

Abstract
Angiotensin-converting enzyme (ACE) — a zinc-dependent dicarboxypeptidase with two catalytic domains — plays a major part in blood pressure regulation by converting angiotensin I to angiotensin II. However, ACE cleaves many peptides besides angiotensin I and thereby affects diverse physiological functions, including renal development and male reproduction. In addition, ACE has a role in both innate and adaptive responses by modulating macrophage and neutrophil function — effects that are magnified when these cells overexpress ACE. Macrophages that overexpress ACE are more effective against tumours and infections. Neutrophils that overexpress ACE have an increased production of superoxide, which increases their ability to kill bacteria. These effects are due to increased ACE activity but are independent of angiotensin II. ACE also affects the display of major histocompatibility complex (MHC) class I and MHC class II peptides, potentially by enzymatically trimming these peptides. Understanding how ACE expression and activity affect myeloid cells may hold great promise for therapeutic manipulation, including the treatment of both infection and malignancy.
Angiotensin-converting enzyme (ACE) was initially discovered in 1953 during the study of the renin-angiotensin system (RAS)1,2. In this system, angiotensinogen is sequentially cleaved by renin and then by ACE to generate the 8-amino acid peptide angiotensin II, which raises blood pressure through effects on the kidneys, brain, adrenal glands, heart and blood vessels. Although ACE is expressed in most tissues of the body, expression levels are particularly high in the lungs, kidneys, testes, duodenum, choroid plexus and placenta3,4. ACE is primarily located on cell membranes via a carboxy-terminal transmembrane domain (BOX 1) and therefore localized to specific tissues, but a cleaved, active form of the enzyme is also present in the circulation. Whereas serum levels among individuals are affected by genetic poly orphisms, individual adult serum ACE levels are thought to be stable5,6. Children generally have higher levels of ACE than adults7. For example, ACE levels in children (6 months to 17 years of age) are 13–100 U/l compared with 9–67 U/l in adults when using an FAPGG-based enzymatic activity assay.
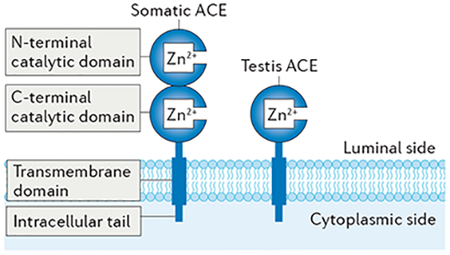
Box 1 |. Structure and substrates of ACE.
Two enzymes — the aspartyl protease renin90 and the zinc-dependent dicarboxypeptidase angiotensin-converting enzyme (ACE) — play a key part in the renin–angiotensin system (RAS). Renin is expressed by granular cells in the juxtaglomerular apparatus (JGA) and cleaves only one chemical bond in a single substrate, angiotensinogen, thereby producing the decapeptide angiotensin I. By contrast, ACE is expressed in multiple cell types (such as endothelial cells, renal tubular epithelial cells, gut epithelial cells and myeloid-derived cells) and cleaves various substrates. ACE is best characterized for its role in cleaving two carboxy-terminal (C-terminal) amino acids of angiotensin I, thereby producing the vasoconstrictor angiotensin II. ACE also cleaves the vasodilator bradykinin, releasing an inactive 7-amino acid product. Although most ACE substrates are 15 amino acids in length or less, the enzyme can cleave substrates as small as 3 amino acids and as large as 42 amino acids (for example, amyloid-β1–42)91.
ACE is a single polypeptide chain that folds into a structure of two independent zinc-containing catalytic domains, a C-terminal transmembrane domain and an intracellular tail (see the figure)92. Both catalytic domains face the luminal side intracellularly and the extracellular space when the enzyme is located on the plasma membrane. Intracellular ACE is likely to be active during trafficking through the endoplasmic reticulum, as ACE has been shown to modify major histocompatibility complex (MHC) class I peptides in this compartment. Catalytically active ACE also circulates in plasma as a result of enzymatic cleavage of the extracellular portions of the enzyme by a still unknown ‘sheddase’. Some authors have suggested that the sheddase is a member of the a disintegrin and metalloproteinase (ADAM) family of proteins93,94.
Some studies indicate that the intracellular tail of ACE can be phosphorylated on Ser1270 as a means of sensing shear stress and regulating ACE expression95,96. The exact physiological role of intracellular signalling mediated by the ACE intracellular tail is an area of ongoing investigation.
In males, two isozymes exist: somatic ACE (1,277 residues in humans and 1,278 in mice), which is present in somatic tissues, such as the lung and kidney, and testis ACE (also known as germinal ACE; 372 residues in both humans and mice), which contains only one catalytic domain identical to that of the C-terminal domain of somatic ACE and is produced by post-meiotic male germ cells owing to a germ cell-specific promoter within the twelfth intron of ACE4,97.
Differences in substrate specificity of the individual catalytic domains in somatic ACE exist: for example, the ACE C-terminal domain cleaves angiotensin I with a threefold higher efficiency (Kcat) than the amino-terminal (N-terminal) domain despite both domains having roughly equal affinities for this substrate98. As a result, most angiotensin I is cleaved by the C-terminal domain in vivo99. By contrast, other peptides, such as the immunosuppressive peptide acetyl-SDKP, are cleaved by the N-terminal domain in vivo100,101.
Both domains in somatic ACE are homologous in amino acid sequence, and ACE-like enzymes are found throughout the animal kingdom, including in species as diverse as the starlet sea anemone (Nematostella vectensis), mosquito (Anopheles gambiae) and the marine invertebrate sea squirt (Ciona intestinalis)4. Phylogenetic analysis of this wide diversity strongly suggests that the two catalytic domains of ACE are the result of a genetic duplication that occurred early in evolutionary history (~700 million years ago) at a time when simple organisms did not require regulation of blood pressure4,102. Taken together, these findings raise the question of why the catalytic activity of the N-terminal domain has been retained. The most obvious explanation would be that a catalytically active N-terminal domain provides a selective advantage apart from the potential to produce angiotensin II.
Given the numerous effects of angiotensin II on blood pressure, ACE inhibitors were originally developed for the treatment of hypertension8. Subsequent clinical studies have also demonstrated the efficacy of these drugs in the treatment of heart failure, diabetic kidney disease and several other diseases, indicating that ACE has broad effects in different systems9–11. In addition, ACE has been increasingly associated with having a role in the immune system. The first connection between ACE and immune function was made in 1975, when 15 of 17 patients with active sarcoidosis were reported to have increased serum ACE levels compared with levels in patients with treated sarcoidosis or in individuals in whom the disease had resolved12. Indeed, in most granulomatous diseases, ACE is expressed by epithelioid macrophages and giant cells making up the granuloma13, raising the question of why ACE expression is upregulated in myeloid-derived cells targeting difficult-to-destroy entities such as Mycobacterium tuberculosis and certain fungi. In this Review, we discuss the effects of ACE expression in neutrophils and macrophages — cells that are central to both the innate and adaptive immune response. Furthermore, we describe how ACE activity taps into a pathway that strongly upregulates myeloid cell function. Such a pathway may hold great promise for therapeutic manipulation in the context of diseases as diverse as cancer and infection or even chronic diseases such as Alzheimer disease.
Functional diversity of ACE
ACE and blood pressure.
ACE plays a part in blood pressure regulation by converting angiotensin I to angiotensin II. However, studies in rodents and computer simulations indicate that angiotensin II production is physiologically regulated by renin4,14. Although a reduction in angiotensin II levels is only accomplished at over 90% ACE inhibition, pharmacological ACE inhibitors are so effective at inhibiting the enzyme that they can reduce blood pressure. The effects of ACE and angiotensin II on blood pressure have been extensively studied and reviewed elsewhere15. ACE also has several other physiological functions owing to the diverse effects of angiotensin II and the actions of other ACE cleavage products (FIG. 1).
Figure 1 |. Functional diversity of ACE.

