

Abstract
Background and Purpose
NO produces arteriolar flow‐induced dilation (FID) in healthy subjects but is replaced by mitochondria‐derived hydrogen peroxide (mtH2O2) in patients with coronary artery disease (CAD). Lysophosphatidic acid (LPA) is elevated in patients with risk factors for CAD, but its functional effect in arterioles is unknown. We tested whether elevated LPA changes the mediator of FID from NO to mtH2O2 in human visceral and subcutaneous adipose arterioles.
Experimental Approach
Arterioles were cannulated on glass micropipettes and pressurized to 60 mmHg. We recorded lumen diameter after graded increases in flow in the presence of either NOS inhibition (L‐NAME) or H2O2 scavenging (Peg‐Cat) ± LPA (10 μM, 30 min), ±LPA1/LPA3 receptor antagonist (Ki16425) or LPA2 receptor antagonist (H2L5186303). We analysed LPA receptor RNA and protein levels in human arterioles and human cultured endothelial cells.
Key Results
FID was inhibited by L‐NAME but not Peg‐Cat in untreated vessels. In vessels treated with LPA, FID was of similar magnitude but inhibited by Peg‐Cat while L‐NAME had no effect. Rotenone attenuated FID in vessels treated with LPA indicating mitochondria as a source of ROS. RNA transcripts from LPA1 and LPA2 but not LPA3 receptors were detected in arterioles. LPA1 but not LPA3 receptor protein was detected by Western blot. Pretreatment of vessels with an LPA1/LPA3, but not LPA2, receptor antagonist prior to LPA preserved NO‐mediated dilation.
Conclusions and Implications
These findings suggest an LPA1 receptor‐dependent pathway by which LPA increases arteriolar release of mtH2O2 as a mediator of FMD.
Abbreviations
- CAD
coronary artery disease
- EC
endothelial cells
- eNOS
endothelial NOS
- ET‐1
endothelin‐1
- FID
flow‐induced dilation
- HMVEC
human microvascular endothelial cells
- LPA
lysophosphatidic acid
- L‐NAME
Nω‐nitro‐L‐arginine methyl ester
- LPP3
lipid phosphate phosphatase 3
- MitoPY1
Mito Peroxy Yellow 1
- mtH2O2
mitochondria‐derived hydrogen peroxide
- NOX
NADPH oxidase
- Non‐CAD
absence of clinically diagnosed coronary artery disease
- Peg‐Cat
polyethylene glycol‐catalase
- SNP
sodium nitroprusside
- VSMC
vascular smooth muscle cells
Introduction
Endothelial cells form an interface between circulating blood and surrounding tissue and actively respond to changes in blood flow. The endothelium‐dependent, flow‐stimulated release of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509 and other vasodilator factors that act on adjacent vascular smooth muscle cells (VSMC) is a key means by which human resistance arterioles dilate and increase tissue perfusion (flow‐induced dilation; FID). Endothelial release of NO promotes vascular health by reducing inflammation and proliferation (Kim et al., 2008). Previous studies demonstrated that the magnitude of FID is preserved in arterioles from patients with coronary artery disease (CAD) or multiple risk factors for CAD, but the mediator switches from NO to pro‐inflammatory and atherogenic mitochondrial hydrogen peroxide (mtH2O2) (Liu et al., 2003; Beyer et al., 2017), providing a potential pathway underlying the microvascular contributions to CAD. We have previously reported intracellular components involved in the transition from NO to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2448, including PGC‐1α (Kadlec et al., 2017) and telomerase (Beyer et al., 2016). The upstream cell‐surface activators of this transition, however, remain incompletely characterized. Understanding what factors and receptors act at the endothelial cell surface of arterioles is essential for constructing a complete signalling pathway and identifying novel targets to reduce H2O2 release in the context of CAD.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=2906 (LPA), a simple phospholipid, is an autacoid found at the cell surface and in the circulation. LPA triggers a plethora of cellular responses ranging from enhanced proliferation, migration and invasiveness to morphological changes, increased permeability or apoptotic cell death in different cells of the vasculature (Siess et al., 1999; van Nieuw Amerongen et al., 2000; Lin et al., 2006; Panchatcharam et al., 2008). Physiological concentrations range from nM to μM levels (Aoki, 2004) and are controlled by a dynamic balance of synthesis (mainly by secreted autotaxin) (Albers et al., 2010; Gierse et al., 2010) and degradation [primarily by membrane‐bound ecto‐activity of lipid phosphate phosphatase 3 (LPP3)] (Tanyi et al., 2003; Panchatcharam et al., 2014). Disruption of these control mechanisms can increase LPA levels by more than 10‐fold, as reported in individuals with cancer (Baker et al., 2002), neurological disorders or risk factors for cardiovascular disease, such as hyperlipidaemia (Dohi et al., 2012) or hypertension (Xu et al., 2003). A rise in LPA has also been implicated in the development and progression of atherosclerosis and cardiovascular disease (Bot et al., 2013; Kurano et al., 2015; Busnelli et al., 2017). These effects are mediated by cognate LPA receptors. Currently, there are six http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=36 that belong to the GPCR family. Each receptor couples to at least one or more heterotrimeric Gα proteins, such as G12/13, Gq/11, Gi/o and Gs and can activate a complex array of downstream signalling pathways with physiological and pathological effects on the vasculature. In particular, signalling through http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=272 is involved in many LPA‐induced vascular effects, such as increased inflammation and endothelial cell migration (Panetti et al., 2000; Panetti et al., 2004). Additionally, inhibition of LPA1 receptor but not others reduces inflammation in cultured endothelial cells (EC) (Rizza et al., 1999). Independent studies have shown that increased LPA, up to 10 μM, decreases http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1249 (eNOS) expression, reduces NO bioavailability and increases ROS production in cultured cells (Chen et al., 1995; Shin et al., 1999; Chen et al., 2012), all hallmarks of endothelial dysfunction. However, LPA's functional influence on intact human arterioles, and the receptor responsible for these effects, has not been explored. Examining the arteriolar effects of elevated LPA is clinically relevant given the prognostic importance of arteriolar dysfunction (van de Hoef et al., 2014).
We hypothesized that exposing arterioles from patients without CAD (non‐CAD) to elevated levels of LPA (10 μM) would shift the mediator of FID away from NO to mtH2O2, reproducing the phenotype observed in arterioles from patients with CAD. Given that LPA signals through cognate LPA receptors, we further examined which receptors are expressed in human arterioles and mediate LPA's effects. We discovered that elevated levels of LPA in vessels from subjects without CAD increases mtH2O2 during FID in a LPA1 receptor‐dependent manner.
Methods
Tissue acquisition
Protocols for tissue acquisition and processing were approved by the Institutional Review Board of the Medical College of Wisconsin. Human adipose (subcutaneous and visceral) was obtained as discarded tissue at the time of surgery and immediately placed in 4°C HEPES buffer [(in mM) NaCl 275, KCL 7.99, MgSO4 4.9, CaCl2 ·2H2O 3.2, KH2PO4 2.35, EDTA 0.07, glucose 11, HEPES acid 20]. Patient demographics were collected and stored in a de‐identified fashion using the Generic Clinical Research Database at the Medical College of Wisconsin. Tissue from subjects without clinically diagnosed CAD and no more than one risk factor (Table 1) were categorized as the non‐CAD group. Vessels defined as CAD were obtained from tissue from subjects with clinically diagnosed CAD.
Table 1.
Patient demographic information
| Patient demographics | Non‐CAD (n = 105) | CAD (n = 6) |
|---|---|---|
| Characteristics | ||
| Sex, male/female | 21/84 | 4/2 |
| Age, years (mean ± SD)* | 51 ± 13 | 70 ± 14 |
| Body mass index (mean ± SD) | 30 ± 6 | 31 ± 3 |
| Underlying conditions | ||
| Coronary artery disease | 0 | 6 |
| Diabetes mellitus | 2 | 1 |
| Hypertension | 5 | 2 |
| Hyperlipidaemia | 3 | 2 |
| Myocardial infarction | 0 | 2 |
| Congestive heart failure | 1 | 0 |
| A‐Fib | 0 | 1 |
| None of the above | 91 | 0 |
Data on patient demographics for adipose CAD and non‐CAD arterioles used in the study. n = number of patients.
P < 0.05 non‐CAD patients versus patients with CAD.
Cannulated arteriole preparation and flow‐induced dilation
Human arterioles were isolated, and excess adipose and connective tissue was removed. The vessels were cannulated onto glass micropipettes of matched impedance (diameters of both pipettes per single chamber preparation did not vary by more than 2 μm and was typically kept between 35 and 45 μm, depending on the size of an arteriole) filled with Krebs buffer [(in mM) NaCl 118, KCl 4.7, CaCl2 2.5, KH2PO4 1.2, MgSO4 1.2, NaHCO3 20, Na2 EDTA 0.026, and dextrose 11, pH 7.4] and secured using sutures in an organ chamber also filled with Krebs buffer solution. The arterioles were kept at 37°C and were pressurized to 60 mmHg in a step‐wise manner (30 min at 30 mmHg followed by 30 min at 60 mmHg) using a dual‐reservoir system, where each micropipette is connected to its own reservoir. Once the equilibration time was reached, the vessels were constricted with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989 (ET‐1) to 30–60% of the passive diameter. With stable constriction lasting ≥10 min, the vessels were exposed to increasing physiological rates of flow (pressure gradient; 5 to 100 cmH2O) with constant intraluminal pressure, as described previously (Kuo et al., 1990; Kuo et al., 1991; Miura et al., 2003). Briefly, in this preparation, flow is initiated by simultaneously moving of the reservoirs in an equal and opposite direction (one down and one up by an equal distance from the starting point) to generate a pressure gradient with shear rates estimated at 5 to 25 g·cm3·s−2 (dyne·cm−2), while preserving intraluminal pressure (Kuo et al., 1990). In some vessels, two studies were performed in sequence separated by a wash (20 mL of Krebs buffer replaced five times) and a 30‐min re‐equilibration period prior to the second curve. At the end of each experiment, papaverine (100 μM) was used to assess maximal dilation. At the end of each experiment, the matched impedance of the pipettes was verified by applying max flow (100 cmH2O) in reverse direction for 5 min and in the presence of papaverine. If vessel diameters were different between flows of opposite direction, vessels were excluded from analysis. Additionally, vessels that did not reach or retain stable constriction for at least 10 min were excluded from the data set, as were arterioles that did not reach 75% of max dilation to papaverine.
