

Abstract
Sulfur(VI) Fluoride Exchange (SuFEx) is a new family of click chemistry transformations which relies on readily available materials to produce compounds bearing the SVI—F motif. The potential of SuFEx in drug discovery has just started to be explored. We report the first method of SuFEx chemistry for the conversion of phenolic compounds to their respective arylfluorosulfate derivatives in situ in 96-well plates. This method is compatible with automated synthesis and screening to quickly assess the biological activities of the in situ generated, crude products. U sing this method, we perform late-stage functionalization of a panel of known anticancer drugs to generate the corresponding arylfluorosulfates. These in situ generated arylfluorosulfates are directly tested in a cancer-cell growth inhibition assay in parallel with their phenolic precursors. We discover three arylfluorosulfates that exhibit improved anticancer cell proliferation activities compared to their phenol precursors. Among these three compounds, the fluorosulfate derivative of Fulvestrant possesses significantly enhanced activity to down-regulate estrogen receptor (ER) expression in ER+ breast cancer cell line M CF-7 and the fluorosulfate derivative of Combretastatin A 4—a general anticancer drug currently being evaluated under clinical trials—exhibits a 70-fold increase in potency in the drug resistant colon cancer cell line HT-29.
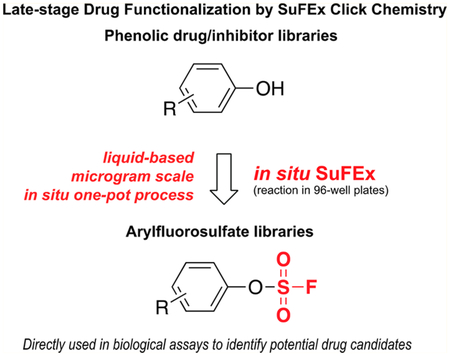
Graphical Abstract
INTRODUCTION
Click chemistry, inspired by Nature’s powerful heteroatom-linkage creating strategy, has found broad applications in materials chemistry, chemical biology, and drug development since the concept was first introduced in 1998.1 The sulfur(VI) fluoride exchange (SuFEx), developed by the Sharpless lab in 2014, represents another set of ideal click chemistry transformations.2 In the manifestation of SuFEx, arylfluorosulfates (Ar—O—SO2—F) and iminosulfur oxydifluorides (R—N= SOF2) are readily synthesized using two gases sulfuryl fluoride (SO2F2) and thionyl tetrafluoride (SOF4), respectively.2,3 Though rarely studied in the past, these two S(VI)—F motifs have since been successfully used as connective linkers in polymer synthesis and for the construction of various functional molecules.2,4,5
However, due to the previous inaccessibility of these com pounds and their assumed high reactivity toward biomolecules as is the case for sulfur(VI) chlorides, the study and application of S(VI)—F in medicinal chemistry remain largely unexplored.6 In the Sharpless lab’s pilot work with the Kelly group, it was discovered that arylfluorosulfates are only reactive toward proximal nucleophilic residues, especially tyrosines, found within specific protein partners.7,8 These unique features make arylfluorosulfates distinct from other classes of known covalent bioprobe groups and herald SuFEx as an emerging invaluable tool in drug development.
Late-stage functionalization (LSF), a strategy for directly introducing functional groups onto a bioactive compound in the late stage of its synthesis,9 enables rapid diversification of drug candidates or drug-like molecules to improve their properties such as potency and metabolic properties.10,11 Many innovative methods have been developed for this endeavor, including late stage C—H functionalization and nucleophilic aromatic substitution, just to name a few.12–14 LSF requires a chemical reaction with high selectivity, high yield, and mild reaction conditions. Converting a phenolic compound with known biological activities to the corresponding arylfluorosulfate via SuFEx is an excellent transformation for LSF waiting to be explored. In fact, the phenolic hydroxyl group is often employed in drug modification and diversification.5–17 We hypothesize that by converting a phenolic molecule with known biological activities to the corresponding arylfluorosulfate will serve as a quick and cost-effective way to identify new hits with improved properties. Currently, there are approximately 120 phenolic compounds within the repertoire of United States Food and Drug Administration (FDA)-approved drugs. In addition, hundreds of drug candidates bearing phenols are under investigation according to the DrugBank database (https://www.drugbank.ca). The wide distribution of phenolic groups makes the hypothesis easy to test. However, the current gas-liquid interface based reaction protocols for the installation of arylfluorosulfates are hurdles for the direct transfer of SuFEx to the drug development pipeline. In a drug discovery process, compounds are usually tested using automated screening to quickly assess their biological activities,18,19 which requires a protocol to achieve the chemoselective and efficient synthesis of screening compounds in situ in multi well plates with low substrate concentrations and small volumes. Ideally, compounds produced in such ways should be subjected to biological assays directly without further purification.