Angiotensin-converting enzyme (ACE) has several peptide substrates, which are involved in multiple physiological functions. The production of the vasoconstrictor angiotensin II and the cleavage of the vasodilator bradykinin result in an increase in blood pressure (BP). In humans, a lack of ACE leads to low BP within the fetus and the development of renal tubular dysgenesis. Many immune effects of ACE are independent of angiotensin II, but the peptide substrate(s) and/or product(s) that mediate these effects are currently unknown. So far, ACE is known to inactivate the 4-amino acid peptide acetyl-SDKP, which has been described as an anti-inflammatory molecule. However, as an inhibitor of acetyl-SDKP formation does not affect the neutrophil immune response of wild-type mice, this molecule is unlikely to play a major part in the improved immune response mediated by ACE overexpression. Male germ cells produce testis ACE, an isozyme of ACE, which is smaller than the somatic form, as it contains only the carboxy-terminal domain. Experiments in mice show that males without testis ACE reproduce very poorly. The substrate and product of testis ACE responsible for normal fertility are not known but are almost certainly not angiotensin I or angiotensin II.
ACE in renal development.
Studies in both mice and humans have revealed a role of ACE in kidney development16–18. Global Ace−/− mice are viable, probably because some renal development in mice takes place after birth16,17. Although blood pressure is profoundly reduced in these mice (a reduction of systolic blood pressure from approximately 110 mmHg to 73 mmHg)17, the kidney structure is identical to that of wild-type (WT) mice at birth. However, by post-natal day 16, Ace−/− mice exhibit expansion of the renal pelvis and underdevelopment of the renal medulla19. An equivalent phenotype is observed in mice with a genetic knockout of any of the RAS components (that is, angiotensinogen, renin or all type 1 angiotensin II receptors (AT1Rs))20–22. Mechanistically, angiotensin II stimulates ureteral smooth muscle growth and uni directional peristaltic waves that originate in the renal pelvis and assist urine transport along the ureters to the bladder23. The absence of such waves in ACE-deficient mice (or any mouse model lacking either angiotensin II or AT1Rs) results in inefficient urine transit that elevates pressures within the renal pelvis and ureter. Hence, the hydronephrosis that occurs in these mice is due to a functional defect and not physical blockage of the outflow tract23. Taken together, ACE and the RAS facilitate the efficient movement of urine away from the kidney and the normal development of the renal outflow tract.
In contrast to mice, human kidney development primarily occurs in utero. Genetic mutations in ACE, or any functional interruption of the RAS, can result in renal tubular dysgenesis (RTD). One study of RTD reported a series of 54 mutations in genes encoding RAS proteins in 48 families, of which ACE mutations were reported in 31 families18. Absence of a functional RAS results in severe hypotension in the fetus, leading to renal hypoperfusion, RTD and oligohydramnios, which cause fetal compression. Severe renal insufficiency and lung hypoplasia usually cause infants to die rapidly after birth.
ACE in male reproduction.
ACE also has a function in male reproduction. Adult testes of Ace−/− mice and WT mice appear to be similar microscopically, and no differences in numbers of mature sperm cells have been reported17,24. However, although Ace−/− mice readily inseminate females, they reproduce poorly compared with WT mice, suggesting that ACE has a role in sperm function. One study showed that selective inactivation of the catalytic activity of testis ACE — which contains only the carboxy-terminal domain of ACE — reduces the offspring number to ~1% of WT levels, whereas basal systolic blood pressure is unaffected owing to the presence of a functional somatic ACE amino-terminal domain in these mice24. Consistent with this finding, evidence in humans suggests that testis ACE is essential for normal fertilization rates by in vitro fertilization25.
Several studies indicate that the functional role of testis ACE in reproduction is independent of angiotensin II production and its effects on blood pressure: both angiotensinogen-deficient mice and mice with catalytically active testis ACE levels but lacking somatic ACE have low blood pressure but reproduce normally26,27. Hence, testis ACE likely affects male reproduction owing to catalytic effects on a substrate other than angiotensin I. However, as the exact substrate of testis ACE is currently unknown, this area warrants further investigation.
ACE in the immune system
Immune effects mediated by angiotensin II.
Angiotensin II mediates several pro-inflammatory responses by signalling through AT1R, a topic that has been extensively reviewed elsewhere28–30. The recruitment of circulating inflammatory cells to the endothelium and subendothelial space is an early step in the inflammatory response. Angiotensin II influences several steps in leukocyte recruitment through AT1R-mediated upregulation of E-selectin, P-selectin, IL-8, CC-chemokine ligand 5 (CCL5, also known as RANTES) and CCL2 (also known as MCP1) expression in endothelial cells29,31. Another major effect of angiotensin II is to increase production of reactive oxygen species (ROS) by AT1R-mediated activation of NADPH oxidase in both endothelial and vascular smooth muscle cells30. ROS in turn have many downstream effects that contribute to inflammation, including the activation of several intracellular kinase pathways and the stimulation of redox-sensitive transcription factors such as nuclear factor-κB (NF-κB) and activator protein 1 (AP1; also known as JUN)30,32,33. Angiotensin II can also trigger Toll-like receptor 4 (TLR4) activation in various cell types, which stimulates the innate immune response34. In addition, angiotensin II has been reported to induce dendritic cell maturation through the NF-κB, extracellular signal-regulated kinase 1 (ERK1)–ERK2 and signal transducer and activator of transcription 1 (STAT1) signalling pathways35. Endogenously produced angiotensin II in T cells has a role in regulating tumour necrosis factor (TNF) expression36. Other studies have reported angiotensin II to be implicated in models of autoimmunity; for example, use of an ACE inhibitor or an AT1R antagonist substantially reduces disease severity in experimental autoimmune encephalomyelitis (EAE)37,38.
Several studies have investigated how angiotensin II affects renal inflammation39. Particularly interesting are experiments that used bone marrow transplantation to create WT mice lacking AT1A either in all bone marrow-derived cells or in selected populations of inflammatory cells such as T cells or macrophages40–42. The lack of AT1A on all bone marrow-derived cells in WT mice was associated with a normal basal blood pressure and, following infusion of angiotensin II, with an increased elevation of mean arterial pressure (58 mmHg increase over basal levels) compared with WT mice receiving WT bone marrow-derived cells that had normal AT1A expression (47 mmHg increase over basal levels)40. The lack of AT1A expression in bone marrow-derived cells also resulted in a 46% increase in albuminuria and roughly a 69% increase in the number of renal macrophages compared with WT mice that received WT bone marrow. A similar result was seen with WT mice transplanted with bone marrow in which AT1A was depleted from T cells only41. In response to angiotensin II-induced hypertension, these mice had increased renal disease severity (40% increase in albuminuria and a 1.8-fold increase in the accumulation of renal CD4+ T cells) compared with mice transplanted with control bone marrow. Mechanistically, these findings have been proposed to be due to an increased propensity of AT1A-deficient CD4+ T cells to differentiate towards pro-inflammatory T helper 1 (TH1) cells41. Similar results were also observed following the transfer of bone marrow from donors in which AT1A was specifically eliminated from myeloid cells; the lack of AT1A in myeloid cells led to increased pro-inflammatory M1 differentiation and a 64% increase in renal tubular interstitial fibrosis42. Thus, at least in these models of hypertension, AT1A expression by WT T cells and myeloid cells suppresses the development of pro-inflammatory TH1 cells and M1 macrophages, respectively. The effect of AT1A on macrophage polarization was also observed in a mouse model of obesity in which AT1A-deficient macrophages had greater expression of M1 markers43. Further studies will be necessary to resolve the conclusions of these studies — an immunomodulatory role of AT1A expression by bone marrow cells — with the traditional view of the AT1 receptor as pro-inflammatory29.
Taken together, these findings show that angiotensin II plays a part in several immune processes. Any attempt to describe the role of ACE in the immune response is complicated by the fact that ACE produces a variety of peptides other than angiotensin II. As outlined below, not all of the immunomodulatory effects of ACE seem to be mediated by angiotensin II.
ACE and granuloma.
