

Abstract
Tryptophan indole 15N–1H signals are well separated in nuclear magnetic resonance (NMR) spectra of proteins. Assignment of the indole 15N–1H signals therefore enables one to obtain site-specific information on complex proteins in supramacromolecular systems, even when extensive assignment of backbone 15N–1H resonances is challenging. Here we exploit the unique indole 15N–1H chemical shift by introducing extrinsic tryptophan reporter residues at judiciously chosen locations in a membrane protein for increased coverage of structure and function by NMR. We demonstrate this approach with three variants of the human A2A adenosine receptor (A2AAR), a class A G protein-coupled receptor, each containing a single extrinsic tryptophan near the receptor intracellular surface, in helix V, VI, or VII, respectively. We show that the native A2AAR global protein fold and ligand binding activity are preserved in these A2AAR variants. The indole 15N–1H signals from the extrinsic tryptophan reporter residues show different responses to variable efficacy of drugs bound to the receptor orthosteric cavity, and the indole 15N–1H chemical shift of the tryptophan introduced at the intracellular end of helix VI is sensitive to conformational changes resulting from interactions with a polypeptide from the carboxy terminus of the GαS intracellular partner protein. Introducing extrinsic tryptophans into proteins in complex supramolecular systems thus opens new avenues for NMR investigations in solution.
Graphical Abstract
■ INTRODUCTION
Early nuclear magnetic resonance (NMR) spectroscopy studies of small globular proteins in solution used the unique proton chemical shift of tryptophan indole hydrogens for investigations at natural isotope abundance.1,2 As the indole proton chemical shift is resolved from the majority of NMR signals in proteins, it has also been used to monitor binding of small molecules to proteins in lead compound screening assays.3 With the introduction of stable-isotope labeling, the separation of indole 15N–1H resonances was further improved in twodimensional heteronuclear correlation experiments, permitting studies of more-complex macromolecules.4,5
Work by Stehle et al.5 and a recent study from our laboratory6 showed that indole 15N–1H correlation signals from tryptophans distributed throughout G protein-coupled receptors (GPCRs) provide a basis also for NMR investigations of membrane proteins in solution. Specifically, in 2D [15N,1H]-TROSY correlation spectra of the human A2A adenosine receptor (A2AAR), a class A GPCR, the tryptophan indole 15N–1H lines are well separated from the other 15N–1H signals in the spectra, thus enabling their unambiguous site-specific assignment and observation of their response to variable efficacy of drugs bound to the extracellular orthosteric site.6
Here, we extend the utility of tryptophan indole signals by introducing exogenous tryptophans in judiciously selected sequence locations of human A2AAR. This new approach is attractive and broadly applicable due to an efficient protocol for production of stable-isotope-labeled wild-type and variant GPCRs for NMR studies in solution. We demonstrate the feasibility of this approach by introducing a single extrinsic tryptophan reporter residue near the receptor intracellular signaling surface of A2AAR in either helix V, VI, or VII. We show that the ligand binding activity of the three A2AAR variants and their overall protein fold are highly similar to those of the native protein. 2D TROSY correlation spectra of the [u-15N, ~70% 2H]-labeled A2AAR variants were recorded, and NMR signals from the extrinsic tryptophan residues were readily identified in 2D spectra, enabling observations of their responses to variable efficacy of bound drugs and to addition of a 21-residue polypeptide that corresponds to the carboxy terminus of the GαS intracellular partner protein. The present data show that introduction of extrinsic tryptophans broadens the choice of available probes for NMR investigations of GPCRs, which should in future work also be applicable with other membrane proteins.
■ RESULTS
As a basis for the work presented here, we developed a protocol for efficient production of stable-isotope-labeled human A2AAR and reconstitution into membrane mimetics that yielded high-quality NMR spectra in solution. This protocol is described in detail in the Experimental Section.
Selection of Sites for Placement of Reporter Tryptophan Residues and Characterization of the A2AAR Variants.
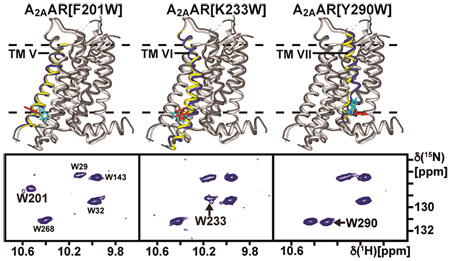
In class A human GPCRs, drug binding to the extracellular surface induces signaling-related changes in the receptor structure near the intracellular surface in a manner that depends on the efficacy of the bound drug.7 To probe these efficacy-dependent changes in structure and dynamics, we sought to introduce extrinsic tryptophans as NMR probes at the tips of three transmembrane helices (TM) near the receptor intracellular surface. Figure 1 shows the location of the extrinsic tryptophan NMR probes in structural models of three A2AAR variants, i.e., A2AAR[F201W], A2AAR[K233W], and A2AAR[Y290W]. As described in the Experimental Section, the selection of the three variants of A2AAR used was guided by model considerations.
Figure 1.

