

Abstract
Objective
Serrated colorectal cancer (CRC) accounts for approximately 25% of cases, and includes tumours that are amongst the most treatment resistant and with worst outcomes. This CRC subtype is associated with activating mutations in the mitogen activated kinase (MAPK) pathway gene, BRAF, and epigenetic modifications termed the CpG Island Methylator Phenotype (CIMP), leading to epigenetic silencing of key tumour suppressor genes. It is still not clear which (epi-)genetic changes are most important in neoplastic progression and we begin to address this knowledge gap herein.
Design
We utilise organoid culture combined with CRISPR/Cas9 genome engineering, to sequentially introduce genetic alterations associated with serrated CRC and which regulate the stem cell niche, senescence and DNA mismatch repair.
Results
Targeted biallelic gene alterations were verified by DNA sequencing. Organoid growth in the absence of niche factors was assessed, as well as analysis of downstream molecular pathway activity. Orthotopic engraftment of complex organoid lines, but not BrafV600E alone, quickly generated adenocarcinoma in vivo with serrated features consistent with human disease. Loss of the essential DNA mismatch repair enzyme, Mlh1, led to microsatellite instability. Sphingolipid metabolism genes are differentially regulated in both our mouse models of serrated CRC and human CRC, with key members of this pathway having prognostic significance in the human setting.
Conclusion
We generate rapid, complex models of serrated CRC to determine the contribution of specific genetic alterations to carcinogenesis. Analysis of our models alongside patient data has led to the identification of a potential susceptibility for this tumour type.
INTRO
Sporadic colorectal cancer (CRC) develops via two main genetic pathways. The conventional pathway described by Fearon & Vogelstein [1] has characteristic chromosomal instability (CIN) with stepwise loss of key tumour suppressors (eg. Adenomatous polyposis coli, APC) and activation of oncogenes (eg. KRASG12D mutation). In the last two decades, an alternate molecular pathway has been identified that accounts for 25% of CRC [2]. This subtype of CRC is named for the serrated, or saw-toothed, morphology of the precursor lesion, including sessile serrated polyps, and comprises a molecularly distinct, somewhat heterogeneous tumour type that forms without CIN. It can be best differentiated from conventional CRC by two characteristic molecular markers; the presence of the BRAFV600E mutation that activates the mitogen-activated protein kinase (MAPK) pathway and the coincident, coordinated epigenetic modification of specific target promoters termed the CpG Island Methylator Phenotype (CIMP) [3]. BRAFV600E/CIMP+ tumours account for 8–20% of all CRC [2, 4]. These are preferentially located in the proximal colon, frequently present with higher grade and contain a group of cancers with the worst prognosis of all CRC [2, 5]. Furthermore, of relevance to the gastrointestinal cancer preventionist, these serrated lesions are less well detected by current CRC screening programs and are overrepresented in colonoscopic interval cancers [6, 7]. The molecular basis for these subtype characteristics are not well understood, but may hold the key to preventing and treating serrated CRC.
The culture of normal and tumour derived organoids pioneered by Hans Clevers and his group [8, 9] allows the indefinite, in vitro propagation of primary gut epithelia and neoplastic specimens. Cells are grown in self-organising aggregates with representative epithelial architecture, suspended within a basement membrane-gel matrix and supported by stem cell niche signalling factors. For the colon these factors, Wnt/R-spondin, Epidermal growth factor (EGF), and Noggin activate the Wnt and MAPK pathways and inhibit the bone morphogenic protein (BMP) pathway, respectively. This culture system supports the stem cell population, as well as more differentiated cell types, allowing long-term primary culture containing many of the representative epithelial cell types of the original tissue.
The next-generation sequencing revolution has armed us with more information than ever before about the (epi-)genetic changes associated with serrated CRC. Mining this static data is relatively simple, yet understanding the contribution of genetic events to carcinogenesis is a real challenge. To this end, we have combined recent advances in stem cell biology [10] with genome editing techniques [11], to produce a system in which multiple genetic alterations can be assessed for their contribution to serrated CRC. This is analogous to the approach taken to model the conventional pathway to CRC [12, 13] and is inspired by the observation that many recurrent CRC mutations lie within genes involved in key signalling pathways that are essential for maintenance of the intestinal stem cell niche [14]. Our work provides new insights into the importance of common (epi-)genetic alterations found in human serrated CRC and highlights a potential vulnerability of this cancer type.
RESULTS
To identify recurrent serrated CRC associated alterations in stem cell niche signalling and senescence pathways, we analysed The Cancer Genome Atlas (TCGA) (epi-)genomic data for 633 CRC cases [15]. BRAFV600E mutation is estimated to mark 50–67% of serrated CRCs [2] and, in the absence of microsatellite instability (MSI), is associated with poor outcome for late stage disease [2, 16]. As only 30% of serrated CRCs retain serrated crypt morphology [2], we use the BRAFV600E molecular marker rather than histological appearance to extract serrated CRC cases from the TCGA set. In the 50 TCGA CRC cases that contain BRAFV600E (depicted in Figure 1), we then used a candidate gene approach to look for frequently co-altered genes that play important roles in regulating the stem cell niche or senescence. p16 INK4A encodes a tumour suppressor protein critical for oncogene induced senescence. Loss of p16 INK4a expression is associated with the conversion of serrated polyps to high-grade dysplasia/CRC and cooperates with BrafV600E in a mouse model of serrated CRC [17, 18, 19]. Consistent with previous reports, we detected p16 INK4A hypermethylation or mutation in 46% of TCGA CRC cases overall and in 84% of BRAFV600E CRC (Figure 1) [20, 21, 22]. The Wnt pathway intricately regulates intestinal stem cell proliferation and differentiation. The classic Wnt-pathway regulator, APC, is the most commonly mutated gene in CRC [15]. However for BRAFV600E CRC, we identified two negative regulators, ZNRF3 and RNF43, as the most commonly altered Wnt pathway genes (Figure 1) [23]. Interestingly, in mice, cooperative inactivation of both Rnf43 and Znrf3 are required for polyp formation [24] and we detect alteration of both genes in 32% of BRAFV600E CRC. This is consistent with reports of differential Wnt pathway disruption in precursor lesions of the serrated pathway to CRC compared to the conventional pathway [25, 26]. The mutation rate of RNF43 is likely under-represented in the TCGA dataset due to incomplete calling of frame-shift mutations because of their similarity to technical DNA polymerase slippage errors [27]. The transforming growth factor-β (TGFβ) pathway is frequently aberrantly regulated in CRC, often through mutation of TGFβ receptor 2 (TGFβR2) [15]. TGFβR2 mutation occurred in 10% of the BRAFV600E CRC cases we examined (Figure 1), again reflecting under calling of the polyadenine repeat tract mutations in the TCGA dataset as use of older sequencing modalities identified TGFBR2 as mutated in 90% of MSI CRC [28]. Lastly, we wanted to model MSI CRC and so examined the key mismatch repair gene, MutL homolog 1 (MLH1). In line with a meta-analysis of MLH1 methylation frequency in CRC, we detected MLH1 hypermethylation in 20% of TCGA CRC cases [29]. This increased to 74% alteration in BRAFV600E CRC (Figure 1).