Pharmacological intervention
All treatments were added to the organ bath (Krebs buffer) and constituted ≤1% of the total circulating volume. Vessels were treated with LPA (Abcam, Cambridge, MA, USA, ab146430; dissolved in PBS, pH 7.4, final concentration in the organ bath 10 μM) for ≥30 min (depending on experiment performed) prior to constriction. To determine whether the mediator of FID was NO or H2O2, we used the NOS inhibitor [http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5213, 100 μM] or a scavenger of H2O2 [polyethylene glycol‐catalase (Peg‐Cat) 500 U·mL−1], respectively, added to the organ chamber for 30 min prior to constriction. Prior studies have shown that these doses effectively block NO and H2O2 components of FID respectively (Beyer et al., 2014; Freed et al., 2014). To further localize the subcellular source of H2O2, we performed FID studies under LPA challenge in the presence of rotenone (mitochondria complex I inhibitor; 1 μM). In separate studies, we assessed endothelial viability; dose–response studies to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=294 (1 nM to 100 μM) with and without LPA were performed. To assess endothelium‐independent, VSMC reactivity, dose–response studies to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9533 (SNP; 1 nM to 100 μM) were performed in the presence and absence of LPA. To determine which LPA receptor is responsible, vessels were pretreated with either http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2907 (Cayman Chemical, Ann Arbor, MI, USA, 10012659, LPA1/3 receptor antagonist; 10 μM) or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?tab=summary&ligandId=6579 (Cayman Chem, 14663, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=273 antagonist; 1 μM or 10 μM) 30 min prior to LPA administration. Concentrations of Ki16425 and H2L5186303 were chosen based on data published by other groups working with these antagonists in the context of vascular biology (Ruisanchez et al., 2014; Wu et al., 2015).
Cell culture
HUVECs were purchased from ATCC (Manassas, VA, USA) and cultured on 100 mm culture plates in endothelial cell growth medium (EGM®‐2; Lonza, Basel, Switzerland) supplemented with growth factors, cytokines and 5% FCS. Human cardiac microvascular endothelial cells (HMVECs) lysates were a gift from Dr Julie Freed and prepared as previously described (Freed et al., 2017). Briefly, cells were purchased from Lonza and cultured in 100 mm culture plates in cell growth medium (EBM‐2; Lonza) supplemented with growth factors, cytokines and 10% FCS. Cells at passage 3–5 with 80–95% confluency were used.
Western blot
Vessels were isolated and connective tissue was carefully removed. Vessels were snap frozen in liquid nitrogen and stored at −80°C until further processing. Cultured endothelial cells (HUVECs) were scraped from culture dishes and centrifuged to form a cell pellet, which was then snap frozen until further processing. Three to five arterioles from each tissue were frozen and constituted one biological replicate. Four culture dishes with HUVECs and three separate cell lysate preparations of HMVECs were used. For lysate preparations, vessels were thawed and homogenized using a glass homogenizer in cold lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% deoxycholic acid, 0.1% SDS, 0.5% NP40) supplemented with a protease and phosphatase inhibitor cocktail (Roche, Basel, Switzerland) and briefly sonicated. Cell pellets were thawed and briefly sonicated. Total protein concentration was determined using BCA protein assay (Thermo Scientific, Waltham, MA, USA). Protein samples of known concentration from each tissue (patient) were subjected to 4–20% SDS‐PAGE and transferred to nitrocellulose membranes. To detect target receptor protein, antibodies to LPA1 (Abcam, ab166903; 1:1000), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=274&familyId=36&familyType=GPCR (LSBio, LS‐A1014; 1:1000), LPA3 [Abcam (Abc), ab23692, 1:1000] and GAPDH (Abcam, ab8245, 1:10 000) were used, followed by peroxidase conjugated secondary antibodies and chemiluminescent substrate (Bio‐Rad, Hercules, CA, USA).
Quantitative real‐time PCR
Isolated and cleaned human adipose vessels were snap frozen in liquid nitrogen and stored in −80°C until further processing. Total RNA was extracted from samples using RNAqueous™‐Micro Total RNA Isolation Kit (Ambion, AM1931); 25 ng of RNA was used to synthesize cDNA using High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA, 4368814). Quantification of target gene expression was performed using Qiagen QuantiTect Primer Assay for LPA1 (Hs_LPAR1_1_SG, QT00021469), LPA2 (Hs_LPAR2_2_SG, QT01851318), LPA3 (Hs_LPAR3_1_SG, QT00092932), PECAM (Hs_PECAM1_1_SG, QT0008172) and normalized to 18 s (Hs_RRN18S_1_SG, QT00199367). Gene expression analysis was performed using BioRad CFX96 Touch Read‐Time PCR Detection System. Each sample was evaluated as an average of a triplicate.
Immunohistochemistry
Vessels were isolated from tissue and immediately placed in zinc formalin buffer followed by paraffin embedding. Immunohistochemistry was performed by the Children's Hospital of Wisconsin Histology Core. Sections were cut at 4 μm and placed on poly‐L‐Lysine coated slides, which were then dried at 45°C overnight and stored in room temperature prior to IHC testing. Slides were then loaded onto the Leica Bond Max immunostainer. All samples were de‐waxed prior to staining on the instrument. Antibodies required antigen retrieval pretreatment as follows: LPA1 receptor H1(20) and LPA3 receptor H1(20) citrate buffer at pH 6 and CD31 H2(10) EDTA retrieval. Antibodies used: LPA1 receptor (Abcam, ab166903, 1:100), LPA3 receptor (LS‐Bio, LS‐A1014/40949, 1:200) and CD31 (Agilent, DAKO, M0823, 1:100). All antibodies were detected and visualized using Leica Bond Polymer Refine Detection System (DS9800) with the addition of a DAB enhancer (AR9432), using the modified versions of the MODF protocol installed by Leica filed service engineers. Slides were removed from the immunostaining platform, dehydrated, cleared and coverslipped with synthetic mounting media. Omission of the primary antibody served as a negative control. Slides were scanned with a NanoZoomer HT slide scanner (Hamamatsu, Japan).
Fluorescence detection of mitochondrial H 2 O 2 in human microvessels
To evaluate mitochondrial H2O2 release in human arterioles challenged with LPA (10 μM, 30 min), Mito Peroxy Yellow 1 (MitoPY1) was used (Dickinson and Chang, 2008). As previously described, vessels were cannulated in a chamber containing HEPES buffer warmed to 37°C and pH 7.4 (Kadlec et al., 2017). The same buffer containing the fluorescent MitoPY1 probe (10 μM, 1 h) was used to perfuse vessels intraluminally. Fluorescence was imaged using a krypton/argon lamp fluorescent microscope (model TE 200 Nikon Eclipse) or X‐cite FIRE LED microscope (model Olympus IX73) and an excitation/emission wavelength of 488 nm/530–590 nm before administration of LPA, 30 min after, and every minute for 5 min once max flow (Pressure Gradient of 100 cmH2O) was applied. Relative fluorescence intensity (arbitrary units) was measured using ImageJ (NIH) by subtracting background fluorescence from vessels fluorescence. The % increase in fluorescence intensity from static values after 5 min of max flow (% increase to flow) was compared between intact control and other treatments of vessels. Endothelial contribution to the fluorescence signal was assessed by denuding a subset of vessels. Briefly, vessels were isolated, cleaned and one end was cannulated onto a glass micropipette. Vessels were then equilibrated for 30 min at 37°C. Once equilibrated, 4 mL of air was passed through the vessel to dislodge the endothelium. After denudation, the loose end of the vessel was cannulated and experimental protocol for fluorescence detection of mitochondrial H2O2 was continued. Viability of the arterioles was assessed by the following protocol: arterioles were allowed to equilibrate without flow for 5–10 min; after equilibration, 0.3 nM of ET‐1 was used to constrict the vessels; the vessels needed to retain constriction for 10 min after which 100 μM of ACh was added followed by 100 μM of papaverine to assess the vessels’ ability to dilate. Vessels that did not constrict to ET‐1 or did not dilate to papaverine were excluded from the study. After completion of the protocol, arterioles were removed from the glass cannulas and snap‐frozen in liquid nitrogen and stored at −80°C until further processing. Denudation efficiency was assessed by measuring expression of the endothelial marker, PECAM, using RT‐qPCR.
