

Abstract
The tRNAHis guanylyltransferase (Thg1) transfers a guanosine triphosphate (GTP) in the 3′–5′ direction onto the 5′-terminal of tRNAHis, opposite adenosine at position 73 (A73). The guanosine at the −1 position (G−1) serves as an identity element for histidyl-tRNA synthetase. To investigate the mechanism of recognition for the insertion of GTP opposite A73, first we constructed a two-stranded tRNAHis molecule composed of a primer and a template strand through division at the D-loop. Next, we evaluated the structural requirements of the incoming GTP from the incorporation efficiencies of GTP analogs into the two-piece tRNAHis. Nitrogen at position 7 and the 6-keto oxygen of the guanine base were important for G−1 addition; however, interestingly, the 2-amino group was found not to be essential from the highest incorporation efficiency of inosine triphosphate. Furthermore, substitution of the conserved A73 in tRNAHis revealed that the G−1 addition reaction was more efficient onto the template containing the opposite A73 than onto the template with cytidine (C73) or other bases forming canonical Watson–Crick base-pairing. Some interaction might occur between incoming GTP and A73, which plays a role in the prevention of continuous templated 3′–5′ polymerization. This study provides important insights into the mechanism of accurate tRNAHis maturation.
Keywords: transfer RNA (tRNA), aminoacyl tRNA synthetase, RNA modification, 3′–5′ polymerase, RNA structure
INTRODUCTION
Nucleotide polymerization generally proceeds in the 5′–3′ direction. This reaction involves nucleophilic attack by the 3′-OH of the terminal nucleotide in the elongating chain on the α-phosphate of an incoming nucleotide. It is likely that polymerization of this nature confers advantages during proofreading (Lehman and Richardson 1964; Uptain et al. 1997) and is thus favored by natural selection pressure. However, previous studies have shown that the tRNAHis guanylyltransferase (Thg1) and members of the Thg1-like protein (TLP) family catalyze nucleotide addition to the 5′-end of tRNA in the reverse (3′–5′) direction (Heinemann et al. 2012; Jackman et al. 2012). Thg1 is a well-characterized protein in this enzyme family. In eukaryotes, Thg1 catalyzes the addition of a guanosine residue to the 5′-end of immature tRNAHis (Gu et al. 2003). This guanosine at position −1 (G−1) of tRNAHis serves as a major recognition element for histidyl-tRNA synthetase (HisRS) (Cooley et al. 1982; Himeno et al. 1989; Rudinger et al. 1994; Rosen and Musier-Forsyth 2004), except in some rare cases (Rao and Jackman 2015). Therefore, Thg1 is essential to the fidelity of protein synthesis in eukaryotes. In addition, yeast Thg1 interacts with the replication origin recognition complex for DNA replication (Rice et al. 2005), and the plant homolog ICA1 was identified as a protein affecting the capacity to repair DNA damage (Zhu et al. 2015). TLPs have also been identified in archaea, bacteria, and mitochondria, and are involved in diverse biological reactions (Heinemann et al. 2010; Abad et al. 2011). TLPs have been shown to catalyze the 5′-end nucleotide addition to truncated tRNAs with a broader substrate specificity range in vitro (Abad et al. 2010; Rao et al. 2011). Furthermore, recent studies have also implicated TLPs in the 5′-end editing of mitochondrial tRNAs in Dictyostelium discoideum (Abad et al. 2011; Long et al. 2016) as well as the incorporation of a nucleotide onto the 5′-end of 5S ribosomal RNA or class 1 noncoding RNA (Long et al. 2016). Taken together, these observations suggest that Thg1s and TLPs have unprecedented reverse-nucleotide polymerization capacity in DNA/RNA repair.
Eukaryotic Thg1 recognizes a 5′-monophosphorylated tRNAHis (p-tRNAHis), which is cleaved by RNase P from pre-tRNAHis through the recognition of the His anticodon (GUG) (Jackman and Phizicky 2006a). The 3′–5′ addition reaction catalyzed by Thg1 involves three chemical reactions (Fig. 1; Jahn and Pande 1991; Gu et al. 2003). In the first step, the p-tRNAHis is activated by ATP, creating a 5′-adenylylated-tRNAHis intermediate. In the second step, the 3′-OH of incoming GTP attacks the 5′–5′ phosphate linkage of the intermediate, yielding pppG−1-tRNAHis. Finally, the pyrophosphate group is removed, and mature G−1-containing tRNAHis is generated. In eukaryotes, Thg1 adds G−1 to the 5′-end of tRNAHis opposite a conserved A73, resulting in a mismatched 3′–5′ addition reaction (G:A mismatch). In addition, when the A73 nucleotide is mutated to cytosine, Thg1 adds multiple guanosines sequentially, extending the 5′-end of tRNA in the 3′–5′ direction in a Watson–Crick template-dependent manner, using the 3′-CCA tail of tRNA as the template (Jackman and Phizicky 2006b). In contrast, TLPs in archaea, bacteria, and mitochondria also catalyze template-dependent 3′–5′ nucleotide addition, but do not catalyze mismatched G−1 addition (Abad et al. 2010; Heinemann et al. 2010; Rao et al. 2011; Jackman et al. 2012).
FIGURE 1.

Reaction scheme for G−1 addition to tRNAHis by eukaryotic Thg1. First, the 5′-monophosphorylated pre-tRNAHis is cleaved by RNase P, and then activated by adenylylation using ATP. Second, GTP is transferred to the activated 5′-end of pre-tRNAHis. Finally, the 5′-pyrophosphate is removed from the G−1 nucleotide to yield the monophosphorylated, G−1-containing tRNAHis that is the determinant for aminoacylation by HisRS.
The crystal structure of human Thg1 (HsThg1) and Bacillus thuringiensis TLP (Hyde et al. 2010, 2013) revealed unexpected structural homology of Thg1/TLP family enzymes with canonical 5′–3′ nucleotide polymerases, such as T7 DNA/RNA polymerases (Doublié et al. 1998; Jeruzalmi and Steitz 1998). Additionally, the structure suggests that the enzyme uses a two-metal-ion catalytic mechanism for nucleotide polymerization. The structure of Thg1 from Candida albicans (CaThg1) in complex with tRNAHis reveals that the tRNA substrate approaches the reaction center in the opposite orientation to those observed in canonical DNA/RNA polymerases (Nakamura et al. 2013). Furthermore, Thg1 tightly binds the anticodon loop of tRNAHis in a sequence-specific manner, while the acceptor stem does not strongly interact with Thg1. Recently, the structure of TLP from Methanosarcina acetivorans (MaTLP) in complex with the 5′-end truncated tRNAPhe and a GTP analog (guanosine 5′-[β,γ-imido triphosphate: GDPNP]) was determined (Kimura et al. 2016). The MaTLP-tRNA-GDPNP complex structure revealed that GDPNP mimics an incoming GTP, and forms a Watson–Crick base-pairing with the opposite C72 residue at the 3′-end region of the tRNA. However, how eukaryotic Thg1 specifically incorporates mismatched GTP onto the 5′-end of tRNAHis opposite A73 in a 3′–5′ addition reaction is still unclear.
