

Abstract
Disulfide‐rich peptides represent an important protein family with broad pharmacological potential. Recent advances in computational methods have made it possible to design new peptides which adopt a stable conformation de novo. Here, we describe a system to produce disulfide‐rich de novo peptides using Escherichia coli as the expression host. The advantage of this system is that it enables production of uniformly 13C‐ and 15N‐labeled peptides for solution nuclear magnetic resonance (NMR) studies. This expression system was used to isotopically label two previously reported de novo designed peptides, and to determine their solution structures using NMR. The ensemble of NMR structures calculated for both peptides agreed well with the design models, further confirming the accuracy of the design protocol. Collection of NMR data on the peptides under reducing conditions revealed a dependency on disulfide bonds to maintain stability. Furthermore, we performed long‐time molecular dynamics (MD) simulations with tempering to assess the stability of two families of de novo designed peptides. Initial designs which exhibited a stable structure during simulations were more likely to adopt a stable structure in vitro, but attempts to utilize this method to redesign unstable peptides to fold into a stable state were unsuccessful. Further work is therefore needed to assess the utility of MD simulation techniques for de novo protein design.
Keywords: disulfide‐rich peptide, computational protein design, rosetta, de novo protein, protein purification, nuclear magnetic resonance, molecular dynamics simulation
Abbreviations
- CD
circular dichroism
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
Introduction
Peptide drugs offer many advantages over traditional small‐molecule therapeutics, including enhanced specificity and reduced off‐target effects.1 Constrained peptides are commonly employed by nature as signaling molecules or toxins, and the incorporation of multiple disulfide covalent crosslinks between pairs of cysteine residues is thought to be responsible for the stability and potent drug‐like properties of these molecules.2 Knottins and short‐chain scorpion toxins are two well‐characterized groups within this protein family, each characterized by conserved structural motifs and highly‐conserved disulfide bonds.3, 4 However, the ability to generate stable structures adopting a wide range of specified sizes and shapes not observed in nature will be necessary to unlock the full pharmacological potential of peptide‐based drugs. Working toward this goal, we previously reported a computational method for the de novo design of genetically encodable cysteine‐rich peptides of various topologies, which were validated by experiments.5, 6 Here, we describe two extensions to this methodology. First, we present an efficient and widely applicable method for the production of de novo designed, disulfide‐rich peptides via genetic fusion to DsbC and expression in the cytoplasm of Escherichia coli. This is demonstrated by producing two peptides with divergent structures and sequences: a mixed α/β topology with a helix packing against a three‐stranded antiparallel β‐sheet stabilized by three disulfide bonds (peptide gHEEE_02), and a helix–turn–helix topology containing a single disulfide bond adjacent to the main chain termini (peptide gHH_44).6 The efficacy of the expression system to successfully fold peptides as designed was confirmed by determining the structure for these two peptides using solution NMR spectroscopy. The NMR structures confirm the accuracy of the two designs, which had not previously been validated by full structure determination. Second, motivated by the observation that some of the computational designs did not appear to fold as expected,6 we developed a computational strategy based on molecular dynamics (MD) simulations to identify misfolded designs prior to synthesis.
Results and Discussion
Production of peptides via fusion to DsbC
The production of small proteins and peptides containing disulfide bonds presents a challenge, as disulfide bond formation does not readily occur under the reducing conditions found in the cytoplasm. Previously, genetically encodable disulfide‐rich peptides were secreted from E. coli via a cleavable genetic fusion to the native E. coli protein OsmY, which promoted folding in the oxidizing periplasmic environment where disulfide bonds can readily form.7 While efficacious, this method proved to be sensitive to unintended (or “leaky”) expression prior to induction and suffered from low yields and consistency issues. An alternative solution described by Nozach et al. utilizes a fusion to the native E. coli disulfide bond isomerase DsbC, followed by intracellular expression from the commonly‐used laboratory expression strain BL21.8 Presumably, the extremely high local concentration of disulfide bond isomerase present during protein purification enables fusion partners to assume the lowest energy pattern of disulfide connectivity.
To facilitate producing many different constructs with a DsbC fusion, we created a new expression vector, pCDB364 (available from Addgene as Plasmid #110275), optimized for gene insertion using the isothermal DNA assembly method developed by Gibson et al.9 In this vector (Fig. 1), the native signal peptide has been truncated from the N‐terminus of DscB to direct cytoplasmic localization. An N‐terminal deca‐histidine tag enables purification of heterologous protein by immobilized metal‐affinity chromatography (IMAC), and a TEV recognition peptide at the C‐terminus of the fusion protein enables removal of the fusion domain by TEV protease. Because the TEV protease cleaves at the C‐terminal end of its recognition peptide, and has remarkable tolerance for amino acid variation at the P1′ and P2′ positions,10 we were able to remove all of the fusion domain from the designed peptides encoded into this construct (i.e., there are no “scar” residues from the fusion domain included in the mature peptide sequence).
Figure 1.
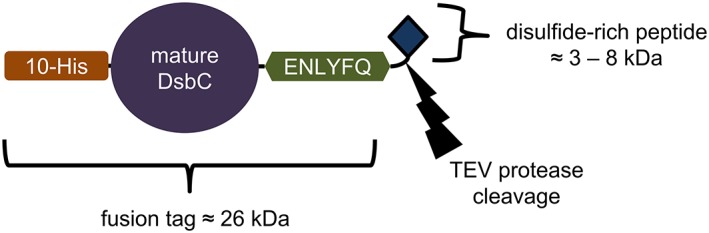
The expression construct created to purify the disulfide‐rich peptides in this study. Peptides were purified via immobilized metal affinity chromatography using the deca‐histidine tag, which is genetically fused to the E. coli disulfide bond isomerase DsbC; the tag is removed from the peptide using TEV protease, which cleaves at the C‐terminal end of the amino acid sequence “ENLYFQ.” The bacterial expression vector for this fusion construct is pCDB364.
To assess the compatibility of this expression system with computationally designed peptides, synthetic genes coding for previously reported de novo peptides gHH_44 and gHEEE_02 were subcloned into the vector pCDB364. After transformation of this expression vector into E. coli cells, uniformly 13C‐ and 15N‐labeled peptides were expressed and purified for detailed structural characterization by solution NMR spectroscopy. All cross peaks in both 2D [15N,1H]HSQC spectra were assigned to NH‐moieties of the designed amino acid sequence, demonstrating that the proteins of interest were cleaved by TEV protease without any remaining “scar” residues.
Solution structure of oxidized gHH_44 and gHEEE_02
The near uniform peak intensities and good signal dispersion in both the proton and nitrogen dimensions of the 2D [15N,1H] HSQC of gHH_44 [Fig. 2(A)] and gHEEE_02 [Fig. 3(A)] are characteristic for folded proteins.11 This is consistent with the previously reported circular dichroism (CD) spectra of both oxidized peptides.6 While CD spectroscopy is a useful method to rapidly probe the secondary structure content of proteins in solution and to monitor their structure and stability under a variety of conditions,12 the method provides no information regarding tertiary structure.13, 14 This challenge is addressed by NMR solution structure determination.15 The designed model of gHEEE_02 has many contacts between the α‐helix and anti‐parallel β‐sheet, while gHH_44 has comparatively fewer contacts between the two α‐helices. By NMR, we detected 301 long range NOEs for gHEEE_02, and 33 for gHH_44 (Table 1). These long range NOEs, together with the other intra‐residue, sequential, and medium range NOEs, dihedral angle restraints, and hydrogen and disulfide bond restraints, resulted in final structural ensembles that converged well (backbone RMSD of 0.41 Å or less) with favorable PSVS scores (all the structure‐quality Z‐scores < −3).16
Figure 2.