Here, we report the first protocol of SuFEx click chemistry for the LSF of phenol-containing drugs or drug candidates and converting them to their respective arylfluorosulfate derivatives in situ in 96-well plates (Scheme 1). The in situ generated crude products of arylfluorosulfates are directly tested in a cancer-cell growth inhibition assay together with their phenolic precursors. Three arylfluorosulfates are discovered that exhibit improved anticancer proliferation activities compared to their phenol precursors. Among these three compounds, the fluorosulfate derivative of Combretastatin A 4—a general anticancer drug currently being evaluated under clinical trials for advanced anaplastic thyroid cancer and platinum-resistant ovarian cancer20,21—exhibits a 70-fold increase in potency on the Combretastatin resistant colon cancer cell line HT-29.22
Scheme 1.

Comparison of the Standard SuFEx and in Situ SuFEx Workflows for Phenolic Drug Functionalization
RESULTS
Development of a Liquid-Based in Situ SuFEx protocol.
A cost-effective protocol for converting a commercial screening library into a new library via LSF should possess the following characteristics: (1) compatible with small reaction scales (e.g., microgram); (2) excellent chemoselectivity; and (3) directly transferable to biological assays. Currently, the established procedure of synthesizing arylfluorosulfates is performed at the gas-liquid interface (Figure S1A): phenol compounds dissolved in organic solvents (e.g., dichloro-methane (DCM)) are subjected to the SO2F2 gas in a sealed reaction vessel in the presence of an organic base (e.g., N,N-diisopropylethylamine (DIPEA)).2 This protocol is suitable for high concentration (>100 mM) reactions in which substrates are present in milligram or greater scales, but becomes impractical for low concentration samples (<10 mM) due to dramatically decreased reaction rates under such conditions.
To develop an in situ SuFEx procedure that could be easily coupled with LSF and quick biological assays, we explored the feasibility of predissolving SO2F2 in an organic solvent to form a saturated solution and use it to react with phenolic compounds in 96-well plates. Such a procedure can be easily conducted by multichannel pipet or a robotic system. Indeed, SO2F2 is known to have good solubility in several organic solvents including carbon tetrachloride and toluene.23 Toward this end, saturated solutions of SO2F2 in various organic solvents (100 μL) were prepared and mixed with Ezetimibe 3, a commercial phenol containing drug that is used to regulate plasma cholesterol levels (0.1 μmol in 10 μL dimethyl sulfoxide (DMSO)), in the presence of TEA or DIPEA (10 equiv., in 10 μL of the solvent) in a sealed Eppendorf tube at room temperature. The reaction progress was monitored by liquid chromatography-mass spectrometry (LC-MS). We found that reactions in CH3CN afforded significantly better yields after 3 h compared to those obtained by using DCM or T H F as the solvent (Figure S2). Next, the efficiency of the interfacial and the liquid-based method was compared in a 96-well plate. Phenol compounds 1, 2, 3 and 4 (0.1 μMmol) were treated either with the SO2F2 gas over an open 96-well plate as shown in Figure S1B or through the addition of a dissolved SO2F2 solution (~ 4 mg/mL in CH3CN) in a sealed 96-well plate as shown in Figure S1C for 12 h (a total volume of 120 μL in each well). All transformations achieved higher yields in the liquid-based system compared to the gas-liquid based method (>20% increase) (Figure 1A).
Figure 1.

Develop and optimize in situ SuFEx reaction and processing conditions. (A) Com parison of gas-liqu id based SuFEx and liquid based SuFEx protocols for reactions in 96-well plates. (B) Removal of fluoride ions in situ after reaction: (1) Volatiles in the reaction mixture were removed in vacuo and19FNMR showed the presence of anionic fluorine (−151 ppm, in CDCI3); (2) after treating with TMSOH, volatiles in the reaction mixture were removed in vacuo and 19FNMR showed no presence of anionic fluorine or TMSF.