Figure 1. Co-occurring molecular events in stem-cell niche, microsatellite instability and senescence pathways in BRAFV600E mutant serrated CRC.

Of the 50 patients with BRAFV600E CRC from the TCGA CRC cohort (n=527 patients total), we depict the co-alteration (non-synonymous mutation and/or hyper-methylation) of selected genes in these pathways. Number of patients with each alterations is indicated by bar graph on right, % of BRAFV600E cases containing the alteration is indicated by numbers on left. Coloured blocks indicate gene is altered in the sample, grey is unaltered.
Carcinogenesis is a story of liberation and, as we genetically engineer successive stages of serrated CRC, we select organoids through their unique biology, microenvironmental requirements and treatment sensitivities. Firstly, the BRAFV600E mutation is an early genetic change in serrated polyps [30]. To first incorporate this mutation, we established colon organoid cultures from BRAFCA/VillinCreERT mice (Figure 2). In vitro treatment with 4-hydroxytamoxifen (4-OHT) induced highly efficient recombination to BrafV600E (Supplementary Figure 1) and activation of ERK1/2 phosphorylation (Figure 2A). This in vitro system mimics the chemoresistance to epidermal growth factor receptor (EGFR) inhibitors observed for MAPK mutant CRC in the clinic [31]. Treatment with EGFR inhibitor results in a significant growth inhibition of wild-type organoids in culture but not BrafV600E organoids (Figure 2B, Supplementary Figure 2).
Figure 2. Introduction of genetic alterations associated with serrated CRC promotes independence from niche factor requirements.
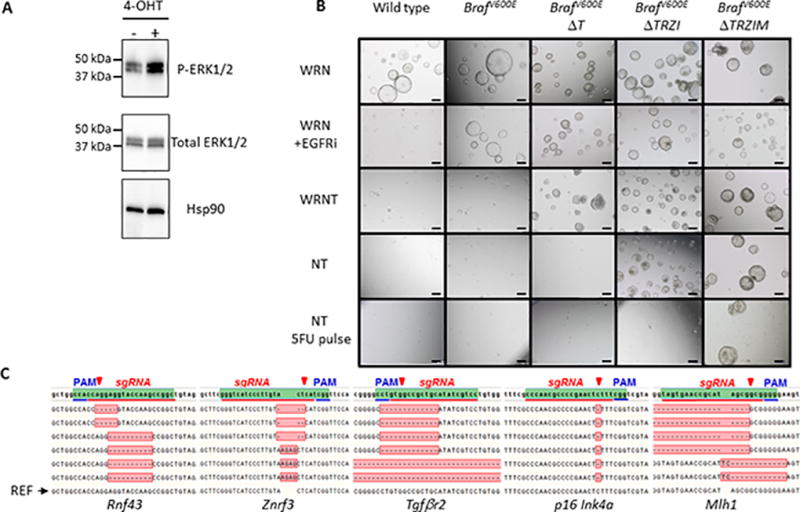
A, activation of MAPK pathway in BrafV600E organoids visualized by phosphorylation of the ERK1/2 effector protein, 4-hydroxytamoxifen (4-OHT). B, Generation of a ‘serratoid’ series from normal mouse colonic organoids through sequential CRISPR/Cas9 targeting and in vitro selection. BrafV600E, BrafV600ETgfbr2Δ/Δ (BrafV600EΔT), BrafV600E Tgfbr2Δ/Δ Rnf43Δ/Δ/Znrf3Δ/Δ p16Ink4aΔ/Δ (BrafV600E ΔTRZI), BrafV600E Tgfbr2Δ/ΔRnf43Δ/Δ/Znrf3Δ/Δ p16Ink4aΔ/ΔMlh1Δ/Δ (BrafV600E ΔTRZIM). Normal media components required as stem cell niche factors Wnt-3a (W), Rspo-2 (R), Noggin (N), additional selection with TGFβ1 (T) and chemotherapeutic agent, 5-Fluorouracil (5FU). C, DNA sequence verification of biallelic insertion/deletion (indel) mutations that result in prematurely truncated proteins.