Locations of extrinsic reporter tryptophans in A2AAR. Ribbon representations are shown of A2AAR structural models, each showing one of the three extrinsic tryptophan reporter residues used here. The structural models are superpositions of crystal structures of A2AAR complexes with the antagonist ZM241385 (dark gray; PDB 3EML) and the agonist UK432097 (light gray; PDB 3QAK). The tryptophans indicated above each structural model are shown in stick representation for the A2AAR complexes with ZM241385 (cyan) and with UK432097 (red), respectively. The transmembrane helix (TM) containing the single extrinsic tryptophan reporter residue is color-coded for A2AAR in complex with ZM241385 (blue) and with UK432097 (yellow). Broken horizontal lines indicate the extracellular (top) and intracellular (bottom) surfaces of the lipid bilayer, as predicated by the “Orientations of Proteins in Membranes” database.44
A first criterion for the selection of the placement of the reporter tryptophan residues in the A2AAR amino acid sequence was to ensure a strong response of the NMR probes to variable drug efficacy. To this end, we considered structural changes between available A2AAR crystal structures for complexes with ligands of different efficacies. Comparison of crystal structures of an A2AAR–agonist complex (PDB 3QAK)8 and an A2AAR–antagonist complex (PDB 3EML)9 revealed outstandingly large structural rearrangements at the intracellular surfaces of TM V, VI and VII. Similar structural rearrangements in these locations were observed with amplified magnitude in comparisons of the same antagonist complex with an A2AAR ternary complex including an agonist and a G protein mimetic (PDB 5G53),10 as well as for other crystal structures of complexes of GPCRs with G proteins11 or G protein mimetics.12,13
The second design consideration was to preserve native-like ligand binding activity for all A2AAR variants containing single tryptophan reporter residues. We measured the ligand binding activities of each variant protein expressed in Pichia pastoris membranes and compared their activity to native A2AAR for a small library of compounds with variable efficacy (Table 1). Radioligand binding assays (Figure S1) demonstrated that the activity of all three single tryptophan variants was nearly identical to the activity of the native protein for all compounds studied (Table 1). This can be rationalized by the fact that all three tryptophan reporter residues were inserted far from the orthosteric drug binding cavity.
Table 1.
Binding Affinities (nM) of Nine Ligands for Wild-Type A2AAR and Variants Containing a Single Extrinsic Tryptophan Reporter Residue
| ligand | Efficacya | wild type | A2AAR[F201W] | A2AAR[K233W] | A2AAR[Y290W] |
|---|---|---|---|---|---|
| CGS21680b | FA | 27.7 (5.3)c | 20.3 (7.7) | 25.8 (1.5) | 32.2 (6.4) |
| NECA | FA | 97.1 (7.8) | 62.3 (12.5) | 98.2 (12.9) | 136.7 (25.8) |
| UK432097 | FA | 34.3 (9.8) | 69.1 (27.5) | 45.4 (9.3) | 64.0 (24.3) |
| LUF5834 | PA | 11.1 (1.6) | 8.4 (1.0) | 11.5 (2.8) | 13.6 (3.4) |
| regadenoson | PA | 405 (55) | 224 (44) | 345 (41) | 501 (29) |
| caffeine | Ant | 15400 (1100) | 15800 (3800) | 10700 (595) | 13500 (2060) |
| XAC | Ant | 24.3 (6.7) | 42.8 (7.2) | 21.1 (1.6) | 30.4 (2.2) |
| ZM241385 | Ant | 3.0 (0.4) | 4.5 (0.7) | 2.4 (0.2) | 3.5 (0.6) |
| preladenant | Ant | 9.4 (1.8) | 14.7 (0.2) | 6.7 (0.2) | 19.9 (1.7) |
aAbbreviations are as follows: “FA” full agonist, “PA” partial agonist, and “Ant” antagonist.
bValues listed for CGS21680 represent KD; for the other ligands, Ki is listed, as determined from radioligand binding experiments (see text).
cData are shown as the mean values, with standard errors given in parentheses.
The third consideration was to preserve the global native fold, homogeneity, and monodispersity of NMR sample preparations for the A2AAR variants containing a tryptophan reporter residue. The amount of expressed protein for each single tryptophan variant in isotopically labeled culture media was similar to the expression of the wild-type protein in the same media. Sample homogeneity was highly similar for all A2AAR variants, as judged by a single resolved peak in the analytical size exclusion chromatograms, indicating monodispersed samples for NMR studies (Figure S2). To characterize the structural folds of the A2AAR variant proteins containing single extrinsic reporter tryptophan residues, we recorded 2D [15N,1H]-TROSY correlation spectra for each [u-15N, 70%-2H]-A2AAR variant. We verified that the native global fold for all variant proteins was preserved by monitoring the chemical shifts of 30 well-resolved signals in the NMR spectra (Figures S3–S5). The chemical shifts for these 30 signals of the variant proteins were all highly similar to the same signals in A2AAR, indicating that the global fold of A2AAR was preserved in the variant proteins. Specifically, among the 30 amide groups, four have previously been assigned to individual glycine backbone 15N–1H signals,6 showing that the structures are preserved at four discrete locations in extracellular loops, on the extracellular surface, and on the intracellular surface.
TROSY Correlation Experiments of Antagonist Complexes with A2AAR Variants Containing Single Extrinsic Reporter Tryptophan Residues.
We recorded 2D [15N/H]-TROSY correlation experiments with the three [u-15N, ~70% 2H]-A2AAR variants containing a single extrinsic tryptophan in complex with the antagonist ZM241385. The indole 15N–1H region of the TROSY spectrum of each variant protein showed a single new resonance compared with A2AAR in complex with the same antagonist, arising from the introduced reporter tryptophan residue (Figure 2). The indole 15N–1H signals arising from each extrinsic tryptophan were separated from the indole 15N–1H signals of the endogenous tryptophans, and for A2AAR[F201W] and A2AAR[Y290W] the new signals could also be resolved in one-dimensional NMR spectra. Variable signal intensities were observed among all three indole 15N–1H signals of the extrinsic tryptophan residues, where W290 had the highest signal-to-noise and W233 was broadest and weakest. Nevertheless, the signal for W233 could be unambiguously identified. The indole 15N–1H signals from the endogenous tryptophans were also observed in the spectra of A2AAR variants with extrinsic tryptophans and exhibited chemical shifts that were highly similar to the corresponding signals observed for A2AAR.
Figure 2.

