited.
In conclusion, genetic approaches to enhance CAR T-cell function have shown promising results in preclinical studies, but so far have not been tested in clinical studies. Clearly, while we have here focused on describing three strategies, others are being actively explored. In addition, the highlighted approaches, like CRISPR/Cas9 gene editing could be applied to other targets.
7. Conclusions
The first preclinical CAR T-cell therapy study targeting IL13Rα2-positive GBM was published in 2004. Since then there has been a dramatic expansion in preclinical studies targeting additional GBM antigens, and exploring additional genetic modification strategies to enhance CAR T-cell effector function. Initial clinical studies with CAR T cells targeting IL13Rα2, HER2, and EGFRvIII has demonstrated safety, however anti-GBM activity was limited. Clinical studies are now in the planning phase to target additional antigens, and/or testing CAR T cells that are further genetically modified to enhance their effector function.
8. Expert commentary
CAR T cells have anti-tumor activity in vitro and in vivo in pre-clinical GBM models. However, in the clinical setting, CAR T cells currently have suboptimal activity against GBM for the majority of patients. Several roadblocks have emerged including heterogeneous antigen expression, and immune escape due to antigen loss variants, and the immunosuppressive tumor microenvironment. Thus, there is a continued need to discover new antigens, model the immunosuppressive tumor microenvironment in preclinical models, and devise strategies to render T cells resistant to it.
Novel antigens are continued to be discovered. For example, since the original submission of this manuscript, targeting chondroitin sulfate proteoglycan 4 with CAR T cells for glioma has been described.[114] In addition to antigen discovery it will be critical to refine current strategies to target multiple antigens. This will be important not only to prevent antigen loss variants, but also to allow targeting of ‘antigen patterns’ present on glioma cells, potentially increasing the repertoire of targetable antigens. However, the repertoire of ‘common shared antigens’ is most likely limited. One approach to ‘tap into private neoantigens’ expressed by glioma[115] is to optimize the ability of CAR T cells to induce antigen spreading. Antigen spreading, the induction of immune responses against ‘non-targeted antigens’, correlates with the clinical activity of cancer vaccines,[116] and also has been demonstrated post cell transfer of a single T-cell clone.[117] Lastly, besides targeting glioma cells with CAR T cells, we believe that it might be critical to target supporting stromal cells with CAR T cells for their optimal clinical efficacy.[118]
As for solid tumor CAR T-cell therapy, the immunosuppressive tumor microenvironment is a significant hurdle. Current preclinical studies have mainly focused on xenograft models, which do not recapitulate the immunosuppressive tumor microenvironment. For example, transgenic expression of cytokines enhance the anti-glioma activity of CAR T cells.[72] However, it will be critical to evaluate this and other approaches in immune competent glioma models. Besides immune competent animal models, evaluating CAR T-cell therapies in patient derived xenograft (pdx) model should become part of preclinical evaluation of CAR T cell therapies, since these models more closely mimic patients’ glioma. While at present not readily accessible, studying CAR T-cell therapy in large animal glioma models[119] holds the promise to improve our ability to realistically model human glioma preclinically.
In addition, to optimizing glioma cell recognition, and improving CAR T-cell function within the hostile glioma microenvironment, the optimal route of CAR T-cell delivery needs to be further refined. Currently, intravenous, intratumoral, and intraventricular routes are being explored. While the best of route delivery most likely depends on the clinical scenario, genetic engineering of T cells has the potential to improve T-cell migration to tumor sites,[120, 121] penetration of tumor masses,[122] and/or migration through the blood brain barrier.