Genome editing using microbial Cas9 nucleases from the clustered regularly interspaced short palindromic repeats (CRISPR) adaptive immune system has revolutionised the field of genome editing [11]. We have modelled serrated CRC using this genome editing technique. Thus, following mutation of Braf we sequentially incorporated 4 further genetic alterations using CRISPR/Cas9 [11] (Figure 2). Similar to previous work [12, 13], the addition of Tgfβ to culture medium selected for organoids with functional loss of the Tgfβ pathway, in this case Tgfβr2 (BrafV600E; Tgfbr2Δ/Δ, abbreviated to BrafV600EΔT) (Figure 2B). Indeed, wild type (WT), BrafV600E and control transfected cultures do not survive in the presence of Tgfβ [32] (Figure 2B). Quantitative real-time PCR (qRT-PCR) analysis of the Tgfβ response gene, Serpin1, showed that Tgfβ can activate this pathway in BrafV600E organoids as shown by an increase in Serpin1 mRNA levels. However, organoids containing loss of function of Tgfβr2 have low basal expression of Serpin1 and are not Tgfβ responsive (Supplementary Figure 3). Next, removal of Wnt-ligands, Wnt3a and Rspo2, kills WT, BrafV600E, BrafV600EΔT and control transfected cultures (Figure 2B). However, transfection of plasmids encoding Cas9 and guideRNAs (gRNAs) targeting the negative Wnt regulators, Rnf43 and Znrf3, allowed outgrowth of organoids with cystic morphology (Figure 2B, growth of organoids in Wnt-ligand deficient NT medium). Loss of the p16Ink4a tumour suppressor was not directly selected for using changed media conditions, but this gene was targeted simultaneously with Rnf43 and Znrf3 and its loss likely provided a growth advantage to BrafV600E mutant cells [17]. This resulted in BrafV600E;Tgfbr2Δ/Δ;Rnf43Δ/Δ/Znrf3Δ/Δ;p16 Ink4aΔ/Δ (BrafV600E ΔTRZI) organoids. qRT-PCR analysis of the Wnt-pathway response genes, Axin2 and Lgr5, showed that activation of this pathway in the BrafV600EΔTRZI organoids in the absence of Wnt ligands is similar to the level in control BrafV600E organoids cultured in the presence of Wnt ligands, i.e. the pathway is super-activated by targeting Rnf43 and Znrf3 (Supplementary Figure 3). qRT-PCR analysis also indicates a reduction in p16Ink4a mRNA levels in BrafV600EΔTRZI compared to BrafV600E organoids (Supplementary Figure 3).
Lastly, to select for loss of Mlh1 we exploited the clinical finding that MSI-High (MSI-H) cancers are reported to show relative resistance to the chemotherapeutic 5-fluorouracil (5-FU) [33]. We used media containing 5-FU to promote DNA mismatches [34]. Mlh1 is an essential component of the DNA mismatch machinery [35] and so we hypothesised that inactivation of Mlh1 would lead to increased survival of cells despite DNA damage-arrest signals, consistent with the poor response of MSI CRC to 5-FU based adjuvant chemotherapy [33]. Organoids that had been transfected with a plasmid encoding Cas9 and a gRNA targeting Mlh1 (BrafV600EΔTRZIM) were better able to withstand a pulse of 5FU treatment than control transfected BrafV600EΔTRZI (Figure 2B). The activity of previously unpublished gRNAs (Tgfβr2 and Mlh1 gRNAs) was evaluated in polyclonal organoids before proceeding to handpicking single clones and expansion of lines (Supplementary Figure 4). We also used bioinformatic prediction of the possible off-target sites for each of these gRNAs and verified that they were not changed in our organoid lines by Sanger sequencing (Supplementary Table 1). Bi-allelic, loss of function insertions and deletions (indels) in target genes were verified using DNA sequencing (Figure 2C, Supplementary Table 2).
To assess the effect of this series of genetic alterations on colorectal tumourigenesis, we transplanted organoids into NOD.Cg-PrkdcscidIl2rgtm1Wjl/Szj (NSG) mice and used in vivo murine colonoscopy to directly visualize and score pathologic changes (Figure 3A, Supplementary Videos). Using a modified needle attachment, we injected organoids into the orthotopic site, the mucosal layer of the colon wall (Figure 3A, Supplementary Figure 5A, 5B) [36]. Injection of BrafV600E mutant organoids failed to induce tumour formation over a 3 month time course as assessed by colonoscopic surveillance, at necropsy and by pathological evaluation (Figure 3B, 4B, Supplementary Figure 6). Introduction of Tgfβ pathway perturbation in BrafV600EΔT organoids results in 57% of organoid injections forming tumours within 3 months (Figure 3B). However, significantly more BrafV600EΔTRZI and BrafV600EΔTRZIM injected organoids formed tumours, 98% and 100% respectively (Figure 3B, p<0.05). Similar to human sessile serrated polyps, 94% (n=11 mice) of BrafV600EΔTRZI and 100% (n=8 mice) of BrafV600EΔTRZIM tumours developed a noticeable mucus cap visible from two weeks post-injection via colonoscopic surveillance [37] (Supplementary Figure 5C). This is in contrast to only 9% (n=12 mice) of the BrafV600EΔT tumours with a mucus cap. The more genetically complex organoids (BrafV600EΔTRZI and BrafV600EΔTRZIM) grew significantly more quickly than BrafV600EΔT tumours (Figure 3C, Supplementary Figure 6, p<0.001) and overall survival was also significantly decreased, with mean time from injection to humane endpoint of the experiment at 6 weeks for BrafV600EΔTRZI and 4 weeks for BrafV600EΔTRZIM (Figure 3D, p<0.001).
Figure 3. BrafV600E alone is not sufficient for colon tumour formation, but with increasing serrated pathway genetic alterations tumour penetrance and growth rate increases.