Assignment of the indole 15N–1H NMR lines of single extrinsic tryptophan residues. Each panel on the left displays the tryptophan indole 15N–1H region of an 800 MHz 2D [15N,1H]-TROSY correlation spectrum of A2AAR in complex with the antagonist ZM241385. On the right are 1D cross sections selected at the 15N chemical shifts indicated by dashed lines in the contour plots. The spectrum of A2AAR is shown in blue, and the spectra of the variant proteins containing a single extrinsic tryptophan reporter residue (Figure 1) are shown in red. The amino acid replacement and its location in the A2AAR structure are indicated by identification of the transmembrane helix (TM) to which it is attached. Comparison of the blue and red spectra shows in each case an additional signal for the A2AAR variant (marked with an asterisk), which arises from the single extrinsic tryptophan.
Indole 15N–1H Signals from A2AAR Variants Containing Single Extrinsic Tryptophan Residues Manifest Response to Variable Drug Efficacy.
To investigate the response of variable efficacy of bound drugs, we recorded 2D [15N,1H]-TROSY correlation spectra of the A2AAR variants with extrinsic tryptophans for ligands of different pharmacological efficacies (Figure 3). The indole 15N–1H signals of the extrinsic tryptophans manifested different responses to variable drug efficacy. For W233, located at the intracellular surface of helix VI, we observed two signals of asymmetric intensities for complexes with full agonists, which had different chemical shifts from the single resonance observed in the complex with the antagonist ZM241385 (Figure 2). For W201 and W290, which are located at the intracellular ends of helices V and VII, respectively, signals that would correspond to the indole 15N–1H signals observed for the antagonist complexes were not observed in complexes with agonists, indicating the presence of exchange broadening. The response of the endogenous indole 15N–1H signals6 was highly similar for A2AAR and the variant proteins.
Figure 3.

NMR response of extrinsic tryptophan indole 15N–1H moieties in A2AAR to variable drug efficacy. Contour plots are shown of 800 MHz 2D [15N,1H]-TROSY correlation spectra of the [u-15N, ~70% 2H]-A2AAR variants identified at the top in complexes with the ligands of different efficacies identified in the panels on the left. Each panel displays the spectrum of one complex. The dashed circles indicate positions of NMR signals observed for W201 and W290 in the spectra of the antagonist complexes, which were so far not identified in spectra of agonist complexes (see text).
Response of Indole 15N–1H Signals to the Addition of a GαS C-terminal Peptide.
To a sample of [u-15N, ~70% 2H]-A2AAR[K233W] in complex with the agonist UK432097, we added a 10-fold molar excess of a 21-residue synthetic polypeptide corresponding to residues 374–394 of the carboxy terminus of the intracellular partner protein GαS. An earlier study reported an increase of the specific binding of A2AAR agonists upon addition of this polypeptide and inability of A2AAR to signal through Gαs after addition of the polypeptide, suggesting that the polypeptide and full length GαS protein bind at similar locations in A2AAR.14
2D TROSY correlation spectra were measured for the A2AAR[K233W] complex with UK432097 in the absence and presence of the polypeptide (Figure 4). A single indole 15N–1H signal was observed for W233 in the ternary complex with UK432097 and the polypeptide, in contrast to the two signals observed for W233 in complex with UK432097 alone (Figure 3). The chemical shift of the single resonance for W233 was slightly shifted in both the 1H and 15N dimensions, by ~0.06 and ~0.2 ppm, respectively, from either of the two resonances observed in the absence of the polypeptide. Several chemical shift changes are also observed for indole 15N–1H signals from endogenous tryptophans upon addition of the polypeptide. W29 and W32, located in helix I at the intracellular surface, both show chemical shift changes between the spectra recorded in the absence and presence of the polypeptide of ~0.15 ppm in the 1H dimension and ~0.4 ppm in the 15N dimension. In contrast, W143 and W268, located near the A2AAR extracellular surface, show at most a very small response to the addition of the polypeptide.
Figure 4.

NMR response of an A2AAR[K233W]–agonist complex to addition of a C-terminal GαS peptide. The left panel shows a superposition of contour plots of 800 MHz 2D [15N,1H]-TROSY correlation spectra of the [u-15N, ~70%2H]-A2AAR[K233W] complex with the agonist UK432097 before (blue) and after (red) addition of the 21-residue GαS C-terminal peptide. The right panel shows a projection along the 15N dimension onto the lower boundary of the region indicated by the dashed box on the left. Assignments for the extrinsic tryptophan indole 15N–1H signals of A2AAR[K233W] are identified, as are additional assignments which have been adapted from A2AAR in complex with UK432097 (see text).
■ DISCUSSION
The indole 15N–1H signals of the three extrinsic tryptophans of Figure 1 exhibit strong responses to variable efficacy of ligands bound to A2AAR (Figure 3). Whereas a single peak for each of these tryptophans was observed for complexes with antagonists, two signals were observed for an agonist complex of W233, and for agonist complexes of W201 and W290 no signal was observed (Figure 3). As all samples were initially purified with the same low-affinity antagonist theophylline, which was later exchanged with other ligands during protein purification (see Experimental Section), the absence of signals for W201 and W290 could not be due to different degrees of back protonation at the three sites. The responses of the indole signals of the extrinsic tryptophans can therefore on the basis of observations with other GPCRs best be rationalized by a situation where A2AAR contains multiple local polymorphisms, with variable rates of exchange between different pairs of corresponding structures at the tips of the TMs V, VI, and VII. The absence of the W201 and W290 signals in the complexes with agonists coincides with similar observations in mammalian rhodopsin upon activation by light,5 which was also related to slowed exchange between two or multiple conformational states. Overall, this suggests high structural plasticity of A2AAR in solution.