ACE is upregulated in several diseases characterized by granulomas, including active sarcoidosis, histoplasmosis, leprosy, silicosis, granulomatosis with polyangiitis and the granulomatous reaction induced by murine schistosomiasis44–48. Whether the formation of a physical barrier is advantageous or disadvantageous to the host is currently unclear49. On the one hand, the physical isolation (‘walling-off’) of bacteria may reduce the rate of disease dissemination50. On the other hand, as oxygen is a major component of antimicrobial activity, reduced access to oxygen for myeloid cells within the granuloma could be a potential disadvantage, despite these cells being in close proximity to the invading pathogen. Although granulomas were first described in 1679, true mechanistic insights into the basic biochemical signals that induce granuloma formation have only emerged in the past 15 years51. Granulomas are composed of mature macrophages that form in response to a chronic stimulus (infection or foreign body). The cytokine TNF seems crucial in this process52,53. These activated macrophages are larger with more cytoplasm and intracellular organelles and a more ruffled membrane than mononuclear phagocytes in blood50,54. Within a granuloma, macrophages can fuse into giant cells, perhaps in response to IL-4 (REF. 55). Macrophages within the granuloma were reported to have an approximately sevenfold increase in ACE mRNA compared with kidney macrophages in a zebrafish model of Mycobacterium marinum infection, in which detailed cellular events that characterize granuloma formation can be visualized56. Fluorescence imaging and electron microscopy showed that forming granulomas undergo macrophage reprogram ming to express adherens-type cellular proteins, such as epithelial cadherin (E-cadherin; also known as CDH1), which are more typical of epithelial cells than myeloid cells, thereby allowing macrophages to form cell–cell contacts. The biochemical signals that induce the ‘epithelial reprogram ming’ described in zebrafish granuloma may also contribute to the observed increase in ACE expression.
In summary, increased ACE expression by myeloid cells within a granuloma has been known for many years. However, why elevated macrophage ACE expression is such a consistent biochemical feature of a granuloma is currently unclear. One possibility is that ACE increases the antibacterial effectiveness of macrophages, as discussed below.
ACE and macrophage function.
Macrophages are important in the initial innate immune response and also have a crucial role as antigen-presenting cells (APCs) by interacting with T cells in the adaptive response. Mice overexpressing ACE in myeloid-derived cells only, known as ACE 10/10 mice, were initially developed to study the role of angiotensin II in the development of atherosclerosis but have proved useful in advancing our understanding of how ACE overexpression can affect innate and adaptive immunity57. Macrophages from ACE 10/10 mice produce 16-fold to 25-fold more ACE protein than do cells from WT littermates (FIG. 2a), whereas neutrophils and dendritic cells mildly overex-press ACE, and expression in T cells and B cells is similar to that of WT cells. By contrast, ACE expression is absent in endothelial cells, which are the main source of ACE in WT mice. Circulating ACE levels in ACE 10/10 mice are similar to those in WT mice and ACE 10/10 mice have a normal basal blood pressure.
Figure 2 |. Macrophage-specific ACE overexpression suppresses tumour growth.

a | ACE 10/10 mice overexpress angiotensin-converting enzyme (ACE) in macrophages. ACE is predominantly located on the cell surface but is also present within the endoplasmic reticulum (ER) and probably within endosomes. b | Analysis of tumour volume 14 days after intradermal implantation of B16-F10 melanoma cells into ACE 10/10 mice, ACE 10/10 mice crossed with ACE wild-type (WT) mice (heterozygous (HZ) mice) and WT mice shows that ACE overexpression in macrophages attenuates tumour growth. Data from individual mice (open blue diamonds) as well as the group means (filled orange circles) and standard error of the mean are shown. c | Representative images of the tumours at day 14 in ACE 10/10 mice and ACE WT mice are presented. Parts b and c are adapted with permission from REF. 57, Elsevier.
Insights into the functional relevance of ACE expression by myeloid cells came from a study of tumour development, in which tumours arising from implantation of B16 melanoma in ACE 10/10 mice were on average only 17% the size of those in WT mice 2 weeks after implantation57 (FIG. 2b,c). Similar differences in tumour size were also observed following implantation of tumour cells in ACE10/10 mice on an outbred genetic background and following intravenous injection of tumour cells57,58. The increased immune response originates from bone marrow-derived cells, as WT recipients of bone marrow from ACE 10/10 mice developed melanoma tumours only 51% the size of those in WT mice receiving bone marrow from WT mice. Importantly, the difference in B16 melanoma tumour size between ACE 10/10 mice and WT mice was abrogated by treatment with the ACE inhibitor captopril. By contrast, treatment of ACE 10/10 mice with the angiotensin receptor blocker losartan did not alter tumour size compared with that of untreated ACE 10/10 mice, indicating that the catalytic activity of ACE is neces sary to inhibit tumour growth but that this effect is not mediated by the effects of angiotensin II on AT1 (REF. 57). Consistent with this finding, when challenged with B16 melanoma, angiotensinogen-deficient mice with ACE-overexpressing myeloid cells develop tumours less than one-third the size of those in mice with WT levels of ACE expression in myeloid cells (Supplementary Figure S1). Taken together, these data suggest that tumour resistance in ACE 10/10 mice is not due to the absence of endothelial ACE but is rather due to overexpression of catalytically active ACE by monocytes and macrophages independent of angiotensin II signalling, thereby potentially facilitating an enhanced immune response57.
In support of this hypothesis, ACE 10/10 macro phages increase the production of pro-inflammatory cytokines — such as IL-12β, TNF or nitric oxide — beyond WT levels in response to lipopolysaccharide (LPS) or IFNγ in vitro or to B16 melanoma in vivo4,57.
To evaluate the innate immune response and test the physiological relevance of the B16 melanoma model, we challenged ACE 10/10 mice with either Listeria monocytogenes or with methicillin-resistant Staphylococcus aureus (MRSA)59. Five days after infection with L. monocytogenes, the spleens and livers of ACE 10/10 mice had 8.0-fold and 5.2-fold less bacteria than WT mice, respectively. MRSA was placed subcutaneously in the skin and, similar to observations in the melanoma model, skin lesions in these mice after 4 days of inoculation were on average 22% the size of those in WT mice and contained ~50-fold fewer viable bacteria than the lesions of WT mice. As in the B16 melanoma model, treatment of ACE 10/10 mice with the ACE inhibitor lisinopril restored the WT phenotype. Thus, ACE 10/10 mice have an enhanced immune response beyond that of WT mice in models of both innate and adaptive immunity.
ACE and antigen processing.
Major histocompatibility complex (MHC) class I antigens have a crucial role in the defence of an organism against intracellular pathogens and perhaps also against tumours. All nucleated cells can present fragments of intracellular proteins on the plasma membrane via the MHC class I complex. Cytoplasmic peptides are imported into the endoplasmic reticulum (ER) through peptide transporter involved in antigen processing (TAP)60,61. In the ER, MHC class I peptides are trimmed and loaded onto MHC class I proteins, from where they undergo vesicular trafficking to the plasma membrane (FIG. 3). These MHC class I-bound peptides can be recognized by CD8+ T cells either as self-peptides or as non-self-peptides (for example, of viral origin), thereby eliciting CD8+ T cell inactivation and activation, respectively. In addition, specialized APCs, such as macrophages and dendritic cells, take up cellular debris and efficiently present MHC class I protein-bound viral peptides to CD8+ T cells.
Figure 3 |. ACE participates in peptide trimming during antigen processing.

Angiotensin-converting enzyme (ACE) overexpression in antigen-presenting cells (APCs) likely changes the display of both major histocompatibility (MHC) class I and MHC class II epitopes. MHC class I and MHC class II proteins bind and display peptides on the surface of cells. As a peptidase, ACE can cut peptides and thereby affect the diversity of peptides that are presented to T cells. a | Cytoplasmic proteins are proteolytically cleaved by the proteasome to become potential MHC class I peptides, which are imported into the endoplasmic reticulum (ER) via peptide transporter involved in antigen processing (TAP). ACE helps trim MHC class I peptides within the ER, which occurs in both wild-type and ACE-overexpressing APCs. b | ACE expression also changes MHC class II presentation of peptide epitopes, although the exact subcellular location and mechanism is somewhat unclear. CLIP, class II-associated invariant chain peptide; Ii, invariant chain; MVB, multivesicular body; TCR, T cell receptor.