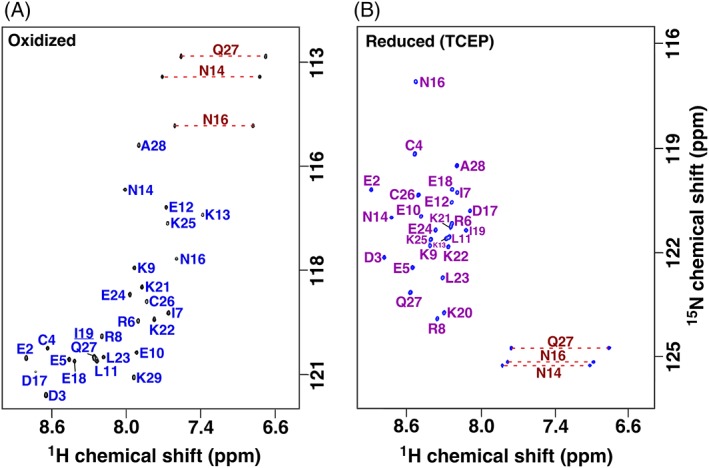
Assigned 2D [15N,1H] HSQC spectra for gHH_44 collected in the oxidized (a) and reduced state (b). Spectra collected in 20 mM sodium acetate, pH 4.8 buffer, 20°C, at 800 MHz 1H resonance frequency without (a) and with (b) 2.5 mM TCEP. Amide side chain resonance pairs are connected by a red dashed line and are folded in the reduced spectrum. Note that the 15N chemical shift range shown is the same in both spectra, while the 15N carrier frequency is different.
Figure 3.
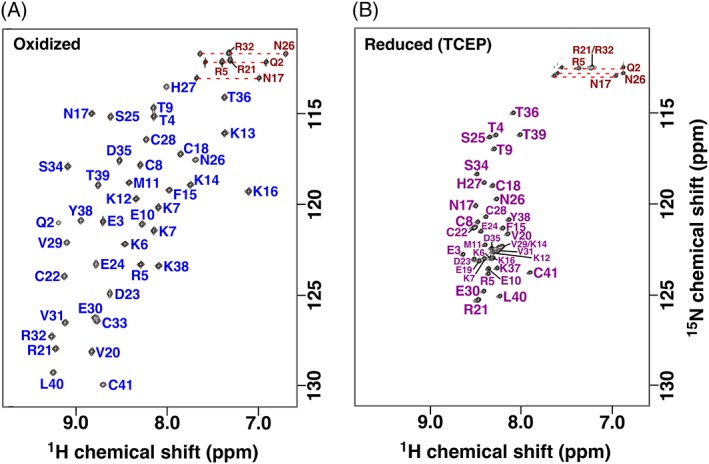
Assigned 2D [15N,1H] HSQC spectra of gHEEE_02 collected in the oxidized (a) and reduced state (b) at 750 MHz 1H resonance frequency. Spectra were recorded in 50 mM sodium phosphate, pH 6.0, 4 μM 4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid, 0.02% sodium azide buffer without (a) or with (b) 5 mM TCEP at 25°C. Amide side chain resonance pairs are connected by a red dashed line.
Table 1.
Summary of the structure statistics
| gHH_44 | gHEEE_02 | |||
|---|---|---|---|---|
| Completeness of 1 H resonance assignments a, b | ||||
| Backbone/side‐chain (%/%) | 100/73 | 99.5/96.5 | ||
| Conformationally restricting constraints c | ||||
| Distance constraints | ||||
| Total | 244 | 1000 | ||
| Intra‐residue (i = j) | 104 | 182 | ||
| Sequential (|i − j| = 1) | 68 | 270 | ||
| Medium range (1 < |i − j| < 5) | 39 | 244 | ||
| Long range (|i − j| ≥ 5) | 33 | 301 | ||
| Dihedral angle constraints | 34 | 36 | ||
| Disulfide bond constraints | 3 | 9 | ||
| Hydrogen bond constraints | 20 | – | ||
| No. of constraints per residue | 19.5 | 25.9 | ||
| No. of long range constraints per residue | 1.3 | 7.5 | ||
| Residual constraint violations c | ||||
| Average no. of distance violations per structure: | ||||
| 0.1–0.2 Å | 0 | 9.2 | ||
| 0.2–0.5 Å | 0 | 2.8 | ||
| >0.5 Å | 0 | 0 | ||
| Average no. of dihedral angle violations per structure: | ||||
| 1–10° | 0 | 6.1 | ||
| Model quality c | ||||
| RMSD backbone atoms (Å) | 0.41 ± 0.10 | 0.36 ± 0.06 | ||
| RMSD heavy atoms (Å) | 1.38 ± 0.13 | 0.90 ± 0.10 | ||
| RMSD bond lengths (Å) | 0.006 | 0.018 | ||
| RMSD bond angles (°) | 0.6 | 1.2 | ||
| MolProbity Ramachandran statistics | ||||
| Most favored regions (%) | 97.6 | 92.3 | ||
| Allowed regions (%) | 2.4 | 7.6 | ||
| Disallowed regions (%) | 0.0 | 0.2 | ||
| Global quality scores (Raw/Z‐score)c | ||||
| Verify3D | 0.41 | −0.80 | 0.37 | −1.44 |
| ProsaII | 1.31 | 2.73 | 0.95 | 1.24 |
| Procheck (phi‐psi) | 0.36 | 1.73 | −0.31 | −0.90 |
| Procheck (all)d | 0.02 | 0.12 | −0.24 | −1.42 |
| MolProbity clash score | 22.78 | −2.38 | 14.40 | −0.95 |
| RPF scores | ||||
| Recall/precision | 0.97 | 0.85 | 0.93 | 0.87 |
| F‐measure/DP‐score | 0.91 | 0.58 | 0.90 | 0.75 |
| BMRB accession number | 30204 | 30312 | ||
| PDB ID | 5TX8 | 5W9F | ||
Structural statistics computed for the ensemble of 20 deposited conformers.
Computed using the expected number of resonances, excluding: highly exchangeable protons (N‐terminal, Lys, and Arg amino groups, hydroxyls of Ser, Thr, Tyr).
Calculated using PSVS 1.5 and based on ordered residue ranges defined by PSVS.
Ordered residues: gHH_44: C4‐N14, D17‐E24. gHEEE_02: E3‐R32, Y38‐L40.
A key component of our genetically encodable, cysteine‐rich, de novo designed peptides are intramolecular disulfide bonds at defined positions. Disulfide bond formation was unambiguously verified from the chemical shifts of the β‐carbon of cysteine residues. Generally, the cysteine 13Cβ chemical shift in the reduced state is <32 ppm and increases to >35 ppm in the oxidized state.17, 18 In the absence of TCEP or other reducing agents, the cysteine 13Cβ chemical shifts for both recombinantly prepared peptides were 39 ppm or greater (Table 2). Because gHH_44 contains 2 cysteine residues, there is only 1 possible disulfide bond combination. On the other hand, gHEEE_02 contains 6 cysteine residues, and therefore, has 15 potential disulfide bond combinations. The pattern of disulfide bond pairs for gHEEE_02 was previously deduced by tandem mass spectrometry,6 and this was further confirmed by the proximity of γ‐sulfur atoms in the preliminary structure calculations. Hence, the following constraints for disulfide bonds were used for the structure calculations: Cys8–Cys26 (gHH_44); Cys8–Cys22, Cys18–Cys33, and Cys28–Cys41 (gHEEE_02).