The impact of a GαS carboxy-terminal oligopeptide (Figure 4) on the NMR data of the A2AAR-UK432097 complex is intriguing in the context of literature data on allosteric coupling in GPCR signaling complexes. We observed two signals for W233 in the A2AAR complex with the agonist UK432097 and a single signal for W233 upon addition of the polypeptide derived from the GαS carboxy terminus to the UK432097 complex. The NMR signals for endogenous tryptophans located at the intracellular surface also showed a response to the addition of the peptide, while signals of endogenous tryptophans located near the extracellular surface showed no measurable response. This is in apparent contrast with NMR studies of the β1-adrenergic receptor (β1AR), which reported changes in the chemical shifts of amide signals of valine residues located near the extracellular surface upon complex formation with a G-protein-mimicking nanobody.15 On the other hand, the data in Figure 4 appear to be consistent with recent crystallographic studies of A2AAR binary and ternary complexes involving an agonist and an engineered “mini GαS” protein, where changes of A2AAR in the ternary complex relative to the complex with agonist alone were observed in the intracellular region but not in the extracellular region.10
Our ligand binding activity data showing highly similar functions for A2AAR and the variant proteins with single extrinsic tryptophan residues suggest that extrinsic tryptophan NMR probes could be broadly applicable to GPCRs, and possibly to other membrane proteins, as long as the NMR probes are introduced near the intracellular surface, as observed in previous 19F-NMR studies of GPCRs.16–21 The use of NMR to monitor resolved signals from extrinsic tryptophans could also be applied to explore protein–ligand interactions for variants where a tryptophan is introduced near the orthosteric ligand binding pocket, providing direct access to structural and functional responses in this molecular region. Additionally, the current number of crystal structures of different human GPCRs has enabled structural homology models to be built for a wide range of human non-olfactory GPCRs, as made available through the GPCRdb.22,23 These models have sufficient resolution to guide the placement of extrinsic tryptophans for NMR studies of GPCRs that have not previously been experimentally studied.
The data presented in Figure S2 document convincingly that protein production for NMR studies may need to differ widely from conditions favoring protein crystallization. Although stable-isotope-labeled human A2AAR using a construct generated for crystal structure determination of an A2AAR– antibody complex (PDB 3VG9; Figure S1A)24 yielded homogeneous protein preparations with wild-type-like ligand binding, the resulting 2D TROSY 15N–1H correlation spectra contained only a small number of narrow signals (Figure S2), which were shown to arise from parts of the 86-residue N-terminal secretion signal peptide that was still covalently attached to the protein. A2AAR was not observed, apparently due to aggregation induced by the covalently attached secretion signal peptide. For A2AAR, as for many other proteins, a major biochemistry effort (Figure 5) was therefore needed to obtain preparations that provided informative, highly dispersed NMR data.
Figure 5.
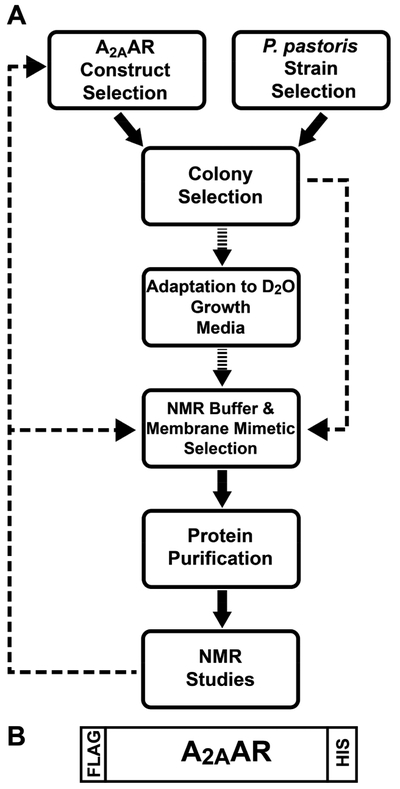
Expression of human A2AAR in Pichia pastoris and purification for NMR studies. (A) Block diagram for efficient production of stable-isotope-labeled human A2AAR and designed variants in P. pastoris for NMR experiments. Broken arrows indicate optimization steps that were carried out at the outset with protonated A2AAR (see text). The feedback loops enabled multistep optimization of the expression construct and the solution conditions. (B) Scheme of the selected A2AAR construct. “HIS” is a 10 X polyhistidine tag, and “FLAG” is a FLAG octapeptide.
■ EXPERIMENTAL SECTION
Materials and Reagents.
15N ammonium sulfate and 99.8%-2H2O were purchased from Cambridge Isotope Labs (Andover, MA). The C-terminal polypeptide GαS(374–394)[C379A] was synthesized by New England Peptide (Gardner, MA). The A2AAR construct that included the N-terminal αMF secretion signal (Figure S6) was provided by Prof. So Iwata. A2AAR and the three variant proteins were cloned into a pPIC9K vector (Invitrogen) containing the AOX1 promoter. Sources for other reagents are given below.
Generation of A2AAR Variant Constructs Containing Single Extrinsic Tryptophan Reporter Residues.
The starting A2AAR construct included the wild-type sequence 1–316, a single amino acid replacement, N154Q to remove a potential glycosylation site, and the addition of an N-terminal FLAG tag and a 10 X C-terminal His tag (Figure 5B). Plasmids for the A2AAR variants containing single extrinsic tryptophans were generated from this construct using PCR-based site-directed mutagenesis (QuikChange II, Stratagene, CA), with primers purchased from Integrated DNA Technologies (Coralville, IA). A list of the primers used to generate the A2AAR variants is provided in Table S1. Amino acid sequences of the three A2AAR variants containing a single extrinsic tryptophan are provided in Figure S7.
Protocol for Production of Stable-Isotope-Labeled Human A2AAR in Pichia pastoris.
We developed a new “in-house” protocol for efficient production of stable-isotope-labeled human A2AAR for NMR studies in solution (Figure 5A), which is described in detail below.
A2AAR Construct Selection.