Unlike low boiling point solvents and organic bases, fluoride ions generated in this transformation could not be removed by vacuo (−151 ppm in CDCl3; Figure 1B). Fluoride ions might have a synergistic or detrimental effect on arylfluorosulfates in the downstream biological assay.24–26 To minimize such influence on the screening results, excess trimethylsilanol (TMSOH, 20 equiv., boiling point 99°C at 1 atm) was used to convert fluoride ions into volatile fluorotrimethylsilane (TMSF, −158 ppm in CDCl3) whose boiling point is 16°C at 1 atm. The subsequent in vacuo treatment could then remove nearly all low boiling point components (TMSF, TMSOH, and TEA), leaving behind only a small amount of unreacted phenol precursors and the arylfluorosulfate products, which could be directly used for the subsequent biological screening tests (Figure 1B; a detailed 19FNMR analysis is shown in Figure S3).
Using this liquid-based protocol in a 96-well plate, we successfully obtained thirty-nine fluorosulfurylation products (0.1 μMmol) in good to quantitative yields, whose parent phenolic compounds27 all possess anticancer activities (Figure S4, the phenol compounds were numbered as 1–39 and the corresponding fluorosulfurylation products were designated 1 F-39F). Notably, some of the compounds bearing multiple phenolic hydroxyl groups (compound 5, 10, 27, and 37) could be fully transformed into the corresponding fluorosulfurylation products. Finally, the crude products were dissolved in DMSO (10 μL; approximately 10 mM) for the direct evaluation of their anticancer activities.
Screening the Anticancer Activity of the in Situ SuFEx Generated 1F–39F.
First, we assessed the cytotoxicity of fluorosulfurylation products 1F-39F and their phenol precursors in cancer cell lines using a double-dose cell viability test. The adenocarcinomic human alveolar basal epithelial cell A549 and the breast cancer cell M CF-7 were treated with vehicle (0.2% DMSO), fluorosulfurylation products or their phenol precursors in a final concentration of 20 μM or 500 nM for 72 h before the cytotoxicity of each compound was assessed according to the viability of the coumpound treated cancer cells relative to that of the vehicle treated control. The difference between the cytotoxicity of a fluorosulfurylation product and that of its phenol precursor reflects the changes in the compound’s anticancer activity after the SuFEx-based derivatization (Figure 2A). A fluorosulfurylation product exhibiting strong cytotoxicity (greater than 50% inhibition at both 20 μM and 500 nM on the growth of M CF-7 or A549 after a three-day treatment) and significantly increased (> 1 0%) cytotoxicity over the parent compound is considered as a “hit”. Based on these two selection criteria, three hits 1F, 11F, and 25F were identified. As shown in Figure 2B, 1F showed significantly enhanced activity than phenol 1 at 500 nM (6 % increase in M CF-7 and 27% increase in A549). The cytotoxicity of 11F was stronger than that of 11 in M CF-7 cell line with an increase of 32% and 20%, respectively, at 20 μM and 500 nM. However, no significant differences were found in A549 cells. Although 25 and 25F showed similar activities in M CF-7 cells at 20 μM, 25F indeed exhibited 8% enhanced cytotoxicity at 500 nM, and its activities were significantly stronger than those of 25 at both concentrations in A549 cells. Notably, the corresponding phenol precursor 1, 11, and 25 either are FDA-approved drugs or are currently under clinical trials. Specifically, 1 (ABT-751), which inhibits polymerization of microtubules, is under a phase 1/2 clinical trial;28,29 11 (Fulvestrant) is the only selective estrogen receptor down regulator (SERD) for the treatment of ER+ metastatic breast cancer in postmenopausal women whose disease has spread after antiestrogen therapy;30–32 25 (Combretastatin A4) is a naturally occurring stilbenoid found in plants. The corresponding phosphate of Combretastatin A4 is a tumor vessel targeting agent and the water-soluble prodrug of Combretastatin A4.33
Figure 2.