Table 2.
Chemical shifts (ppm) for the 13Cβ carbons of the cysteine residues in gHH_44 and gHEEE_02 in the oxidized and reduced states
| gHH_44 C4 |
gHH_44 C26 |
gHEEE_02 C8 |
gHEEE_02 C18 |
gHEEE_02 C22 |
gHEEE_02 C28 |
gHEEE_02 C33 |
gHEEE_02 C41 |
|
|---|---|---|---|---|---|---|---|---|
| Oxidized | 39.2 | 41.6 | 39.7 | 48.4 | 42.2 | 47.9 | 42.8 | 45.8 |
| Reduced | 27.6 | 27.7 | 27.9 | 27.9 | 28.1 | 27.7 | 28.0 | 29.0 |
| Δδ13 C β | 11.6 | 13.9 | 11.8 | 20.5 | 14.1 | 20.2 | 14.8 | 16.8 |
As with previously determined structures,6 these new models confirm the atomic‐resolution accuracy of the de novo design method used to create them (Fig. 4). For gHH_44, the mean Cα RMSD between the design model and each member of the ensemble is 2.3 Å. The largest conformational deviations occur at the main chain termini, outside of the geometric constraints imposed by the disulfide bond, and the mean Cα RMSD to the design model drops to 0.7 Å when these five terminal residues are excluded from the calculation. Thus, for very small systems, such as the disulfide rich peptides studied here, it may be advantageous to place the disulfide bonds as close to the termini as possible, or to engineer additional contacts to restrain flexibility. For gHEEE_02, the mean Cα RMSD between the design model and each member of the ensemble is 1.3 Å. In both cases, core side chain rotameric states exhibit excellent agreement with the design model, and all disulfide bonds are formed in the designed pattern of connectivity.
Figure 4.
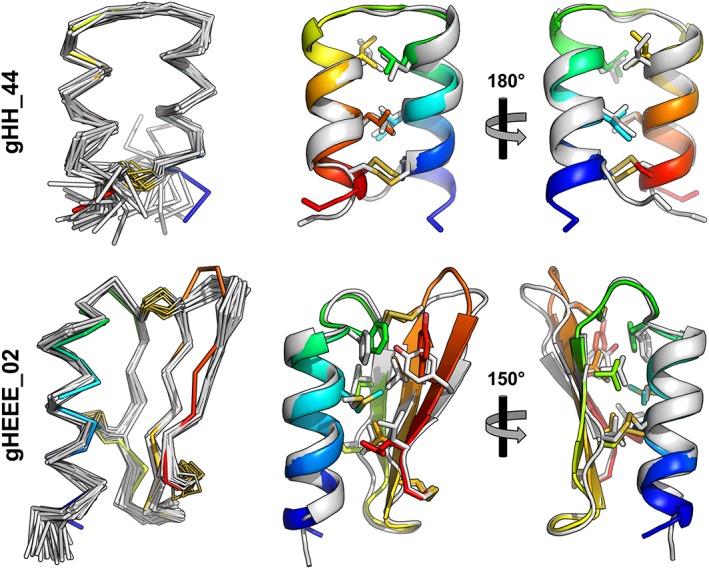
Structural alignment of determined structures (gray) with computationally designed models (rainbow). On the left, the Cα trace is shown for each member of the NMR ensemble as well as the design. On the right are cartoon representations showing the lowest‐energy model from the NMR ensemble aligned to the design model; side chains for hydrophobic core residues and disulfide bonds are shown as sticks.
Solution structure of reduced gHH_44 and gHEEE_02
We previously reported that the presence of an approximately 100‐fold molar excess of the reductant tris(2‐carboxyethyl)phosphine (TCEP) results in a loss of regular secondary structure for gHH_44 and gHEEE_02 as measured by CD spectroscopy. The reduced‐state CD spectra were dominated by a feature common to unfolded proteins: a negative maximum below 200 nm.19 While the double minima at 208 and 222 nm (which indicate the presence α‐helical structure) were ablated by TCEP treatment for gHEEE_02, these negative peaks were merely diminished for gHH_44. Hence, to unambiguously verify that TCEP completely reduced the disulfide bonds, and to assess structural character of each peptide in the reduced state, we also assigned the NMR chemical shifts in the presence of TCEP.
The assigned 2D [15N, 1H] HSQC spectra for reduced gHH_44 and gHEEE_02 [Figs. 2(B) and 3(B)] differ markedly relative to the spectra measured under oxidizing conditions [Figs. 2(A) and 3(A)]. For both peptides, the backbone amide chemical shift dispersion is narrower in the presence of TCEP: gHH_44: δ1H, ~1.6 → ~1.0 ppm (δΔ1H = ~0.6 ppm), δ15N, ~7 → ~6 ppm (δΔ15N = ~1 ppm); gHEEE_02: δ1H, ~2.1 → ~0.7 ppm (δΔ1H = ~1.4 ppm), δ15N, ~16.5 → ~10.4 ppm (δΔ15N = ~6 ppm). These chemical shift dispersion trends indicate that both peptides are less structured in the presence of TCEP.11 The differences in chemical shift dispersion between redox states are smaller for gHH_44 than gHEEE_02, and this is likely due to the differences in secondary structure content between these peptides: gHH_44 is completely helical, while gHEEE_02 is a mixture of helical and β‐sheet structure. In the proton dimension, the chemical shift dispersion is generally narrower for helical regions than for β‐strand regions, and this is likely the major contributor to the observed chemical shift dispersion differences. A minor contributor is that β‐strands involve hydrogen bonding to non‐local residues, while α‐helices are stabilized by hydrogen bonding to residues adjacent in the primary structure. Consequently, because gHH_44 only contains α‐helices, it is more likely to exhibit transient secondary structure in the absence of a stable tertiary structure than gHEEE_02.
As illustrated in the 2D [15N, 1H] HSQC spectra in Figures 2(B) and 3(B), the backbone moieties and 13Cα and 13Cβ chemical shifts of both peptides in the reduced state were completely assigned (Fig. 5). Analysis of the 13Cβ chemical shifts for the cysteine residues enables unambiguous verification of the oxidation state of each cysteine in the peptide: all cysteine 13Cβ chemical shifts of both peptides were smaller than 29 ppm in the presence of TCEP (Table 2), indicating the ablation of all disulfide bonds. Furthermore, deviations of the 13Cα and 13Cβ chemical shifts from their random coil values (Δδ13Cα/β = δ13Cα/β observed − δ13Cα/β random coil) are highly predictive for secondary structure in polypeptide segments.20, 21 These deviations from random coil values are plotted for both peptides in Figure 5. In the oxidized state (blue), the 13Cα and 13Cβ chemical shift deviations from random coil values generally follow the pattern expected given the calculated structures. In the reduced state (cyan), the 13Cα and 13Cβ chemical shifts do not significantly differ from random coil values, indicating both peptides were unfolded with little transient structure. The absence of structure in the reduced state was corroborated by absence of medium and long range NOEs in 3D 15N‐resolved [1H,1H] NOESY (gHH_44) and 3D 15N/13Caliphatic/13Caromatic‐resolved [1H,1H] NOESY spectra (gHEEE_02).
Figure 5.