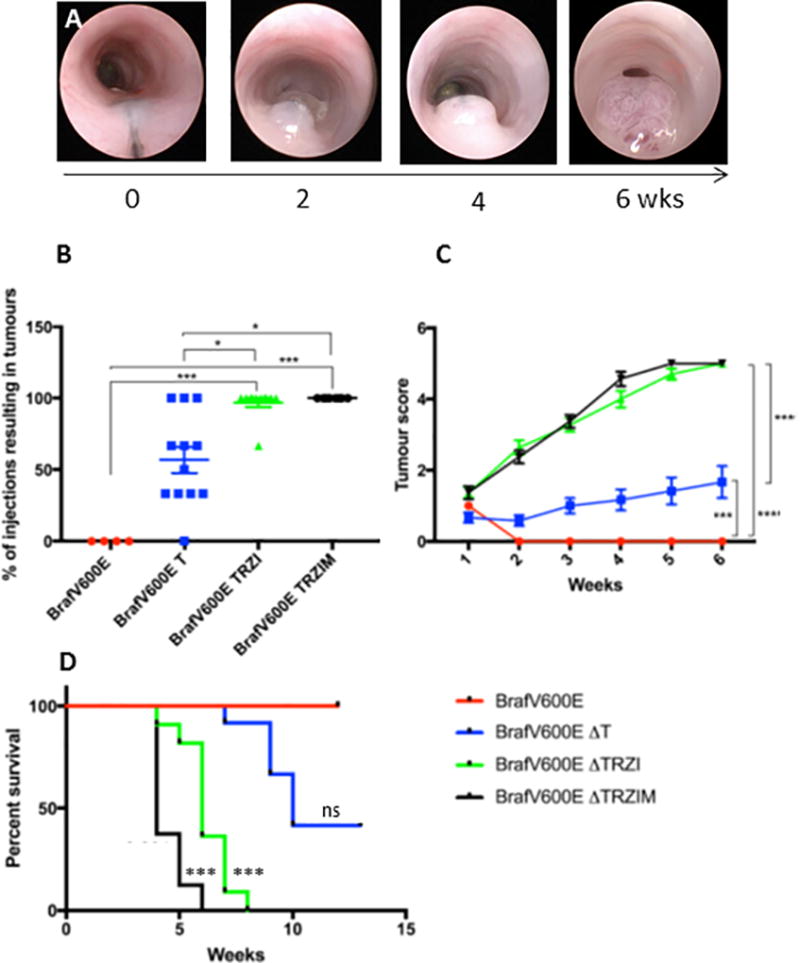
A, colonoscopic images showing injection and rapid growth of serratoid BrafV600E ΔTRZI line. B, Tumour penetrance as a percentage of the number of organoid injections that gave rise to a tumour/mouse in 3 months for each line, with 1–3 injections/mouse. C, Colonoscopic scoring of largest tumour in each mouse (n=5 mice per group, Becker scale). D, Kaplan-Meier plot showing overall survival post-injection with organoid lines BrafV600E n=4 mice, BrafV600E ΔT n=12 mice, BrafV600E ΔTRZI n=11 mice, BrafV600E ΔTRZIM n=8 mice, compared to BrafV600E using a Bonferroni adjustment for multiple comparisons. ns=not significant, *=p≤0.05, **=p≤0.01, ***=p≤0.001.
Figure 4. Multi-hit ‘serratoids’ generate invasive adenocarcinoma with features of human serrated CRC.
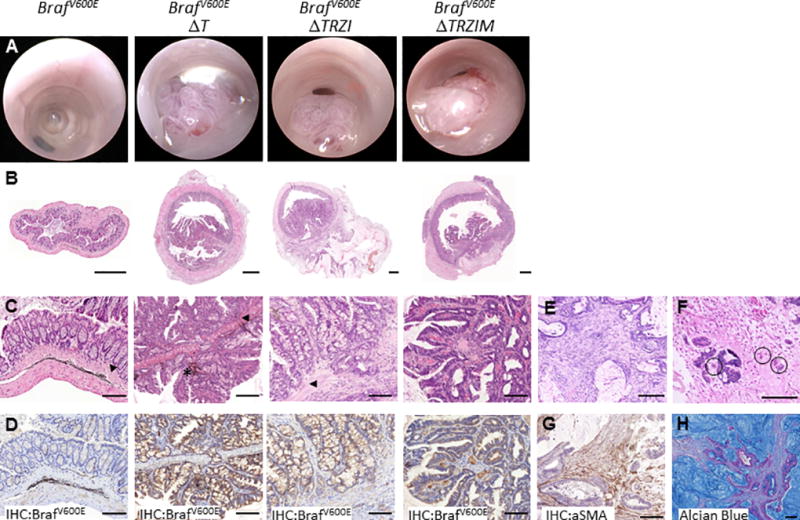
Colonoscopy (A) and histology (B–H) images of mouse colon orthotopically injected with mutant organoid lines BrafV600E, BrafV600E ΔT, BrafV600E ΔTRZI, BrafV600E ΔTRZIM. B, H&E stained sections of whole colon. C, higher magnification H&E, arrows denote position of muscularis mucosae, * denotes remnant ink from injection. D, immunohistochemical staining for mutant BrafV600E protein clearly delineates serratoid derived tumour cells. E, representative images of desmoplastic stromal response and F, tumour budding (circled). G, tumour stromal response stains positive for alpha-smooth muscle actin (aSMA). H, mucin lakes present in mucinous adenocarcinoma visualised using Alcian Blue stain. Scale bars are (B) 500um, (C–H) 100um.
Histologically, tumours generated using BrafV600EΔT, BrafV600EΔTRZI and BrafV600EΔTRZIM organoids are invasive, adenocarcinomas with features of human serrated CRC (Figure 4, Supplementary Table 3) [2, 38]. We observed infrequent liver metastasis (9% or 1 out of 11) mice), with no macro-metastasis to other sites (lung, peritoneum) after injection with BrafV600EΔTRZI (Supplementary Figure 5D). Organoid-derived cells were readily visualised by morphology and using immunohistochemical detection of BrafV600E (Figure 4D, Supplementary Figure 5E). The tumours display a substantial desmoplastic stromal reaction (Figure 4E), particularly with BrafV600EΔTRZI compared to the less genetically complex BrafV600EΔT (85% vs. 33% p<0.05, Supplementary Table 3). Thus the genetics of the tumour in this model was important in the co-evolution of the stroma. This stromal reaction is composed of smooth muscle actin expressing cancer activated fibroblasts amongst other cell types (Figure 4G). The more complex BrafV600EΔTRZI and BrafV600EΔTRZIM, but not BrafV600EΔT tumours, featured tumour budding (Figure 4F, Supplementary Figure 5F) and approximately half were categorised as mucinous adenocarcinoma [38], indicating a change to the amount of mucin produced by tumours with more complex genetic alterations, (42–50% vs. 0% p<0.01, Supplementary Table 3). This could also be visualised using Alcian Blue mucin staining (Figure 4G). Muc2 mRNA, encoding a core component of mucus, was increased in the BrafV600EΔTRZI but not the BrafV600E or BrafV600EΔT organoids (p<0.05, Supplementary Figure 7).