Besides ‘preclinlcal optimization’ of CAR T-cell therapies, it will critical to improve our ability to track T cells post infusion, and monitor their antiglioma activity in glioma patients. While proof-of-principle studies with PET imaging have shown promise, these studies have relied on genetically modifying T cells with HSV-tk, an immunogenic protein.[79, 123] Thus, there is an urgent need to develop novel approaches that would enable us to track CAR T cells post infusion. Assessment of the anti-glioma activity of immunotherapies by imaging using standard response criteria is difficult. In this regard, the Response Assessment for Neuro-Oncology working group published their recommendation for immunotherapy studies.[124] However, these criteria were developed based on results of vaccine studies, and at present it is not clear if these criteria require further modifications for CAR T-cell therapies. Besides imaging, the use of circulating biomarkers hold the promise to improve our ability to assess the anti-glioma activity of CAR T cells.[125] In addition, these biomarkers might also shed light if CAR T cells are able to reverse the immunosuppressive environment induced by glioma.
Cancer therapeutics rely on combinatorial strategies. Thus, combining CAR T-cell therapies with other therapeutic approaches will most likely be key for their success. Besides other immunotherapeutic approaches such as checkpoint blockade and vaccines, epigenetic modifiers, radiation, oncolytics, and drugs that alter the metabolism of cancer cells should be explored.
In conclusion, we believe that immunotherapy for GBM holds the promise to improve outcomes for patients affected by this devastating disease. While this review has focused on cell therapy with genetically modified T cells, other approaches are actively being explored including cell therapies with (1) conventional T cells,[126] (2) T cells that are genetically modified to express TCRs,[127] and (3) other cellular platforms including unmodified or genetically-engineered gamma delta T cells, natural killed (NK), invariant NKT, or NK-92 cells.[128–132] Lastly, multiple preclinical and clinical studies remain focused on GBM vaccines.[133, 134]
9. 5-year view
Over the next 5 years we are optimistic that CAR T-cell therapies will have an impact on the outcome of GBM patients. To benefit GBM patients, CAR T cells most likely need to be further genetically modified to enhance their effector function, or combined with other targeted approaches including vaccines, MAbs, or small molecules. Critical for advancing this field will be the development of preclinical animal models that allow us to study the interactions on adoptive transferred CAR T cells, tumor cells, and resident immune cells. Lastly, performing carefully crafted correlative studies in early phase clinical studies will be the key to advance the field of CAR T-cell therapy for GBM.
Key issues.
Currently, there is a limited array of targetable GBM antigens and their expression is heterogeneous
CAR design is empiric requiring careful optimization for the targeted antigens
Selection of the appropriate T-cell subset for genetic modification is critical for optimal CAR function
The GBM microenvironment is immunosuppressive and actively inhibits CAR T cells
Carefully planned correlative studies are needed on ongoing and future CAR T-cell therapies for GBM to get mechanistic insights into successes and failures
The complexity and cost of evaluating CAR T cells in the clinic has the potential to impede progress
Acknowledgments
Funding
The authors report receiving financial support of their preclinical and clinical GBM studies by the American Brain Tumor Association Basic Research Fellowship in honor of Joel A. Gingras, Jr. (BRF160004), Alex Lemonade Stand Foundation, National Institute of Health grant CA203270, and the James McDonald Foundation and Cancer Prevention and Research Institute of Texas grant RP110553.