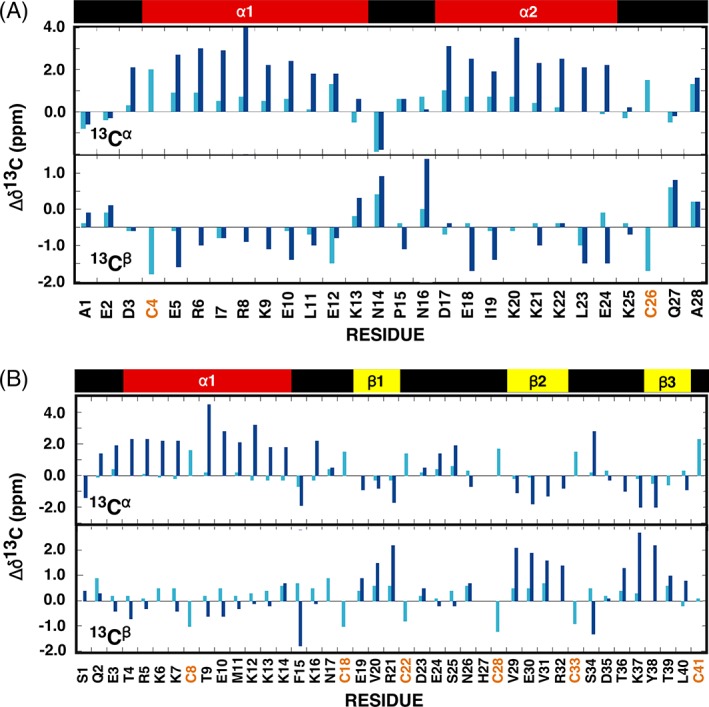
Analysis of the observed 13Cα and 13Cβ chemical shift deviations from random coil values for gHH_44 (a) and gHEEE_02 (b) where Δδ13C = δ13CObserved − δ13CRandom coil. The random coil carbon values were taken from CNS (cns_solve_1.1) with no calculations for histidine and oxidized cysteine residues. Cyan = reduced (TCEP). Blue = oxidized. On top of the graph is a schematic illustration of the elements of secondary structure observed in the NMR‐derived structure for each oxidized peptide with the α‐helices colored red and β‐strands colored yellow.
Figure 6.
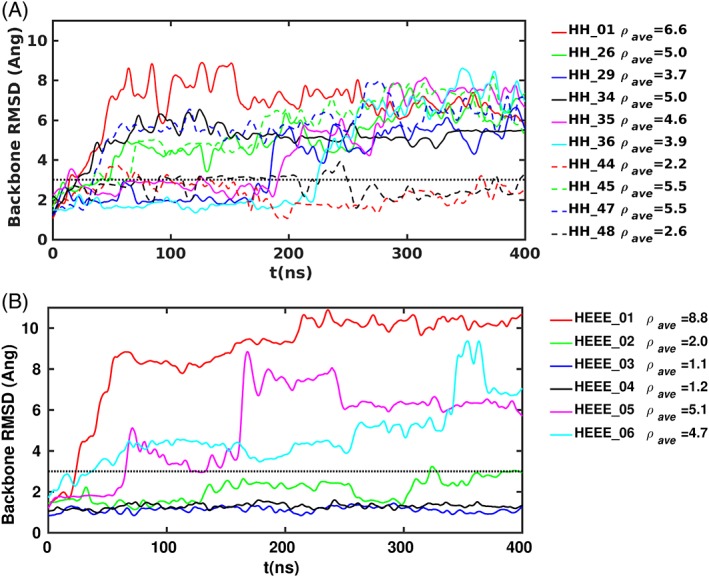
RMSD between the simulation structures and the corresponding initial structure of designs with the gHH topology (a) and designs with the gHEEE topology (b). ρ ave indicates the average RMSD over the simulation in Å and the horizontal dotted line marks 3 Å.
Thermostability of gHH_44
As previously reported, CD data indicate that gHH_44 is susceptible to chemical denaturation with guanidinium hydrochloride, but largely resistant to thermal denaturation up to 95°C.6 We confirmed the CD thermal denaturation observations for gHH_44 by NMR spectroscopy to 80°C, the temperature limit of the probe. At this temperature, the 2D [15N, 1H] HSQC spectrum for gHH_44 still contained 21 broad amide cross peaks with chemical shift dispersion in both the 1H and 15N dimensions, characteristic of a folded protein.https://paperpile.com/c/qRdOtt/8iI4Z Upon returning the sample to 20°C, the 2D [15N, 1H] HSQC spectrum for gHH_44 was indistinguishable from the pre‐heated spectrum, confirming thermal resistance.
Evaluation of designs by MD simulation
In the previous study, many designs for each backbone topology were generated, then assessed in silico, and produced and tested in vitro. 6 As part of the in silico screening, ensembles of short MD simulations were incorporated into the computational design protocol. This was to ensure that the designed structures were not significantly perturbed by local conformational relaxation at room temperature. However, even small proteins can populate relatively long‐lived metastable basins with escape times far beyond the timescale of current MD simulation. The conformational mobility in such MD simulations is reduced further by the presence of disulfide bonds or cyclization. In an attempt to improve the design protocol, we evaluated the use of long‐time MD simulations with enhanced sampling. Briefly, the MD protocol involves simulating each design for up to 400 ns using an extended canonical ensemble in which the temperature is varied adaptively within a prescribed range,22 which we chose here to be 300–450 K. The use of tempering has previously been found to accelerate sampling by 1–3 orders of magnitude.23 For details on the simulation protocol see “Materials and Methods.”
First, we evaluated the predictive ability of the simulation protocol using the previously reported designs belonging to the families gHH_{x} and gHEEE_{y}, with x in the range {01–06}, and y in the set {01, 26, 29, 34, 35, 36, 44, 45, 47, 48} (also reported under the names: HH_2.0_{y} and HEEE_2.2_{x}).6 These families contain the structurally validated designs gHH_44 and gHEEE_02. Additionally, design gHEEE_05 exhibited room temperature steady‐state CD spectra consistent with the designed topology. Designs gHEEE_01, gHEEE_06, gHH_01, gHH_26, gHH_29, gHH_34, gHH_35, gHH_36, gHH_45, and gHH_47 mainly adopted a random coil conformation at room temperature as determined by CD spectroscopy, and designs gHEEE_03, gHEEE_04, and gHH_48 were not able to be produced in sufficient quantity and/or purity for definitive assessment.6 When evaluated by MD simulation, all the designs could be clustered into two categories: one group appeared relatively stable and remained near the starting conformation, while the other group deviated from the starting conformation. These groups can be demarcated by a final RMSD from the starting model at or below 3 Å; models that deviated more than this amount were considered “unstable” in solution. By this metric, designs gHH_44, gHH_48, gHEEE_02, gHEEE_03, and gHEEE_04 were stable, while all other designs appeared to be unstable by MD. These simulations correctly predicted a stable conformation for all of the designs which successfully folded following expression in bacteria, while also correctly predicting an unstable conformation for all designs that adopted a random coil state at room temperature. The simulations also predicted that two of the three designs for which a folding assessment could not be performed would be stable in solution. It is noteworthy that some of the unstable designs unfold only after more than 100 ns of simulation, indicating that extensive sampling is important for the MD protocol. We also performed the simulations at 300 K without tempering, but stopped them after 200 ns because none of the designs deviated by more than 3.5 Å from the initial structures, underscoring the need for enhanced sampling.