The expression construct of Figure 5B provided high overall yield of monodispersed, stable-isotope-labeled A2AAR. Protein produced from this construct exhibited nearly identical ligand-binding activity to that of native A2AAR expressed in mammalian or insect cells6 both in isolated Pichia membranes and in mixed detergent micelles. These data were obtained without using ligand affinity purification,6 which can significantly reduce the final protein yield.25
This expression construct was selected by comparing the yield and homogeneity of A2AAR samples produced from different expression construct designs, which included variations in the peptide leader sequence, the protease cleavage sites, and the positioning of polyhistidine tags (Figure S8). An important feature of the construct in Figure 5B is the absence of the N-terminal 86-residue α-mating factor (αMF) secretion signal sequence, which has previously been used to express membrane proteins,26,27 including GPCRs,24,28 in P. pastoris. In our initial attempts to express stable-isotope-labeled A2AAR, we also used a construct containing the N-terminal αMF secretion signal sequence, as it was assumed to be needed for high protein expression yields. However, the αMF secretion signal was not processed by the cell and remained covalently attached to purified A2AAR, thereby corrupting the NMR signals of A2AAR (Figure S8). Subsequent attempts to benefit from the αMF secretion sequence for A2AAR expression were made by insertion of a protease recognition site between the αMF secretion sequence and A2AAR, which resulted in an overall low yield of purified, cleaved protein. Surprisingly, the αMF secretion signal was thus found not to be needed for obtaining high levels of expressed A2AAR, and since A2AAR expressed from a construct lacking the αMF secretion signal required fewer purification steps, the highest overall yield of stable-isotope-labeled A2AAR was obtained with the construct of Figure 5B.
Selection of Pichia Strain.
Selection of the Pichia strain was critical for obtaining high levels of stable-isotope-labeled A2AAR. A commercially available “wild-type” strain, Bg12 (Biogrammatics, Carlsbad CA), provided the highest yield of stable-isotope-labeled A2AAR and also exhibited the fastest growth rates in cell culture media containing deuterium oxide (D2O). Overall, the yield of stable-isotope-labeled A2AAR expressed in the Bg12 strain was several-fold higher than in the strain SMD1163, which has been a popular choice for expression of human GPCRs in earlier studies.24,28–32 In addition, the growth rate of the Bg12 strain was significantly faster in both protonated and deuterated cell culture media, with doubling rates in deuterated media about twice as fast as for SMD1163.
Colony Selection for Production of A2AAR.
As the copy number of DNA integrated into transformed Pichia varies among transformed colonies, selection of colonies with high protein expression was another key to maximizing production of stable-isotope-labeled A2AAR. In our hands, directly testing the amount of protein produced among colonies in small scale cultures of BMGY and BMMY media provided better results when compared with selection using antibiotics. We did not observe a direct correlation between better growth on plates with agar containing antibiotics and higher protein expression, as has been noted in other systems.33
Plasmids containing A2AAR variants were transformed via electroporation into the P. pastoris strain Bg12 (Biogrammatics, Carlsbad, CA) or the strain SMD1163 (provided by Professor So Iwata). The transformed cells were spread onto yeast extract peptone dextrose (YPD) plates and allowed to mature for 48 h at 30 °C. For each transformed construct, 20–22 isolated colonies were indiscriminately selected and spread onto new YPD plates over a larger area and allowed to grow for 48 h at 30 °C. Cells from these plates were used to inoculate 4 mL cultures in buffered minimal glycerol (BMGY) media, which were incubated at 30 °C and 250 rpm for 2 days until the cell density reached an OD600 between 15 and 20. Cultures were centrifuged at 2000g for 15 min, media was removed, and each cell pellet was resuspended in 4 mL of buffered minimal methanol (BMMY) media containing 0.5% (w/v) methanol and incubated at 27 °C and 250 rpm. 0.5% methanol was added after 12 h and again after 24 h for a total expression time of 36 h. Cells were harvested by centrifugation at 2000g for 15 min, frozen in liquid nitrogen and stored at −80 °C until processed.
Cell pellets from the 4 mL cultures were resuspended in buffer containing 50 mM HEPES pH 7, 1 M NaCl, protease inhibitor solution, 20 mM MgCl2, and 10 mM KCl, and lysed. To extract A2AAR, broken cells were mixed at a 1:1 ratio with buffer containing 1% DDM, 0.2% CHS, 100 mM HEPES pH 7, and 500 mM NaCl for 3 h. This mixture was centrifuged at 15000g for 30 min and at 4 °C. Aliquots from the soluble fraction were used for running Western blots with the mouse monoclonal anti-FLAG M2 alkaline phosphatase (Sigma-Aldrich) to detect expressed protein.
To maximize expression of stable-isotope-labeled A2AAR, the three highest-expressing clones were selected and subjected to further expression characterization in larger cultures of 500 mL of BMGY/BMMY media. Protein expressed from these colonies was purified and the yields were quantitatively compared by analytical size exclusion chromatography (aSEC). By comparison of the intensities of purified protein with aSEC, the highest expressing clone was selected for expressing stable-isotope-labeled A2AAR.
Adaptation of Pichia to D2O-Containing Growth Media.
For production of [u-15N, ~70% 2H]-A2AAR, it was necessary to adapt Pichia cells to media in D2O prior to large-scale protein production in ~99% D2O. We found that the Bg12 strain could be adapted with a two-step approach by inoculation of cultures in fully protonated media into media containing 85–90% D2O, followed by two inoculations into media containing ~99% D2O, which has also been done for protein production in the KM71H strain.34,35 We found no difference in cell densities and amount of produced protein between this method and an earlier method of adaption to D2O in smaller increments.36 In contrast, production of A2AAR in the SMD1163 strain would have required adaptation to D2O media in smaller steps.
Optimal Buffer Selection.
Optimal selection of components in the buffer used for NMR studies was important to obtaining high-quality NMR data and maintaining the protein fold and monodispersity during NMR experiments. We found an optimal balance between conditions for preserving the protein fold for longer periods of time and conditions for NMR experiments (i.e., salt concentration, pH, and selection of an appropriate buffer) by monitoring changes in the fraction of monodispersed protein versus total purified protein with aSEC. The choice of a membrane mimetic for solubilization of the membrane protein is always critical.37–41 We found that mixed micelles of lauryl maltose neopentyl glycol (LMNG, Anatrace, Maumee, OH) and cholesteryl hemisuccinate (CHS, Sigma-Aldrich) provided the optimal conditions for sample stability. This system also provided good resolution of NMR data, when compared with the widely used mixed micelles of n-dodecyl-β-D-maltopyrano-side (DDM) and CHS42 (Figure S9). The amount of purified, monodispersed A2AAR was maximized by using the same detergent mixture throughout the entire protein extraction and purification process, rather than exchanging detergents during purification.