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Reference annotations
* Of interest
** Of considerable interest
- 1.Ostrom QT, Gittleman H, Liao P, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-oncology. 2017;19(suppl_5):v1–v88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruella M, June CH. Chimeric Antigen Receptor T cells for B Cell Neoplasms: Choose the Right CAR for You. Current hematologic malignancy reports. 2016;11(5):368–84. [DOI] [PubMed] [Google Scholar]
- 3.Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(6):540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. NEnglJMed. 2014;371(16):1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turtle CJ, Hanafi LA, Berger C, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Science translational medicine. 2016;8(355):355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turtle CJ, Riddell SR, Maloney DG. CD19-Targeted chimeric antigen receptor-modified T-cell immunotherapy for B-cell malignancies. Clinical pharmacology and therapeutics. 2016;100(3):252–8. [DOI] [PubMed] [Google Scholar]
- 10.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brentjens RJ, Davila ML, Riviere I, et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. SciTranslMed. 2013;5(177):177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JH, Riviere I, Gonen M, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. The New England journal of medicine. 2018;378(5):449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. **.O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science translational medicine. 2017;9(399). Detailed correlative studies highlighting how GBMs evade CAR T cells in humans [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed N, Brawley V, Hegde M, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA oncology. 2017;3(8):1094–101. *Clinical study of HER2-CAR T cells in GBM demonstrating safety [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown CE, Badie B, Barish ME, et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(18):4062–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heczey A, Louis CU, Savoldo B, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Molecular therapy : the journal of the American Society of Gene Therapy. 2017;25(9):2214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro-oncology. 2015;17 Suppl 7:vii9–vii14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perng P, Lim M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front Oncol. 2015;5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257(1):107–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Current opinion in immunology. 2015;33:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadelain M Chimeric antigen receptors: driving immunology towards synthetic biology. Current opinion in immunology. 2016;41:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbanska K, Lanitis E, Poussin M, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72(7):1844–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rafiq S, Purdon TJ, Daniyan AF, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia. 2017;31(8):1788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. *.Kawalekar OU, O’Connor RS, Fraietta JA, et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44(2):380–90. *Detailed comparison between CD28 and 41BB signaling domain in CAR T cells [DOI] [PubMed] [Google Scholar]
- 25.Zhao Z, Condomines M, van der Stegen SJC, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28(4):415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. *.Kahlon KS, Brown C, Cooper LJ, et al. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–6. First preclinical study to demonstrate the anti-glioma activity of CAR T cells [DOI] [PubMed] [Google Scholar]
- 27.Brown CE, Aguilar B, Starr R, et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Molecular therapy : the journal of the American Society of Gene Therapy. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed N, Salsman VS, Kew Y, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. ClinCancer Res. 2010;16(2):474–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chow KK, Naik S, Kakarla S, et al. T Cells Redirected to EphA2 for the Immunotherapy of Glioblastoma. MolTher. 2013;21(3):629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krenciute G, Krebs S, Torres D, et al. Characterization and Functional Analysis of scFv-based Chimeric Antigen Receptors to Redirect T Cells to IL13Ralpha2-positive Glioma. Molecular therapy : the journal of the American Society of Gene Therapy. 2016;24(2):354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krebs S, Chow KK, Yi Z, et al. T cells redirected to interleukin-13Ralpha2 with interleukin-13 mutein-chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Ralpha1. Cytotherapy. 2014;16(8):1121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caruso HG, Hurton LV, Najjar A, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer research. 2015;75(17):3505–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bullain SS, Sahin A, Szentirmai O, et al. Genetically engineered T cells to target EGFRvIII expressing glioblastoma. J Neurooncol. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan RA, Johnson LA, Davis JL, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. HumGeneTher. 