Statistical analysis
All data are expressed as mean ± SEM with statistical significance threshold set at P < 0.05. For vascular studies, data are expressed as a % of maximal dilation from ET‐1‐induced constriction, where 100% represents full relaxation to the maximal diameter noted throughout the experiment. To compare flow‐response relationship, a two‐way ANOVA was used with pressure gradient (or dose–response to SNP or ACh) and treatment as parameters. When a significant difference was observed between curves (P < 0.05), responses at individual pressure gradients were compared using a Holm–Sidak multiple comparison test. To compare the concentration of SNP required to cause 50% of the maximum response (EC50), a three‐parameter nonlinear regression using least squares (ordinary) fit analysis was performed. One‐way ANOVA was used to compare fluorescence (MitoPY1 probe) in microvessels before and after interventions. Wilcoxon test was used to compare PECAM expression in control versus denuded arterioles. All analyses were performed using GraphPad Prism 7.04 (GraphPad Software, San Diego, CA, USA) and SAS 9.4 (SAS, Cary, NC, USA) with statistical significance defined at P < 0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Results
Discarded adipose tissue (subcutaneous and visceral) was collected from a total of 111 patients, six with CAD and 105 categorized as non‐CAD. Patient demographics are detailed in Table 1. Mean diameter prior to constriction, after constriction and concentration of ET‐1 needed to achieve desired constriction across all experiments are summarized in Table 2.
Table 2.
Basic parameters for vascular studies
| Vessel diameter prior to constriction (μm) | Vessel diameter post constriction (μm) | ET‐1 needed to achieve desired constriction (nM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SEM | n | Mean | SEM | n | Mean | SEM | n | |
| Control | 111.2 | 15.6 | 6 | 62.1 | 6.2 | 6 | 0.25 | 0.05 | 6 |
| L‐NAME | 86.9 | 11.5 | 7 | 59.5 | 5.4 | 7 | 0.15 | 0.04 | 7 |
| Peg‐Cat | 122.4 | 8.5 | 6 | 58.4 | 6.6 | 6 | 0.28 | 0.06 | 6 |
| LPA | 181.4 | 16.3 | 10 | 106.3 | 11.0 | 10 | 0.39 | 0.06 | 10 |
| LPA + L‐NAME | 184.9 | 22.9 | 7 | 121.3 | 20.6 | 7 | 0.48* | 0.12 | 7 |
| LPA + Peg‐Cat | 165.3 | 11.8 | 7 | 74.6 | 5.9 | 7 | 0.31 | 0.06 | 7 |
| SNP Control | 123.3 | 8.8 | 6 | 62.8 | 6.3 | 6 | 0.39 | 0.09 | 6 |
| SNP + LPA | 130.9 | 9.0 | 5 | 61.0 | 8.9 | 5 | 0.40 | 0.05 | 5 |
| Ach control | 135.0 | 18.0 | 5 | 70.0 | 9.0 | 5 | 0.27 | 0.04 | 5 |
| Ach + LPA | 109.5 | 20.0 | 5 | 58.1 | 11.5 | 5 | 0.15 | 0.03 | 5 |
| Ki16425 + LPA | 165.1 | 12.5 | 6 | 90.8 | 9.5 | 6 | 0.54* | 0.11 | 6 |
| Ki16425 + LPA + L‐NAME | 133.4 | 13.8 | 7 | 86.0 | 13.0 | 7 | 0.39* | 0.04 | 7 |
| Ki16425 + LPA + Peg‐Cat | 122.8 | 10.7 | 7 | 71.5 | 8.2 | 7 | 0.41 | 0.04 | 7 |
| H2L5186303 + LPA | 117.7 | 14.6 | 6 | 76.5 | 9.7 | 6 | 0.48* | 0.08 | 6 |
| H2L5186303 + LPA + L‐NAME | 123.6 | 14.7 | 6 | 73.0 | 10.4 | 6 | 0.30 | 0.07 | 6 |
| H2L5186303 + LPA + Peg‐Cat | 145.9 | 22.0 | 6 | 82.4 | 13.3 | 6 | 0.43 | 0.11 | 6 |
| Rotenone + LPA | 115.7 | 4.7 | 7 | 68.4 | 5.4 | 7 | 0.52 | 0.08 | 7 |
Mean diameter prior to constriction, post constriction and concentration of ET‐1 needed to achieve desired constriction across all vascular experiments. n = number of patients. Comparisons were made between studies without LPA (greyed out) and corresponding studies with LPA + intervention.
P < 0.05 for L‐NAME versus LPA + L‐NAME, control versus Ki16425 + LPA, L‐NAME versus Ki16425 + LPA + L‐NAME, control versus H2L5186303 + LPA, L‐NAME versus H2L5186303 + LPA + L‐NAME.
LPA shifts the mediator of FID from NO to H 2 O 2 in human arterioles
To confirm NO as the baseline mediator of FID in human adipose arterioles, vessels were treated with either L‐NAME or Peg‐Cat in the absence of LPA. The presence of L‐NAME blunted the peak FID, but Peg‐Cat had no effect on the dilation in untreated arterioles (Figure 1A). Treatment of non‐CAD vessels with LPA preserved the magnitude of dilation but shifted the meditator of FID to H2O2 (dilation inhibited by Peg‐Cat) (Figure 1B). In contrast to its inhibitory effect on dilation in untreated vessels (Figure 1A), pre‐incubation with L‐NAME in LPA‐treated vessels did not inhibit but instead slightly increased dilation at lower levels of flow (Figure 1B).
Figure 1.
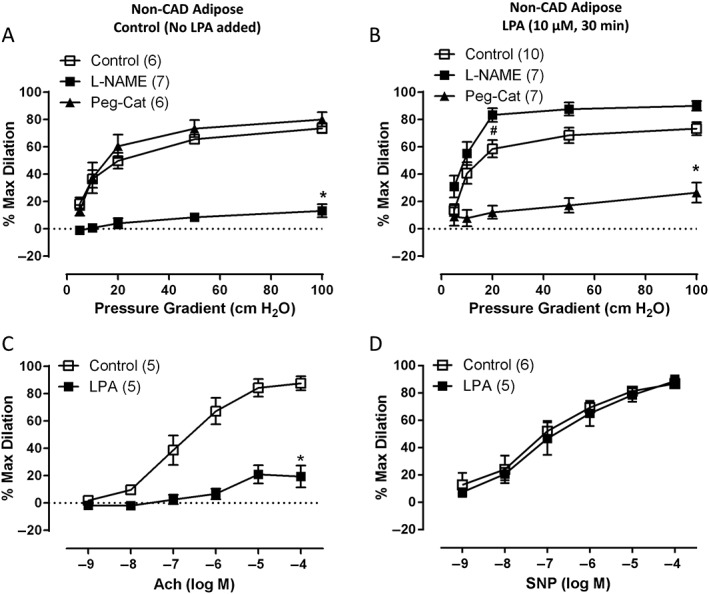
Effects of LPA on the mediator of FID in non‐CAD human adipose arterioles. (A) L‐NAME inhibits peak FID in control vessels, but Peg‐Cat has no effect. (B) In vessels treated with LPA (10 μM, 30 min), Peg‐Cat significantly reduces peak FID. Presence of L‐NAME in LPA‐treated vessels significantly improved FID but only at the 20 cmH2O of pressure gradient. (C) Dilation to ACh is significantly decreased in arterioles treated with LPA in comparison to control arterioles. (D) Dilation to SNP is not different between control and LPA‐treated vessels. *P < 0.05 versus control at specific pressure gradients, # P < 0.05 versus control at 20 cmH2O pressure gradient (L‐NAME curve), P = NS between 1st and 2nd curve at each pressure gradient; n is shown in parentheses.
Arterioles treated with LPA also showed reduced dilation to an endothelium‐dependent dilator (ACh) with differences in peak value (Figure 1C) and EC50 (−log EC50: 6.83 ± 0.18 for control and −log EC50: 5.86 ± 0.44 for LPA, P < 0.05). To determine whether treatment with LPA affected endothelium‐independent arteriolar responses, dose–response curves to SNP (a direct NO donor) were performed in LPA‐treated and untreated arterioles. No significant differences were observed in EC50 (−log EC50: 7.03 ± 0.24 for control and −log EC50: 7.03 ± 0.23 for LPA, Figure 1D) or peak values (Figure 1D). Additionally, because two videomicroscopy studies (control vs. L‐NAME or Peg‐Cat) were often performed in succession on a single arteriole, we also assessed whether LPA treatment had a time‐dependent effect on the overall magnitude of FID and found no differences in these time control experiments (Supporting Information Figure S1).
Signalling through LPA 1 receptor leads to the loss of NO‐mediated FID in non‐CAD human arterioles
We next examined which receptor might be responsible for the LPA‐induced shift in the mediator of FID. Figure 2A shows that non‐CAD arterioles treated with the LPA1/3 antagonist, Ki16425, maintained NO‐mediated FID after the acute LPA challenge. Peg‐Cat, which inhibits FID after LPA treatment alone, had no effect on the dilation after combined treatment with LPA and Ki16425 (Figure 2A). Blocking LPA2 (Figure 2B) did not prevent the shift to H2O2‐mediated dilation (Peg‐Cat abolished FID). To ensure that the shift to H2O2‐mediated dilation is not caused by the LPA2 receptor antagonist alone, we tested the effects of the antagonist in the absence of LPA and observed no effect on NO‐mediated dilation (Supporting Information Figure S2). H2L5186303 is an LPA2 receptor antagonist when used at a nM concentrations, but at higher, μM concentrations, it can also act as an LPA3 receptor antagonist (Fells et al., 2009). We, therefore, tested a higher concentration of H2L5186303 (10 μM) before LPA challenge. As before, Peg‐Cat inhibited the dilation (% maximal dilation at 100 cm H2O: H2L5186303 + LPA 80.6 ± 4.6, n = 3 vs. H2L5186303 + LPA + Peg‐Cat −26.3 ± 1.2, n = 3) while L‐NAME had no effect (Supporting Information Figure S3).