Evidence from bone marrow transplantation and other experiments in mice suggests that ACE enzymatically trims both the self and non-self MHC class I peptides before they are bound to MHC class I proteins and displayed by cells62,63 (FIG. 3a). Donor splenocytes from ACE-deficient mice were recognized as foreign by CD8+ T cells in WT syngeneic mice, and vice versa, splenocytes from WT donor mice expressing ACE were also recognized as non-self in ACE-deficient mice. These findings imply that ACE-deficient APCs process peptides differently from those that express ACE.
ACE is normally a cell surface protein and traffics through the ER. To examine whether ACE is active in the ER, we used an approach that functionally isolated the ER from the cell cytoplasm by using a cell line lacking TAP, which is thus unable to import cytoplasmic peptides into the ER63. The cell line was modified to express a short peptide from chicken ovalbumin (OVA) with a signal sequence that allowed it to enter the ER in the absence of TAP and without passing through the cytoplasm. When comparing the MHC class I peptide-processing ability of truncated ACE — which lacks the signal peptide and thereby does not enter the ER — with that of full-length ACE, which does enter the ER, we found that carboxy-terminal peptide cleavage of the OVA peptide within the ER only occurred in cells with full-length ACE. These data suggest that ACE is catalytically active in the ER, a conclusion consistent with in vivo analysis63. Additional experiments using purified cellular ER may serve to further support this conclusion.
Moreover, ACE 10/10 mice — when challenged with B16 melanoma cells constitutively expressing OVA — develop elevated levels of CD8+ T cells directed against both an intrinsic B16 melanoma MHC class I epitope and an OVA MHC class I epitope57. Thus, ACE over-expression in myeloid cells facilitates a greater CD8+ T cell response than do WT levels of ACE expression in myeloid cells. This finding is particularly interesting, given that the immune pathway(s) by which myeloid ACE overexpression increases the immune response are currently unknown. Taken together, however, these findings strongly suggest that ACE plays a crucial role in peptide antigen processing and presentation.
The presentation of MHC class I-bound and MHC class II-bound peptides is important for initial immunization to an antigen but also for re-stimulating memory T cells formed during previous antigen exposure. We compared the anti-viral memory CD8+ T cell response in ACE 10/10 and WT mice that were challenged with a vaccinia virus strain modified to express a major polyoma MHC class I epitope 1 month after an initial polyoma infection. This approach tests the ability of anti-polyoma memory T cells to recognize and kill cells infected with the modified vaccinia virus. Four days after vaccinia virus infection, viral particles could not be detected in six of eight ACE 10/10 mice compared with only one of eight WT mice64. By contrast, an equivalent experiment performed with unmodi fied vaccinia virus (a strain that does not express the polyoma epitope) showed no difference between the two groups. These experiments suggest that ACE over-expression potentiates the anti-viral memory CD8+ T cell response. Mechanistically, we do not yet know whether this potentiation is due to changes in antigen presentation or alterations in how APCs interact with T cells. Understanding the precise mechanism may contribute to novel immunization strategies. Antigen processing also has a role in antibody production, as the humoral immune response depends on antigen presentation by APCs, B cells that recognize the antigen and CD4+ T helper cells (either TH1 or TH2) that help B cells mature and proliferate. To test whether ACE expression levels in macrophages affect the humoral immune response, we used an enzyme-linked immunosorbent assay (ELISA) to measure antibody concentrations in plasma in ACE-deficient, WT and ACE 10/10 mice immunized with OVA65. We also compared the ability of these macrophages to stimulate CD4+ T cells. ACE 10/10 mice consistently produced more anti-OVA antibody than WT mice; for example, IgG1 levels in ACE 10/10 mice were over 20-fold higher than those in WT mice. Furthermore, ACE expression levels in macrophages positively correlated with their ability to stimulate OVA-sensitive CD4+ T cells ex vivo. ACE overexpression in these cells therefore facilitates the processing and display of OVA peptides bound to MHC class II proteins, which stimulate CD4+ T cells (FIG. 3b).
The extent to which ACE trimming of both MHC class I and MHC class II peptides affects antigen presentation depends on the amino acid sequence of the individual peptides. For example, increased ACE expression increases MHC class I OVA epitope (SIINFEKL) presentation but decreases presentation of the MHC class I polyoma virus middle T antigen (RRLGRTLLL)62. Similarly, ACE overexpression differentially affected the display of four hen egg lysozyme MHC class II epitopes: peptide presentation was increased for one peptide, decreased for two peptides and had no effect on the fourth peptide65. Given these data, one cannot conclude that ACE always increases the immunogenicity of a particular peptide. Rather, it seems likely that as cells change their production of ACE, the peptides they display will also change. This may ultimately increase the immune response through the simple mechanism of displaying different epitopes and engaging a different subset of CD4+ TH1 or TH2 cell clones.
ACE and neutrophil function.
Neutrophils are among the first cells to respond to a bacterial infection, and they use a variety of mechanisms — such as phagocytosis, ROS generation, antibacterial peptides and neutrophil extracellular trap (NET) formation (see below) — to destroy the invading bacteria66,67. WT neutrophils increase ACE expression levels in response to an MRSA-mediated immune challenge68. To study the effect of neutrophil ACE overexpression in response to bacterial infection, we generated a transgenic mouse model (NeuACE mice) in which so-called NeuACE neutrophils overexpress ACE by 12-fold to 18-fold compared with cells from WT mice68 (FIG. 4a). NeuACE mice have substantially increased resistance to infections of MRSA, Klebsiella pneumoniae and Pseudomonas aeruginosa. For example, when challenged with MRSA infection, the lesion size and bacterial counts in NeuACE mice were only ~25% of those observed in WT mice (FIG. 4b–d). This phenotype is dependent on neutrophil function, as neutrophil elimination with anti-neutrophil antisera eliminates the differences in both lesion size and bacterial counts between NeuACE and WT animals68. In addition, NeuACE neutrophils kill bacteria more efficiently than do WT neutrophils in vitro. By contrast, ACE-deficient neutrophils are significantly less effective at killing bacteria than are WT cells in vitro and in vivo: for example, at the end of an in vitro whole-blood killing assay, bacterial counts in samples with ACE-deficient neutrophils were 6-fold greater than in those using WT neutrophils and 30-fold greater than in those using NeuACE neutrophils68. These data suggest that a direct relationship between ACE production in neutrophils and the ability to kill bacteria exists.
Figure 4 |. ACE overexpression in neutrophils reduces skin lesions caused by MRSA infection.