2012;23(10):1043–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen CJ, Yang YX, Han EQ, et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. JHematolOncol. 2013;6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Science translational medicine. 2015;7(275):275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaffer DR, Savoldo B, Yi Z, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117(16):4304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin L, Ge H, Long Y, et al. CD70, a novel target of CAR-T-cell therapy for gliomas. Neuro-oncology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J ClinInvest. 2011;121(5):1822–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watanabe N, Bajgain P, Sukumaran S, et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. 2016;5(12):e1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hudecek M, Sommermeyer D, Kosasih PL, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer immunology research. 2015;3(2):125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461–72. [DOI] [PubMed] [Google Scholar]
- 43.Jonnalagadda M, Mardiros A, Urak R, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23(4):757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bridgeman JS, Hawkins RE, Bagley S, et al. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184(12):6938–49. [DOI] [PubMed] [Google Scholar]
- 45.Gomes-Silva D, Mukherjee M, Srinivasan M, et al. Tonic 4–1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell reports. 2017;21(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sabatino M, Hu J, Sommariva M, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Held-Feindt J, Mentlein R. CD70/CD27 ligand, a member of the TNF family, is expressed in human brain tumors. IntJCancer. 2002;98(3):352–6. [DOI] [PubMed] [Google Scholar]
- 49.Wischhusen J, Jung G, Radovanovic I, et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer research. 2002;62(9):2592–9. [PubMed] [Google Scholar]
- 50.Aulwurm S, Wischhusen J, Friese M, et al. Immune stimulatory effects of CD70 override CD70-mediated immune cell apoptosis in rodent glioma models and confer long-lasting antiglioma immunity in vivo. Int J Cancer. 2006;118(7):1728–35. [DOI] [PubMed] [Google Scholar]
- 51.Pratt D, Pittaluga S, Palisoc M, et al. Expression of CD70 (CD27L) Is Associated With Epithelioid and Sarcomatous Features in IDH-Wild-Type Glioblastoma. J Neuropathol Exp Neurol. 2017;76(8):697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ge H, Mu L, Jin L, et al. Tumor associated CD70 expression is involved in promoting tumor migration and macrophage infiltration in GBM. Int J Cancer. 2017;141(7):1434–44. [DOI] [PubMed] [Google Scholar]
- 53.Zadeh G, Bhat KP, Aldape K. EGFR and EGFRvIII in glioblastoma: partners in crime. Cancer Cell. 2013;24(4):403–4. [DOI] [PubMed] [Google Scholar]
- 54.Padfield E, Ellis HP, Kurian KM. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front Oncol. 2015;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montano N, Cenci T, Martini M, et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia. 2011;13(12):1113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Felsberg J, Hentschel B, Kaulich K, et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017. [DOI] [PubMed] [Google Scholar]
- 57.Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. MolCancer Res. 2005;3(10):541–51. [DOI] [PubMed] [Google Scholar]
- 58.Wykosky J, Gibo DM, Stanton C, Debinski W. Interleukin-13 receptor alpha 2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin Cancer Res. 2008;14(1):199–208. [DOI] [PubMed] [Google Scholar]
- 59.Hernan R, Fasheh R, Calabrese C, et al. ERBB2 up-regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res. 2003;63(1):140–8. [PubMed] [Google Scholar]
- 60.Koka V, Potti A, Forseen SE, et al. Role of Her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am JClin Oncol. 2003;26(4):332–5. [DOI] [PubMed] [Google Scholar]
- 61.Liu G, Ying H, Zeng G, et al. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64(14):4980–6. [DOI] [PubMed] [Google Scholar]
- 62.Zhang JG, Kruse CA, Driggers L, et al. Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. JNeurooncol. 2008;88(1):65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Debinski W, Obiri NI, Powers SK, et al. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. ClinCancer Res. 1995;1(11):1253–8. [PubMed] [Google Scholar]
- 64.Saikali S, Avril T, Collet B, et al. Expression of nine tumour antigens in a series of human glioblastoma multiforme: interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for immunotherapy. Journal of neuro-oncology. 2007;81(2):139–48. [DOI] [PubMed] [Google Scholar]
- 65.Kawakami M, Kawakami K, Takahashi S, et al. Analysis of interleukin-13 receptor alpha2 expression in human pediatric brain tumors. Cancer. 2004;101(5):1036–42. [DOI] [PubMed] [Google Scholar]