In the present study, first, we constructed and optimized a new substrate RNA for Thg1, a two-piece tRNA composed of a 5′-short primer and the remaining 3′ template RNA strands that allows the 3′–5′ nucleotide addition reaction of Thg1 to be detected by a primer/template assay similar to canonical DNA/RNA polymerases. Next, we examined the nucleotide addition reaction with various GTP analogs using the developed two-piece tRNA to reveal the substrate specificity of Thg1 for GTP incorporation opposite A73 in a 3′–5′ addition reaction.
RESULTS AND DISCUSSION
Division of tRNA molecules into primer and template strands
Recently, the crystal structure of Thg1 from C. albicans in complex with S. cerevisiae (Sc) tRNAPhe with the phenylalanine anticodon substituted for the histidine anticodon GUG (SctRNAPheGUG) was determined. The report shows that the D-loop of tRNA is not directly bound with Thg1, and U17 in the loop is flipped out from the tertiary core region of tRNA without interacting with any other nucleotide in the tRNA (Supplemental Fig. S1; Nakamura et al. 2013). It has been demonstrated that fragmentation of tRNA molecules allows for direct measurement of the fraction of aminoacylated tRNA by its altered mobility on denaturing polyacrylamide gel (Wolfson et al. 1998). To apply Thg1 catalysis in various biochemical assays similar to typical DNA/RNA polymerases or aminoacyl-tRNA synthetases, we constructed two-stranded RNA substrates for use in Thg1 reactions.
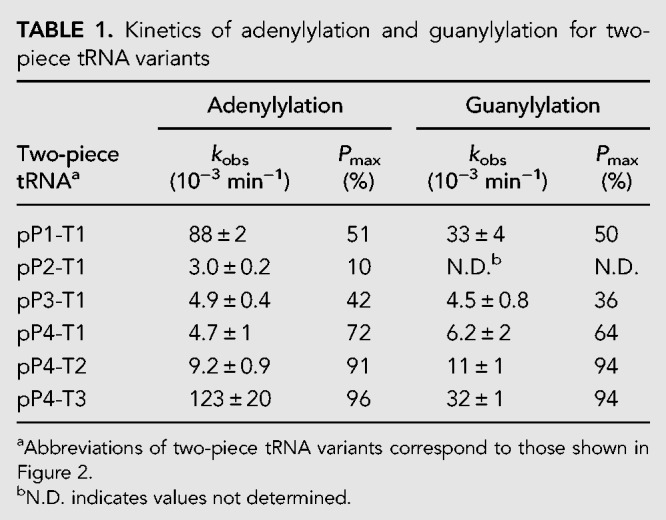
First, we prepared a two-piece tRNA composed of 17 nt on the 5′-side (pP1) and 59 nt on the 3′-side (T1) of the U17 residue of SctRNAPheGUG (Fig. 2A). 5′-phosphorylated P1 (pP1) was hybridized with T1, followed by the addition of Thg1. The 5′–3′ guanylylated (pG−1pP1) product could not be separated from 5′–5′ adenylylated P1 (AppP1) in the denaturing gel analysis because they had almost the same molecular weight. Phosphatase treatment producing 5′-dephosphorylated G−1pP1 (Supplemental Fig. S2) enabled the determination of product percentages of AppP1 and G−1pP1 from the polyacrylamide gel analysis (Fig. 2B). Under single-turnover conditions, observed rate constants (kobs) of the adenylylation and guanylylation reactions for pP1-T1 were 8.8 × 10−2 and 3.3 × 10−2 min−1, respectively (Table 1; Fig. 2C). However, pP2 formed a long stem with T1 (pP2-T1; Fig. 2A), and showed a remarkable decrease in both adenylylation and guanylylation reactions (Table 1; Fig. 2C; Supplemental Fig. S3). Interestingly, the shorter primers (pP3 and pP4) generated single-stranded regions in the D-loop in complex with T1, and both adenylylation and guanylylation activities were improved compared with those for pP2 (Table 1; Fig. 2C; Supplemental Fig. S3). Moreover, adenylylation and guanylylation activities were increased when pP4 was complexed with T2 containing the whole D-loop sequence (Table 1; Fig. 2A,D; Supplemental Fig. S3). Additionally, the maximal product formation (Pmax) of pP4-T2 (91%) greatly increased from that of pP1-T1 (51%; Table 1; Fig. 2D; Supplemental Fig. S3). These results indicate that the entire D-loop sequence and structure in the template strand are required for efficient Thg1 activity using the two-piece tRNA. To improve Thg1 activity on the two-piece tRNAs further, we predicted the secondary structure of T2 by using Mfold (Zuker 2003) which showed two possible secondary structures of T2 (Supplemental Fig. S4). Then, we synthesized the T3 strand to promote proper folding into the cloverleaf-like structure (T3; Fig. 2A). As the results of adenylylation and guanylylation of pP4 show, the reaction products increased by 13-fold and threefold, respectively, compared with those from the reaction using T2 (Table 1; Fig. 2D; Supplemental Fig. S3). This indicates that thermodynamic stabilization of the anticodon stem in the template strand improves both adenylylation and guanylylation activities using the two-piece tRNA. We changed the anticodon GUG of the T3 strand essential for the Thg1 recognition (Jackman and Phizicky 2006b) with GAA, and both adenylylation and guanylylation activities were entirely abolished (Supplemental Fig. S3), confirming that the modification of pP4-T3 was mediated by Thg1. Taken together, maintenance of the D-loop and stabilization of the anticodon stem in the template strand are important for efficient Thg1 activity using the two-piece tRNA, thus suggesting that the formation of an L-shaped tRNA-like structure would be required for two-piece tRNA recognition by Thg1.
FIGURE 2.