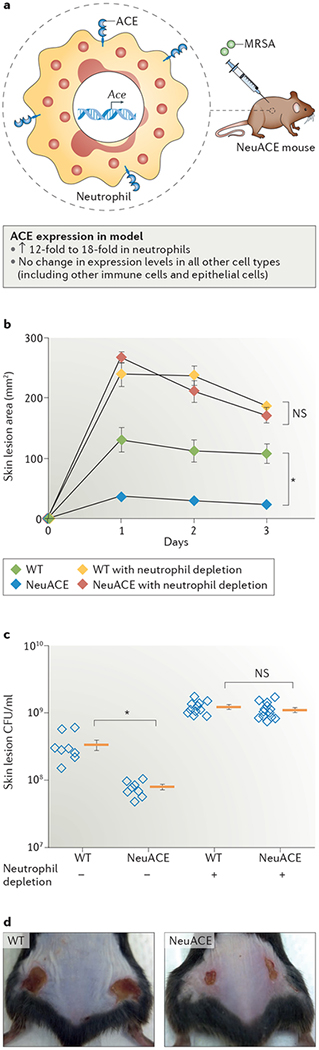
a | NeuACE mice overexpress angiotensin-converting enzyme (ACE) in neutrophils, but the enzyme is present in other cell types as well. b | Skin lesion area sizes (and standard error of the mean) of NeuACE and wild-type (WT) mice that were subcutaneously injected with methicillin-resistant Staphylococcus aureus (MRSA) on day 0, including mice that had previously been depleted of neutrophils using anti-neutrophil antibodies. The data show that overexpression of ACE in neutrophils provides resistance against MRSA infection. c | Bacterial numbers within the skin lesions after 3 days in NeuACE and WT mice show that overexpression of ACE in neutrophils attenuates bacterial numbers following MRSA infection. Data from individual mice (open blue diamonds) as well as the group means (orange bars) and standard error of the mean are shown. d | Representative images of NeuACE and WT mice 3 days after infection with MRSA, illustrating the difference in lesion size. *P < 0.001. CFU, colony forming unit; NS, not significant. Parts b–d are republished with permission of American Society of Hematology, from Khan, Z. et al. Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 130, 328–339 (2017); permission conveyed through Copyright Clearance Center, Inc. (REF. 68).
ACE and superoxide production.
One of the central ways by which neutrophils destroy bacteria is through phagocytosis, followed by fusion of the phagocyte with cytoplasmic granules. The enclosed bacteria are killed by several different mechanisms, but one of the most powerful is the generation of superoxide (O2−) by NADPH oxidase. O2− is then converted to hydrogen peroxide (H2O2) and hypochlorous acid (HClO), which are bactericidal69. To evaluate whether ACE has a role in O2− production, we challenged neutrophils from NeuACE and WT mice with MRSA and found that, although both cell populations had increased O2− production, levels from NeuACE cells were >2-fold higher than those from WT cells68. By contrast, O2− production by ACE-deficient neutrophils was ~50% that of WT neutrophils68. These changes correspond to the increased bacterial killing by NeuACE neutrophils and the decreased bacterial killing by ACE-deficient neutrophils described above. To verify that the increased bacterial killing in NeuACE neutrophils is due to increased O2−, O2− production was blocked in NeuACE and WT neutrophils using the NADPH oxidase inhibitor gp91 ds-tat. This treatment resulted in bacterial counts that were comparable between the two cell types. O2− production in ACE 10/10 macrophages was also over twofold greater than in macrophages from WT mice and thus appears to be a common feature of ACE over-expression in myeloid cells (FIG. 5). Taken together, these data suggest that increased O2− production is a major mechanism that enables improved killing of bacteria by NeuACE neutrophils.
Figure 5 |. ACE overexpression enhances the adaptive and innate immune response.

a | In ACE 10/10 mice, angiotensin-converting enzyme (ACE) overexpression in monocytes and macrophages enhances the immune response in several ways. Resistance to bacterial infection is increased through elevated production of superoxide (O2−), nitric oxide, tumour necrosis factor (TNF) and IL-12β; resistance to tumour growth is due to direct cytotoxic effects on tumour cells through an enhanced CD8+ T cell response and an increase in pro-inflammatory cytokines. In addition, ACE 10/10 mice produce more antibodies than do wild-type animals, presumably owing to an enhanced display of cell surface epitopes that increase the response of CD4+ T helper 1 (TH1) or TH2 cells, which have a key role in B cell maturation. b | In NeuACE mice, neutrophils overexpress ACE, leading to increased production of O2− in response to bacterial infection via phosphorylation of p47-PHOX, the regulatory subunit of the NADPH complex. Increased O2− production improves cytokine and neutrophil extracellular trap (NET) formation. IgG, immunoglobulin-γ; MHC, major histocompatibility complex; TCR, T cell receptor.
Neutrophils kill bacteria using mechanisms besides phagocytic killing, such as by releasing extracellular fibres termed NETs70–72, which is stimulated by ROS generation73. These fibres are composed of DNA and proteins that coalesce and act to entrap and consequently kill bacteria. NET release by NeuACE neutrophils (as measured by extracellular release of neutrophil elastase) is increased by tenfold in vitro compared with WT neutrophils68. The increase was reverted to WT levels with inhibitors of either ROS generation or ACE activity. These data show that the ACE-mediated increase in ROS production improves bacterial killing by several mechanisms.
As indicated, neutrophil O2− is produced by NADPH oxidase, a membrane-bound enzyme composed of several protein chains. This enzyme is activated when one of the protein chains called p47-PHOX (also known as NCF1) — normally located in the cytoplasm — is phosphorylated and recruited to the cell or phagocytic membrane, where it assembles with other protein chains, forming the active enzyme. We have verified, using western blots, that in NeuACE neutrophils, the increased O2− production is due to increased phosphorylation of the p47-PHOX subunit. However, the exact biochemical link between increased cellular ACE activity and increased phosphorylation of p47-PHOX is still under active investigation. One approach focuses on identifying the kinase(s) activated by ACE that phosphorylates p47-PHOX. Another approach is to investigate which peptide(s) cleaved by ACE can affect NADPH oxidase activity. Finally, studies in NeuACE mice support the conclusion from ACE 10/10 mice that the observed phenotype is independent of angiotensin II, the inflammatory mediator bradykinin or the anti-inflammatory peptide acetyl-SDKP68. Currently, the peptide(s) responsible for the increased immune effect in ACE 10/10 and NeuACE mice are not known.
Effect of ACE inhibitors on immune response.
Pharmacological ACE inhibitors, which are well tolerated by the majority of patients with hyper-tension and cardiovascular disease, do not induce immunosuppression — a finding that may not come as a surprise, given the many different and over lapping systems that make up the normal human immune response. Some circumstances in which ACE inhibitors have been implicated as negatively affecting the immune response — for example, potentially increasing the risk of urinary tract infection and sepsis74–76 — have been suggested. However, other studies have not found a deleterious effect of ACE inhibitors on the immune response77,78. Taken together, this area has not been extensively studied and requires further investigation. Others have investigated whether blocking the RAS can either change the incidence of tumours or positively affect tumour treatment79,80. Given the large number of different tumours and tumour treatments, this is still an evolving area of investigation. Extensive evidence from animal models of autoimmune disease (EAE, arthritis, autoimmune myocarditis and other diseases) indicates that inhibitors of ACE and the RAS typically suppress the autoimmune process37,38,81–85. These findings are consistent with Ace−/− mice having a less vigorous immune response to MRSA infection68. In humans, there is little information on the effectiveness of RAS blockade in treating autoimmune diseases such as rheumatoid arthritis, though one small-cohort study (n = 15) suggests a positive effect of ACE inhibition in 66% of patients86,87. Additional progress in human disease probably awaits a better mechanistic understanding of how exactly ACE affects the immune response.
Conclusion and outlook
ACE is naturally overexpressed in response to an immune challenge in the context of granulomas. Studies of macrophages and neutrophils in which ACE is over-expressed now indicate that their immune function is substantially augmented beyond that of stimulated WT cells57,59,68. Whether ACE also has a role in the migration of these cells is currently unknown and warrants further investigation.
Furthermore, the effects of ACE overexpression in human macrophages and neutrophils are currently unknown. The differentiation of human monocytes into either macrophages or dendritic cells by in vitro culture was reported to be associated with an increase in ACE expression (40-fold to 55-fold for macrophages and 150-fold for dendritic cells)88,89. In part, these drastic increases are due to the very low basal ACE expression levels in monocytes. Nonetheless, this pattern of increasing ACE expression with human myeloid differentiation is similar to what has been observed in mice63. Further experiments are needed to study whether ACE over-expression in human myeloid cells has effects similar to those in mice.