Figure 2.
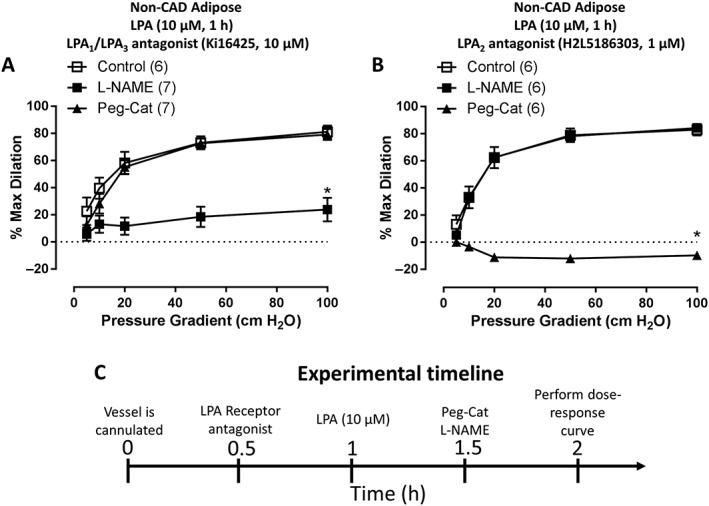
LPA acts through LPA receptors to shift the mediator of FID. (A) Treatment of arterioles with Ki16425, LPA1 and LPA3 receptor inhibitor, prior to LPA challenge preserves NO‐mediated FID as indicated by L‐NAME but not Peg‐Cat inhibitable dilation (n = 8, 7 and 7 respectively). (B) Inhibition of LPA2 receptor with H2L5186303 prior to LPA challenge had no effect on LPA‐induced shift to H2O2‐mediated FID (n = 6, 6 and 6 respectively). (C) Experimental timeline. *P < 0.05 control.
LPA1 and LPA2 but not LPA3 receptors are expressed in human adipose microvessels
To investigate which LPA receptors are responsible for the arteriolar effects of LPA, we examined LPA1–3 receptor expression in intact human adipose arterioles. As shown in Figure 3, transcripts for LPA1 and LPA2 receptors but not LPA3 were abundantly detectable by PCR. The pattern of expression was not different between CAD versus non‐CAD tissue for any of the three receptors (Figure 3A). Because our functional studies using an LPA2 receptor antagonist demonstrated no involvement of that receptor in the LPA‐induced shift of the mediator of FID, the remaining expression studies focused on LPA1 and LPA3 receptors. Western blot analysis was performed on LPA1 and LPA3 receptors to examine protein levels. Figures 3B show robust detection of LPA1 in both CAD and non‐CAD vessels as well as HUVECs and left ventricle of the human heart, which was used as a positive control. The LPA3 receptor was not detected in arterioles from patients with or without CAD, and only faint expression was detected in HUVECs (Figure 3C). The LPA3 receptor was also detected in human heart samples with some additional non‐specific bands. Immunohistochemical studies of human non‐CAD arterioles also demonstrated strong staining for LPA1 (Figure 4A) but not LPA3 receptors (Figure 4B). A similar pattern of expression was observed in CAD arterioles, with the LPA1 receptor but not LPA3 being expressed in the vessels. Sections of human pancreas were used to verify efficiency of the LPA3 receptor antibody (Supporting Information Figure S6).
Figure 3.
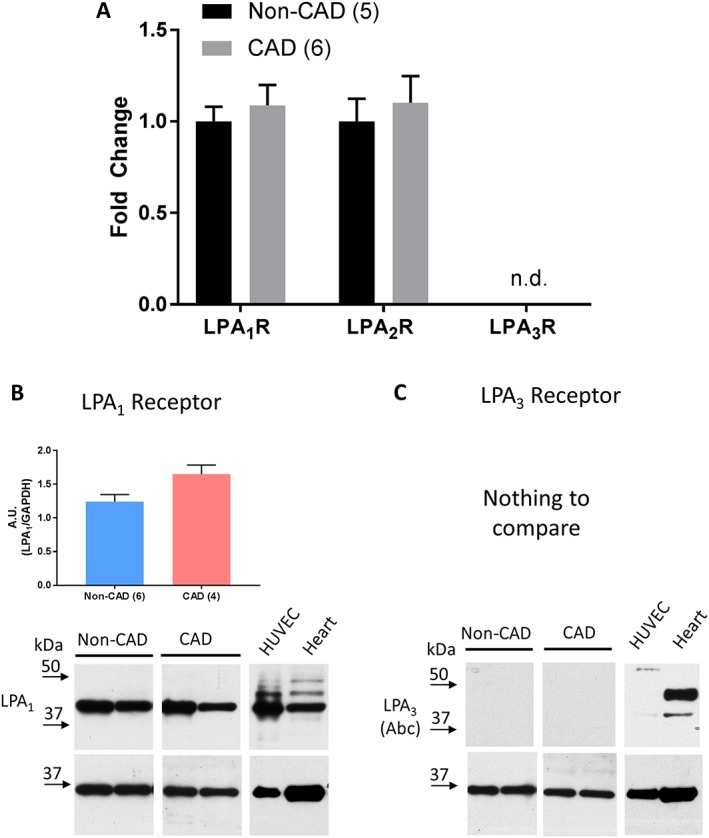
Reverse‐transcriptase PCR analysis of LPA receptors in arterioles. (A) RNA from intact arterioles from patients with and without CAD was analysed for transcript levels of LPA1, LPA2 and LPA3 receptors. The presence of CAD had no effect on expression of LPA1 receptor or LPA2 receptor but no target sequences for LPA3 receptor were detected in either non‐CAD or CAD (n.d. – not detected). CAD expression is represented as fold change from non‐CAD. The number of transcripts was normalized to 18 s in the same sample. (B) Western blot quantification of LPA1 receptor in non‐CAD versus CAD arterioles. (C) No quantification was possible for the LPA3 receptor. Representative images for each receptor show two non‐CAD and two CAD patients, HUVECs and human heart left ventricle. GAPDH used for loading control. A.U. – arbitrary units, (n) indicates number of subjects.
Figure 4.
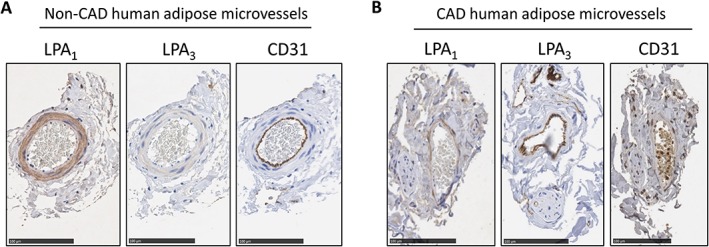
LPA1 and LPA3 receptor protein expression in intact non‐CAD and CAD arterioles. (A) Immunohistochemical analyses for LPA1 and LPA3 receptors from vessels isolated from an individual without CAD. GAPDH used for loading control. (B) Immunohistochemistry analyses for LPA1 and LPA3 receptors in vessels from an individual with CAD. CD31 staining used to identify endothelial cell layer. Specificity of the antibody was determined by removal of the primary antibody (not shown). Bar for all images = 100 μm.
Immunohistochemical analyses appeared to localize LPA1 receptor expression predominantly to VSMCs, with little to no staining in ECs (Figure 4A,B). To further investigate whether LPA receptors are expressed in human ECs, we examined the expression of LPA1 and LPA3 receptors in two types of cultured endothelial cells, HUVECs and HMVECs. There was robust expression of LPA1 receptors in HUVECs and HMVECs as shown in Figure 5A,B respectively. LPA3 was detected in HUVECs, albeit at a low level, and no expression of the LPA3 receptor was detected in HMVECs (Figure 5C,D respectively). Two different antibodies (see Methods section) targeting different epitopes were used to ensure validity of the expression patterns of the LPA3 receptor in arterioles and cultured endothelial cells. In both cases, we found the expression of the receptor to be lower or absent across different preparations. Supporting Information Figure S4 shows a lower expression of the LPA3 receptor in two types of cultured endothelial cells, with robust expression in the heart using Abc antibody, while Supporting Information Figure S5 validates lower expression of LPA3 receptor in non‐CAD arterioles and immunohistochemical preparations using LSBio antibody. These data suggest that LPA1 receptors are expressed in the arteriolar medial layer and, to a much lower extent in vascular endothelium, even though expression is robust in culture microvascular endothelial cells. LPA3 receptors are minimally expressed in human coronary arterioles or cultured endothelial cells.
Figure 5.
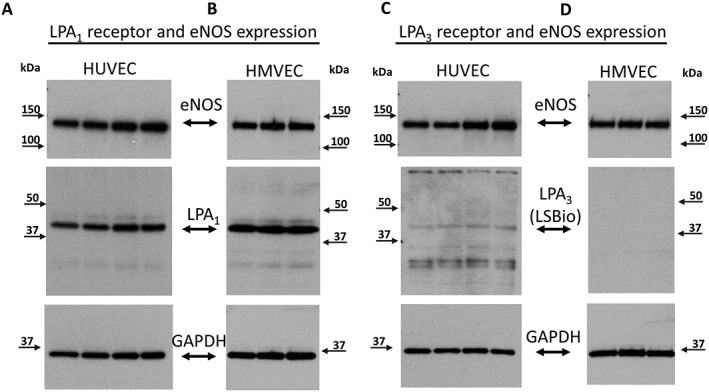
LPA1 and LPA3 receptor protein expression in endothelial cells. LPA1 receptor protein expression in HUVEC and HMVEC (A and B respectively). LPA3 receptor expression in HUVEC (C) and HMVEC (D). eNOS was used as an endothelial marker and GAPDH as loading control.