To investigate the consequences of mutating the essential DNA damage response gene, Mlh1, we analysed microsatellite marker length in our organoid series. BrafV600EΔTRZIM, but not BrafV600EΔTRZI organoids have altered microsatellite length (MSI) when compared to the donor mouse from which organoids were derived, in 4 mononucleotide markers previously validated for use in mouse [39, 40]. These cells accrued further MSI in vivo, with BrafV600EΔTRZIM, but not BrafV600EΔTRZI tumours containing additional microsatellite tract length variability (Supplementary Figure 8).
Discrete transcript profiles of serrated and conventional pathway tumours
In addition to our series of serrated pathway tumours, we also generated tumours driven by alteration of commonly mutated conventional CRC pathway genes, Apc and KrasG12D. Similar to previous reports [36], colonic organoids expressing adeno-virally delivered Cre, derived from KrasLSL-G12D/+;Apcfl/fl mice survived culture in the absence of Wnt3a or Rspo2 (Supplementary Figure 9). Adenocarcinomas arising from orthotopic injection of KrasG12D/+;Apcfl/fl organoids, were verified as KrasG12D/+;ApcΔ580l/Δ580 (KrasG12D;ApcΔ/Δ) genotype and displayed nuclear β-catenin localisation (Supplementary Figure 9). We isolated RNA from our series of serrated and conventional pathway tumours and compared mRNAseq expression profiles between these different tumours and normal colonic mucosa from tumour bearing animals (n=4 biological replicates per group). The multidimensional scaling (MDS) plot generated from our RNAseq data showed that, as expected, the normal colon samples had quite different transcript profiles to the tumour samples (Figure 5A). In comparison, the BrafV600E, serrated subtype tumours all clustered closely together and separated from the KrasG12D;ApcΔ/Δ tumours (Figure 5A). Consistent with alterations to the MAPK pathway in our BrafV600E organoid series, gene set enrichment analysis (GSEA) identified differential expression of MAPK-pathway transcripts as enriched in the BrafV600E tumours compared to normal colon (Supplementary Figure 10A). Validating our attempt to model MSI CRC, the tumours derived from BrafV600EΔTRZIM organoids were also significantly enriched for differential expression of a human MSI CRC-associated gene set (Supplementary Figure 10B) [41].
Figure 5. Serrated pathway tumours are molecularly distinct from conventional pathway tumours.

A, multi-dimensional scaling plot of RNA expression data from normal mouse colon (black), serrated pathway BrafV600E ΔT (blue), BrafV600E ΔTRZI (red) and BrafV600E ΔTRZIM (yellow) and conventional pathway tumours KrasG12D;ApcΔ/Δ (grey), n=4 samples per group. B, gene set enrichment analysis (GSEA) for Sphingolipid de novo biosynthesis Reactome between BrafV600EΔTRZI serrated tumour and normal mouse colon. Enrichment score (ES), normalised enrichment score (NES), false discovery rate (FDR). C, Expression of Sphk1 is increased and Sgpp1 is decreased in mouse BrafV600E serrated CRC compared to normal mouse colon. Fold induction of mRNA expression is normalized to Gapdh, with transcript level in normal colon set to 1. Results from at least four animals with triplicate technical replicates are shown, error bars denote standard deviation. Two-tailed t-test used for pair-wise statistical analysis. D, Violin plots depicting z-score values for normalized expression of SPHK1 (top) and SGPP1 (bottom) transcripts in 622 human TCGA CRC and normal colon samples separated into wild-type BRAF/KRAS, KRAS mutant and BRAFV600E CRC. E, Kaplan-Meier plots showing TCGA patient survival probability based on expression level of SPHK1 (top, SPHK1 high group n=139 in red, low group n=483 in green) or SGPP1 (bottom, SGPP1 low group n=145 in green, high group n=477 in red). *=p≤0.05, **=p≤0.01, ***=p≤0.001, NS= not significant.
The top enriched gene set based on normalised enrichment score in our analyses across all BrafV600E, but not KrasG12D;ApcΔ/Δ, tumours when compared to normal colon was the sphingolipid (SP) de novo biosynthesis pathway (Figure 5B, Supplementary Table 4). We validated the differential expression of SP pathway transcripts from the leading edge of the GSEA by real-time qPCR in BrafV600E and conventional pathway tumours (Figure 5C, Supplementary Figure 11). Two key enzymes regulating S1P levels in this pathway are sphingosine kinase 1 (SPHK1) and the counter-acting sphingosine-1-phosphate phosphatase 1 (SGPP1) [42]. Consistent with previous reports in human CRC [43, 44], we observed increased expression of Sphk1 transcripts in our series of BrafV600E tumours compared to normal mouse colon and a converse decrease in Sgpp1 transcripts (Figure 5C). This suggested at the transcriptomic level that the pathway may be primed for increased production of the pro-survival molecule sphingosine 1-phosphate (S1P) in our model of serrated CRC [42]. We next wished to determine if this shift at the transcript level was also observed in human CRC samples. SPHK1 was significantly upregulated in 622 TCGA CRC samples compared to matched normal controls, with greatest expression levels in BRAFV600E tumours (Figure 5D). Conversely, SGPP1 expression was significantly decreased in all CRC samples compared to matched normal controls. Increased expression of SPHK1 and decreased expression of SGPP1 were both separately associated with poor survival in the TCGA cohort (Figure 5E).