MD‐guided redesign of gHH_47 and gHEEE_06
Generally, globular proteins are not soluble in the unfolded state and precipitate in aqueous solution. These de novo designed peptides present a unique opportunity to study protein folding, as they possess a hydrophobic core, and they can be expressed, purified, and remain soluble in aqueous buffer while exhibiting a random coil conformation at room temperature. Our goal was to identify structural elements that may contribute to poor stability of the designed conformation, and then to modify the peptide sequence to promote folding. For these experiments, we chose a soluble, unstable design to represent each topology: gHH_47 and gHEEE_06.
For gHH_47, the trajectory indicated that unfolding was preceded by the formation of salt bridges between residue pairs Arg/Lys and Glu from distant regions of the structure that were not present in the designed conformation [Fig. 7(A)]. In order to systematically investigate this phenomenon, we replaced each long‐chain, charged residue with a polar, non‐charged residue using ROSETTA design in order to eliminate the possibility of forming a particular salt bridge. We repeated the tempering simulations for each mutation, as well as a design where all mutations were combined [Fig. 8(A)]. While the original gHH_47 exceeded the 3 Å RMSD threshold by 40 ns, 10 different mutations induced stability below this threshold for up to 100 ns. Out of these 10 mutations, four conferred stability up to 300 ns, and one (K28Q) remained stable out to 400 ns. The design where all mutations were combined was not stable.
Figure 7.
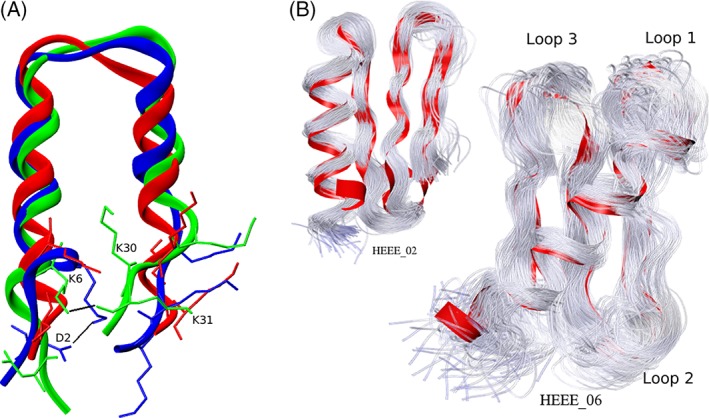
(a) Three simulation structures of the design gHH_47 are overlaid in red, green, and blue, corresponding to the simulation at 0, 30, and 40 ns, respectively, to illustrate the stabilization of two slightly unfolded structures (green and blue) by salt bridges (black lines) involving residue K6 with neighboring side chains. In the design, K6 does not make any salt bridge; at 30 ns, its side chain hydrogen‐bonds to the partially unfolded backbone of K31 on the opposing strand; at 40 ns, the K6 side chain has moved to the other side of the protein (into the page) and hydrogen bonds to the D2 side chain. (b) Illustration of conformational flexibility differences between the stable design gHEEE_02 (small panel on the left) and the unstable design gHEEE_06 (main panel). One hundred backbone traces are overlaid onto the initial simulation structure (red ribbon) spanning the time interval 0–60 ns to illustrate the particularly large flexibility of the loops in the unstable design prior to unfolding.
Figure 8.
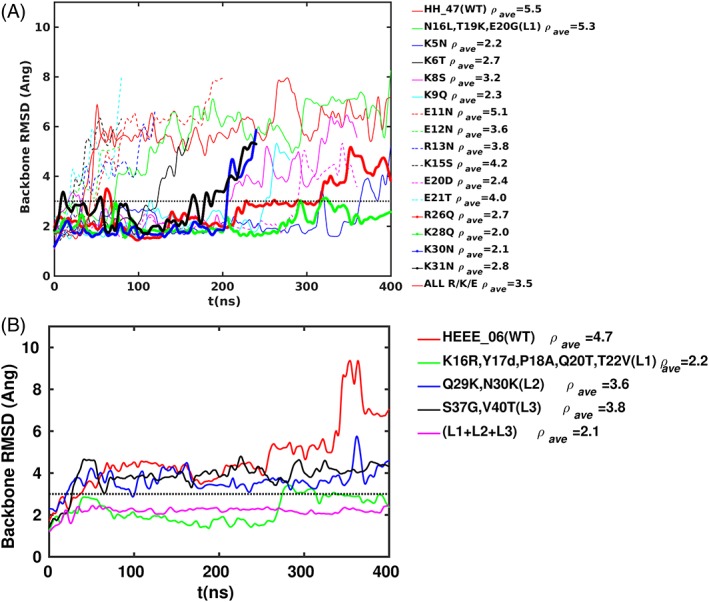
RMSD between the simulation structures and the corresponding initial structure of the mutant designs based on gHH_47 (a) and the mutant designs based on gHEEE_06 (b). ρ ave indicates the average RMSD over the simulation and the horizontal dotted line marks the stability threshold of 3 Å. The WT sequence of HH_47 is 1 ADCDKKLKK 10 VEERSKNGLT 20 EEVQQLRDKV 30 KKC, and WT sequence of gHEEE_06 is 1 SESEKMCRQ 10 CEEERKKYPT 20 QETSVRLPKQ 30 NCECRVGSTT 40 VDCDC (sites of trial mutations are underlined and colored red). In the HEEE mutant with all three loops mutated (panel B), the sequence for loop 3 (36 LDDGY) is not identical to that in the single loop 3 mutant (36 GGTGT); the sequence differs because we optimized loops 1 and 3 simultaneously, and residues in these loops interact because of their close proximity [see Fig. 7(B)].
The gHEEE_06 trajectory indicated that unfolding was preceded by large‐amplitude fluctuations in the loops connecting the secondary structure elements [Fig. 7(B)]. To stabilize the design, we trimmed each of the three loops from the peptide structure and replaced the starting sequence and conformation with a fragment obtained from the PDB. We tested the replacement of each loop individually, as well as all three loops together, using the tempering MD simulations [Fig. 8(B)]. Each loop replacement resulted in a stability enhancement, and the greatest stabilization was achieved in the design with all three loops replaced, which was the only design to pass the 3 Å RMSD threshold at 400 ns. The greatest individual contribution to stability was conferred by the replacement of the first loop in the sequence, which became one residue shorter than the starting loop. Because of the stability gain we observed with replacing loops for gHEEE_06, we also tried replacing the only loop for gHH_47 using the same protocol; however, this redesign also remained unstable [Fig. 8(A)].
Next, we sought to determine whether the improvements in stability observed by MD would translate into conformational stability in vitro. We selected the single stable redesigned construct gHH_47 with the point mutation K28Q. For gHEEE_06, we chose the most stabilizing loop replacement (loop 1), as well as the replacement of all loops (loops 1–3). These three peptides were expressed and purified as before. All peptides were soluble upon cleavage of the fusion protein, and exhibited a CD steady‐state wavelength spectrum consistent with a random coil structure at room temperature (Supporting Information Fig. S1). Although MD simulation suggested the redesigned models were more stable than the initial design, it seems this was insufficient to maintain the designed state in solution (an obvious limitation of MD based assessment of stability is the limited time scale that can be simulated, which precludes rigorous determination of the folded state population at equilibrium). Multiple destabilizing elements may be present in the peptides that fail to fold as designed, and these elements function together (additively or cooperatively) to determine solution behavior. This hypothesis is supported by a recent study of the de novo design and folding of disulfide‐free peptides where it was shown that different factors, such as buried non‐polar surface area and side chain electrostatics, contribute to design stability.24
While the ability of MD simulations to predict stability of the initial gHH and gHEEE designs is encouraging, the use of MD to guide rational redesign and optimization of unstable designs was unsuccessful. One possible cause of failure is that the MD simulations are not sufficiently long, that is, if the redesigned peptides are able to transiently populate the designed conformation, the unfolding could be too slow to observe in the MD simulations conducted here. Further improvements in MD simulation hardware, software, and sampling methodology will be needed before this question can be answered fully. More generally, follow‐up studies in which MD simulations are used in a blindly predictive fashion to design proteins with various topologies will be necessary.