Beyond its role in immune function, ACE has been linked to neurodegenerative disease (BOX 2), and further understanding of its pleiotropic effects could therefore have additional benefits. We do not believe there are many systems that improve the immune cell response beyond that achieved by maximally stimulating WT cells, yet ACE overexpression seems to supercharge the immune response. The improved immune response induced by ACE overexpression is dependent on the catalytic activity of the enzyme. Either the elimination of currently unknown ACE substrate(s) or the production of undefined ACE product(s) must profoundly influence myeloid cell function, presumably through an unknown series of downstream biochemical events. Determining the detailed biochemical pathway by which ACE augments myeloid cell function could lead to the design of pharmacological agents to either mimic the effect of ACE overexpression (to improve the immune response) or block the effect (to achieve immuno suppression). Thus, the ACE substrate(s) that elicit an increased immune response and the downstream pathway(s) that instigate these effects are the key areas of interest and hold great promise for novel therapies.
Box 2 |. ACE in Alzheimer disease.
Alzheimer disease is the most common neurodegenerative disease in the US with an estimated prevalence of 5.5 million people103,104. Although the pathogenesis of Alzheimer disease is complex, substantial evidence suggests that the accumulation of amyloid plaques within the central nervous system is a major pathological feature of the disease105. Some authors have speculated whether immunotherapy might be advantageous in the treatment of this disease106. To examine whether an improved adaptive immune response mediated by angiotensin-converting enzyme (ACE) overexpression would affect amyloid plaque formation, we compared Alzheimer-prone mice crossed with ACE 10/10 mice to Alzheimer-prone mice with wild-type (WT) ACE expression and found that increased ACE levels in macrophages conferred a 70% reduction in plaque size in mice aged 8 months (Supplementary Figure S2)107. More importantly, cognitive function, as assessed using a Barnes maze test, in the Alzheimer-prone mice overexpressing ACE in macrophages was comparable to that of WT mice (non-Alzheimer-prone control mice) with WT ACE levels. Although the clinical relevance of these results is limited because the experiments were only conducted in mice, this study indicates the potential of manipulating ACE expression and/or activity in Alzheimer disease.
Supplementary Material
Key points.
Angiotensin-converting enzyme (ACE) expression by myeloid cells is increased in response to infection.
Forced ACE overexpression in mouse macrophages increases their ability to respond to infection and some tumour models, which is in part mediated by increased levels of nitric oxide, superoxide (O2−) and pro-inflammatory cytokines.
Forced ACE overexpression in neutrophils increases their response to infection by increasing NADPH oxidase-dependent production of O2−.
The effects of ACE overexpression are not mediated by angiotensin II, any other angiotensin peptides or the type 1 angiotensin II receptor but instead by unknown ACE substrate(s) or product(s).
No known immunological framework can currently explain the effects of ACE overexpression, but an as yet unrecognized pathway capable of stimulating myeloid function beyond levels achievable by wild-type cells must exist.
ACE inhibitors.
Compounds that block the formation of angiotensin II and all other angiotensin-converting enzyme (ACE) products by inhibiting ACE activity.
Sarcoidosis.
A granulomatous disease characterized by abnormal foci of inflammation. This disease is often associated with high plasma levels of angiotensin-converting enzyme.
Type 1 angiotensin II receptors.
(AT1Rs). Receptors for angiotensin II that mediate a variety of functions, such as decreased renal blood flow. Whereas humans have a single gene for this receptor (AGTR1), mice have two genes (Agtr1a and Agtr1b), which encode proteins that are referred to as AT1A and AT1B, respectively. Studies in Agtr1a–/– mice have demonstrated that most angiotensin II-mediated effects in the kidney are mediated by AT1A.
ACE-like enzymes.
Enzymes that have protein sequence similarity to human or mouse angiotensin-converting enzyme (ACE). Such enzymes usually bind zinc and have two catalytic domains.
Granulomas.
Compact aggregates of immune cells in response to infection or another form of chronic stimulation, resulting in a physical barrier surrounding the enclosed bacteria and hindering the spread of the pathogen throughout the organism. Tuberculosis is most commonly associated with granulomas, but a number of other infectious and noninfectious diseases induce granulomas, as do difficult-tophagocytize inert foreign bodies such as those in berylliosis.
Histoplasmosis.
A granulomatous disease, often primary of the lung, caused by the fungus Histoplasma capsulatum.
Leprosy.
A granulomatous disease caused by Mycobacterium leprae; also known as Hansen disease.
Silicosis.
A granulomatous disease, often primary of the lung, caused by exposure to silica dust.
Granulomatosis with polyangiitis.
A systemic form of vasculitis (inflammation of blood vessels) often referred to as Wegener granulomatosis. The vasculitis typically involves small-to-medium-sized blood vessels that become inflamed and can develop granulomas. The origin of the disease is thought to be due to anti-neutrophil cytoplasmic antibodies (ANCAs).
Schistosomiasis.
A parasitic infection caused by the flatworm genus Schistosoma. Eggs of these worms can induce a granulomatous inflammatory response.
ACE 10/10 mice.
Mice homozygous for the angiotensin-converting enzyme (ACE) 10 mutation, resulting in ACE overexpression in myeloid cells, particularly in monocytes and macrophages. These animals develop normally and have normal blood pressure because ACE present on the surface of monocytes and macrophages, as well as circulating ACE shed from these cells, maintains normal angiotensin II plasma levels.
B16 melanoma.
An aggressive mouse tumour cell line that, when implanted into the skin, develops into a 600 mm3 lesion in ~2 weeks.
Syng4eneic mice.
Mice that can donate tissue or cells without triggering an immune response in the recipient mice because cellular proteins and the derived cell surface major histocompatibility complex (MHC) class I peptides are identical.
Ovalbumin.
(OVA). A main protein found in egg white. OVA is often used in immunology studies, as many reagents are commercially available, and many details are known as to how the protein stimulates an immune response.
NeuACE mice.
Mice that have been genetically modified to overexpress angiotensin-converting enzyme (ACE) in neutrophils. Unlike ACE 10/10 mice, NeuACE mice retain normal levels of ACE expression in all other tissues and cells, including lungs, kidneys, monocytes and macrophages. These animals also develop normally and have normal blood pressure.
Anti-neutrophil antisera.
Antibodies that recognize and attach to neutrophils, leading to neutrophil depletion.
Amyloid plaques.
Extracellular collections of amyloid protein in the brain. Such plaques are typical of Alzheimer disease.
Alzheimer-prone mice.
Mice that have been genetically modified to produce mutant versions of amyloid precursor protein and presenilin owing to expression of transgenic APPK595N,M596L and PS1ΔE9. These mice develop amyloid plaques in the brain over time and show a decreased ability to learn new tasks.
Barnes maze test.
A test involving a white platform with 20 equally spaced holes. Only one hole leads to an escape box, whereas the other holes lead to boxes that are too small to enter. The mice are trained to enter the escape box over several days. By measuring the time an animal takes to enter the escape box, and by measuring how rapidly knowledge of the location of the escape box is lost over several days, one can estimate learning and memory retention.
Acknowledgements
The authors thank B. Taylor of Cedars-Sinai Medical Center for help in preparing the manuscript before submission. The authors’ work described in this Review was supported by US National Institute of Health grants P01HL129941, R21AI114965 and R03DK101592, and American Heart Association grants 16SDG30130015 and 17GRNT33661206.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/nrneph.2018.15.