Construction of two-piece tRNAs. (A) Two-piece tRNA variants are drawn as cloverleaf structures. S. cerevisiae tRNAPhe with GAA anticodon altered to GUG (SctRNAPheGUG) was separated into the 5′-side primer fragment (pP) and the 3′-side template fragment (T) shown in red and gray, respectively. The altered bases of T3 are indicated in blue. (B) A primer/template assay for simultaneously measuring the kinetics of adenylylation and G−1 addition reactions by Thg1 with pP1-T1. Representative single-turnover assays with pP1-T1 for determining kobs for adenylylation in the presence of ATP and for G−1 addition in the presence of both ATP and GTP. The reactions shown are time courses of activity with 10 µM CaThg1 in excess over pP1-T1; aliquots from each time point were treated with phosphatase (CIP), followed by resolution on urea-PAGE gel. RNA fragments were detected by SybrGold staining. (C) Time course experiments of G−1 addition reaction with D-stem loop variants of two-piece tRNA; pP1-T1 (♦), pP2-T1 (▴), pP3-T1 (•), pP4-T1 (▪). Lines represent each time course fitted to a single-exponential equation (Equation 1) to yield kobs. (D) Time course experiments of G−1 addition reaction by using short primer (pP4) and template fragment variants (T1, T2, and T3); pP4-T1 (▪), pP4-T2 (♦), pP4-T3 (•). The bars in the graphs are SD of more than two independent experiments.
TABLE 1.
Kinetics of adenylylation and guanylylation for two-piece tRNA variants
Recently, Desai and coworkers reported the engineering of TLP enzymes and tRNAHis to apply their unique 3′–5′ polymerization activity to the labeling of RNA molecules (Desai et al. 2017). To apply the 3′–5′ polymerization activity to non-tRNA molecules, they divided E. coli tRNAHis into two RNA fragments at the same position as our two-piece tRNA (pP4-T2); however, their fragmented tRNAHis could not act as a substrate for archaeal Pyrobaculum aerophilum TLP. These observations suggest that the structural requirements of tRNAHis recognition may differ between eukaryotic Thg1 and archaeal TLP.
Structural requirements of GTP for the G−1 addition reaction by Thg1
GTP is incorporated at the −1 position opposite the highly conserved A73 residue in eukaryotic tRNAHis. It should be noted that the incorporation efficiency of GTP is 20-times higher than that of UTP (Abad et al. 2010); it is still unclear why GTP rather than UTP is preferentially attached to the −1 position opposite adenosine. To investigate the structural requirements of GTP addition, we measured the incorporation efficiency of GTP analogs (XTPs; Fig. 3A,B) using the aforementioned two-piece tRNA platform that allows precise identification of a single-nucleotide-elongated product. Because GTP can be directly conjugated with 5′-triphosphorylated tRNAHis without ATP activation (Jackman and Phizicky 2006b), 5′-triphosphorylated P4 (pppP4) was prepared as a primer. Observed rate constants (kobs) of natural NTP or GTP analogs to pppP4 complexed with T3 (Y = A) were measured under single-turnover conditions (Supplemental Fig. S5).
FIGURE 3.

Nucleotide addition reaction for natural NTPs and various GTP analogs onto the developed two-piece tRNA. (A) Reaction scheme of nucleotide addition reaction with natural NTPs and various GTP analogs (XTP) into 5′-triphosphorylated P4-T3. (B) Natural NTPs and GTP analogs used in the nucleotide addition reaction. (C–F) The relative kobs values of nucleotide addition reactions with various GTP analogs for the template Y = A (C), Y = C (D), Y = U (E), and Y = G (F). Values are relative to the kobs value of wild-type activity (GTP addition in Y = A), which was set at 1.0. The bars in the graphs are SD of more than two independent experiments.
GTP showed the kobs value of 6.5 × 10−2 min−1, and that of UTP was one-twentieth that of GTP (kobs = 3.0 × 10−3 min−1). Neither ATP nor CTP was incorporated onto pppP4. This result is consistent with the previous result obtained from wild-type tRNAHis (Abad et al. 2010). Next, we examined incorporation of the GTP analogs shown in Figure 3B; kobs values are listed in Table 2 and Figure 3C. 8-Br-GTP (8BrG) and 8-oxo-GTP (8OG) that adopt a syn-conformation (Kapuler and Reich 1971; Uno et al. 1971) did not yield product formation, indicating that the anti-conformation is recognized for GTP addition by Thg1. 7-Deazaguanosine (7DG) triphosphate showed a rate constant one-seventh that of GTP, indicating that a nitrogen atom at position 7 (N7) is important for Thg1 catalysis. The kobs value of 2-aminopurine triphosphate (2AP) was one-half that of GTP. In addition, isoguanine triphosphate (isoG) with a 6-amino group was completely inactive. These data suggest that an exocyclic 6-keto group is important for the efficient GTP addition. However, surprisingly, inosine triphosphate (ITP) exhibited a kobs that was 2.8 times higher than that of GTP, thus indicating that the 2-amino group is nonessential in the reaction. This reaction profile was also confirmed in the full-length tRNA substrate (Supplemental Fig. S6). The kobs of two-piece tRNA for GTP addition was 15 times slower than that of the full length tRNA.
TABLE 2.
Kinetics of nucleotide addition with NTP and various GTP analogs into the two-piece tRNA

The G−1 addition opposite A73 is a characteristic feature of eukaryotic Thg1. TLPs, Thg1 homologs in archaea, bacteria, and mitochondria, only catalyze template-dependent 3′–5′ nucleotide addition (Abad et al. 2010; Heinemann et al. 2010; Rao et al. 2011; Jackman et al. 2012). Recently, the structural analysis of the TLP-tRNA-GTP analog (GDPNP) complex has revealed that the base moiety of GDPNP forms Watson–Crick (WC) base-pairing with the opposite C on the substrate tRNA (Fig. 4; Kimura et al. 2016). The structural comparison of Thg1 and TLP in the complex with tRNA revealed that the position of the 5′-ends of both tRNAs is almost the same, and the reaction center and incoming nucleotide binding site are well conserved. Furthermore, the superposing of Thg1-tRNA and TLP-tRNA-GDPNP structures suggests that only Thg1 contains a highly conserved N-terminal α-helix close to the major groove side of WC base-pairing on the TLP complex structure (Fig. 4). As mentioned above, the N7 and 6-keto group at the major groove side of incoming GTP are important for Thg1 catalysis. The E7 and K10 on the helix are positioned near the N7 and 6-keto group (4–6 Å) on the model structure, suggesting that these hydrophilic residues may form hydrogen bonds or water-mediated interactions between incoming GTP (Fig. 4B). Interestingly, the eukaryote Thg1-specific HINNLYN sequence (Jackman et al. 2012) is located at the minor groove side of the WC base-pairing, and the N156 in the motif may form hydrogen bonds with the incoming GTP (3–4 Å). Thus, we propose that the N-terminal α-helix and the specific motif in Thg1 may participate in GTP recognition via hydrophilic interaction with the G−1 addition opposite A73.