Conclusion
We have developed a bacterial system for the intracellular expression and purification of computationally designed, disulfide‐rich peptides. This method is easily adaptable to production of peptides harboring specific isotopic enrichment, which we leveraged to label two de novo designed peptides for solution structure determination by NMR. The capability to isotopically label peptides for NMR is advantageous for structural characterization because it circumvents difficulties in finding conditions to generate diffraction quality crystals, which can be challenging for disulfide‐rich peptides.6 Furthermore, isotope labeling provides opportunities to characterize the disulfide‐rich peptides under a more dynamic range of solution conditions, such as the presence of heat or reductant. Thus, the experimental methods detailed in this report are broadly useful for the production and study of peptides featuring multiple disulfide bonds which form a specific pattern of connectivity.
The oxidized peptides prepared recombinantly in this study each follow a primary folding pathway that leads to the computationally designed structure. CD and NMR spectroscopy demonstrate these peptides do not predominantly occupy the designed state at biologically relevant temperatures without disulfide crosslinks; gHH_44 and gHEEE_02 are unfolded, random coils in the presence of reductant. During folding, sampling the designed conformation can result in disulfide bond formation, kinetically trapping the protein main chain. If the ROSETTA design calculations are accurate, formation of disulfide bonds between incorrect cysteine residues will result in a state of higher free energy than the designed state (which also includes intermolecular disulfide bonds). Fusing the peptides directly to disulfide bond isomerase during purification allows the peptides to escape from these metastable states, and drives them toward the designed global free energy minima. Additionally, some peptides may sample insoluble conformations during folding; the isomerase is highly soluble and significantly larger than the peptide fused to it, enabling the peptide to remain soluble throughout the folding process.
As a step toward enhancing in silico screening of designed peptides, we developed a strategy based on MD simulations to identify misfolded peptides prior to expression, and we leveraged this in an attempt to improve the stability of misfolded designs. While we were not successfully able to use the MD predictions to aid in the design of stability enhancing mutations, our simulations were able to identify unstable peptides with high reliability (in fact, all designs predicted to be unstable by MD adopted a random coil conformation in solution). This result is encouraging and suggests that MD simulations could be useful in de novo protein design. However, future studies are necessary where MD is used more systematically in a predictive manner.
Materials and Methods
Peptide expression and purification
Genes of designed disulfide‐rich peptides were cloned into the vector pCDB364 using Gibson Assembly.9 Expression vectors containing gHH_44 (pCDB368) and gHEEE_02 (pCDB367) were transformed into BL21*(DE3) E. coli (Invitrogen), and all media recipes were supplemented with 100 μg/mL kanamycin. We have made these vectors available via Addgene. Transformations were plated on MDAG‐11 agar25 and incubated ≤16 h at 37°C. Next, a single colony was subcultured into 50 mL of MDAG‐135 broth25 and incubated for ≤16 h at 37°C with shaking at 225 rpm. This culture was then diluted 1:100 to inoculate a 0.5 L culture of the expression media (15N)M(13C)G, which consists of: 0.35% [wt/vol] 13C‐D‐glucose, 25 mM Na2HPO4, 25 mM KH2PO4, 50 mM 15NH4Cl, 5 mM Na2SO4, 2 mM MgSO4, 0.2× Studier Trace Metals,25 and 1 μM thiamine. Cultures were grown at 37°C to an OD600 of approximately 0.8, and then induced by adding isopropyl β‐D‐1‐thiogalactopyranoside to 1 mM. The temperature was reduced to 20°C and cultures were incubated with shaking at 225 rpm for an additional 12–16 h. Following expression, the fusion protein was purified from soluble cellular lysate by immobilized metal affinity chromatography. Purified fusion proteins were cleaved using the site‐specific protease superTEV,https://paperpile.com/c/qRdOtt/sSkp and mature peptides were purified to homogeneity by reverse‐phase high‐performance liquid chromatography on an Agilent 1260 HPLC equipped with a C‐18 Zorbax SB‐C18 4.6 × 150 mm column. Solvent A (water + 0.1%TFA) and solvent B (acetonitrile + 0.1%TFA) were run at a flow rate of 5 mL/min using the following gradient: 0%–5% solvent B (5 min), 5%–45% solvent B (40 min).
NMR analysis and structure determination
Agilent NMR spectrometers operating at 1H resonance frequencies between 500 and 800 MHz equipped with 1H{13C,15N} probes were used to acquire NMR data for gHH_44 and gHEEE_02. Uniformly 13C,15N‐labeled peptides (1–1.5 mM) were dissolved in a buffer containing 50 mM sodium chloride and 20 mM sodium acetate at pH 4.8 (gHH_44) or 50 mM sodium phosphate at pH 6.0, 4 μM 4,4‐dimetlyl‐4‐silapentane‐1‐sulfonic acid, 0.02% sodium azide (gHEEE_02). Isotropic overall rotational correlation times of about 4 ns were inferred from averaged backbone 15N spin relaxation times (http://cns.chem.buffalo.edu/nesg.wiki) for gHH_44 and gHEEE_02.
Multidimensional NMR spectra were processed with Felix2007 (MSI) or PROSA.26 Visualization and analysis of all spectra were performed with Sparky (v3.115), XEASY,27 or CARA. Proton chemical shifts were referenced to internal DSS, while 13C and 15N chemical shifts were referenced indirectly via gyromagnetic ratios. Sequence‐specific backbone (HN, N, Cα) and C′ resonance assignments under oxidizing conditions were obtained by analyzing 3D HNCACB, CBCA(CO)NH, HNCO, and HN(CA)CO spectra for both gHH_44 and gHEEE_02. Resonance assignment of side chains was performed by using 3D 15N‐resolved [1H,1H] TOCSY and H(C‐TOCSY‐CO)NH for gHH_44, and 3D HBHA(CO)NH, aliphatic (H)CCH‐COSY, aliphatic (H)CCH‐TOCSY, and 15N/13Caliphatic/13Caromatic‐resolved NOESY (τ mix = 200 ms) for gHEEE_02. Chemical shifts, NOESY peak lists and time‐domain NMR data were deposited in the BioMagResBank with accession codes 30204 (gHH_44) and 30312 (gHEEE_02).
Samples under reducing conditions were prepared in buffers containing 2.5 mM TCEP (gHH_44) or 5 mM TCEP (gHEEE_02). Sequence‐specific backbone and 13Cβ resonance assignments were obtained by analyzing 3D HNCACB, CBCA(CO)NH, HNCO, and HN(CA)CO spectra for gHH_44 and gHEEE_02. Resonance assignment of side chains was performed by using 15N‐resolved [1H,1H] TOCSY and H(CC‐TOCSY)NH for gHH_44, and HBHA(CO)NH, aliphatic (H)C(C‐TOCSY‐CO)NH, H(C‐TOCSY‐CO)NH, aliphatic (H)CCH‐TOCSY, and 15N/13Caliphatic/13Caromatic‐resolved NOESY (τ mix = 70 ms) spectra for gHEEE_02. Chemical shifts were deposited in the BioMagResBank with accession codes 27420 (gHH_44) and 27269 (gHEEE_02).