References
- 1.Skeggs LT Jr Discovery of the two angiotensin peptides and the angiotensin converting enzyme. Hypertension 21, 259–260 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Skeggs LT Jr., Marsh WH, Kahn JR & Shumway NP The existence of two forms of hypertensin. J. Exp. Med 99, 275–282 (1954). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metzger R et al. Heterogeneous distribution of angiotensin I-converting enzyme (CD143) in the human and rat vascular systems: vessel, organ and species specificity. Microvasc. Res 81, 206–215 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Bernstein KE A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol. Rev 65, 1–46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rigat B et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Invest 86, 1343–1346 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dux S et al. Serum angiotensin converting enzyme activity in normal adults and patients with different types of hypertension. Isr. J. Med. Sci 20, 1138–1142 (1984). [PubMed] [Google Scholar]
- 7.Bénéteau-Burnat B, Baudin B, Morgant G, Baumann FC & Giboudeau J Serum angiotensin-converting enzyme in healthy and sarcoidotic children: comparison with the reference interval for adults. J. Clin. Chem 36, 344–346 (1990). [PubMed] [Google Scholar]
- 8.Cushman DW & Ondetti MA History of the design of captopril and related inhibitors of angiotensin converting enzyme. Hypertension 17, 589–592 (1991). [DOI] [PubMed] [Google Scholar]
- 9.Mentz RJ et al. The past, present and future of renin-angiotensin aldosterone system inhibition. Int. J. Cardiol 167, 1677–1687 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med 316, 1429–1435 (1987). [DOI] [PubMed] [Google Scholar]
- 11.The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N. Engl. J. Med 325, 293–302 (1991). [DOI] [PubMed] [Google Scholar]
- 12.Lieberman J Elevation of serum angiotensin-converting-enzyme (ACE) level in sarcoidosis. Am. J. Med 59, 365–372 (1975). [DOI] [PubMed] [Google Scholar]
- 13.Silverstein E, Pertschuk LP & Friedland J Immunofluorescent localization of angiotensin converting enzyme in epithelioid and giant cells of sarcoidosis granulomas. Proc. Natl Acad. Sci. USA 76, 6646–6648 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smithies O, Kim. HS, Takahashi N & Edgell MH Importance of quantitative genetic variations in the etiology of hypertension. Kidney Int 58, 2265–2280 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Kim S & Iwao H Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol. Rev 52, 11–34 (2000). [PubMed] [Google Scholar]
- 16.Krege JH et al. Male-female differences in fertility and blood pressure in ACE deficient mice. Nature 375, 146–148 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Esther CR Jr et al. Mice lacking angiotensin-converting enzyme have low blood pressure, renal pathology, and reduced male fertility. Lab. Invest 74, 953–965 (1996). [PubMed] [Google Scholar]
- 18.Gribouval O et al. Spectrum of mutations in the reninangiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum. Mutat 33, 316–326 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Ertoy D & Bernstein KE in Drugs, Enzymes and Receptors of the Renin-Angiotensin System: Celebrating a Century of Discovery (eds Husain A & Graham R) 205–223 (Harwood Academic Publishers, The Netherlands, 2000). [Google Scholar]
- 20.Kim HS et al. Genetic control of blood pressure and the angiotensinogen locus. Proc. Natl Acad. Sci. USA 92, 2735–2739 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanai K et al. Renin-dependent cardiovascular functions and renin-independent blood-brain barrier functions revealed by renin-deficient mice. J. Biol. Chem 275, 5–8 (2000). [DOI] [PubMed] [Google Scholar]
- 22.Tsuchida S et al. Murine double nullizygotes of the angiotensin type 1A and 1B receptor genes duplicate severe abnormal phenotypes of angiotensinogen nullizygotes. J. Clin. Invest 101, 755–760 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazaki Y et al. Angiotensin induces the urinary peristaltic machinery during the perinatal period. J. Clin. Invest 102, 1489–1497 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs S et al. Male fertility is dependent on dipeptidase activity of testis ACE. Nat. Med 11, 1140–1142 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Li LJ et al. Human sperm devoid of germinal angiotensin-converting enzyme is responsible for total fertilization failure and lower fertilization rates by conventional in vitro fertilization. Biol. Reprod 90 125, 1–7 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Hagaman JR et al. Angiotensin-converting enzyme and male fertility. Proc. Natl Acad. Sci. USA 95, 2552–2557 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramaraj P, Kessler SP, Colmenares C & Sen GC Selective restoration of male fertility in mice lacking angiotensin-converting enzymes by sperm-specific expression of the testicular isozyme. J. Clin. Invest 102, 371–378 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marchesi C, Paradis P & Schiffrin EL Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci 29, 367–374 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Benigni A, Cassis P & Remuzzi G Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol. Med 2, 247–257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montezano AC, Nguyen Dinh Cat A, Rios FJ & Touyz RM Angiotensin II and vascular injury. Curr. Hypertens. Rep 16, 431 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Mateo T et al. Angiotensin II-induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J. Immunol 176, 5577–5586 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Nathan C Specificity of a third kind: reactive oxygen and nitrogen intermediates in cell signaling. J. Clin. Invest 111, 769–778 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torres M & Forman HJ Redox signaling and the MAP kinase pathways. Biofactors 17, 287–296 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Biancardi VC, Bomfim GF, Reis WL, Al-Gassimi S & Nunes KP The interplay between Angiotensin II, TLR4 and hypertension. Pharmacol. Res 120, 88–96 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Meng Y, Chen C, Liu Y, Tian C & Li HH Angiotensin II regulates dendritic cells through activation of NF-κB /p65, ERK1/2 and STAT1 pathways. Cell Physiol. Biochem 42, 1550–1558 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Hoch NE et al. Regulation of T cell function by endogenously produced angiotensin II. Am. J. Physiol. Reful Integr. Comp. Physiol 296, R208–R216 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Platten M et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl Acad. Sci. USA 106, 14948–14953 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lanz TV et al. Angiotensin II sustains brain inflammation in mice via TGF-beta. J. Clin. Invest 120, 2782–2794 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crowley SD, & Rudemiller NP. Immunologic Effects of the Renin-Angiotensin System. J. Am. Soc. Nephrol 28, 1350–1361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowley SD et al. A role for angiotensin II type 1 receptors on bone marrow-derived cells in the pathogenesis of angiotensin II-dependent hypertension. Hypertension 55, 99–108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang JD et al. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ. Res 110, 1604–1617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang JD et al. Type 1 angiotensin receptors on macrophages ameliorate IL-1 receptor-mediated kidney fibrosis. J. Clin. Invest 124, 2198–2203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma LJ et al. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am. J. Physiol. Renal Physiol 300, F1203–F1213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brice EA, Friedlander W, Bateman ED & Kirsch RE Serum angiotensin-converting enzyme activity, concentration, and specific activity in granulomatous interstitial lung disease, tuberculosis, and COPD. Chest 107, 706–710 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Lieberman J & Rea TH Serum angiotensin converting enzyme in leprosy and coccidioidomycosis. Ann. Intern. Med 87, 422–425 (1977). [DOI] [PubMed] [Google Scholar]
- 46.Olle EW et al. Screening of serum samples from Wegener’s granulomatosis patients using antibody microarrays. Proteom. Clin. Appl 1, 1212–1220 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Weinstock JV Production of neuropeptides by inflammatory cells within the granulomas of murine schistosomiasis mansoni. Eur. J. Clin. Invest 21, 145–153 (1991). [DOI] [PubMed] [Google Scholar]
- 48.Williams GT & Williams WJ Granulomatous inflammation - a review. J. Clin. Pathol 36, 723–733 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nathan C Macrophages’ choice: take it in or keep it out. Immunity 45, 710–711 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Ramakrishnan L Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol 12, 352–366 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Sylvius F Opera Medica (Wolfgang A, 1679).