FIGURE 4.
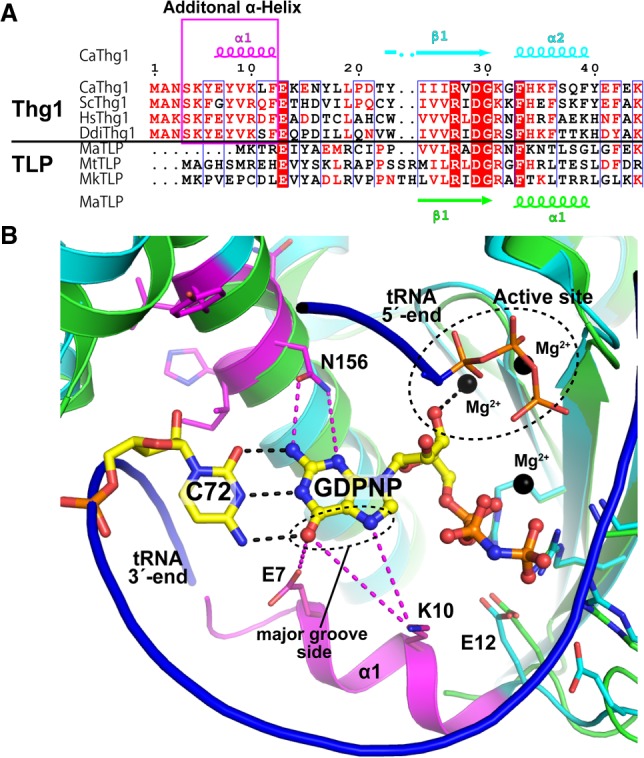
Structural comparison between the active sites of Thg1 and TLP. (A) Multiple sequence alignment of Thg1 and TLP enzymes. Secondary structures of CaThg1 and M. acetivorans TLP (MaTLP) are indicated at the top (α, α-helix; β, β-strand; η, 310-turn). The additional N-terminal helix is highlighted with a pink box. The species aligned are as follows: C. albicans, CaThg1; S. cerevisiae, ScThg1; Human, HsThg1; D. discoideum, DdiThg1; M. acetivorans, MaTLP; M. thermautotrophicus, MtTLP; and M. kandleri, MkTLP. Each protein sequence was aligned by CLUSTALW (Thompson et al. 1994) and ESPript (Robert and Gouet 2014) was used to prepare the figure. (B) Superposition of CaThg1-tRNA complex without a tRNA molecule (PDB ID: 3WC2, cyan and magenta) and MaTLP-tRNA-GDPNP complex (PDB ID: 5AXN, green), where the tRNA molecule bound to MaTLP is shown as a blue ribbon model. GDPNP and the opposite C72 with Watson–Crick hydrogen bonds (black dashed lines) in the tertiary complex structure of MaTLP are shown as yellow sticks, and Mg2+ ions are indicated as black spheres. The residues around the active site of CaThg1 and MaTLP are shown as stick models. The N-terminal helix and the eukaryotic Thg1-specifc HINNLYN sequence are indicated in magenta. Possible interactions between CaThg1 and incoming GTP are indicated as magenta dashed lines. Pymol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC) was used to generate the figure.
Comparison of GTP and ITP addition reaction by Thg1
It should be noted that the incorporation of ITP is accelerated. To investigate the incorporation of ITP further, maximal first-order rate constants (kmax) and apparent equilibrium dissociation constants (KDapp) of GTP and ITP were calculated from the kobs values obtained at various nucleotide concentrations (Fig. 5; Smith and Jackman 2012, 2014). Both GTP and ITP showed almost the same KDapp values (278 µM and 336 µM, respectively), showing that there is no difference in the affinity of Thg1 for either ITP or GTP. In contrast, the kmax value of ITP was approximately 2.8 times higher than that of GTP. This result indicates that, upon ITP addition, the conjugation step is accelerated in the ITP addition.
FIGURE 5.
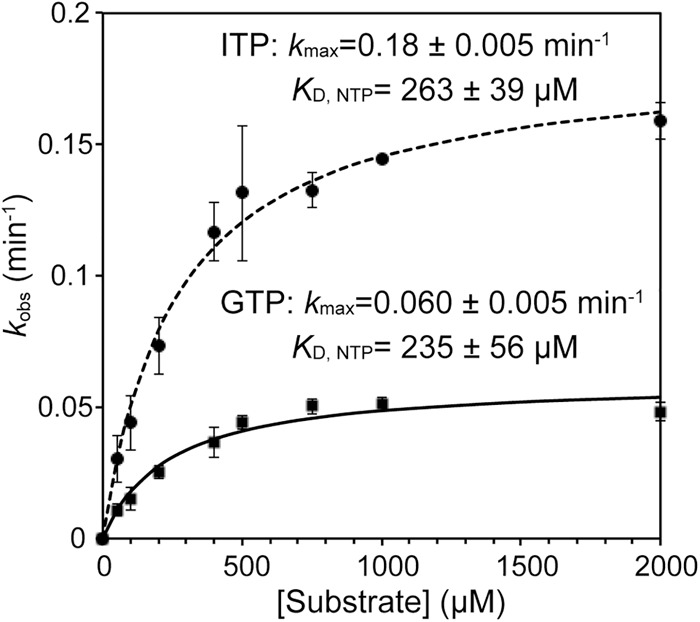
Comparison of the maximum rate constants (kmax) and apparent dissociation constants (KDapp) of GTP and ITP in the nucleotide addition reaction. Observed rates of nucleotide addition reaction determined in the presence of 10 µM Thg1 and various concentrations of GTP or ITP (50–2000 µM) were plotted as a function of (substrate NTP) and fit to Equation 2 to determine the kmax and KDapp for ITP or GTP. The bars in the graphs are SD of more than two independent experiments.
The structural analysis of TLP-tRNA-GDPNP complex revealed that tRNA binding to TLP induces the large movement of 3′-OH of GDPNP and coordination with Mg2+ at the active site to activate 3′-OH for the conjugation step in the nucleotide addition (Fig. 4; Kimura et al. 2016). Based on the sequential and structural conservation of Thg1 and TLP, it is likely that the 3′-OH of incoming GTP would also be coordinated with Mg2+ at the reaction center on Thg1. This suggests that the differences in the base structure between GTP and ITP may affect coordination with Mg2+ of incoming nucleotides through base interaction with the opposite A73 of tRNAHis.