Mitochondria are the source of LPA‐induced H 2 O 2
We previously showed that the mitochondria are the main source of flow‐induced H2O2 release in CAD arterioles (Liu et al., 2003; Freed et al., 2014). To assess whether LPA specifically increases mitochondria‐derived H2O2 in non‐CAD arterioles, we applied mitoPY1, a fluorescent probe specifically designed to image mitochondrial H2O2 in our cannulated arteriole experiments. Figure 6A shows representative images of the cannulated vessels in the bright field, under static conditions and after 5 min of maximal (100 cmH2O) flow, in intact untreated and LPA‐treated (10 μmol·L−1, 30 min) and as well as denuded untreated and LPA‐treated arterioles. Quantification of the relative fluorescence intensity is shown in Figure 6B. Untreated, non‐CAD vessels (control) showed minimal increases in fluorescence as compared to LPA‐treated, non‐CAD arterioles. Denuded controls and LPA‐treated arterioles showed a trend towards an increase in fluorescence in comparison to intact control vessels (Figure 6B). More importantly, denuded arterioles treated with LPA do not show a significant increase in fluorescence signal in response to flow when compared to intact arterioles treated with LPA. Denudation efficiency was analysed by examining expression of endothelium‐specific target protein, PECAM (Supporting Information Figure S7). To further test the mitochondria as the source of H2O2, we measured FID in the presence of Rotenone, an electron transport chain complex I inhibitor that has been shown previously to reduce mtH2O2 production and block FID in arterioles from patients with CAD (Liu et al., 2003). Figure 6C shows that rotenone significantly reduces FID after LPA treatment. Rotenone alone has been known not to affect either the overall magnitude of the dilation or the mediator of FID (Freed et al., 2014; Beyer et al., 2016).
Figure 6.
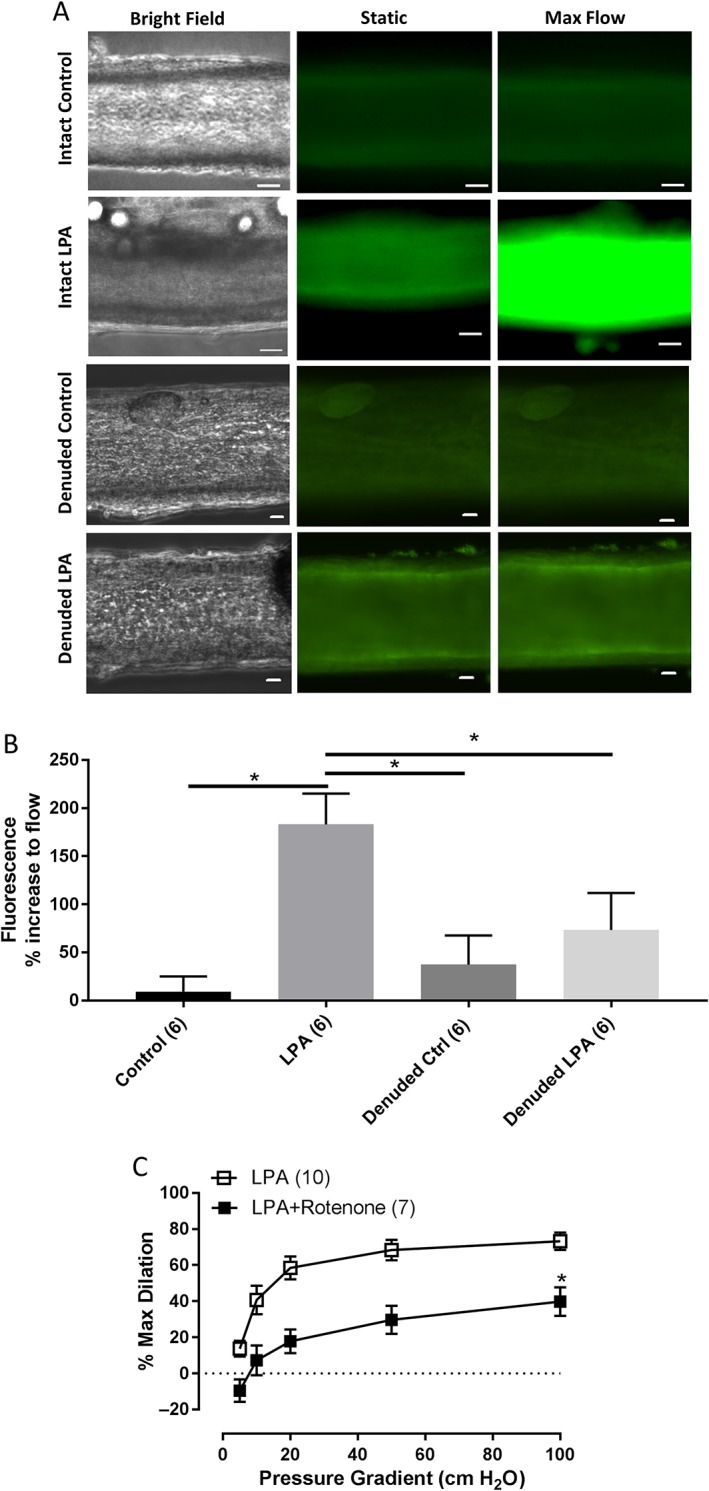
Intracellular source of H2O2 in arterioles after LPA challenge. (A) Representative images of cannulated vessels perfused intraluminally with mito peroxy yellow (MitoPY1) under intact control (n = 6), intact LPA treatment (n = 6), denuded control (6) and denuded LPA treatment (6). Images show bright field, static (no flow) and 5 min of flow (100 cm H2O gradient). (B) Quantification of mitochondrial H2O2 expressed as % increase in fluorescence from baseline (static) to 5 min of maximal (100 cmH2O) flow. *P < 0.05 intact control versus intact LPA, intact LPA versus denuded control and intact LPA versus denuded LPA. (C) Source of dilator H2O2 was determined by incubating vessels with a mitochondria‐targeted inhibitor, rotenone (1 μM) for 30 min after LPA challenge. n = 10 and 7 respectively. *P < 0.05 versus LPA. Bar for all images = 20 μm.
Discussion
The major novel findings of the present study are as follows: first, acute (30 min) exposure of non‐CAD human arterioles to 10 μM LPA induces a switch in the mechanism of FID from NO to H2O2, recapitulating the CAD phenotype; second, LPA1 receptor is involved in the LPA‐induced shift to H2O2‐mediated FID; third, LPA1 and LPA2 receptors but not LPA3 are expressed in human arterioles from patients without CAD; and fourth, mitochondria are the source of LPA‐induced H2O2 production. These findings show for the first time that elevated concentrations of LPA act in an LPA1 receptor‐dependent manner to induce an arteriolar endothelial phenotype characteristic of that seen in patients with CAD.
LPA receptor‐dependent effects on the mediator of FID
Elevated LPA is associated with vascular pathology and is known to exert multiple harmful effects on vascular endothelial (Rizza et al., 1999; Panetti et al., 2000; van Nieuw Amerongen et al., 2000; Panetti, 2002; Panetti et al., 2004; Lin et al., 2006; Avraamides et al., 2007; Lin et al., 2007) and smooth muscle (Hayashi et al., 2001; Panchatcharam et al., 2008) integrity. In particular, studies have shown that LPA can lead to increased levels of ROS either independent of (Brault et al., 2007; Staiculescu et al., 2014) or in conjunction with decreased eNOS expression (Chen et al., 2012), effectively decreasing endothelium‐dependent dilation to bradykinin in porcine arterioles (Chen et al., 2012). We extend these findings and demonstrate that acutely elevated levels of LPA disrupt endothelium‐dependent FID in human arterioles by shifting the mechanism away from NO to mtH2O2. There was a noticeable improvement in dilation at lower pressure gradients after L‐NAME treatment (Figure 1B), which could potentially be ascribed to elimination of uncoupled eNOS‐derived reactive nitrogen/oxygen species, which scavenge the dilatory ROS (Yang et al., 2009) or the fact that quenching any remaining NO releases the block on mtH2O2 (Beyer et al., 2017). LPA significantly reduced dilation to ACH, potentially by increasing intracellular ROS, which can compromise endothelial NO bioavailability. This observation is consistent with others from our lab (Beyer et al., 2016; Durand et al., 2016) where ACh‐induced dilation is more sensitive to ROS than FID. LPA did not negatively affect smooth muscle responses to NO as indicated by lack of difference in dilation to SNP between treated and untreated vessels. Additionally, the amount of constrictor agent, ET‐1, did not vary between control versus LPA‐treated vessel. Presence of LPA and L‐NAME did require more ET‐1. Although LPA has been described to affect vessel tone (Tokumura et al., 1981; Tokumura et al., 1995), our data argue against either a direct vasoconstrictor or vasodilator effect of LPA, but rather suggest that LPA‐induced production of the dilator H2O2 might be responsible for the higher dose required for constriction since presence of Peg‐Cat (H2O2 scavenger) eliminated that effect.