DISCUSSION
Here we have focused on the serrated pathway to CRC that accounts for up to 25% of all CRC, and for microsatellite stable cases has very poor outcome [2]. We have developed a panel of CRISPR/Cas9 genome engineered organoids that model DNA alterations found in BRAF mutant serrated CRC. In this way we are producing novel, preclinical models of colorectal adenocarcinoma with features of serrated CRC, and most notably desmoplastic stromal responses, infiltrative growth, tumour budding and mucinous differentiation [2, 45, 46] (see Figure 4, Supplementary Table 3 & Figure 5). Mucous caps characterise human serrated polyps at colonoscopy. When we look at an early time point in tumour development in our serrated CRC mouse models with complex genetics (BrafV600EΔTRZI and BrafV600EΔTRZIM) we also observe mucous caps. Previous studies using CRISPR/Cas9 to model the conventional molecular pathway to CRC using human intestinal organoids were either unable to generate invasive CRC [12], or required 4 gene alterations for invasive tumour growth [13]. We find here that alterations to just two genes, Braf and Tgfbr2, results in transmurally invasive adenocarcinoma. Further altering the oncogene-induced senescence factor, p16 Ink4a, and the Wnt pathway leads to increased tumour initiation and decreased survival time (Figure 3 & 4). These more genetically complex tumours had increased stromal response, tumour budding and mucinous differentiation. We also model MSI “hypermutator” CRC by inducing loss-of-function mutations in the DNA mismatch repair gene, Mlh1. Similar to recent work describing a 6-fold higher DNA mutation rate in CRISPR/Cas9 engineered MLH1Δ/ Δ human colon organoid lines [47], we have successfully generated the MSI phenotype as judged by instability in microsatellite repeat tract length (Supplementary Figure 8).
We utilise the recently adapted small animal colonoscopy technique [36, 48] to enable rapid, orthotopic injection of engineered organoids to the mucosal layer of the colon wall and tumour monitoring thereafter (Supplementary Figure 5 & videos). This is especially salient given the observed differences in metastatic potential for engineered conventional pathway CRC organoids delivered either subcutaneously or using a rectal prolapse model, where only tumours growing at the orthotopic colon site produced liver metastases [49]. A second report using similar conventional pathway mutant organoids injected using a colonoscope resulted in metastasis in 30% of mice [36]. We observe liver metastasis in the serrated CRC models presented here, albeit infrequently, and no lung or peritoneal spread of the disease. This may be due to the model not containing strong metastasis promoting alterations or alternately, in the future, luminal preservation through colonoscopic biopsy may allow a more complete evaluation of disease progression.
These models will advance this field by providing a rapid, inexpensive and pathologically faithful approach to studying the biology of serrated CRC. Other models, whilst valuable in their own right, are slow, require complicated in vivo breeding and have a prescribed number and sequence of genetic alterations [18, 19, 50, 51, 52, 53]. Our models can be readily adapted to investigate the many leads generated by next generation sequencing of human serrated CRC and will be particularly valuable for testing potential therapeutics in the setting of complex, but known, genetic landscapes. Transcriptome analysis of our genome engineered models of serrated and conventional CRC have identified putative changes to sphingolipid metabolism in these tumours. The differential expression of transcripts encoding two key enzymes in this pathway are mirrored in human CRC and are associated with worse prognosis. Future work will investigate the therapeutic efficacy of targeting the sphingolipid biosynthesis pathway for BRAFV600E CRC.
METHODS
Mouse experiments
This study was approved by the MIT Institutional Animal Care and Use Committee, QIMR Berghofer and SAHMRI Animal Ethics committees (P1208, SAM174, SAM205). Mice heterozygous for both the conditionally active BrafCA and Villin-CreERT2 alleles to induce the mutation analogous to human BRAFV600E specifically in the intestine were generated as described [52]. Similarly, mice containing the KrasLSL-G12D/+ [54] and ApcCKO/CKO [55] alleles were interbred to produce double transgenic offspring that were used to generate KrasG12D/+; ApcΔ580l/Δ580 colonic organoids [36].
Orthotopic Injection
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (male and female, 6–12 weeks old) were obtained from the SAHMRI Bioresources facility and housed under pathogen-free conditions. Organoids were isolated from matrigel and dissociated to single cells and small clusters using TrypLE. The cell clusters (equivalent to ~150 organoids) were washed three times with cold PBS containing 10uM Y-27632 and then resuspended in 20ul 10% GFR matrigel 1:1000 india ink, 10uM Y-27632 in PBS and injected into the mucosa of the distal colon of anaesthetised NSG mice. Colonoscopy-guided orthotopic injection into the colon wall was undertaken as previously described [36] with modifications described in Supplementary methods.
Genome engineering
gRNAs specific for each target gene were either published sequences targeting Rnf43, Znrf3 or p16 Ink4a [56, 57] or designed de novo using the CRISPR design Tool to target the first exon immediately downstream of the translation start codon [11] (see Supplementary Methods table 2). gRNA oligos were cloned into px-459, which expresses both the Cas9 nuclease and single gRNAs [11]. Organoid transfection and culture details are included in the Supplementary methods section.
Nucleic acid analyses, bioinformatics, western blotting and immunohistochemistry
Please see the Supplementary Methods section.
Supplementary Material
SUMMARY BOX.
What is already known about this subject
25% of colorectal cancers (CRC) form via an alternate molecular pathway typified by activating mutation in the BRAF kinase and accrual of epigenetic modifications at specific promoter locations.
The molecular evolution of these serrated CRCs and how this relates to natural history is poorly understood and may hold the key to better treatment and prevention options for this form of CRC.
What are the new findings
Here we use next generation sequence information from human serrated CRC, combined with organoid culture, gene editing and orthotopic transplantation techniques to rapidly generate complex, preclinical models of serrated CRC.
How might it impact on clinical practice in the foreseeable future?
These preclinical models will allow therapeutic evaluation in known, previously untested genetic landscapes.
Transcriptomic analysis of our models, combined with patient data, have suggested potential vulnerabilities for this tumour type that we will test in the future.
Acknowledgments
The Rspo-2 expression plasmid was a kind gift from Prof. Antony Burgess (Walter & Eliza Hall Institute of Medical Research, Australia), L-Wnt3a cells were a kind gift of Prof. Hans Clevers (Hubrecht Institute, The Netherlands). Dr. Mark Van der Hoek and staff at the David R. Gunn Genomics Facility at SAHMRI, John Toubia, Andreas Schreiber, Joel Geoghegan and staff at the Adelaide Australian Cancer Research Foundation Genomics Facility for useful discussions.