Cytotoxicity profiling of the in situ generated 1F to 39F using a double-dose cell-viability assay. (A) Thirty-nine in situ SuFEx generated fluorosulfurylation products 1F-39F were tested in a cell viability assay with MCF-7 and A549 cells at the final concentrations of 20 μM and 500 nM. After a 72 h treatment, the cytotoxicity of fluorosulfurylation products 1F-39F and their phenol precursors 1–39 were evaluated according to the viability of the compound treated cancer cells relative to that of the vehicle (DMSO) treated control. The color of each unit in the heat maps reflects the increase (red) or decrease (blue) of the cytotoxicity of the corresponding fluorosulfurylation product compared to its phenol precursor (n = 3). (B) Cancer cell viabilities under the treatment of in situ generated 1F, 11F, and 25F and their phenol parents at a final concentration of 20 and 500 nM for 72 h. Cells treated with DMSO (0.2%) as the vehicle control. The structures of 1F, 11F, and 25F were confirmed by NMR after flash column chromatography purification. P-Values were calculated using two-way ANOVA. Error bars represent the mean ± SEM (n = 3); ns: P ≥ 0.05; *P ≤ 0.1; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
IC50 Evaluation of 1F, 11F, and 25F in Cancer Cell Lines.
To quantitatively compare the potency of 1F, 11F, and 25F with their parent phenolic drugs, we synthesized and purified these compounds in gram quantities and determined their IC50 values in a panel of cancer cell lines.
The antitumor potencies of arylfluorosulfate 1F and its phenol precursor 1 were evaluated against lung cancer cell line A549, breast cancer cell line MCF-7, SKBR3, and colon cancer cell line HT-29. As shown in Table 1 and Figure S6, compound 1 showed IC50 values of 632, 266, 13.5, and 236 nM in A549, MCF-7, SKBR3, and HT-29 cells, respectively. In comparison, 1F exhibited 2–5 fold lower IC50 values in these three cancer cell lines: 89 nM in A549, 134 nM in MCF-7, 6.9 nM in SKBR3, and 82 nM in HT-29. Similarly, 11F exhibited strong inhibition of the proliferation of ER+ breast cancer cell lines MCF-7 and T47D, with IC50 values of 5.5 and 4.8 nM, respectively, which were at the same level comparing with those of 11 (7.7 and 14.8 nM, respectively) (Table 1 and Figure S7). In addition, both 11 and 11F had no activities in ER-MCF-7 cells and lung cancer A549 cells, suggesting 11F may still target estrogen receptors. Although the measured IC50 values of arylfluorosulfate 25F on A549, MCF-7, and SKBR3 cells were slightly higher compared to those of its parent drug 25 (Table 1), 25F showed stronger activities to inhibit these three cells at high concentrations (20 μM and 2 μM) according to dose - response studies (Figure S8), which was consistent with the two-dose screening results (Figure 2). Surprisingly, the IC50 value of 25F in HT-29 cells, a drug resistant colon cancer cell line, was 70 nM, which was 70-fold lower than compound 25’s IC 50 (4947 nM). After confirming the antitumor potency of these three compounds, two of them (11F and 25F) were selected for further mechanistic studies.
Table 1.
IC50 Values of Arylfluorosulfates and Their Phenol Precursors in Different Cancer Cell Lines
| IC50 (nM) in different cancer cell lines | ||||
|---|---|---|---|---|
| A549 | MCF-7 | SKBR3 | HT-29 | |
| 1 (ABT-751) | 632 | 266 | 13.5 | 236 |
| 1F | 89 | 134 | 6.9 | 82 |
| A549 | MCF-7 | ER− MCF-7 | T47D | |
| 11 (Fulvenstrant) | NIa | 7.7 | NI | 14.8 |
| 11F | NI | 5.5 | NI | 4.8 |
| A549 | MCF-7 | SKBR3 | HT-29 | |
| 25 (combretastatin A4) | 28 | 6.6 | 0.48 | 4947 |
| 25F | 113 | 15.6 | 1.8 | 70 |
NI: No inhibition (less than 50% inhibition at 10000 nM).
11F Is a New SERD with Strong Potency.
As 11F had stronger anticancer potency than the commercial drug 11, we investigated whether this new compound shared the same cellular target with 11. As described previously, 11 is the only SERD on the market for the treatment of ER+ breast cancer. It competitively binds to estrogen receptors, a crucial regulator of ER+ breast cancer growth, inhibiting its dimerization and leading to its degradation.34,35 As shown in Figure 3A, 11F inhibited ER+ MCF-7 proliferation but had no effect on ER - MCF-7 cells, suggesting that the anticancer activity of 11F is still ER dependent.
Figure 3.