To our knowledge, this is also the first study examining the function and expression of LPA1–3 receptors in human resistance‐size arterioles. Functional data from vessels treated with LPA2 receptor antagonist indicated that unlike LPA1 and/or LPA3, LPA2 fails to shift the mediator of FID. Instead, LPA1 receptor appears to be mechanistically involved. qPCR analysis showed robust expression of LPA1 and LPA2 but not LPA3, with no difference between arterioles from subjects with and without CAD. Protein analysis from human arterioles identified LPA1 receptor as the dominant isoform with no difference between CAD and non‐CAD phenotype. LPA3 receptor protein was detected in human heart, albeit at a lower level than LPA1 receptor. Faint expression of LPA3 receptor protein was also detected in HUVECs. Absence or minimal expression of LPA3 has been observed in endothelial (Lee et al., 2000; Lin et al., 2007; Ruisanchez et al., 2014) and smooth muscle (Dancs et al., 2017) cells from animal models or cultured vascular human cell lines. We confirmed previously published data using HUVEC and HMVECs. Immunohistochemistry not only confirmed robust staining for LPA1 in comparison to LPA3 in arterioles but also localized the signal predominantly to SMC with little to no expression in arteriolar endothelial cells.
Lack of pronounced expression of LPA receptors, in particular LPA1 receptor, in arteriolar tunica intima was an unexpected finding. Prior findings in animal models and endothelial cell cultures show majority of LPA effects being mediated through LPA1 receptor (Gobeil et al., 2003; Brault et al., 2007; Cui, 2011; Ruisanchez et al., 2014; Dancs et al., 2017). Additionally, we and others observe strong protein and RNA expression in cultured endothelial cells (Lin et al., 2007; Ptaszynska et al., 2010; Ren et al., 2011). Collectively, we interpret these findings to indicate that LPA1 are likely present in human arteriolar endothelial cells, albeit at a much lower level of expression than in SMC, yet sufficient to cause an LPA‐induced shift in the mediator of FID. An alternative explanation of these findings is that LPA acts on SMC to release a factor that acts on the endothelium in a way that changes the mediator of FID. Future efforts should be centred on evaluating the LPA‐mediated bidirectional crosstalk between these two vascular layers as well as cell‐specific expression of these receptors in their native vascular environment.
The source of ROS production is not identified in most LPA‐related vascular studies, but mitochondria and NADPH Oxidases (NOX) are prominent candidates in ECs (Widlansky and Gutterman, 2011; Breton‐Romero and Lamas, 2014; Panieri and Santoro, 2015). We tested whether mitochondria were a source of H2O2 under LPA challenge using the mitochondrial complex I inhibitor, rotenone. Rotenone reduced dilation to the same extent as the general H2O2 scavenger, Peg‐Cat. A mitochondrial source of H2O2 was further suggested using a mitochondria‐specific fluorescent probe. In some settings, NOX is the source of LPA‐stimulated ROS (Lin et al., 2013). We cannot exclude a role for NOX in FID following LPA treatment, which could occur via ROS‐induced ROS release where small amounts of NOX‐related ROS stimulate release of mitochondrial ROS or vice versa (Zinkevich and Gutterman, 2011; Zinkevich et al., 2017). In fact, a recent study showed LPA1 and LPA3 receptor‐dependent activation of PLC, PKC and eventual stimulation of NOX to generate ROS in cell culture model (Lin et al., 2013). Our current findings align with previously published observations demonstrating that the mitochondria are the main source of the dilator H2O2 in human arterioles affected by CAD (Freed et al., 2014; Beyer et al., 2016; Kadlec et al., 2017). Acute LPA exposure thus establishes a CAD phenotype in non‐CAD arterioles.
Details of the signalling pathway by which LPA induces the shift to H2O2‐mediated FID through LPA1 necessitate further investigation. One compelling possibility is the well‐documented crosstalk between Rho‐ and Rac‐dependent cytoskeleton rearrangement and ROS signalling. In fact, recent publication from Chandra and colleagues, using real‐time measurements, demonstrated that increasing levels of LPA or down‐regulating expression of LPP3, one of the main enzymes that degrades LPA, leads to dysfunctional mitochondria with increased production of superoxide in a Rho‐ and ERK‐dependent manner (Chandra et al., 2018). LPA induces changes to the endothelial cytoskeleton in an LPA1 receptor‐dependent manner by phosphorylating myosin light chain (Wu et al., 2015), forming stress fibres and RhoA‐ and Rac‐dependent cytoskeletal rearrangements (Siess et al., 1999). Studies examining mitochondrial dynamics identified LPA, amongst other glycerol‐based lipids, to regulate mitochondrial fission and fusion (Baba et al., 2014; Ha and Frohman, 2014; Frohman, 2015). Together, these studies suggest that LPA may affect mitochondrial structural dynamics, loss of which results in increase in mitochondria‐derived ROS production. Future studies will focus on examining the potential downstream mediators of the observed LPA‐induced effects.
Preliminary studies from our laboratory examined the ability of arterioles to recover and regain NO‐mediated FID by incubating LPA‐treated arterioles in fresh media lacking LPA. Even overnight (~18 h) incubation in fresh media of arterioles treated with LPA for 1 h did not restore NO as the dilator (Supporting Information Figure S8). We therefore suggest that once the LPA‐activated signalling cascade is initiated, damage is prolonged, predisposing the vasculature to further damage caused by increased ROS. This observation sheds light on an exciting clinical opportunity, whereupon assessing the expression of enzymes responsible for both synthesis and degradation of LPA may prove to be a novel preventive or early intervention strategy. In fact, a risk allele leading to decreased expression of LPP3, the enzyme that degrades LPA, has been identified by genome‐wide association studies in nearly 80% of the population, and shown to be highly predictive of CAD, independent of traditional risk factors (Schunkert et al., 2011; Erbilgin et al., 2013). Therefore, the effects of LPA revealed in this study may affect a large portion of the population and represent an underappreciated pathophysiological pathway in cardiovascular disease.
Study limitations
We obtained samples that are surgical discards and are therefore unable to control for all patient characteristics. Vessels are isolated from different fat depots, but we have not observed major differences in vascular responses to flow as evident in our previous publications. The mechanism of FID seems to be preserved across these vascular beds, including the phenotypic switch in tissue acquired from patients with clinically diagnosed CAD. The non‐CAD arterioles are isolated from individuals whose clinical profiles (medications, demographics, etc) are not fully known. However, we do acquire information on prior diagnosis of CAD and presence of risk factors. By defining our non‐CAD cohort strictly as having one or no CV risk factors, we limit the degree to which non‐CAD samples are classified falsely. Females represent most of our non‐CAD vessels. Our sample sizes are too small to detect sex differences in vascular function within and between the phenotypes, but this should be addressed in a larger study. Although we used receptor antagonists well‐documented in the literature, it is possible that they may be non‐specific or have off‐target effects. We addressed such possibilities by testing antagonists for different receptors as well as examining receptor expression. In regard to LPA1 receptor expression, although we and others show expression in cultured endothelial cells, we were unable to detect the receptor in arteriolar ECs using immunohistochemical approach. This suggests at least three possibilities. (i) Arteriolar ECs may have LPA1 receptors sufficient to elicit the switch, but the immunohistochemical approach is not sufficiently sensitive to detect them; (ii) the epitope recognized by the antibody is not accessible in IHC preparations; or (iii) LPA may be acting on LPA1 receptor in SMC and stimulate release of a factor that acts on the endothelium in a way that changes the mediator of FID. Additionally, we have made attempts to knockdown LPA1 receptor using siRNA at two different concentrations with incubations periods as long as 48 h (time limit for isolated arterioles). However, these attempts have been unsuccessful. siRNA transfected arterioles do not show a decrease in LPA1 receptor protein expression. Lastly, because we did not perform histological analysis on all the arterioles used throughout our studies, we cannot exclude the possibility of denudation in some of our vessels. However, because FID is an endothelium‐dependent process (in the presence or absence of LPA), maintained dilation suggests that the endothelium was intact during our studies.
Author contributions
D.S.C., A.O.K., K.A. and J.C.H. planned and performed the experiments. D.S.C., A.M.B. and D.D.G. analysed the data. D.S.C., A.O.K., K.A., P.J.P., A.M.B. and D.D.G. designed the study and wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Supporting info item
Figure S1 Time control study for vascular experiments. To ensure the experimental design of performing two flow studies dose–responses per vessel does not interfere with observed outcomes, overall dilation was assessed on first vs second response curve under LPA challenge. (n) indicates number of subjects.
Figure S2 Effects of LPA2 receptor antagonist alone on the mediator of FID. (A) Treatment with H2L5186303 in the absence of LPA did not induce a shift to Peg‐Cat inhibitable (H2O2‐mediated) dilation. (B) Experimental timeline.
Figure S3 Effects of equimolar dose of LPA2 receptor antagonist on the mechanism of FID. (A) H2L5186303 at equimolar dose (10 μM) to LPA (10 μM) did not prevent LPA from causing the switch to H2O2‐mediated FID. Additionally, higher dose of this antagonist can selectively inhibit LPA3 receptor. (B) Experimental timeline.
Figure S4 LPA3 receptor protein expression immunoblot analysis using a second LPA3 receptor‐targeting antibody.Human umbilical vein endothelial cells (HUVEC) and human microvascular endothelial cells (HMVEC) showed low LPA3 receptor protein expression, while human heart samples (from three different non‐CAD subjects) revealed robust expression of the protein. GAPDH was used as loading control.
Figure S5 LPA3 receptor protein expression in intact non‐CAD arterioles using LSBio antibody. (A) Immunoblot analyses for LPA3 receptor from vessels isolated from individuals without CAD. GAPDH used for loading control. (B) Immunohistochemistry analyses for LPA3 receptor in vessels from an individual without CAD. CD31 staining used to identify endothelial cell layer. Specificity of the antibody was determined by removal of the primary antibody (not shown). Bar for all images = 100 μm.