ITPs are present at low levels in the cell as by-products generated either from deamination of purine bases or from phosphorylation of inosine monophosphate (IMP) (Zamzami et al. 2013). However, ITP is converted to IMP and pyrophosphate by the housecleaning enzyme inosine triphosphatase (ITPA). Reduced ITPA activity produces the accumulation of the rogue nucleotides ITP and dITP (88–533 µM in erythrocytes), which may be incorporated into RNA and DNA, posing a risk for mutagenesis, and potentially resulting in DNA damage, cancer, and cell death (Holmes et al. 1979; Behmanesh et al. 2009; Menezes et al. 2012; Zamzami et al. 2013). These observations indicate the possibility that ITP incorporation into tRNAHis by Thg1 may occur in the cells. Further studies are required to reveal the biological function of efficient ITP incorporation by Thg1 within the inosine metabolism in human cells.
The reaction efficiency of GTP addition is affected by the structure of the opposite base at position 73
It has been reported that the alteration of A73 of tRNAHis into cytidine, uridine, and guanine induced WC-dependent nucleotide addition by Thg1 at position −1 (Jackman and Phizicky 2006b). However, there has been no quantitative evaluation of how Thg1 efficiently adds GTP opposite A73 rather than a WC base-pairing at position −1 in tRNAHis mutants at position 73. We changed A73 of T3 to cytidine, uridine, and guanine (T3C73, T3U73, and T3G73; Fig. 3A), and examined the incorporation efficacies of NTPs into pppP4 (Fig. 3A; Supplemental Fig. S5). Although a single GTP was incorporated into the pppP4-T3G73, three GTP molecules were continuously incorporated opposite C73, C74, and C75 of pppP4-T3C73, as reported previously (Jackman and Phizicky 2006b), and also into the pppP4-T3U73 through the G:U73 pair (Supplemental Fig. S7). To determine the kobs values from single nucleotide addition activity, we changed C74 in T3C73 and T3U73 into A74 (T3C73A74 and T3U73A74; Fig. 3A) to prevent multiple GTP incorporation. All kobs values of pppP4-T3A73 (Y = A), -T3C73A74 (Y = C), -T3U73A74 (Y = U), and -T3G73 (Y = G) are summarized in Table 2 and Figure 3C–F.
Thg1 incorporated GTP or CTP into the Y = C or G template in a template-dependent manner; kobs values were 5.6 × 10−2 min−1 and 2.0 × 10−2 min−1, respectively. These observed kobs values were lower than the activity of GTP incorporation opposite A73 (kobs = 6.5 × 10−2 min−1). Thg1 also added ATP into the Y = U template, but with significantly lower activity (kobs = 2.5 × 10−3 min−1), similar to the activity of UTP incorporation into the Y = A template (kobs = 3.5 × 10−3 min−1). A comparison of these observed kobs values revealed that Thg1 catalyzes the G−1 addition reaction opposite A73 more efficiently than canonical WC base paring at position −1 of tRNAHis. Furthermore, GTP was most efficiently incorporated onto the 5′-end of pppP4 opposite all templates (Y = A, C, U, or G) among natural NTPs, suggesting that Thg1 would exhibit specific interaction for a GTP in all templates.
Then, we examined the incorporation of GTP analogs into pppP4 complexed with all templates. The incorporation efficiency of 7DG was significantly lower than that of GTP with all the templates, indicating that the N7 of GTP is important for GTP addition opposite all the templates and would be recognized by the Thg1 template independently as described above. Although ITP was added in the Y = A template, the kobs value of ITP was decreased 14-fold in the Y = C or G template than that of the Y = A. It is interesting that the high reactivity of ITP is specific to the Y = A template. 2AP was incorporated into the Y = A or U template, but not into the Y = C or G template. These results demonstrate that the incorporation profile of GTP analogs depends on the opposite base in the template, suggesting that the template base is involved in GTP analog recognition by Thg1. Taken together with the fact that the reaction efficiency of GTP addition was slightly changed by base substitution of A73, these results imply that GTP addition may also be affected by the structure of the opposite base, and only GTP but not GTP analogs can perform some interaction with all template bases. Thus incoming GTP may be recognized not only by specific interaction with Thg1, but also by an interaction with A73 of tRNAHis.
One possible reason for this result is that incoming GTP forms a non-WC G:A base pair with A73. Although four types of non-WC base pairs are proposed for the G:A pairing (Kretulskie and Spratt 2006), two of them can be estimated from the results of GTP analog reactions, as shown in Figure 6. In one G:A base pair (Fig. 6A), both nucleotides have anti-conformations; and in the other, A forms a syn-conformation, resulting in a Hoogsteen base pair (Fig. 6B; Brown et al. 1986). As mentioned above, N7 is necessary for G−1 addition, but it is not involved in both types of G:A base pairs. Thus, N7 of GTP may be directly recognized by Thg1.
FIGURE 6.
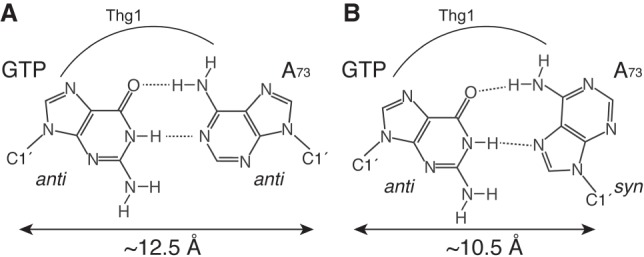
Potential structures of a G:A base pair. The structure of a G:A base pair with both bases in the anti-conformation with respect to the ribose (A), and the Hoogsteen G:A base pair with the incoming GTP in the anti-conformation and opposite A73 in the syn-conformation (B).