11F is a new SERD that binds and downregulates estrogen receptor. (A) 11F specifically inhibits the proliferation of ER+ MCF-7 cells. Error bars represent the mean ± SEM (n = 3). (B) Competitive binding curves of estrogen, 11 and 11F to ERα evaluated by a PolarScreen ERα competitor assay kit (Green) from Life Technologies. Error bars represent the mean ± SEM (n = 3). (C) 11 and 11F induce ER down-regulation in MCF-7 cells. P-Values were calculated using two-way ANOVA. Error bars represent the mean ± SEM (n = 4); ns: P ≥ 0.05; *P ≤ 0.1; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
To seek further evidence that ER is a target of 11F, we performed a competitive displacement ER binding assay, in which we compared the binding affinity of 11 and 11F to ER a protein in competition with a fluormone ligand. Notably, we observed that 11F bound to ERα with a relative EC50 of 1.5 nM, which was 3fold stronger than that of 11 (EC50 = 4.8 nM) (Figure 3B). Finally, to determine whether 11F was capable of downregulating the ERα expression level, MCF-7 cells were treated with 11 or 11F at concentrations of 3, 9, or 27 nM for 5 days. The ERα expression levels of the treated cells were then determined by Western blot. Both 11 and 11F downregulated ERα in a dose dependent manner, with 11F showing significantly stronger ERα downregulation activity (Figure 3C). These results confirmed that 11F acts as a potent SERD.
25F Overcomes the Drug Resistance of HT-29.
Combretastatin A4 (25) is one of the most potent antivascular agents that targets the colchicine-binding site of β-tubulin and hence disrupts tubulin polymerization. To assess if 25F also acted by the same mechanism, we treated HT-29 cells with 25F followed by staining with anti-tubulin-FITC antibody. Confocal imaging revealed that cells treated with 25F (1 and 0.1 μM) for 24h completely lost microtubule structure, consistent with tubulin depolymerization (Figure 4). By contrast, HT-29 cells treated with vehicle (0.2% DMSO) or 25 at various concentrations clearly showed the presence of the microtubule network (Figure 4). Previous investigations of the in vitro and in vivo phase II metabolism revealed that 25 is rapidly converted into the corresponding glucuronide and sulfate metabolites via glucuronidation and sulfation, respectively.36,37 In fact, glucuronidation of 25 on the phenol group by uridine 5-diphosphoglucuronosyl transferases (U G T s) has been previously identified as a mechanism of resistance evolved by HT-29 colon cancer cells and hepatocellular cancer cells (Figure S9).22 Indeed, the expression level of UGT1 in H T-29 cells is significantly higher than those of nondrug-resistant A549 and M CF-7 cells (Figure S9). Combined together, the above observations strongly suggested that 25F inhibits microtubule formation and overcomes Combretastatin A4 (25) resistance in HT-29 cells.
Figure 4.

25F exhibited significantly higher activity than 25 to disrupt the microtubule network of drug resistant HT-29 cells. HT-29 cells were treated with vehicle control (0.2% DMSO), 25 (1 and 0.1 μM), and 25F (1, 0.1, and 0.01 μM) for 24 h before cells were stained with anti-tubulin-FITC antibody (clone DM1A) (green) and counterstained with DAPI (blue). Scale bar: 20 μm.
DISCUSSION
In the past decade, LSF has attracted increased attention as a powerful method to diversify the pool of drug candidates or druglike molecules. Its potentials to facilitate the development of structure—activity relationships, the optimization of pharmacokinetic properties, and the improvement of physical properties have gradually been realized.9 In the current study, we developed the first in situ protocol for building arylfluorosulfate libraries from phenolic compounds with known biological activities, thereby enabling SuFEx click chemistry directly transferable to the LSF of drugs and drug candidates. This liquid-based protocol features several advantages, including: (1) only 0.1 μmol of phenol precursors is required; (2) a simple workup procedure enables the library to be directly subjected to biological assays without further purification; and (3) compatible with commercial lab automation systems.