Figure S6 Expression of LPA3 receptor in human pancreas. (A) Human pancreas was used to test the efficiency of LPA3 antibody. Islets of Langerhans stained positive for LPA3 receptor. (B) H&E staining of the section of pancreas. Bar 100 μm.
Figure S7 Assessment of the denudation efficiency in arterioles. RNA from intact and denuded arterioles from patients without CAD was analyzed for PECAM transcript levels. Denuded expression is represented as fold change from intact. The number of transcripts was normalized to 18 s in the same sample.
Figure S8 Prolonged effects of LPA on the mediator of FID. (A) Peg‐Cat continued to inhibit FID after LPA stimulation (10 μM, 1 hr) and 16–18 hours of fresh media recovery period. L‐NAME had no effect. (B) Experimental timeline.
Acknowledgements
The authors want to thank Christine Duris at the Children's Hospital of Wisconsin Histology Core for her assistance with immunohistochemistry studies, Dr Paula North for her assistance in reading the histology slides and Dr Julie Freed for providing cell lysates. We also thank the following locations for their assistance in providing tissue for this study: Wisconsin Donor Network, Wheaton Franciscan Healthcare's Elmbrook Memorial Hospital, St. Joseph's Hospital, Froedtert Memorial Lutheran Hospital and Aurora St. Luke's Medical Center. This work was supported by the National Institute of Health Grants R01 HL113612 and R01 HL135901 (to D.D.G.) and 1R01 HL133029‐01 (to A.M.B.), the American Heart Association Postdoctoral Fellowship Grant 16POST26430075 (to K.A.‐A.) and the Predoctoral Fellowship Grant 17PRE33410986 (to D.S.C.).
Chabowski, D. S. , Kadlec, A. O. , Ait‐Aissa, K. , Hockenberry, J. C. , Pearson, P. J. , Beyer, A. M. , and Gutterman, D. D. (2018) Lysophosphatidic acid acts on LPA1 receptor to increase H2O2 during flow‐induced dilation in human adipose arterioles. British Journal of Pharmacology, 175: 4266–4280. 10.1111/bph.14492.
References
- Albers HM, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW et al (2010). Boronic acid‐based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc Natl Acad Sci U S A 107: 7257–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki J (2004). Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol 15: 477–489. [DOI] [PubMed] [Google Scholar]
- Avraamides C, Bromberg ME, Gaughan JP, Thomas SM, Tsygankov AY, Panetti TS (2007). Hic‐5 promotes endothelial cell migration to lysophosphatidic acid. Am J Physiol Heart Circ Physiol 293: H193–H203. [DOI] [PubMed] [Google Scholar]
- Baba T, Kashiwagi Y, Arimitsu N, Kogure T, Edo A, Maruyama T et al (2014). Phosphatidic acid (PA)‐preferring phospholipase A1 regulates mitochondrial dynamics. J Biol Chem 289: 11497–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DL, Morrison P, Miller B, Riely CA, Tolley B, Westermann AM et al (2002). Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA 287: 3081–3082. [DOI] [PubMed] [Google Scholar]
- Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD (2014). An acute rise in intraluminal pressure shifts the mediator of flow‐mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circ Physiol . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer AM, Freed JK, Durand MJ, Riedel M, Ait‐Aissa K, Green P et al (2016). Critical role for telomerase in the mechanism of flow‐mediated dilation in the human microcirculation. Circ Res 118: 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer AM, Zinkevich N, Miller B, Liu Y, Wittenburg AL, Mitchell M et al (2017). Transition in the mechanism of flow‐mediated dilation with aging and development of coronary artery disease. Basic Res Cardiol 112: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bot M, de Jager SC, MacAleese L, Lagraauw HM, van Berkel TJ, Quax PH et al (2013). Lysophosphatidic acid triggers mast cell‐driven atherosclerotic plaque destabilization by increasing vascular inflammation. J Lipid Res 54: 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault S, Gobeil F Jr, Fortier A, Honore JC, Joyal JS, Sapieha PS et al (2007). Lysophosphatidic acid induces endothelial cell death by modulating the redox environment. Am J Physiol Regul Integr Comp Physiol 292: R1174–R1183. [DOI] [PubMed] [Google Scholar]
- Breton‐Romero R, Lamas S (2014). Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol 2: 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnelli M, Manzini S, Hilvo M, Parolini C, Ganzetti GS, Dellera F et al (2017). Liver‐specific deletion of the Plpp3 gene alters plasma lipid composition and worsens atherosclerosis in apoE−/− mice. Sci Rep 7: 44503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra M, Escalante‐Alcalde D, Bhuiyan MS, Orr AW, Kevil C, Morris AJ et al (2018). Cardiac‐specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox Biol 14: 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ochoa LN, Kagan A, Chai H, Liang Z, Lin PH et al (2012). Lysophosphatidic acid causes endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Atherosclerosis 222: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Olashaw N, Wu J (1995). Participation of reactive oxygen species in the lysophosphatidic acid‐stimulated mitogen‐activated protein kinase activation pathway. J Biol Chem 270: 28499–28502. [DOI] [PubMed] [Google Scholar]
- Cui MZ (2011). Lysophosphatidic acid effects on atherosclerosis and thrombosis. Clin Lipidol 6: 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancs PT, Ruisanchez E, Balogh A, Panta CR, Miklos Z, Nusing RM et al (2017). LPA1 receptor‐mediated thromboxane A2 release is responsible for lysophosphatidic acid‐induced vascular smooth muscle contraction. FASEB J 31: 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, Chang CJ (2008). A targetable fluorescent probe for imaging hydrogen peroxide in the mitochondria of living cells. J Am Chem Soc 130: 9638–9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi T, Miyauchi K, Ohkawa R, Nakamura K, Kishimoto T, Miyazaki T et al (2012). Increased circulating plasma lysophosphatidic acid in patients with acute coronary syndrome. Clin Chim Acta 413: 207–212. [DOI] [PubMed] [Google Scholar]
- Durand MJ, Zinkevich NS, Riedel M, Gutterman DD, Nasci VL, Salato VK et al (2016). Vascular actions of angiotensin 1‐7 in the human microcirculation: novel role for telomerase. Arterioscler Thromb Vasc Biol 36: 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbilgin A, Civelek M, Romanoski CE, Pan C, Hagopian R, Berliner JA et al (2013). Identification of CAD candidate genes in GWAS loci and their expression in vascular cells. J Lipid Res 54: 1894–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fells JI, Tsukahara R, Liu J, Tigyi G, Parrill AL (2009). Structure‐based drug design identifies novel LPA3 antagonists. Bioorg Med Chem 17: 7457–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed JK, Beyer AM, LoGiudice JA, Hockenberry JC, Gutterman DD (2014). Ceramide changes the mediator of flow‐induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res 115: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed JK, Durand MJ, Hoffmann BR, Densmore JC, Greene AS, Gutterman DD (2017). Mitochondria‐regulated formation of endothelium‐derived extracellular vesicles shifts the mediator of flow‐induced vasodilation. Am J Physiol Heart Circ Physiol 312: H1096–H1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA (2015). Role of mitochondrial lipids in guiding fission and fusion. J Mol Med (Berl) 93: 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gierse J, Thorarensen A, Beltey K, Bradshaw‐Pierce E, Cortes‐Burgos L, Hall T et al (2010). A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J Pharmacol Exp Ther 334: 310–317. [DOI] [PubMed] [Google Scholar]
- Gobeil F Jr, Bernier SG, Vazquez‐Tello A, Brault S, Beauchamp MH, Quiniou C et al (2003). Modulation of pro‐inflammatory gene expression by nuclear lysophosphatidic acid receptor type‐1. J Biol Chem 278: 38875–38883. [DOI] [PubMed] [Google Scholar]
- Ha EE, Frohman MA (2014). Regulation of mitochondrial morphology by lipids. Biofactors 40: 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Takahashi M, Nishida W, Yoshida K, Ohkawa Y, Kitabatake A et al (2001). Phenotypic modulation of vascular smooth muscle cells induced by unsaturated lysophosphatidic acids. Circ Res 89: 251–258. [DOI] [PubMed] [Google Scholar]
- Kadlec AO, Chabowski DS, Ait‐Aissa K, Hockenberry JC, Otterson MF, Durand MJ et al (2017). PGC‐1α (peroxisome proliferator‐activated receptor γ coactivator 1‐α) overexpression in coronary artery disease recruits NO and hydrogen peroxide during flow‐mediated dilation and protects against increased intraluminal pressure. Hypertension 70: 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE et al (2008). Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol 28: 1982–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo L, Chilian WM, Davis MJ (1991). Interaction of pressure‐ and flow‐induced responses in porcine coronary resistance vessels. Am J Physiol 261: H1706–H1715. [DOI] [PubMed] [Google Scholar]
- Kuo L, Davis MJ, Chilian WM (1990). Endothelium‐dependent, flow‐induced dilation of isolated coronary arterioles. Am J Physiol 259: H1063–H1070. [DOI] [PubMed] [Google Scholar]
- Kurano M, Suzuki A, Inoue A, Tokuhara Y, Kano K, Matsumoto H et al (2015). Possible involvement of minor lysophospholipids in the increase in plasma lysophosphatidic acid in acute coronary syndrome. Arterioscler Thromb Vasc Biol 35: 463–470. [DOI] [PubMed] [Google Scholar]