Accurate tRNAHis maturation would be achieved by the mismatch G:A pairing to prevent the possibility of continuous templated 3′–5′ polymerization
The G−1 residue of tRNAHis is a crucial identity element for the corresponding HisRS in bacteria (Himeno et al. 1989; Fromant et al. 2000; Connolly et al. 2004; Rosen and Musier-Forsyth 2004) and eukaryotes (Rudinger et al. 1994; Nameki et al. 1995; Rosen et al. 2006). Most prokaryotes encode both G−1 and C73 in their tRNAHis gene to form a WC base pair. In eukaryotes, Thg1 precisely catalyzes the single G−1 addition opposite A73 at the 5′-end of tRNAHis (Gu et al. 2003). Interestingly, the alteration of conserved A73 of tRNAHis into C73, G73, and U73 induced a templated multiple-nucleotide addition by Thg1 using the conserved CCA-end of tRNAHis as a template (Supplemental Fig. S7; Sprinzl et al. 1998; Marck and Grosjean 2002; Jackman and Phizicky 2006b; Preston and Phizicky 2010). Therefore, the single G−1 addition to tRNAHis can be achieved only with an A residue in position 73. In the case of canonical DNA/RNA polymerases, WC base-pairing is selected for and mismatched bases are strongly discriminated against (Echols and Goodman 1991; Thomas et al. 1998). When these polymerases make a mismatched base pair, they stall and proofread the mismatch using editing exonucleases, or “backtracking” (Johnson 1993; Thomas et al. 1998; Kunkel and Bebenek 2000; Johnson and Beese 2004; James et al. 2017). However, Thg1 catalyzes the formation of the mismatch G:A pairing with high efficiency, and the 3′–5′ polymerization reaction is discontinued. These observations indicate that eukaryotes may have evolved Thg1 and tRNAHis with the mismatch G:A pairing to prevent the possibility of continuous templated 3′–5′ polymerization, resulting in accurate single G−1 addition.
Conclusion
In the present research, first, we constructed a two-stranded tRNA molecule composed of a primer and a template strand by dividing the parent tRNAHis at the D-loop. As the results of adenylylation and G−1 addition demonstrate, the two-piece tRNA acts as a substrate for Thg1, and maintenance of the D-loop and stabilization of the anticodon stem in the template strand improved Thg1 activity. The structure–function relationship analysis of incoming GTP suggested the possibility that a non-WC G:A base pair is formed in the G−1 addition. Future structural study of Thg1 complexed with tRNAHis and incoming GTP should shed light on the molecular mechanism by which Thg1 accommodates the formation of the mismatch base-pairing in contrast to canonical DNA/RNA polymerases. In addition, the two-piece tRNA developed in the present study allows Thg1 to incorporate a nucleotide triphosphate onto the 5′-end of single-stranded RNA in the 3′–5′ direction. We also revealed the structural requirements of GTP in the addition reaction, which provides information about the acceptable position to introduce functional groups, such as fluorophores or biotin, into incoming GTP. These findings provide important insights into the application of Thg1 to the 5′-terminal modification of RNAs.
MATERIALS AND METHODS
Nucleotides and oligonucleotides
NTPs used for enzyme assays were purchased from Thermo Scientific. Inosine-5′-triphosphate (ITP), 2-Aminopurine-riboside-5′-triphosphate (2AP), 7-Deazaguanosine-5′-triphosphate (7DG), Isoguanosine-5′-triphosphate (isoG), and 8-Oxoguanosine-5′-triphosphate (8OG) were purchased from Trilink Biotechnologies. 8-bromo-5′-triphosphate (8BrG) was purchased from Abcam. 5′-monophosphorylated primer RNA (pP) were purchased from GeneDesign. DNA oligonucleotides were purchased from Hokkaido System Science.
Preparation of CaThg1
The N-terminally His6-tagged CaThg1 was overexpressed in E. coli BL21 (DE3)-pRARE2 (Novagen), and purified as previously described with slight modifications (Nakamura et al. 2013). CaThg1 overexpressed cells were disrupted by sonication in buffer A (50 mM HEPES-Na, pH 7.5, 500 mM NaCl, 10 mM MgCl2, 10% [v/v] glycerol, 4 mM imidazole) with 0.5 mg/mL lysozyme, and 0.1 mg/mL DNase I. Cell debris was removed by centrifugation (20000g, 1 h), and clarified supernatant was loaded onto a HIS-Select Nickel Affinity Gel (Sigma) preequilibrated with buffer A. The column was washed with buffer A, and proteins were eluted with buffer A containg 250 mM imidazole. Eluton fractions were dialyzed against buffer B (25 mM HEPES-Na pH 7.5, 500 mM NaCl, 4 mM MgCl2, 1 mM DTT, 50% [v/v] glycerol). Dialyzed samples were concentrated by ultrafiltration to a final concentration of 190 µM, and stored at −30°C.
Preparation of template RNA
S. cerevisiae tRNAPheGUG (phenylalanine anticodon GAA was replaced with the histidine anticodon GUG) and its 5′-truncated tRNA fragments (template RNAs) were transcribed using T7 RNA polymerase. Double-stranded DNAs encoding the T7 promoter and target RNA sequences were amplified by PCR with three overlapping primers, and cloned into the BamHI/HindIII site of pUC19. The inserted sequences were verified by DNA sequencing. Double-stranded DNA transcription templates were obtained by PCR and purified using a GenElute PCR Clean-Up kit (Sigma). The in vitro transcription was performed using a DuraScribe T7 Transcription kit (Epicentre) at 37°C for 6 h. The reaction mixture was subsequently treated with DNase I at 37°C for 30 min to degrade the template DNA, and purified using 10% denaturing urea-polyacrylamide gel electrophoresis (Urea-PAGE). RNAs were extracted from gel slices and refolded simultaneously in H2O at 4°C for 18 h. The extracted RNA samples were precipitated with ethanol, dissolved in TE buffer pH8.0, and stored at −80°C.
Preparation of 5′-triphospholylated primer RNA
The 5′-triphosphorylated primer RNA fragment (pppP, 13 nt) was synthesized by in vitro transcription (Milligan et al. 1987; Korencic et al. 2002). The following DNA oligonucleotides were used for preparation of a transcription template: T7 promoter D, 5′-GGAATTGGATCCTAATACGACTCACTATAG; template strand, 5′-GAGCTAAATCCGCTATAGTGAGTCGTATTAGGATCCAATTCC. The transcription template was prepared by heating the two DNA strands together to 95°C for 5 min and allowing to gradually cool to 25°C. The in vitro transcription was performed by using a DuraScribe T7 Transcription kit at 37°C for 6 h. The transcribed RNA was purified by 20% (w/v) Urea-PAGE, and extracted in H2O at 4°C for 18 h. The extracted RNA was loaded onto a reverse-phase column (YMC) preequilibrated with H2O. The column was washed with H2O, and the RNA was eluted with 50% acetonitrile. Elution fractions were dried up and disolved in H2O, and stored at −80°C.