Using this protocol, we efficiently converted 39 anticancer phenolic compounds into the corresponding fluorosulfurylation products in a 96-well plate. A total of three arylfluorosulfates with enhanced anticancer activities were identified, among which we further studied the mechanism of actions of 11F and 25F. We found that like its parent compound 25F inhibited tubulin polymerization. It also overcame the resistance of 25 in HT-29 colon cancer cells presumably via blocking glucuronidation of the phenol moiety of the parent compound. Significantly, 11F had stronger binding affinity toward E R a than its phenol precursor Fulvestrant 11, and it down-regulated the ERα level in M CF-7 cancer cells in a dose dependent manner, indicating that 11F functions as a new SERD with strong potency. In 2015, Fulvestrant registered sales of $704 million. However, this drug has poor bioavailability, and the only administration method is through intramuscular injection at a dose of 250 mg/month. It takes over 3 months to reach a steady serum concentration due to its quick clearance, significantly limiting its clinical value.32,38 Therefore, new SERDs with improved bioavailability are needed. In a recent report, Wang et al. demonstrated that the phenol moiety in 11 could be converted into a borate in a five-step synthesis.39 The resulting compound has excellent bioavailability, albeit slightly weaker potency. The cause of its improved bioavailability may result from blocked phase II metabolism in the bloodstream. Based on this reasoning, we hypothesize that 11F is likely to have better bioavailability in vivo compared to Fulvestrant. Importantly, this compound can be synthesized from 11 using a one-step quantitative “click” procedure, making it readily accessible for the future pharmacokinetics evaluation in animal models.
Based on the results presented in this study and our previous reports,7,8 we reason that converting a phenol group into an arylfluorosulfate may not change the protein targeting specificity of the parent compound, but instead this transformation may confer the resulting arylfluorosulfate new properties to enact in distinct mechanisms. First, an arylfluorosulfate may form a covalent bond with the target protein via SVI—F exchange given the presence of Tyr or Ser in the binding pocket and the presence of amino acid side chains to facilitate the F departure. Representative examples include an arylfluorosulfate probe for the inactivation of intracellular lipid binding protein(s)7 and an arylfluorosulfate inhibitor of the m RNA decapping scavenger enzyme, DcpS.40 Alternatively, a fluorosulfate-containing molecule may bind to its target via noncovalent interactions stronger than that of the parent phenolic compound. Under this circumstance, there is a possibility that the fluorosulfate formation may block the phase II metabolism pathway of the phenol moiety, i.e., sulfation, glucuronidation, and oxidation, key routes changing a compound’s pharmacokinetic properties. No matter which of the above scenarios apply, a drug candidate with improved properties may be engendered.
Moreover, this liquid-based in situ SuFEx protocol can be readily extended to the functionalization of amine-containing drug-like molecules. Using saturated SO F4 solution, highly selective transformation of amine groups to tetrahedral iminosulfur oxydifluorides (R - N = SOF2) is achieved (examples are shown in Figure S10). To date, click chemistry based on azide-alkyne cycloaddition has been successfully employed as a robust conjugation reaction, through which fragment- or peptide-based libraries were constructed.41–44 Now, with the newly developed SuFEx transformations, the click chemistry repertoire is expanded to enable in situ late-stage drug functionalization, which would significantly speed up the drug discovery process.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by the NIH (R01GM 117145 to K.B.S. and R01GM 111938 and R01GM 113046 to P.W.). We are grateful to the Christopher K. Glass group (University of California, San Diego) for providing T47D and ER MCF-7 cancer cells.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b12788.
Experimental section; photographs of gas-based and liquid-based SuFEx systems; solvent/base screening; demonstration of fluoride ions rem oval through TMSOH workup; structures of phenol compounds; representative examples of cancer cell viability assay results; proliferation inhibition curves of 1/1F, 11/11F, and 25/25F on different cancer cell lines; combretastatin drug resistance on HT-29; expansion of liquid-based SuFEx protocols; NMR spectra and LC traces (PDF)
REFERENCES