- 66.Okada H, Low KL, Kohanbash G, et al. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol. 2008;88(3):245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joshi BH, Puri RA, Leland P, et al. Identification of interleukin-13 receptor alpha2 chain overexpression in situ in high-grade diffusely infiltrative pediatric brainstem glioma. NeuroOncol. 2008;10(3):265–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lanitis E, Poussin M, Klattenhoff AW, et al. Chimeric antigen receptor T Cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer ImmunolRes. 2013;1(1):43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown CE, Starr R, Aguilar B, et al. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(8):2199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coffman KT, Hu M, Carles-Kinch K, et al. Differential EphA2 epitope display on normal versus malignant cells. Cancer Res. 2003;63(22):7907–12. [PubMed] [Google Scholar]
- 71.Damschroder MM, Widjaja L, Gill PS, et al. Framework shuffling of antibodies to reduce immunogenicity and manipulate functional and biophysical properties. MolImmunol. 2007;44(11):3049–60. [DOI] [PubMed] [Google Scholar]
- 72. *.Krenciute G, Prinzing BL, Yi Z, et al. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer immunology research. 2017;5(7):571–81. *Demonstrate that transgenic expression of IL15 enhance the anti-glioma activity of CAR T cells [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hegde M, Corder A, Chow KK, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21(11):2087–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. *.Hegde M, Mukherjee M, Grada Z, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. The Journal of clinical investigation. 2016;126(8):3036–52. Design and characterization of CAR T cells that simultanouesly traget two GBM antigens [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. The Journal of clinical investigation. 2016;126(10):3814–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T-cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro-oncology. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. *.Sampson JH, Choi BD, Sanchez-Perez L, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(4):972–84. Preclinical study in immune competent murine GBM model with CAR T cells [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Chang WC, Wong CW, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. **.Keu KV, Witney TH, Yaghoubi S, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Science translational medicine. 2017;9(373). In vivo imaging of CAR T cells in GBM patients [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. **.Brown CE, Alizadeh D, Starr R, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. The New England journal of medicine. 2016;375(26):2561–9. Sustained regression of GBM post intraventricular CAR T-cell delivery [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lucas KG, Bao L, Bruggeman R, et al. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neurooncol. 2010. [DOI] [PubMed] [Google Scholar]
- 82.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002;8(8):793–800. [DOI] [PubMed] [Google Scholar]
- 84.Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol. 2013;94(1):25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu VF, Yang J, Lebrun DG, Li M. Understanding the role of cytokines in Glioblastoma Multiforme pathogenesis. Cancer Lett. 2012;316(2):139–50. [DOI] [PubMed] [Google Scholar]
- 86.Kesarwani P, Kant S, Prabhu A, Chinnaiyan P. The interplay between metabolic remodeling and immune regulation in glioblastoma. Neuro-oncology. 2017;19(10):1308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hurton LV, Singh H, Najjar AM, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(48):E7788–E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoyos V, Savoldo B, Quintarelli C, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koneru M, Purdon TJ, Spriggs D, et al. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4(3):e994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yeku OO, Purdon TJ, Koneru M, et al. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Scientific reports. 2017;7(1):10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71(17):5697–706. [DOI] [PubMed] [Google Scholar]
- 92.Hu B, Ren J, Luo Y, et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell reports. 2017;20(13):3025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang L, Morgan RA, Beane JD, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(10):2278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hunter MR, Prosser ME, Mahadev V, et al. Chimeric gammac cytokine receptors confer cytokine independent engraftment of human T lymphocytes. Molecular immunology. 2013;56(1–2):1–11. [DOI] [PubMed] [Google Scholar]
- 95.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shum T, Omer B, Tashiro H, et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer discovery. 2017;7(11):1238–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–80. [DOI] [PubMed] [Google Scholar]
- 98.Bollard CM, Rossig C, Calonge MJ, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99(9):3179–87. [DOI] [PubMed] [Google Scholar]
- 99.Bollard CM, Gottschalk S, Torrano V, et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J Clin Oncol. 2014;32(8):798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilkie S, Burbridge SE, Chiapero-Stanke L, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J BiolChem. 2010;285(33):25538–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Leen AM, Sukumaran S, Watanabe N, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. MolTher. 2014;22(6):1211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Watanabe N, Anurathapan U, Brenner M, et al. Transgenic Expression of a Novel Immunosuppressive Signal Converter on T Cells. MolTher. 2013;22(S1):S153. [Google Scholar]