Proton‐proton upper distance constraints for structure calculations were obtained from 3D 15N‐edited NOESY and 13C‐resolved [1H,1H] NOESY (τ mix = 90 ms) spectra for gHH_44 and from 3D 15N/13Caliphatic/13Caromatic‐resolved NOESY (τ mix = 200 ms) spectra for gHEEE_02. Slowly exchanging amides were identified for gHH_44 by lyophilizing a [U‐13C,15N]‐labeled peptide sample, re‐dissolving in D2O, and collecting a 2D [15N,1H] HSQC spectrum about 10 min after re‐dissolving the peptide. Chemical shifts were used to derive dihedral Φ and Ψ angle constraints using the program TALOS+28 for residues located in well defined regular secondary structure elements. Stereo‐specific assignments of methylene protons and isopropyl groups of valine and leucine residues were performed with the GLOMSA module of CYANA29, 30 for gHEEE_02. The 1H, 13C, and 15N chemical shift assignments and NOESY peak lists were used for iterative structure calculations using the program CYANA (v2.1 or v3.97). The final 20 conformers out of 100 with the lowest target function were further refined by restrained molecular dynamics in explicit water31 using the program CNS.32 Structure validation was accomplished using the Protein Structure Validation Software (PSVS) server 1.516 and agreement of structures and NOESY peak lists was verified using RPF scores obtained from the PSVS server.33 The structural statistics are summarized in Table 1. The atomic coordinates for all 20 of these conformers for gHH_44 and gHEEE_02 were deposited in the Protein Data Bank with PDB IDs 5TX8 and 5W9F, respectively.
Molecular dynamics simulations
Structures modeled by ROSETTA34 were energy‐minimized in vacuum using the CHARMM35 force field with restraints on the protein heavy atoms, and immersed in a cubic box of TIP3P water of sufficient size that the smallest distance between the peptide and the box boundary was 11 Å. Ions were added to maintain system neutrality and achieve an approximate salt concentration of 100 mM. The final system sizes were approximately 13,000 and 17,000 atoms for the HH and HEEE protein designs, respectively. The MD simulations were performed using GPU hardware with the program ACEMD36 and the CHARMM36 energy function. Each system was equilibrated for 2 ns with positional restraints on the heavy atoms, with a restraint constant of 1 kcal/mol/Å.2 The Berendsen barostat was used with a relaxation time of 10,000 steps to allow the simulation box to gradually resize according to the target pressure. A Langevin thermostat was used to maintain temperature at 300 K with a friction constant of 1 ps−1. The timestep was 1 fs for the first 100 ps, and 2 fs for the remainder of the equilibration. The nonbonded cutoff was 11 Å. After equilibration, each system was simulated using adaptive tempering (AD),22 a method that samples molecular configurations which form an extended canonical (NVT) ensemble. The AD temperature is treated as a dynamic variable that evolves stochastically according to a prescribed temperature distribution.22 The AD method can be considered as a continuous version of the well‐known simulated tempering method.37, 38 In the AD simulations, the temperature was allowed to vary between 300 and 450 K according to a linear distribution (i.e., biased toward higher temperatures to accelerate sampling at high temperatures), as suggested by the original authors.22 Temperature was advanced using a nondimensional time step of Δtγ = 0.00002 (where γ is a friction constant with units of inverse time). The temperature distribution was represented on a discretized grid of 1000 points. As suggested by Zhang and Ma,22 the energy interpolation width was 50 grid points, and the damping constant for energy averaging was 0.1. To accelerate the simulations, the nonbonded cutoff was set to 9 Å, the switching function was made nonzero at 7.5 Å, long‐range electrostatics were evaluated at every other simulation step, and hydrogen repartitioning was used, increasing hydrogen masses to 4 a.m.u., which allowed the use of a 4 fs time step. For the AD simulations, the barostat was turned off to prevent the systems from boiling at high temperatures. Covalent bonds to hydrogens were constrained using SHAKE22, 39 in all simulations that used a time step greater than 1 fs. For the validation simulations (see Results), each AD simulation was performed for 400 ns. For the rescue mutation simulations, each simulation was performed in 10 stages of 40 ns. After each stage, the simulation was continued up to a maximum time of 400 ns only if the RMSD from the initial structure remained below 4.5 Å.
Circular dichroism spectroscopy
The CD spectroscopy was performed using a Jasco J‐1500 CD spectrometer. Peptides samples were prepared in 10 mM sodium phosphate buffer (pH 7.0) with steady‐state wavelength scans from 190 to 260 nm recorded at 20°C for each sample using a quartz cuvette with a 1 mm light path.
Statement of importance
Genetically encodable disulfide‐rich peptides are a promising class of biologic drug scaffolds that possess many of the beneficial properties of traditional small‐molecule therapeutics. We took steps toward understanding the sequence‐structure relationship of these peptides through solution structure determination and molecular dynamics simulation of peptides that were computationally designed de novo.
Supporting information
Supplementary Figure 1. Steady state circular dichroism spectra for re‐designed peptides reveals random‐coil structural signatures.
Acknowledgements
GWB and PJM were supported by the National Institute of Allergy and Infectious Diseases under Federal Contract No. HHSN2722001200025C and HHSN272201700059C. Part of this research was performed at the W.R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility located at Pacific Northwest National Laboratory (PNNL) and sponsored by U.S. Department of Energy's Office of Biological and Environmental Research (BER) program. Battelle operates PNNL for the U.S. Department of Energy. Financial support to VO and MK was provided from NIH grant No. 5R03AI111416 and from the CHARMM Development Project at Harvard. SVSRKP and TS were supported by NSF grant MCB‐1615570. CDB was supported by NIH grant T32‐H600035. DB and CDB are inventors on a patent application assigned to the University of Washington for the computational design of peptides.