Large-Scale Production and Purification of Stable-Isotope-Labeled A2AAR.
The D2O-adapted cells were used to inoculate culture tubes containing 4 mL of 2H-BMGY media, which were incubated at 30 °C and 250 rpm for 2 days. The cultures were centrifuged at 2000g, and cells were resuspended in 50 mL of BMGY media and allowed to incubate for another 2 days at 30 °C and 250 rpm. These cultures were centrifuged at 2000g for 15 min and resuspended in baffled flasks containing 500 mL of BMGY and allowed to incubate for 2 days at 30 °C and 250 rpm. Cells were centrifuged at 2000g and resuspended in BMMY media without methanol to permit digestion of any remaining glycerol. 0.5% (w/v) methanol was added 6 h after resuspension in BMMY, then again 18 and 30 h after resuspension for a total expression time of 40 h. Cells were harvested at 2000g, frozen in liquid nitrogen and stored at −80 °C for future processing.
A2AAR-containing P. pastoris membranes were isolated by a cell distruptor (Constant Systems), and A2AAR was extracted by incubating membranes with buffer containing 0.5% lauryl LMNG and 0.025% CHS, 1 mM theophylline (Sigma-Aldrich), protease inhibitor solution (prepared in-house), and 2 mg/mL iodoacetamide (Sigma) for 6 h. For experiments using DDM in the NMR buffer, A2AAR was extracted with the same buffer by replacing LMNG with 0.5% DDM. Solid debris was removed by centrifugation at 45000g for 30 min, and the soluble fraction incubated overnight with Co2+ Talon resin (Clontech) and 30 mM imidazole. A2AAR was purified via three consecutive 20 x column volume batch washes of the resin in buffer containing detergent (either LMNG/CHS or DDM/CHS), 50 mM HEPES pH 7, 150 mM NaCl, and ligand. A2AAR was eluted in buffer containing 300 mM imidazole, 25 mM HEPES pH 7, 150 mM NaCl, detergent, and ligand. Purified protein was transferred to the final NMR buffer (20 mM HEPES pH 7.0, 75 mM NaCl, 0.025% LMNG, 0.00125% CHS, and ligand), using a PD-10 desalting column (GE Healthcare Life Sciences). A saturating concentration of ligand was used for all experiments. For NMR experiments, samples were concentrated to 280 μL, 20 μL of 99.8% D2O was added, and the solution was transferred to a 5 mm Shigemi NMR tube. Yields of purified [u-15N, ~70% 2H]-A2AAR were ~4 mg per 1 L of cell culture, and yields of [u-15N, ~70% 2H]-A2AAR variants containing a single extrinsic tryptophan reporter were nearly identical to A2AAR, as determined by analytical SEC. For NMR studies, 2 L of cell culture was used to prepare each sample.
Generation of Structural Models of A2AAR Variants.
Structural models of A2AAR[F201W], A2AAR[K233W], and A2AAR-[Y290W] complexes with the antagonist ZM241385 and agonist UK432097 were computationally generated in ICM-Pro43 (Molsoft, San Diego, CA), starting from the crystal structures of A2AAR in complex with ZM241385 (PDB 3EML) and in complex with UK432097 (PDB 3QAK). Boundaries of the lipid bilayer were predicted from the Orientations of Proteins in Membrane database.44 After amino acid replacement, structural models were energy minimized with side-chain conformational sampling. Energy refinements were performed using Biased Probability Monte Carlo simulations45 with flexible side chains for the replaced amino acid and residues located within 6.0 Å in the structures. Refined models were evaluated for steric clashes and penalties in free energy, ΔΔG (kcal/mol), and solubility values. The introduced amino acid replacements were found not to have significant destabilization effects on the receptors (Table S1). This information was an important part of the selection of the positions of the extrinsic tryptophan NMR probes.
Ligand Binding Activity Measurements.
Radioligand binding assays were performed using membranes prepared from P. pastoris expressing either native-like human A2AAR or the tryptophan variants. For saturation binding experiments, increasing concentrations (ranging from 2 nM to 80 nM) of [3H]CGS21680 (35.2 Ci/mmol, American Radiolabeled Chemicals, Inc., St. Louis, MO) or increasing concentrations of [3H]ZM241385 (0.2 nM to 12 nM) were incubated with membranes (5 μg protein) at 25 °C for 60 min in a total of 200 μL of Tris-HCl buffer (50 mM, pH 7.5) containing 10 mM MgCl2. NECA (100 μM) or XAC (10 μM) was used to determine nonspecific binding. For ligand displacement experiments, increasing concentrations of ligands were incubated with [3H]CGS21680 (5 nM) and membrane preparations at 25 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using an MT-24 cell harvester (Brandell, Gaithersburg, MD) followed by washing twice with 5 mL of cold Tris-HCl buffer. Radioactivity was measured using a scintillation counter (Tri-Carb 2810TR). Binding parameters were calculated using Prism 6 software (GraphPAD, San Diego, CA). IC50 values obtained from displacement curves were converted to Ki values using the Cheng– Prusoff equation.46 All data are expressed as the mean ± standard error from three independent experiments.
NMR Spectroscopy.
Two-dimensional transverse relaxation-optimized spectroscopy (TROSY)47 correlation spectra were measured on a Bruker Avance II 800 MHz spectrometer equipped with a 5 mm TXI HCN probe, running Topspin 3.1 (Bruker Biospin, Billerica, MA). Experiments were measured at 307 K, and the temperature was calibrated using a standard sample of 4% methanol in methanol-d4. Chemical shifts were referenced to an internal DSS standard. All data were processed identically in Topspin 3.5pl2.