- (1).(a) Sharpless KB; Kolb HC Book of Abstracts, 217th ACS National Meeting, Anaheim, CA, 1999; O RGN-105, Accession Num ber 199:145537. [Google Scholar]; (b) Kolb HC; Finn MG; Sharpless KB Angew. Chem, Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (2).Dong J; Krasnova L; Finn MG; Sharpless KB Angew. Chem., Int. Ed 2014, 53, 9430–9448. [DOI] [PubMed] [Google Scholar]
- (3).Li S; Wu P; M oses JE; Sharpless KB Angew. Chem., Int. Ed 2017, 56, 2903–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Wang H; Zhou F; Ren G; Zheng Q; Chen H; Gao B; Klivansky L; Liu Y; Wu B; Xu Q; Lu J; Sharpless KB; Wu P Angew. Chem., Int. Ed 2017, 56, 11203–11208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dong J; Sharpless KB; Kwisnek L; Oakdale JS; Fokin VV Angew. Chem., Int. Ed 2014, 53, 9466–9470. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhu H; Chen D; Li N; Xu Q; Li H; He J; W ang H; Wu P; Lu J Chem. -Eur. J 2017, 23, 14712–14717. [DOI] [PubMed] [Google Scholar]; (d) Yatvin J; Brooks K; Locklin J Angew. Chem., Int. Ed 2015, 54, 13370–13373. [DOI] [PubMed] [Google Scholar]; (e) Yatvin J; Brooks K; Locklin J Chem. - Eur. J 2016, 22, 16348–16354. [DOI] [PubMed] [Google Scholar]; (f) Brooks K; Yatvin J; M cNitt CD; Reese RA; Jung C; Popik VV; Locklin J Langmuir 2016, 32, 6600–6605. [DOI] [PubMed] [Google Scholar]; (g) Li S; Beringer LT; Chen S; Averick S Polymer 2015, 78, 37–41. [Google Scholar]; (h) Oakdale JS; Kwisnek L; Fokin VV Macromolecules 2016, 49, 4473–4479. [Google Scholar]; (i) Brendel JC; Martin L; Zhang J; Perrier S Polym. Chem 2017, 8, 7475–7485. [Google Scholar]
- (5).Chen W; Dong J; Plate L; M ortenson DE; Brighty GJ; Li S; Liu Y; Galmozzi A; Lee PS; Hulce JJ; Cravatt BF; Saez E; Powers ET; Wilson IA; Sharpless KB; Kelly JW J. Am. Chem. Soc 2016, 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Baker BR Annu. Rev. Pharmacol 1970, 10, 35–50. [DOI] [PubMed] [Google Scholar]; (b) Baker BR Ann. N. Y. Acad. Sci 1971, 186, 214–226. [DOI] [PubMed] [Google Scholar]
- (7).Chen W; Dong J; Plate L; M ortenson DE; Brighty GJ; Li S; Liu Y; Galmozzi A; Lee PS; Hulce JJ; Cravatt BF; Saez E; Powers ET; Wilson IA; Sharpless KB; Kelly JW J. Am. Chem. Soc 2016, 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Baranczak A; Liu Y; Connelly S; Du W-GH; Greiner ER; Genereux JC; W iseman RL; Eisele YS; Bradbury NC; Dong J; N oodlem an L; Sharpless KB; Wilson IA; Encalada SE; Kelly JW J. Am. Chem. Soc 2015, 137, 7404–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (10).O’Hara F; Burns AC; Collins MR; Dalvie D; Ornelas MA; Vaz ADN; Fujiwara Y; Baran PS J. Med. Chem 2014, 57, 1616–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Li C; W ang J; Barton LM; Yu S; Tian M; Peters DS; Kumar M; Yu AW; Johnson KA; Chatterjee AK; Yan M; Baran PS Science 2017, 356, eaam7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sladojevich F; Arlow SI; Tang P; Ritter T J. Am. Chem. Soc 2013, 135, 2470–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fier PS; Hartwig JF J. Am. Chem. Soc 2014, 136, 1013910147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang P; Verma P; Xia G; Shi J; Qiao JX; Tao S; Cheng PTW; Poss MA; Farmer ME; Yeung K-S; Yu J-Q Nature 2017, 551, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Carocho M; Ferreira IC Anti-Cancer Agents Med. Chem 2013, 13, 1236–1258. [DOI] [PubMed] [Google Scholar]
- (16).Huang W-Y; Cai Y-Z; Zhang Y Nutr. Cancer 2009, 62, 120. [DOI] [PubMed] [Google Scholar]
- (17).Ferriz JM; Vinsova J Curr. Pharm. Des 2010, 16, 2033–2052. [DOI] [PubMed] [Google Scholar]
- (18).White RE Annu. Rev. Pharmacol. Toxicol 2000, 40, 133–157. [DOI] [PubMed] [Google Scholar]
- (19).Hughes JP; Rees S; Kalindjian SB; Philpott KL Br. J. Pharmacol 2011, 162, 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Simoni D; Romagnoli R; Baruchello R; Rondanin R; Grisolia G; Eleopra M; Rizzi M; Tolomeo M; Giannini G; Alloatti D; Castorina M; Marcellini M; Pisano CJ Med. Chem 2008, 51, 6211–6215. [DOI] [PubMed] [Google Scholar]
- (21).Tozer GM; Kanthou C; Parkins CS; Hill SA Int. J. Exp. Pathol 2002, 83, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Malebari AM; Greene LM; Nathwani SM; Fayne D; O ‘Boyle NM; W ang S; Twamley B; Zisterer DM; Meegan MJ Eur. J. Med. Chem 2017, 130, 261–285. [DOI] [PubMed] [Google Scholar]
- (23).O’Neil MJ et al. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 13th ed.; Merck Research Laboratories: 2001; p 1002. [Google Scholar]
- (24).Arakawa Y; Bhawal UK; Ikoma T; Kimoto K; Kuroha K; Kubota T; Hamada N; Kubota E; Arakawa H Biomed. Res 2009, 30, 271–277. [DOI] [PubMed] [Google Scholar]
- (25).Thrane EV; Refsnes M; Thoresen GH; Lag M; Schwarze PE Toxicol. Sci 2001, 61, 83–91. [DOI] [PubMed] [Google Scholar]
- (26).Ying J; Xu J; Shen L; M ao Z; Liang J; Lin S; Yu X; Pan R; Yan C; Li S; Bao Q; Li P Biol. Trace Elem. Res 2017, 179, 5969. [DOI] [PubMed] [Google Scholar]
- (27).Phenolic com pounds were purchased from Selleck Chemicals (http://www.selleckchem.com/).