References
- 1. Leader B, Benjamin L, Baca QJ, Golan DE (2008) Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov 7:21–39. [DOI] [PubMed] [Google Scholar]
- 2. Kolmar H, Harald K (2011) Natural and engineered cystine knot miniproteins for diagnostic and therapeutic applications. Curr Pharm Des 17:4329–4336. [DOI] [PubMed] [Google Scholar]
- 3. Combelles C, Gracy J, Heitz A, Craik DJ, Chiche L (2008) Structure and folding of disulfide‐rich miniproteins: insights from molecular dynamics simulations and MM‐PBSA free energy calculations. Proteins 73:87–103. [DOI] [PubMed] [Google Scholar]
- 4. Correnti CE, Gewe MM, Mehlin C, Bandaranayake AD, Johnsen WA, Rupert PB, Brusniak M‐Y, Clarke M, Burke SE, De Van Der Schueren W, Pilat K, Turnbaugh SM, May D, Watson A, Chan MK, Bahl CD, Olson JM, Strong RK (2018) Screening, large‐scale production and structure‐based classification of cystine‐dense peptides. Nat Struct Mol Biol 25:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chevalier A, Silva D‐A, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam K‐H, Yao G, Bahl CD, Miyashita S‐I, Goreshnik I, Fuller JT, Koday MT, Jenkins CM, Colvin T, Carter L, Bohn A, Bryan CM, Fernandez‐Velasco DA, Stewart L, Dong M, Huang X, Jin R, Wilson IA, Fuller DH, Baker D (2017) Massively parallel de novo protein design targeted therapeutics. Nature 550:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SVSRK, Kaas Q, Eletsky A, Huang P‐S, Johnsen WA, Greisen PJ, Rocklin GJ, Song Y, Linsky TW, Watkins A, Rettie SA, Xu X, Carter LP, Bonneau R, Olson JM, Coutsias E, Correnti CE, Szyperski T, Craik DJ, Baker D (2016) Accurate de novo design of hyperstable constrained peptides. Nature 538:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qian Z‐G, Xia X‐X, Choi JH, Lee SY (2008) Proteome‐based identification of fusion partner for high‐level extracellular production of recombinant proteins in Escherichia coli . Biotechnol Bioeng 101:587–601. [DOI] [PubMed] [Google Scholar]
- 8. Nozach H, Fruchart‐Gaillard C, Fenaille F, Beau F, Pereira Ramos OH, Douzi B, Saez NJ, Moutiez M, Servent D, Gondry M, Thai R, Cuniasse P, Vincentelli R, Dive V (2013) High throughput screening identifies disulfide isomerase DsbC as a very efficient partner for recombinant expression of small disulfide‐rich proteins in E. coli . Microb Cell Fact 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gibson DG, Young L, Chuang R‐Y, Venter JC, Hutchison CA 3rd, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Meth 6:343–345. [DOI] [PubMed] [Google Scholar]
- 10. Waugh DS (2011) An overview of enzymatic reagents for the removal of affinity tags. Protein Expr Purif 80:283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yee A, Chang X, Pineda‐Lucena A, Wu B, Semesi A, Le B, Ramelot T, Lee GM, Bhattacharyya S, Gutierrez P, Denisov A, Lee C‐H, Cort JR, Kozlov G, Liao J, Finak G, Chen L, Wishart D, Lee W, McIntosh LP, Gehring K, Kennedy MA, Edwards AM, Arrowsmith CH (2002) An NMR approach to structural proteomics. Proc Natl Acad Sci U S A 99:1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kelly SM, Jess TJ, Price NC (2005) How to study proteins by circular dichroism. Biochim Biophys Acta 1751:119–139. [DOI] [PubMed] [Google Scholar]
- 13. Buchko GW, Niemann G, Baker ES, Belov ME, Smith RD, Heffron F, Adkins JN, McDermott JE (2010) A multi‐pronged search for a common structural motif in the secretion signal of Salmonella enterica serovar Typhimurium type III effector proteins. Mol Biosyst 6:2448–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dyson HJ, Wright PE (1991) Defining solution conformations of small linear peptides. Annu Rev Biophys Biophys Chem 20:519–538. [DOI] [PubMed] [Google Scholar]
- 15. Wüthrich K. NMR of Proteins and Nucleic Acids. New York, NY: Wiley‐Interscience, 1986. [Google Scholar]
- 16. Bhattacharya A, Tejero R, Montelione GT (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66:778–795. [DOI] [PubMed] [Google Scholar]
- 17. Sharma D, Rajarathnam K (2000) 13C NMR chemical shifts can predict disulfide bond formation. J Biomol NMR 18:165–171. [DOI] [PubMed] [Google Scholar]
- 18. Kornhaber GJ, Snyder D, Moseley HNB, Montelione GT (2006) Identification of zinc‐ligated cysteine residues based on 13Cα and 13Cβ chemical shift data. J Biomol NMR 34:259–269. [DOI] [PubMed] [Google Scholar]
- 19. Woody RW (1995) Circular dichroism. Methods Enzymol 246:34–71. [DOI] [PubMed] [Google Scholar]
- 20. Buchko GW, Hewitt SN, Napuli AJ, Van Voorhis WC, Myler PJ (2009) Backbone and side chain (1)H, (13)C, and (15)N NMR assignments for the organic hydroperoxide resistance protein (Ohr) from Burkholderia pseudomallei . Biomol NMR Assign 3:163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Szilagyi L (1995) Chemical shifts in proteins come of age. Prog Nucl Magn Reson Spectrosc 27:325–443. [Google Scholar]
- 22. Zhang C, Ma J (2010) Enhanced sampling and applications in protein folding in explicit solvent. J Chem Phys 132:244101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pan AC, Weinreich TM, Piana S, Shaw DE (2016) Demonstrating an order‐of‐magnitude sampling enhancement in molecular dynamics simulations of complex protein systems. J Chem Theory Comput 12:1360–1367. [DOI] [PubMed] [Google Scholar]
- 24. Rocklin GJ, Chidyausiku TM, Goreshnik I, Ford A, Houliston S, Lemak A, Carter L, Ravichandran R, Mulligan VK, Chevalier A, Arrowsmith CH, Baker D (2017) Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 357:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Studier FW (2005) Protein production by auto‐induction in high‐density shaking cultures. Protein Expr Purif 41:207–234. [DOI] [PubMed] [Google Scholar]
- 26. Güntert P, Dötsch V, Wider G, Wüthrich K (1992) Processing of multi‐dimensional NMR data with the new software PROSA. J Biomol NMR 2:619–629. [Google Scholar]
- 27. Bartels C, Xia TH, Billeter M, Güntert P, Wüthrich K (1995) The program XEASY for computer‐supported NMR spectral analysis of biological macromolecules. J Biomol NMR 6:1–10. [DOI] [PubMed] [Google Scholar]
- 28. Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302. [DOI] [PubMed] [Google Scholar]
- 29. Güntert P, Mumenthaler C, Wüthrich K (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273:283–298. [DOI] [PubMed] [Google Scholar]
- 30. Herrmann T, Güntert P, Wüthrich K (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319:209–227. [DOI] [PubMed] [Google Scholar]
- 31. Linge JP, Williams MA, Spronk CAEM, Alexandre MJ, Michael N (2003) Refinement of protein structures in explicit solvent. Proteins 50:496–506. [DOI] [PubMed] [Google Scholar]
- 32. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse‐Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Cryst D 54:905–921. [DOI] [PubMed] [Google Scholar]
- 33. Huang YJ, Powers R, Montelione GT (2005) Protein NMR recall, precision, and F‐measure scores (RPF scores): structure quality assessment measures based on information retrieval statistics. J Am Chem Soc 127:1665–1674. [DOI] [PubMed] [Google Scholar]
- 34. Leaver‐Fay A, Tyka M, Lewis SM, Lange AF, Thompson J, Jacak R, Kaufmann KW, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban Y‐EA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P (2011) ROSETTA3: An object‐oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brooks BR, Brooks CL III, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaeffer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M (2009) CHARMM: The biomolecular simulation program. J Comput Chem 30:1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harvey MJ, Giupponi G, De Fabritiis G (2009) ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput 5:1632–1639. [DOI] [PubMed] [Google Scholar]
- 37. Marinari E, Parisi G (1992) Simulated tempering: a new Monte Carlo scheme. Europhys Lett 19:451–458. [Google Scholar]
- 38. Lyubartsev AP, Martsinovski AA, Shevkunov SV, Vorontsov‐Velyaminov PN (1992) New approach to Monte Carlo calculation of the free energy: Method of expanded ensembles. J Chem Phys 96:1776–1783. [Google Scholar]
- 39. Ryckaert J‐P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n‐alkanes. J Comput Phys 23:327–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Steady state circular dichroism spectra for re‐designed peptides reveals random‐coil structural signatures.