- 52.Bean AG et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol 162, 3504–3511 (1999). [PubMed] [Google Scholar]
- 53.Roach DR et al. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J. Immunol 168, 4620–4627 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Bouley DM, Ghori N, Mercer KL, Falkow S & Ramakrishnan L Dynamic nature of host-pathogen interactions in Mycobacterium marinum granulomas. Infect. Immun 69, 7820–7831 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helming L & Gordon S The molecular basis of macrophage fusion. Immunobiology 212, 785–793 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Cronan et al. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity 45, 861–876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen XZ et al. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am. J. Pathol 170, 2122–2134 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shen XZ et al. Myeloid expression of angiotensin-converting enzyme facilitates myeloid maturation and inhibits the development of myeloid-derived suppressor cells. Lab Invest 94, 536–544 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okwan-Duodu D et al. Angiotensin-converting enzyme overexpression in mouse myelomonocytic cells augments resistance to Listeria and methicillin-resistant Staphylococcus aureus. J. Biol. Chem 285, 39051–39060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pamer E & Cresswell P Mechanism of MHC class I− resticted antigen processing. Annu. Rev. Immunol 16, 323–358 (1998). [DOI] [PubMed] [Google Scholar]
- 61.Purcell AW & Elliott T Molecular machinations of the MHC-I peptide loading complex. Curr. Opin. Immunol 20, 75–81 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Shen XZ, Lukacher AE, Billet S, Williams IR & Bernstein KE Expression of angiotensin-converting enzyme changes major histocompatibility complex class I peptide presentation by modifying C termini of peptide precursors. J. Biol. Chem 283, 9957–9965 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shen XZ et al. The carboxypeptidase ACE shapes the MHC class I peptide repertoire. Nat. Immunol 12, 1078–1085 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gonzalez-Villalobos RA et al. Rediscovering ACE: novel insights into the many roles of the angiotensin converting enzyme. J. Mol. Med 91, 1143–1154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao T, Bernstein KE, Fang J & Shen XZ Angiotensin-converting enzyme affects the presentation of MHC class II antigens. Lab. Invest 97, 764–771 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Oliveira S, Rosowski EE & Huttenlocher A Neutrophil migration in infection and wound repair: going forward in reverse. Nat. Rev. Immunol 16, 378–391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guerra FE, Borgogna TR, Patel DM, Sward EW & Voyich JM Epic immune battles of history: neutrophils versus Staphylococcus aureus. Front. Cell. Infect. Microbiol 7, 286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Khan Z et al. Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 130, 328–339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Winterbourn CC, Kettle AJ & Hampton MB Reactive oxygen species and neutrophil function. Annu. Rev. Biochem 85, 765–792 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Li P et al. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med 207, 1853–1862 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Remijsen Q et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 18, 581–588 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Branzk N et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol 15, 1017–1025 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Branzk N & Papayannopoulos V Molecular mechanisms regulating NETosis in infection and disease. Semin. Immunopathol 35, 513–530 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pouwels KB, Visser ST & Hak E Effect of pravastatin and fosinopril on recurrent urinary tract infections. J. Antimicrob. Chemother 68, 708–714 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pouwels KB, Bos JH & Hak E ACE inhibitors and urinary tract infections. Epidemiology 25, 466–467 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Dial S, Nessim SJ, Kezouh A, Benisty J & Suissa S Antihypertensive agents acting on the reninangiotensin system and the risk of sepsis. Br. J. Clin. Pharmacol 78, 1151–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mortensen EM, Restrepo MI, Anzueto A & Pugh J The impact of prior outpatient ACE inhibitor use on 30-day mortality for patients hospitalized with community-acquired pneumonia. BMC Pulm. Med 5, 12 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mortensen EM et al. Impact of statins and angiotensin converting enzyme inhibitors on mortality of subjects hospitalised with pneumonia. Eur. Respir. J 31, 611–617 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Sobczuk P, Szczylik C, Porta C & Czarnecka AM Renin angiotensin system deregulation as renal cancer risk factor. Oncol. Lett 14, 5059–5068 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pinter M & Jain RK Targeting the reninangiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl Med 9, eaan5616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Steinman L Development of therapies for autoimmune disease at Stanford: a tale of multiple shots and one goal. Immunol. Res 58, 307–314 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Lühder F, Lee DH, Gold R, Stegbauer J & Linker RA Small but powerful: short peptide hormones and their role in autoimmune inflammation. J. Neuroimmunol 217, 1–7 (2009). [DOI] [PubMed] [Google Scholar]
- 83.Fahmy Wahba MG, Shehata Messiha BA & Abo-Saif AA Ramipril and haloperidol as promising approaches in managing rheumatoid arthritis in rats. Eur. J. Pharmacol 765, 307–315 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Sagawa K, Nagatani K, Komagata Y & Yamamoto K Angiotensin receptor blockers suppress antigen-specific T-cell responses and ameliorate collagen-induced arthritis in mice. Arthr. Rheumat 52, 1920–1928 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Bahk TJ, Daniels MD, Leon JS, Wang K & Engman DM Comparison of angiotensin converting enzyme inhibition and angiotensin II receptor blockade for the prevention of experimental autoimmune myocarditis. Int. J. Cardiol 125, 85–93 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chang Y & Wei W Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clin. Exp. Immunol 179, 137–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martin MF et al. Captopril: a new treatment for rheumatoid arthritis? Lancet 1, 1325–1328 (1984). [DOI] [PubMed] [Google Scholar]
- 88.Danilov SM et al. Angiotensin-converting enzyme (CD143) is abundantly expressed by dendritic cells and discriminates human monocyte-derived dendritic cells from acute myeloid leukemia-derived dendritic cells. Exp. Hematol 31, 1301–1309 (2003). [DOI] [PubMed] [Google Scholar]
- 89.Viinikainen A, Nyman T, Fyhrquist F & Saijonmaa O Downregulation of angiotensin converting enzyme by TNF-alpha in differentiating human macrophages. Cytokine 18, 304–310 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Inagami T in Biochemical Regulation of Blood Pressure (ed. Soffer RL) 39–72 (John Wiley and Sons, New York, 1981). [Google Scholar]
- 91.Sun X et al. Catabolic attacks of membrane-bound angiotensin-converting enzyme on the N-terminal part of species-specific amyloid-beta peptides. Eur. J. Pharmacol 588, 18–25 (2008). [DOI] [PubMed] [Google Scholar]
- 92.Chen HL, Lünsdorf H, Hecht HJ & Tsai H Porcine pulmonary angiotensin I-converting enzyme—biochemical characterization and spatial arrangement of the N- and C-domains by three-dimensional electron microscopic reconstruction. Micron 41, 674–685 (2010). [DOI] [PubMed] [Google Scholar]
- 93.Allinson TM et al. The role of ADAM10 and ADAM17 in the ectodomain shedding of angiotensin converting enzyme and the amyloid precursor protein. Eur. J. Biochem 271, 2539–2547 (2004). [DOI] [PubMed] [Google Scholar]
- 94.Parkin ET, Turner AJ & Hooper NM Secretase-mediated cell surface shedding of the angiotensin-converting enzyme. Protein Pept. Lett 11, 423–432 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Barauna VG, Campos LC, Miyakawa AA & Krieger JE ACE as a mechanosensor to shear stress influences the control of its own regulation via phosphorylation of cytoplasmic Ser(1270). PLoS ONE 6, e22803 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fleming I Signaling by the angiotensin-converting enzyme. Circ. Res 98, 887–896 (2006). [DOI] [PubMed] [Google Scholar]
- 97.Howard TE, Shai S-Y, Langford KG & Martin BM & Bernstein KE Transcription of testicular angiotensin-converting enzyme (ACE) is initiated within the 12th intron of the somatic ACE gene. Mol. Cell. Biol 10, 4294–4302 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wei L, Alhenc-Gelas F, Corvol P & Clauser E The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem 266, 9002–9008 (1991). [PubMed] [Google Scholar]
- 99.Fuchs S et al. Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension 51, 267–274 (2008). [DOI] [PubMed] [Google Scholar]
- 100.Rousseau A et al. The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J. Biol. Chem 270, 3656–3661 (1995). [DOI] [PubMed] [Google Scholar]
- 101.Zhu L et al. Ac-SDKP suppresses TNF-α-induced ICAM-1 expression in endothelial cells via inhibition of IκB kinase and NF-κB activation. Am. J. Physiol. Heart Circ. Physiol 310, H1176–H1183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hubert C, Houot AM, Corvol P & Soubrier F Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem 266, 15377–15383 (1991). [PubMed] [Google Scholar]
- 103.[No authors listed.] Alzheimer’s Disease Facts and Figures Alzheimer’s Association https://www.alz.org/facts/ (2017)
- 104.Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 12, 459–509 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Selkoe DJ & Hardy J The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schwartz M Can immunotherapy treat neurodegeneration? Science 357, 254–255 (2017). [DOI] [PubMed] [Google Scholar]
- 107.Bernstein KE et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Invest 124, 1000–1012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.