This work was supported by Cure Cancer Australia/Cancer Australia (APP1102534), the Cancer Council SA Beat Cancer Project on behalf of its donors and the State Government of South Australia through the Department of Health SA, by Pathology Queensland, by the Australian National Health and Medical Research Council (NHMRC) through (APP1081852, APP1140236), by the National Institutes of Health (NIH) (K08 CA198002, R00 AG045144, R01 CA211184), the Department of Defense (CA120198), the V Foundation V Scholar Award, the Sidney Kimmel Scholar Award, the Pew-Stewart Trust Scholar Award, the Koch Institute Frontier Research Program through the Kathy and Curt Marble Cancer Research Fund, the American Federation of Aging Research, as well as by the Koch Institute Support Grant P30-CA14051 from the National Cancer Institute. DLW is supported by a NHMRC Career Development Fellowship, VLW by a Gastroenterological Society of Australia Senior Research Fellowship.
Abbreviations
- CRC
colorectal cancer
- MAPK
mitogen activated kinase
- CIMP
CpG Island Methylator Phenotype
- CIN
chromosomal instability
- APC
Adenomatous polyposis coli
- EGF
Epidermal growth factor
- BMP
bone morphogenic protein
- TCGA
The Cancer Genome Atlas
- MSI
microsatellite instability
- TGFβ
transforming growth factor-β
- MLH1
MutL homolog 1
- 4-OHT
4-hydroxytamoxifen
- EGFR
epidermal growth factor receptor
- CRISPR
clustered regularly interspaced short palindromic repeats
- WT
wild type
- qRT-PCR
Quantitative real-time PCR
- gRNAs
guideRNAs
- MSI-H
MSI-High
- NSG
NOD.Cg-PrkdcscidIl2rgtm1Wjl/Szj
- MDS
multidimensional scaling
- GSEA
gene set enrichment analysis
- SP
sphingolipid
- SPHK1
sphingosine kinase 1
- SGPP1
sphingosine-1-phosphate phosphatase 1
- S1P
sphingosine 1-phosphate
- BrafV600EΔT
BrafV600ETgfbr2Δ/Δ
- BrafV600EΔTRZI
BrafV600E Tgfbr2Δ/Δ Rnf43Δ/Δ/Znrf3Δ/Δ p16Ink4aΔ/Δ
- BrafV600E ΔTRZIM
BrafV600E Tgfbr2Δ/ΔRnf43Δ/Δ/Znrf3Δ/Δ p16Ink4aΔ/ΔMlh1Δ/Δ
- W
Wnt-3a
- R
Rspo-2
- N
Noggin
- T
TGFβ1
- 5FU
5-Fluorouracil
- ES
Enrichment score
- NES
normalised enrichment score
- FDR
false discovery rate
References
- 1.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 2.Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–86. doi: 10.1111/his.12055. [DOI] [PubMed] [Google Scholar]
- 3.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature genetics. 2006;38:787–93. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 4.Tejpar S, Bertagnolli M, Bosman F, Lenz HJ, Garraway L, Waldman F, et al. Prognostic and predictive biomarkers in resected colon cancer: current status and future perspectives for integrating genomics into biomarker discovery. Oncologist. 2010;15:390–404. doi: 10.1634/theoncologist.2009-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pai RK, Jayachandran P, Koong AC, Chang DT, Kwok S, Ma L, et al. BRAF-mutated, microsatellite-stable adenocarcinoma of the proximal colon: an aggressive adenocarcinoma with poor survival, mucinous differentiation, and adverse morphologic features. Am J Surg Pathol. 2012;36:744–52. doi: 10.1097/PAS.0b013e31824430d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol. 2012;107:1315–29. doi: 10.1038/ajg.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson JC, Robertson DJ. Serrated Polyp Detection by the Fecal Immunochemical Test: An Imperfect FIT. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2017;15:880–2. doi: 10.1016/j.cgh.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762–72. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 9.Barker N. Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nature reviews Molecular cell biology. 2014;15:19–33. doi: 10.1038/nrm3721. [DOI] [PubMed] [Google Scholar]
- 10.Clevers H. Modeling Development and Disease with Organoids. Cell. 2016;165:1586–97. doi: 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- 11.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–62. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 13.Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521:43–7. doi: 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 14.Fujii M, Shimokawa M, Date S, Takano A, Matano M, Nanki K, et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell. 2016;18:827–38. doi: 10.1016/j.stem.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen R, Cervera P, Svrcek M, Pellat A, Dreyer C, de Gramont A, et al. BRAF-Mutated Colorectal Cancer: What Is the Optimal Strategy for Treatment? Curr Treat Options Oncol. 2017;18:9. doi: 10.1007/s11864-017-0453-5. [DOI] [PubMed] [Google Scholar]
- 17.Kriegl L, Neumann J, Vieth M, Greten FR, Reu S, Jung A, et al. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24:1015–22. doi: 10.1038/modpathol.2011.43. [DOI] [PubMed] [Google Scholar]
- 18.Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, et al. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–46. doi: 10.1016/j.ccr.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 19.Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell. 2013;24:15–29. doi: 10.1016/j.ccr.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krtolica K, Krajnovic M, Usaj-Knezevic S, Babic D, Jovanovic D, Dimitrijevic B. Comethylation of p16 and MGMT genes in colorectal carcinoma: correlation with clinicopathological features and prognostic value. World J Gastroenterol. 2007;13:1187–94. doi: 10.3748/wjg.v13.i8.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma X, Wang YW, Zhang MQ, Gazdar AF. DNA methylation data analysis and its application to cancer research. Epigenomics. 2013;5:301–16. doi: 10.2217/epi.13.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–30. [PubMed] [Google Scholar]
- 23.Bond CE, McKeone DM, Kalimutho M, Bettington ML, Pearson SA, Dumenil TD, et al. RNF43 and ZNRF3 are commonly altered in serrated pathway colorectal tumorigenesis. Oncotarget. 2016;7:70589–600. doi: 10.18632/oncotarget.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488:665–9. doi: 10.1038/nature11308. [DOI] [PubMed] [Google Scholar]