- (28).Meany HJ; Sackett DL; Maris JM; Ward Y; Krivoshik A; Cohn SL; Steinberg SM; Balis FM; Fox E Pediatr. Blood Cancer 2010, 54, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jorgensen TJ; Tian H; Joseph IB; M enon K; Frost D Cancer Chemother. Pharmacol 2007, 59, 725–732. [DOI] [PubMed] [Google Scholar]
- (30).Vergote I; Robertson J Br. J. Cancer 2004, 90, S11–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pritchard KI; Rolski J; Papai Z; Mauriac L; Cardoso F; Chang J; Panasci L; Ianuli C; Kahan Z; Fukase K; Lindemann JPO; MacPherson MP; Neven P Breast Cancer Res. Treat 2010, 123, 453–461. [DOI] [PubMed] [Google Scholar]
- (32).Robertson JF; Lindemann J; Garnett S; Anderson E; Nicholson RI; Kuter I; Gee JM W. Clin. Breast Cancer 2014, 14, 381–389. [DOI] [PubMed] [Google Scholar]
- (33).Kanthou C; Tozer GM Blood 2002, 99, 2060–2069. [DOI] [PubMed] [Google Scholar]
- (34).Long X; Nephew KP J. Biol. Chem 2006, 281, 9607–15. [DOI] [PubMed] [Google Scholar]
- (35).Osborne CK; Wakeling A; Nicholson RI Br. J. Cancer 2004, 90, S2–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Aprile S; Del Grosso E; Grosa G Xenobiotica 2009, 39, 148161. [DOI] [PubMed] [Google Scholar]
- (37).Aprile S; Del Grosso E; Grosa G Drug Metab. Dispos 2010, 38, 1141–1146. [DOI] [PubMed] [Google Scholar]
- (38).Robertson JF Oncologist 2007, 12, 774–784. [DOI] [PubMed] [Google Scholar]
- (39).Liu J; Zheng S; Akerstrom VL; Yuan C; Ma Y; Zhong Q; Zhang C; Zhang Q; Guo S; Ma P; Skripnikova EV; Bratton MR; Pannuti A; Miele L; Wiese TE; Wang GJ Med. Chem 2016, 59, 8134–8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fadeyi OO; Hoth LR; Choi C; Feng X; Gopalsamy A; Hett EC; Kyne RE Jr.; Robinson RP; Jones LH ACS Chem. Biol 2017, 12, 2015–2020. [DOI] [PubMed] [Google Scholar]
- (41).Thirumurugan P; M atosiuk D; Jozwiak K Chem. Rev 2013, 113, 4905–4979. [DOI] [PubMed] [Google Scholar]
- (42).Kolb HC; Sharpless KB Drug Discovery Today 2003, 8, 1128–1137. [DOI] [PubMed] [Google Scholar]
- (43).Lewis WG; Green LG; Grynszpan F; Radic Z; Carlier PR; Taylor P; Finn MG; Sharpless KB Angew. Chem., Int. Ed 2002, 41, 1053–1057. [DOI] [PubMed] [Google Scholar]
- (44).Li H; Aneja R; Chaiken I Molecules 2013, 18, 9797–9817. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.