Adenylylation and nucleotide addition assay using two-piece tRNAs
Two-piece tRNAs for adenylylation and nucleotide addition assays were constructed by annealing of a 5′-pP and a template RNA fragment. Annealing conditions for all the two-piece tRNAs were as follows: A mixture of 1 µM of the primer RNA and 2.5 µM of template RNA in 25 mM HEPES-Na pH 7.5, 125 mM NaCl, 10 mM MgCl2, 2 mM spermidin HCl was incubated at 65°C for 2 min, then gradually cooled to 25°C. Reactions contained the annealed two-piece tRNA in 25 mM HEPES-Na pH 7.5, 125 mM NaCl, 10 mM MgCl2, 3 mM DTT, 2 mM spermidine HCl, and 1 mM ATP for adenylylation reactions, or 1 mM ATP and 1 mM GTP for nucleotide addition reactions, and were preincubated at 25°C for 5 min, and initiated by addition of a saturating concentration of enzyme (10 µM). At various time points, aliquots (1 µL) were mixed with 0.5 µL of 5 U/µL calf alkaline intestinal phosphatase (CIP, New England Biolabs) and incubated at 30°C for 10 min to quench the reactions. The quenched sample was mixed with an equal volume of loading buffer (10 M urea, 50 mM EDTA, and 0.1% bromophenol blue), and analyzed by 20% Urea-PAGE. The gels were stained with SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher Scientific), and visualized and quantified using Typhoon and Image Quant software (GE Healthcare). To evaluate substrate specificity of Thg1, 5′-pppP and the template RNA fragment were used to construct the two-piece tRNA, and incubated with Thg1 and 1 mM NTP or GTP analogs under the same conditions as described above, without phosphatase treatment. Single-turnover rate constants (kobs) and maximal product formation (Pmax) were determined as previously reported (Smith and Jackman 2012). Time courses of product formation were plotted and fit to a single-exponential rate equation:
| (1) |
where Pt is the fraction of product formed at each time, and Pmax is the maximum amount of product conversion observed during each time course. The resulting kobs values determined for each concentration of GTP or ITP were plotted and fit to an equation:
| (2) |
to yield the first-order maximal rate constants (kmax) and dissociation constants (KDapp). All reported parameters were determined from at least two independent experiments.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dr. Eiko Ohtsuka, Dr. Yasuhiro Mie, Dr. Yu Hirano, and Dr. Mashiki Ikegami for helpful insights. This work was supported by Grant-in-Aid for Young Scientists (B) (16K18511, to A.N.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.067330.118.
REFERENCES
- Abad MG, Rao BS, Jackman JE. 2010. Template-dependent 3′–5′ nucleotide addition is a shared feature of tRNAHis guanylyltransferase enzymes from multiple domains of life. Proc Natl Acad Sci 107: 674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad MG, Long Y, Willcox A, Gott JM, Gray MW, Jackman JE. 2011. A role for tRNAHis guanylyltransferase (Thg1)-like proteins from Dictyostelium discoideum in mitochondrial 5′-tRNA editing. RNA 17: 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behmanesh M, Sakumi K, Abolhassani N, Toyokuni S, Oka S, Ohnishi YN, Tsuchimoto D, Nakabeppu Y. 2009. ITPase-deficient mice show growth retardation and die before weaning. Cell Death Differ 16: 1315–1322. [DOI] [PubMed] [Google Scholar]
- Brown T, Hunter WN, Kneale G, Kennard O. 1986. Molecular structure of the G.A base pair in DNA and its implications for the mechanism of transversion mutations. Proc Nat Acad Sci 83: 2402–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Rosen AE, Musier-Forsyth K, Francklyn CS. 2004. G-1:C73 recognition by an arginine cluster in the active site of Escherichia coli histidyl-tRNA synthetase. Biochemistry 43: 962–969. [DOI] [PubMed] [Google Scholar]
- Cooley L, Appel B, Söll D. 1982. Post-transcriptional nucleotide addition is responsible for the formation of the 5′ terminus of histidine tRNA. Proc Natl Acad Sci 79: 6475–6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai R, Kim K, Buchsenschutz HC, Chen AW, Bi Y, Mann MR, Turk MA, Chung CZ, Heinemann IU. 2017. Minimal requirements for reverse polymerization and tRNA repair by tRNAHis guanylyltransferase. RNA Biol 15: 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S, Tabor S, Long AM, Richardson CC, Ellenberger T. 1998. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391: 251–258. [DOI] [PubMed] [Google Scholar]
- Echols H, Goodman MF. 1991. Fidelity mechanisms in DNA replication. Annu Rev Biochem 60: 477–511. [DOI] [PubMed] [Google Scholar]
- Fromant M, Plateau P, Blanquet S. 2000. Function of the extra 5′-phosphate carried by histidine tRNA. Biochemistry 39: 4062–4067. [DOI] [PubMed] [Google Scholar]
- Gu W, Jackman JE, Lohan AJ, Gray MW, Phizicky EM. 2003. tRNAHis maturation: an essential yeast protein catalyzes addition of a guanine nucleotide to the 5′ end of tRNAHis. Genes Dev 17: 2889–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann IU, Randau L, Tomko RJ Jr, Söll D. 2010. 3′–5′ tRNAHis guanylyltransferase in bacteria. FEBS Lett 584: 3567–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann IU, Nakamura A, O'Donoghue P, Eiler D, Söll D. 2012. tRNAHis-guanylyltransferase establishes tRNAHis identity. Nucleic Acids Res 40: 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeno H, Hasegawa T, Ueda T, Watanabe K, Miura K, Shimizu M. 1989. Role of the extra G-C pair at the end of the acceptor stem of tRNAHis in aminoacylation. Nucleic Acids Res 17: 7855–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes SL, Turner BM, Hirschhorn K. 1979. Human inosine triphosphatase: catalytic properties and population studies. Clin Chim Acta 97: 143–153. [DOI] [PubMed] [Google Scholar]
- Hyde SJ, Eckenroth BE, Smith BA, Eberley WA, Heintz NH, Jackman JE, Doublié S. 2010. tRNAHis guanylyltransferase (THG1), a unique 3′–5′ nucleotidyl transferase, shares unexpected structural homology with canonical 5′–3′ DNA polymerases. Proc Natl Acad Sci 107: 20305–20310. [Google Scholar]
- Hyde SJ, Rao BS, Eckenroth BE, Jackman JE, Doublié S. 2013. Structural studies of a bacterial tRNAHis guanylyltransferase (Thg1)-like protein, with nucleotide in the activation and nucleotidyl transfer sites. PLoS one 8: e67465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Phizicky EM. 2006a. tRNAHis guanylyltransferase adds G−1 to the 5′ end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA 12: 1007–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Phizicky EM. 2006b. tRNAHis guanylyltransferase catalyzes a 3′–5′ polymerization reaction that is distinct from G−1 addition. Proc Natl Acad Sci 103: 8640–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Gott JM, Gray MW. 2012. Doing it in reverse: 3′-to-5′ polymerization by the Thg1 superfamily. RNA 18: 886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn D, Pande S. 1991. Histidine tRNA guanylyltransferase from Saccharomyces cerevisiae. II. Catalytic mechanism. J Biol Chem 266: 22832–22836. [PubMed] [Google Scholar]