- Lee H, Goetzl EJ, An S (2000). Lysophosphatidic acid and sphingosine 1‐phosphate stimulate endothelial cell wound healing. Am J Physiol Cell Physiol 278: C612–C618. [DOI] [PubMed] [Google Scholar]
- Lin CC, Lin CE, Lin YC, Ju TK, Huang YL, Lee MS et al (2013). Lysophosphatidic acid induces reactive oxygen species generation by activating protein kinase C in PC‐3 human prostate cancer cells. Biochem Biophys Res Commun 440: 564–569. [DOI] [PubMed] [Google Scholar]
- Lin CI, Chen CN, Chen JH, Lee H (2006). Lysophospholipids increase IL‐8 and MCP‐1 expressions in human umbilical cord vein endothelial cells through an IL‐1‐dependent mechanism. J Cell Biochem 99: 1216–1232. [DOI] [PubMed] [Google Scholar]
- Lin CI, Chen CN, Lin PW, Chang KJ, Hsieh FJ, Lee H (2007). Lysophosphatidic acid regulates inflammation‐related genes in human endothelial cells through LPA1 and LPA3. Biochem Biophys Res Commun 363: 1001–1008. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD (2003). Mitochondrial sources of H2O2 generation play a key role in flow‐mediated dilation in human coronary resistance arteries. Circ Res 93: 573–580. [DOI] [PubMed] [Google Scholar]
- Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD (2003). Role for hydrogen peroxide in flow‐induced dilation of human coronary arterioles. Circ Res 92: e31–e40. [DOI] [PubMed] [Google Scholar]
- Panchatcharam M, Miriyala S, Yang F, Rojas M, End C, Vallant C et al (2008). Lysophosphatidic acid receptors 1 and 2 play roles in regulation of vascular injury responses but not blood pressure. Circ Res 103: 662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchatcharam M, Salous AK, Brandon J, Miriyala S, Wheeler J, Patil P et al (2014). Mice with targeted inactivation of ppap2b in endothelial and hematopoietic cells display enhanced vascular inflammation and permeability. Arterioscler Thromb Vasc Biol 34: 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panetti TS (2002). Differential effects of sphingosine 1‐phosphate and lysophosphatidic acid on endothelial cells. Biochim Biophys Acta 1582: 190–196. [DOI] [PubMed] [Google Scholar]
- Panetti TS, Hannah DF, Avraamides C, Gaughan JP, Marcinkiewicz C, Huttenlocher A et al (2004). Extracellular matrix molecules regulate endothelial cell migration stimulated by lysophosphatidic acid. J Thromb Haemost 2: 1645–1656. [DOI] [PubMed] [Google Scholar]
- Panetti TS, Nowlen J, Mosher DF (2000). Sphingosine‐1‐phosphate and lysophosphatidic acid stimulate endothelial cell migration. Arterioscler Thromb Vasc Biol 20: 1013–1019. [DOI] [PubMed] [Google Scholar]
- Panieri E, Santoro MM (2015). ROS signaling and redox biology in endothelial cells. Cell Mol Life Sci 72: 3281–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptaszynska MM, Pendrak ML, Stracke ML, Roberts DD (2010). Autotaxin signaling via lysophosphatidic acid receptors contributes to vascular endothelial growth factor‐induced endothelial cell migration. Mol Cancer Res 8: 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Hale J, Srikanthan S, Silverstein RL (2011). Lysophosphatidic acid suppresses endothelial cell CD36 expression and promotes angiogenesis via a PKD‐1‐dependent signaling pathway. Blood 117: 6036–6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza C, Leitinger N, Yue J, Fischer DJ, Wang DA, Shih PT et al (1999). Lysophosphatidic acid as a regulator of endothelial/leukocyte interaction. Lab Invest 79: 1227–1235. [PubMed] [Google Scholar]
- Ruisanchez E, Dancs P, Kerek M, Nemeth T, Farago B, Balogh A et al (2014). Lysophosphatidic acid induces vasodilation mediated by LPA1 receptors, phospholipase C, and endothelial nitric oxide synthase. FASEB J 28: 880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H et al (2011). Large‐scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43: 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin I, Kweon SM, Lee ZW, Kim SI, Joe CO, Kim JH et al (1999). Lysophosphatidic acid increases intracellular H2O2 by phospholipase D and RhoA in rat‐2 fibroblasts. Mol Cells 9: 292–299. [PubMed] [Google Scholar]
- Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C et al (1999). Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc Natl Acad Sci U S A 96: 6931–6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staiculescu MC, Ramirez‐Perez FI, Castorena‐Gonzalez JA, Hong Z, Sun Z, Meininger GA et al (2014). Lysophosphatidic acid induces integrin activation in vascular smooth muscle and alters arteriolar myogenic vasoconstriction. Front Physiol 5: 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanyi JL, Morris AJ, Wolf JK, Fang X, Hasegawa Y, Lapushin R et al (2003). The human lipid phosphate phosphatase‐3 decreases the growth, survival, and tumorigenesis of ovarian cancer cells: validation of the lysophosphatidic acid signaling cascade as a target for therapy in ovarian cancer. Cancer Res 63: 1073–1082. [PubMed] [Google Scholar]
- Tokumura A, Kume T, Fukuzawa K, Tsukatani H (1981). Cardiovascular effects of lysophosphatidic acid and its structural analogs in rats. J Pharmacol Exp Ther 219: 219–224. [PubMed] [Google Scholar]
- Tokumura A, Yotsumoto T, Masuda Y, Tanaka S (1995). Vasopressor effect of lysophosphatidic acid on spontaneously hypertensive rats and Wistar Kyoto rats. Res Commun Mol Pathol Pharmacol 90: 96–102. [PubMed] [Google Scholar]
- van de Hoef TP, van Lavieren MA, Damman P, Delewi R, Piek MA, Chamuleau SA et al (2014). Physiological basis and long‐term clinical outcome of discordance between fractional flow reserve and coronary flow velocity reserve in coronary stenoses of intermediate severity. Circ Cardiovasc Interv 7: 301–311. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, Vermeer MA, van Hinsbergh VW (2000). Role of RhoA and Rho kinase in lysophosphatidic acid‐induced endothelial barrier dysfunction. Arterioscler Thromb Vasc Biol 20: E127–E133. [DOI] [PubMed] [Google Scholar]
- Widlansky ME, Gutterman DD (2011). Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal 15: 1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Huang RT, Kuo CH, Kumar S, Kim CW, Lin YC et al (2015). Mechanosensitive PPAP2B regulates endothelial responses to atherorelevant hemodynamic forces. Circ Res 117: e41–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YJ, Aziz OA, Bhugra P, Arneja AS, Mendis MR, Dhalla NS (2003). Potential role of lysophosphatidic acid in hypertension and atherosclerosis. Can J Cardiol 19: 1525–1536. [PubMed] [Google Scholar]
- Yang YM, Huang A, Kaley G, Sun D (2009). eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol 297: H1829–H1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkevich NS, Fancher IS, Gutterman DD, Phillips SA (2017). Roles of NADPH oxidase and mitochondria in flow‐induced vasodilation of human adipose arterioles: ROS‐induced ROS release in coronary artery disease. Microcirculation (New York, NY: 1994) 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkevich NS, Gutterman DD (2011). ROS‐induced ROS release in vascular biology: redox‐redox signaling. Am J Physiol Heart Circ Physiol 301: H647–H653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Figure S1 Time control study for vascular experiments. To ensure the experimental design of performing two flow studies dose–responses per vessel does not interfere with observed outcomes, overall dilation was assessed on first vs second response curve under LPA challenge. (n) indicates number of subjects.
Figure S2 Effects of LPA2 receptor antagonist alone on the mediator of FID. (A) Treatment with H2L5186303 in the absence of LPA did not induce a shift to Peg‐Cat inhibitable (H2O2‐mediated) dilation. (B) Experimental timeline.
Figure S3 Effects of equimolar dose of LPA2 receptor antagonist on the mechanism of FID. (A) H2L5186303 at equimolar dose (10 μM) to LPA (10 μM) did not prevent LPA from causing the switch to H2O2‐mediated FID. Additionally, higher dose of this antagonist can selectively inhibit LPA3 receptor. (B) Experimental timeline.
Figure S4 LPA3 receptor protein expression immunoblot analysis using a second LPA3 receptor‐targeting antibody.Human umbilical vein endothelial cells (HUVEC) and human microvascular endothelial cells (HMVEC) showed low LPA3 receptor protein expression, while human heart samples (from three different non‐CAD subjects) revealed robust expression of the protein. GAPDH was used as loading control.
Figure S5 LPA3 receptor protein expression in intact non‐CAD arterioles using LSBio antibody. (A) Immunoblot analyses for LPA3 receptor from vessels isolated from individuals without CAD. GAPDH used for loading control. (B) Immunohistochemistry analyses for LPA3 receptor in vessels from an individual without CAD. CD31 staining used to identify endothelial cell layer. Specificity of the antibody was determined by removal of the primary antibody (not shown). Bar for all images = 100 μm.
Figure S6 Expression of LPA3 receptor in human pancreas. (A) Human pancreas was used to test the efficiency of LPA3 antibody. Islets of Langerhans stained positive for LPA3 receptor. (B) H&E staining of the section of pancreas. Bar 100 μm.
Figure S7 Assessment of the denudation efficiency in arterioles. RNA from intact and denuded arterioles from patients without CAD was analyzed for PECAM transcript levels. Denuded expression is represented as fold change from intact. The number of transcripts was normalized to 18 s in the same sample.
Figure S8 Prolonged effects of LPA on the mediator of FID. (A) Peg‐Cat continued to inhibit FID after LPA stimulation (10 μM, 1 hr) and 16–18 hours of fresh media recovery period. L‐NAME had no effect. (B) Experimental timeline.