- 25.Murakami T, Mitomi H, Saito T, Takahashi M, Sakamoto N, Fukui N, et al. Distinct WNT/beta-catenin signaling activation in the serrated neoplasia pathway and the adenoma-carcinoma sequence of the colorectum. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2015;28:146–58. doi: 10.1038/modpathol.2014.41. [DOI] [PubMed] [Google Scholar]
- 26.Borowsky J, Dumenil T, Bettington M, Pearson SA, Bond C, Fennell L, et al. The role of APC in WNT pathway activation in serrated neoplasia. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2017 doi: 10.1038/modpathol.2017.150. [DOI] [PubMed] [Google Scholar]
- 27.Giannakis M, Hodis E, Jasmine Mu X, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nature genetics. 2014;46:1264–6. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, et al. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer research. 1995;55:5548. [PubMed] [Google Scholar]
- 29.Li X, Yao X, Wang Y, Hu F, Wang F, Jiang L, et al. MLH1 promoter methylation frequency in colorectal cancer patients and related clinicopathological and molecular features. PLoS One. 2013;8:e59064. doi: 10.1371/journal.pone.0059064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.IJspeert J, Vermeulen L, Meijer GA, Dekker E. Serrated neoplasia-role in colorectal carcinogenesis and clinical implications. Nat Rev Gastroenterol Hepatol. 2015;12:401–9. doi: 10.1038/nrgastro.2015.73. [DOI] [PubMed] [Google Scholar]
- 31.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 32.Fessler E, Drost J, van Hooff SR, Linnekamp JF, Wang X, Jansen M, et al. TGFbeta signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol Med. 2016;8:745–60. doi: 10.15252/emmm.201606184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. New England Journal of Medicine. 2003;349:247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 35.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 36.Roper J, Tammela T, Cetinbas NM, Akkad A, Roghanian A, Rickelt S, et al. In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat Biotechnol. 2017;35:569–76. doi: 10.1038/nbt.3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saito S, Tajiri H, Ikegami M. Serrated polyps of the colon and rectum: Endoscopic features including image enhanced endoscopy. World J Gastrointest Endosc. 2015;7:860–71. doi: 10.4253/wjge.v7.i9.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumours of the Digestive System. 2010 [Google Scholar]
- 39.Bacher JW, Abdel Megid WM, Kent-First MG, Halberg RB. Use of mononucleotide repeat markers for detection of microsatellite instability in mouse tumors. Mol Carcinog. 2005;44:285–92. doi: 10.1002/mc.20146. [DOI] [PubMed] [Google Scholar]
- 40.Kabbarah O, Mallon MA, Pfeifer JD, Edelmann W, Kucherlapati R, Goodfellow PJ. A panel of repeat markers for detection of microsatellite instability in murine tumors. Mol Carcinog. 2003;38:155–9. doi: 10.1002/mc.10157. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe T, Kobunai T, Toda E, Yamamoto Y, Kanazawa T, Kazama Y, et al. Distal colorectal cancers with microsatellite instability (MSI) display distinct gene expression profiles that are different from proximal MSI cancers. Cancer Res. 2006;66:9804–8. doi: 10.1158/0008-5472.CAN-06-1163. [DOI] [PubMed] [Google Scholar]
- 42.Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci. 2011;36:97–107. doi: 10.1016/j.tibs.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T, et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006;20:386–8. doi: 10.1096/fj.05-4331fje. [DOI] [PubMed] [Google Scholar]
- 44.Oskouian B, Saba J. Sphingosine-1-phosphate metabolism and intestinal tumorigenesis: lipid signaling strikes again. Cell Cycle. 2007;6:522–7. doi: 10.4161/cc.6.5.3903. [DOI] [PubMed] [Google Scholar]
- 45.Chen D, Huang JF, Liu K, Zhang LQ, Yang Z, Chuai ZR, et al. BRAFV600E mutation and its association with clinicopathological features of colorectal cancer: a systematic review and meta-analysis. PLoS One. 2014;9:e90607. doi: 10.1371/journal.pone.0090607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Solano J, Conesa-Zamora P, Trujillo-Santos J, Makinen MJ, Perez-Guillermo M. Tumour budding and other prognostic pathological features at invasive margins in serrated colorectal adenocarcinoma: a comparative study with conventional carcinoma. Histopathology. 2011;59:1046–56. doi: 10.1111/j.1365-2559.2011.04043.x. [DOI] [PubMed] [Google Scholar]
- 47.Drost J, van Boxtel R, Blokzijl F, Mizutani T, Sasaki N, Sasselli V, et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science. 2017;358:234–8. doi: 10.1126/science.aao3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zigmond E, Halpern Z, Elinav E, Brazowski E, Jung S, Varol C. Utilization of murine colonoscopy for orthotopic implantation of colorectal cancer. PLoS One. 2011;6:e28858. doi: 10.1371/journal.pone.0028858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Sousa e Melo F, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature. 2017;543:676–80. doi: 10.1038/nature21713. [DOI] [PubMed] [Google Scholar]
- 50.Carragher LA, Snell KR, Giblett SM, Aldridge VS, Patel B, Cook SJ, et al. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO molecular medicine. 2010;2:458–71. doi: 10.1002/emmm.201000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davies EJ, Marsh Durban V, Meniel V, Williams GT, Clarke AR. PTEN loss and KRAS activation leads to the formation of serrated adenomas and metastatic carcinoma in the mouse intestine. The Journal of pathology. 2014;233:27–38. doi: 10.1002/path.4312. [DOI] [PubMed] [Google Scholar]
- 52.Bond CE, Liu C, Kawamata F, McKeone DM, Fernando W, Jamieson S, et al. Oncogenic BRAF mutation induces DNA methylation changes in a murine model for human serrated colorectal neoplasia. Epigenetics. 2017:01–20. doi: 10.1080/15592294.2017.1411446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, et al. Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:2688–98. doi: 10.1158/1078-0432.CCR-12-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ, et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006;2:e146. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–8. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 57.Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–60. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.