- James K, Gamba P, Cockell SJ, Zenkin N. 2017. Misincorporation by RNA polymerase is a major source of transcription pausing in vivo. Nucleic Acids Res 45: 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeruzalmi D, Steitz TA. 1998. Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J 17: 4101–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA. 1993. Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem 62: 685–713. [DOI] [PubMed] [Google Scholar]
- Johnson SJ, Beese LS. 2004. Structures of mismatch replication errors observed in a DNA polymerase. Cell 116: 803–816. [DOI] [PubMed] [Google Scholar]
- Kapuler AM, Reich E. 1971. Some stereochemical requirements of Escherichia coli ribonucleic acid polymerase. Interaction with conformationally restricted ribonucleoside 5′-triphosphates: 8-bromoguanosine, 8-ketoguanosine, and 6-methylcytidine triphosphates. Biochemistry 10: 4050–4061. [DOI] [PubMed] [Google Scholar]
- Kimura S, Suzuki T, Chen M, Kato K, Yu J, Nakamura A, Tanaka I, Yao M. 2016. Template-dependent nucleotide addition in the reverse (3′–5′) direction by Thg1-like protein. Sci Adv 2: e1501397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korencic D, Söll D, Ambrogelly A. 2002. A one-step method for in vitro production of tRNA transcripts. Nucleic Acids Res 30: e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretulskie AM, Spratt TE. 2006. Structure of purine-purine mispairs during misincorporation and extension by Escherichia coli DNA polymerase I. Biochemistry 45: 3740–3746. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K. 2000. DNA replication fidelity. Annu Rev Biochem 69: 497–529. [DOI] [PubMed] [Google Scholar]
- Lehman IR, Richardson CC. 1964. The deoxyribonucleases of Escherichia coli. IV. An exonuclease activity present in purified preparations of deoxyribonucleic acid polymerase. J Biol Chem 239: 233–241. [PubMed] [Google Scholar]
- Long Y, Abad MG, Olson ED, Carrillo EY, Jackman JE. 2016. Identification of distinct biological functions for four 3′–5′ RNA polymerases. Nucleic Acids Res 44: 8395–8406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marck C, Grosjean H. 2002. tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 8: 1189–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes MR, Waisertreiger IS, Lopez-Bertoni H, Luo X, Pavlov YI. 2012. Pivotal role of inosine triphosphate pyrophosphatase in maintaining genome stability and the prevention of apoptosis in human cells. PLoS One 7: e32313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A, Nemoto T, Heinemann IU, Yamashita K, Sonoda T, Komoda K, Tanaka I, Söll D, Yao M. 2013. Structural basis of reverse nucleotide polymerization. Proc Natl Acad Sci 110: 20970–20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nameki N, Asahara H, Shimizu M, Okada N, Himeno H. 1995. Identity elements of Saccharomyces cerevisiae tRNAHis. Nucleic Acids Res 23: 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston MA, Phizicky EM. 2010. The requirement for the highly conserved G−1 residue of Saccharomyces cerevisiae tRNAHis can be circumvented by overexpression of tRNAHis and its synthetase. RNA 16: 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao BS, Jackman JE. 2015. Life without post-transcriptional addition of G−1: two alternatives for tRNAHis identity in Eukarya. RNA 21: 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao BS, Maris EL, Jackman JE. 2011. tRNA 5′-end repair activities of tRNAHis guanylyltransferase (Thg1)-like proteins from Bacteria and Archaea. Nucleic Acids Res 39: 1833–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice TS, Ding M, Pederson DS, Heintz NH. 2005. The highly conserved tRNAHis guanylyltransferase Thg1p interacts with the origin recognition complex and is required for the G2/M phase transition in the yeast Saccharomyces cerevisiae. Eukaryot Cell 4: 832–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42: W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen AE, Musier-Forsyth K. 2004. Recognition of G-1:C73 atomic groups by Escherichia coli histidyl-tRNA synthetase. J Am Chem Soc 126: 64–65. [DOI] [PubMed] [Google Scholar]
- Rosen AE, Brooks BS, Guth E, Francklyn CS, Musier-Forsyth K. 2006. Evolutionary conservation of a functionally important backbone phosphate group critical for aminoacylation of histidine tRNAs. RNA 12: 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudinger J, Florentz C, Giegé R. 1994. Histidylation by yeast HisRS of tRNA or tRNA-like structure relies on residues −1 and 73 but is dependent on the RNA context. Nucleic Acids Res 22: 5031–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BA, Jackman JE. 2012. Kinetic analysis of 3′–5′ nucleotide addition catalyzed by eukaryotic tRNAHis guanylyltransferase. Biochemistry 51: 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BA, Jackman JE. 2014. Saccharomyces cerevisiae Thg1 uses 5′-pyrophosphate removal to control addition of nucleotides to tRNAHis. Biochemistry 53: 1380–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S. 1998. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 26: 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Platas AA, Hawley DK. 1998. Transcriptional fidelity and proofreading by RNA polymerase II. Cell 93: 627–637. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno H, Oyabu S, Otsuka E, Ikehara M. 1971. The effect of guanosine 5′-triphosphate analogues on protein synthesis. Biochim Biophys Acta 228: 282–288. [DOI] [PubMed] [Google Scholar]
- Uptain SM, Kane CM, Chamberlin MJ. 1997. Basic mechanisms of transcript elongation and its regulation. Annu Rev Biochem. 66: 117–172. [DOI] [PubMed] [Google Scholar]
- Wolfson AD, Pleiss JA, Uhlenbeck OC. 1998. A new assay for tRNA aminoacylation kinetics. RNA 4: 1019–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami MA, Duley JA, Price GR, Venter DJ, Yarham JW, Taylor RW, Catley LP, Florin TH, Marinaki AM, Bowling F. 2013. Inosine triphosphate pyrophosphohydrolase (ITPA) polymorphic sequence variants in adult hematological malignancy patients and possible association with mitochondrial DNA defects. J Hematol Oncol 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Ausin I, Seleznev A, Mendez-Vigo B, Pico FX, Sureshkumar S, Sundaramoorthi V, Bulach D, Powell D, Seemann T, et al. 2015. Natural variation identifies ICARUS1, a universal gene required for cell proliferation and growth at high temperatures in Arabidopsis thaliana. PLoS Genet 11: e1005085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.