Supplementary Material
Table S1, sequences of primers used for generation of A2AAR variants; Figure S1, radioligand binding data for A2AAR and A2AAR variants; Figure S2, analytical size exclusion chromatograms of A2AAR and three A2AAR variants containing a single extrinsic tryptophan residue; Figures S3–S5, NMR spectra of [u-15N, ~70% 2H]- A2AAR[F201W], [u-15N, ~70% 2H]-A2AAR[K233W], and [u-15N, ~70% 2H]-A2AAR[Y290W] in complex with ZM241385; Table S2, evaluated penalties in free energy and solubility for structural models of A2AAR variants; Figure S6, schemes of A2AAR constructs examined in the context of Figure 1; Figure S7, amino acid sequences of A2AAR variants containing a single extrinsic tryptophan residue; Figure S8, NMR comparison of [u-15N, ~70% 2H]-A2AAR with and without a covalently attached peptide leader sequence; Figure S9, comparison of NMR spectra of A2AAR in different mixed micelles (PDF)
ACKNOWLEDGMENTS
The authors acknowledge support from the NIH/NIGMS (PSI Biology grant U54 GM094618 and R01GM115825) and the NIDDK Intramural Research Program (ZIA DK031117). M.T.E. acknowledges an American Cancer Society postdoctoral research fellowship. K.W. is the Cecil H. and Ida M. Green Professor of Structural Biology at The Scripps Research Institute.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b03805.
REFERENCES
- (1).McDonald CC; Phillips WD J. Am. Chem. Soc 1969, 91, 1513–1521. [DOI] [PubMed] [Google Scholar]
- (2).McDonald CC; Phillips WD J. Am. Chem. Soc 1967, 89, 6332–6341. [DOI] [PubMed] [Google Scholar]
- (3).Bista M; Kowalska K; Janczyk W; Dömling A; Holak TA J. Am. Chem. Soc 2009, 131, 7500–7501. [DOI] [PubMed] [Google Scholar]
- (4).Wedin RE; Delepierre M; Dobson CM; Poulsen FM Biochemistry 1982, 21, 1098–1103. [DOI] [PubMed] [Google Scholar]
- (5).Stehle J; Silvers R; Werner K; Chatterjee D; Gande S; Scholz F; Dutta A; Wachtveitl J; Klein-Seetharaman J; Schwalbe H Angew. Chem 2014, 126, 2110–2116. [DOI] [PubMed] [Google Scholar]
- (6).Eddy MT; Lee M-Y; Gao Z-G; White KL; Didenko T; Horst R; Audet M; Stanczak P; McClary KM; Han GW; Jacobson KA; Stevens RC; Wüthrich K Cell 2018, 172, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kenakin T Annu. Rev. Pharmacol. Toxicol 2002, 42, 349–379. [DOI] [PubMed] [Google Scholar]
- (8).Xu F; Wu H; Katritch V; Han GW; Jacobson KA; Gao Z-G; Cherezov V; Stevens RC Science 2011, 332, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jaakola V-P; Griffith MT; Hanson MA; Cherezov V; Chien EYT; Lane JR; IJzerman AP; Stevens RC Science 2008, 322, 1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Carpenter B; Nehmé R; Warne T; Leslie AGW; Tate CG Nature 2016, 536, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rasmussen SGF; DeVree BT; Zou Y; Kruse AC; Chung KY; Kobilka TS; Thian FS; Chae PS; Pardon E; Calinski D; Mathiesen JM; Shah STA; Lyons JA; Caffrey M; Gellman SH; Steyaert J; Skiniotis G; Weis WI; Sunahara RK; Kobilka BK Nature 2011, 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huang W; Manglik A; Venkatakrishnan AJ; Laeremans T; Feinberg EN; Sanborn AL; Kato HE; Livingston KE; Thorsen TS; Kling RC; Granier S; Gmeiner P; Husbands SM; Traynor JR; Weis WI; Steyaert J; Dror RO; Kobilka BK Nature 2015, 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Che T; Majumdar S; Zaidi SA; Ondachi P; McCorvy JD; Wang S; Mosier PD; Uprety R; Vardy E; Krumm BE; Han GW; Lee M-Y; Pardon E; Steyaert J; Huang X-P; Strachan RT; Tribo AR; Pasternak GW; Carroll FI; Stevens RC; Cherezov V; Katritch V; Wacker D; Roth BL Cell 2018, 172, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mazzoni MR; Taddei S; Giusti L; Rovero P; Galoppini C; D’Ursi A; Albrizio S; Triolo A; Novellino E; Greco G; Lucacchini A; Hamm HE Mol. Pharmacol 2000, 58, 226–236. [DOI] [PubMed] [Google Scholar]
- (15).Isogai S; Deupi X; Opitz C; Heydenreich FM; Tsai C-J; Brueckner F; Schertler GF; Veprintsev DB; Grzesiek S Nature 2016, 530, 237–241. [DOI] [PubMed] [Google Scholar]
- (16).Eddy MT; Didenko T; Stevens RC; Wüthrich K Structure 2016, 24, 2190–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sušac L; O’Connor C; Stevens RC; Wüthrich K Angew. Chem 2015, 127, 15461–15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Horst R; Liu JJ; Stevens RC; Wüthrich K Angew. Chem., Int. Ed 2013, 52, 10762–10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Liu JJ; Horst R; Katritch V; Stevens RC; Wüthrich K Science 2012, 335, 1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim TH; Chung KY; Manglik A; Hansen AL; Dror RO; Mildorf TJ; Shaw DE; Kobilka BK; Prosser RS J. Am. Chem. Soc 2013, 135, 9465–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Manglik A; Kim TH; Masureel M; Altenbach C; Yang Z; Hilger D; Lerch MT; Kobilka TS; Thian FS; Hubbell WL; Prosser RS; Kobilka BK Cell 2015, 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pándy-Szekeres