- 103.Berghoff AS, Kiesel B, Widhalm G, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-oncology. 2015;17(8):1064–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nduom EK, Wei J, Yaghi NK, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-oncology. 2016;18(2):195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zeng J, Zhang XK, Chen HD, et al. Expression of programmed cell death-ligand 1 and its correlation with clinical outcomes in gliomas. Oncotarget. 2016;7(8):8944–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(20):5290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reardon DA, Omuro A, Brandes AA, et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro-oncology. 2017;19(suppl_3):iii21–iii. [Google Scholar]
- 108.Bouffet E, Larouche V, Campbell BB, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016;34(19):2206–11. [DOI] [PubMed] [Google Scholar]
- 109.Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Scientific reports. 2017;7(1):737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li S, Siriwon N, Zhang X, et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(22):6982–92. [DOI] [PubMed] [Google Scholar]
- 111.Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. The Journal of clinical investigation. 2016;126(8):3130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ankri C, Shamalov K, Horovitz-Fried M, et al. Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J Immunol. 2013;191(8):4121–9. [DOI] [PubMed] [Google Scholar]
- 113.Liu X, Ranganathan R, Jiang S, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer research. 2016;76(6):1578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pellegatta S, Savoldo B, Di Ianni N, et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Science translational medicine. 2018;10(430). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Johanns TM, Dunn GP. Applied Cancer Immunogenomics: Leveraging Neoantigen Discovery in Glioblastoma. Cancer journal. 2017;23(2):125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Butterfield LH, Ribas A, Dissette VB, et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. ClinCancer Res. 2003;9(3):998–1008. [PubMed] [Google Scholar]
- 117.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. NEnglJMed. 2008;358(25):2698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kakarla S, Wang L, Rowley D, et al. Improving T-Cell Immunotherapies for Solid Tumors by Targeting the Tumor Stroma. Biology of Blood and Marrow Transplantation. 2011;17(2 (supplement 1)):S270. [Google Scholar]
- 119.Bentley RT, Ahmed AU, Yanke AB, et al. Dogs are man’s best friend: in sickness and in health. Neuro-oncology. 2017;19(3):312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Di Stasi A, De Angelis B, Rooney CM, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Craddock JA, Lu A, Bear A, et al. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nature medicine. 2015;21(5):524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107(6):2294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Okada H, Weller M, Huang R, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. The Lancet Oncology. 2015;16(15):e534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Westphal M, Lamszus K. Circulating biomarkers for gliomas. Nature reviews Neurology. 2015;11(10):556–66. [DOI] [PubMed] [Google Scholar]
- 126.Schuessler A, Smith C, Beagley L, et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014;74(13):3466–76. [DOI] [PubMed] [Google Scholar]
- 127.Chheda ZS, Kohanbash G, Okada K, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215(1):141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Suck G, Odendahl M, Nowakowska P, et al. NK-92: an ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer immunology, immunotherapy : CII. 2016;65(4):485–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Heczey A, Liu D, Tian G, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang C, Burger MC, Jennewein L, et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst. 2016;108(5). [DOI] [PubMed] [Google Scholar]
- 131.Baker GJ, Chockley P, Yadav VN, et al. Natural killer cells eradicate galectin-1-deficient glioma in the absence of adaptive immunity. Cancer research. 2014;74(18):5079–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pereboeva L, Harkins L, Wong S, Lamb LS. The safety of allogeneic innate lymphocyte therapy for glioma patients with prior cranial irradiation. Cancer immunology, immunotherapy : CII. 2015;64(5):551–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Iwami K, Shimato S, Ohno M, et al. Peptide-pulsed dendritic cell vaccination targeting interleukin-13 receptor alpha2 chain in recurrent malignant glioma patients with HLA-A*24/A*02 allele. Cytotherapy. 2012;14(6):733–42. [DOI] [PubMed] [Google Scholar]
- 134.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J ClinOncol. 2011;29(3):330–6. [DOI] [PMC free article] [PubMed] [Google Scholar]