G; Munk C; Tsonkov TM; Mordalski S; Harpsøe K; Hauser AS; Bojarski AJ; Gloriam DE Nucleic Acids Res. 2018, 46, D440–D446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Munk C; Isberg V; Mordalski S; Harpsøe K; Rataj K; Hauser AS; Kolb P; Bojarski AJ; Vriend G; Gloriam DE Br. J. Pharmacol 2016, 173, 2195–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hino T; Arakawa T; Iwanari H; Yurugi-Kobayashi T; Ikeda-Suno C; Nakada-Nakura Y; Kusano-Arai O; Weyand S; Shimamura T; Nomura N; Cameron AD; Kobayashi T; Hamakubo T; Iwata S; Murata T Nature 2012, 482, 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ye L; Orazietti AP; Pandey A; Prosser RS, High-Efficiency Expression of Yeast-Derived G-Protein Coupled Receptors and 19F Labeling for Dynamical Studies In Protein NMR; Springer: 2018; pp 407–421. [DOI] [PubMed] [Google Scholar]
- (26).Emami S; Fan Y; Munro R; Ladizhansky V; Brown LS J. Biomol. NMR 2013, 55, 147–155. [DOI] [PubMed] [Google Scholar]
- (27).Fan Y; Shi L; Ladizhansky V; Brown LS J. Biomol. NMR 2011, 49, 151–161. [DOI] [PubMed] [Google Scholar]
- (28).Noguchi S; Satow YJ Biochem. 2006, 140, 799–804. [DOI] [PubMed] [Google Scholar]
- (29).Shimamura T; Shiroishi M; Weyand S; Tsujimoto H; Winter G; Katritch V; Abagyan R; Cherezov V; Liu W; Han GW; Kobayashi T; Stevens RC; Iwata S Nature 2011, 475, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Asada H; Uemura T; Yurugi-Kobayashi T; Shiroishi M; Shimamura T; Tsujimoto H; Ito K; Sugawara T; Nakane T; Nomura N; Murata T; Haga T; Iwata S; Kobayashi T Microb. Cell Fact 2011, 10, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yurugi-Kobayashi T; Asada H; Shiroishi M; Shimamura T; Funamoto S; Katsuta N; Ito K; Sugawara T; Tokuda N; Tsujimoto H; Murata T; Nomura N; Haga K; Haga T; Iwata S; Kobayashi T Biochem. Biophys. Res. Commun 2009, 380, 271–276. [DOI] [PubMed] [Google Scholar]
- (32).Ye L; Van Eps N; Zimmer M; Ernst OP; Prosser RS Nature 2016, 533, 265–268. [DOI] [PubMed] [Google Scholar]
- (33).Weiss HM; Haase W; Michel H; Reiländer H Biochem. J 1998, 330, 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Clark LD; Dikiy I; Chapman K; Rödström KE; Aramini J; LeVine MV; Khelashvili G; Rasmussen SG; Gardner KH; Rosenbaum DM eLife 2017, 6, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Clark L; Zahm JA; Ali R; Kukula M; Bian L; Patrie SM; Gardner KH; Rosen MK; Rosenbaum DM J. Biomol. NMR 2015, 62, 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Morgan WD; Kragt A; Feeney J J. Biomol. NMR 2000, 17, 337–347. [DOI] [PubMed] [Google Scholar]
- (37).Horst R; Stanczak P; Stevens RC; Wüthrich K Angew. Chem., Int. Ed 2013, 52, 331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Stanczak P; Zhang Q; Horst R; Serrano P; Wuthrich K J. Biomol. NMR 2012, 54, 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Stanczak P; Horst R; Serrano P; Wüthrich K J. Am. Chem. Soc 2009, 131, 18450–18456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Krueger-Koplin RD; Sorgen PL; Krueger-Koplin ST; Rivera-Torres IO; Cahill SM; Hicks DB; Grinius L; Krulwich TA; Girvin ME J. Biomol. NMR 2004, 28, 43–57. [DOI] [PubMed] [Google Scholar]
- (41).Vinogradova O; Sönnichsen F; Sanders CR II. J. Biomol. NMR 1998, 11, 381–386. [DOI] [PubMed] [Google Scholar]
- (42).Thompson AA; Liu JJ; Chun E; Wacker D; Wu H; Cherezov V; Stevens RC Methods 2011, 55, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Abagyan R; Totrov M; Kuznetsov D J. Comput. Chem 1994, 15, 488–506. [Google Scholar]
- (44).Lomize MA; Pogozheva ID; Joo H; Mosberg HI; Lomize AL Nucleic Acids Res. 2012, 40, D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Abagyan R; Totrov M J. Mol. Biol. 1994, 235, 983–1002. [DOI] [PubMed] [Google Scholar]
- (46).Cheng YC; Prusoff WH Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (47).Pervushin K; Riek R; Wider G; Wüthrich K Proc. Natl. Acad. Sci U. S. A 1997, 94, 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, sequences of primers used for generation of A2AAR variants; Figure S1, radioligand binding data for A2AAR and A2AAR variants; Figure S2, analytical size exclusion chromatograms of A2AAR and three A2AAR variants containing a single extrinsic tryptophan residue; Figures S3–S5, NMR spectra of [u-15N, ~70% 2H]- A2AAR[F201W], [u-15N, ~70% 2H]-A2AAR[K233W], and [u-15N, ~70% 2H]-A2AAR[Y290W] in complex with ZM241385; Table S2, evaluated penalties in free energy and solubility for structural models of A2AAR variants; Figure S6, schemes of A2AAR constructs examined in the context of Figure 1; Figure S7, amino acid sequences of A2AAR variants containing a single extrinsic tryptophan residue; Figure S8, NMR comparison of [u-15N, ~70% 2H]-A2AAR with and without a covalently attached peptide leader sequence; Figure S9, comparison of NMR spectra of A2AAR in different mixed micelles (PDF)