

Abstract
In biology, polymorphism is a well-known phenomenon by which a discrete biomacromolecule can adopt multiple specific conformations in response to its environment. The controlled incorporation of polymorphism into noncovalent aqueous assemblies of synthetic small molecules is an important step toward the development of bioinspired responsive materials. Herein, we report on a family of carboxylic acid functionalized water-soluble benzene-1,3,5-tricarboxamides (BTAs) that self-assemble in water to form one-dimensional fibers, membranes, and hollow nanotubes. Interestingly, one of the BTAs with the optimized position of the carboxylic group in the hydrophobic domain yields nanotubes that undergo reversible temperature-dependent dynamic reorganizations. SAXS and Cryo-TEM data show the formation of elongated, well-ordered nanotubes at elevated temperatures. At these temperatures, increased dynamics, as measured by hydrogen–deuterium exchange, provide enough flexibility to the system to form well-defined nanotube structures with apparently defect-free tube walls. Without this flexibility, the assemblies are frozen into a variety of structures that are very similar at the supramolecular level, but less defined at the mesoscopic level.
Introduction
Inspired by supramolecular assemblies in nature, noncovalent synthesis of complex architectures from discrete small molecules in aqueous solution offers great potential for the development of advanced materials.1,2 By carefully balancing solubility and noncovalent attractive forces (hydrophobic effect, H-bonding, π–π stacking, etc.) a variety of small molecule amphiphiles (peptide amphiphiles,3,4 steroidal amphiphiles,5,6 extended π-conjugated systems,7,8 discotics,9,10 lipids,11,12 etc.) have been assembled into a diverse array of supramolecular structures in water, including fibrous aggregates,10,13 membranes,14,15 and tubes.12,16 The dynamic nature of such noncovalent assemblies in aqueous solution are ideal for the preparation of responsive materials for applications such as tissue regeneration,17,18 drug delivery,19,20 biosensing,21,22 and signal processing.23,24 However, to achieve control over the morphologies of dynamic assemblies, a thorough understanding of molecular design principles on the impact of pathway complexity in water is necessary.25 The question of how to balance the on–off rates of the assembly with the robustness of the morphology of the assembly is both intriguing and open. Only a perfect molecular fit will yield defect-free architectures; whereas a fit that is just off ideal will require a certain degree of dynamics to allow the molecules to form well-defined architectures. Where nature has optimized this trade-off at 37 °C, artificial systems in water will have a molecule-dependent optimal temperature. Hence, different structures often arise from the same molecules in solution due to this pathway-dependent process.
Numerous proteins in nature can assemble into different types of supramolecular polymers, in some cases through the change of chemical structures of the protein monomers, but in many more cases by transforming into a different packing morphology while retaining identical chemical structures of monomers.26,27 For example, microtubules in cilia and flagella can respond to ATP to take different organization and shapes;28 tropomyosin can form two-dimensional crystals with either a square or hexagonal lattice by varying the charge pattern on the surface;29 tubular polymers from self-association of tobacco mosaic virus capsid protein transform between different forms when pH or ionic strength changes.30 Such polymorphism from the same type of monomers is important for the protein supramolecular structures to remain adaptable to the environmental conditions and is a remarkable phenomenon.
Polymorphism of protein polymers originates either from the different conformation of states of protein subunits or a change in the state of interaction sites.27 For instance, simply changing the charge states of ionizable groups in a monomer can alter the interaction energy between monomers and induce a change in the mode of interactions.31 Particularly, regulating the charge states of side chain carboxylic acid groups of amino acids is prevalent in many plant viruses and other biological supramolecular structures. In one example, carboxylate groups on the surface of capsid proteins of tobacco mosaic virus (TMV) particles, when in close proximity, can bind protons with unusually high affinity and form carboxyl-carboxylate pairs.32,33 In another example, strong binding of the proton can be realized by burying of the carboxyl group in a low dielectric constant environment of the protein core.34 Moreover, the pairwise interaction of carboxylic acids, via hydrogen bonds, in a hydrophobic local environment is a well-known motif in supramolecular chemistry.35−37 Polymorphism of synthetic assemblies in aqueous solution is similarly intriguing and may have profound implications on the development of stimuli-responsive materials. Many self-assembled polymeric and small molecule aggregates are known to display morphological transitions in response to external stimuli,38,39 such as pH,40,41 temperature,42,43 light, molecular cues,40,44,45 etc. Furthermore, polymorphism has been studied in out-of-equilibrium synthetic supramolecular assemblies.46,47 However, the general principles on how to design the synthetic subunits to introduce polymorphism into a given supramolecular system remain elusive.
Herein, inspired by the intricate role of the carboxyl group in polymorphism of protein complexes, we introduce carboxylic acid groups at the end of solubilizing chains of discotic molecules derived from the benzene-1,3,5-tricarboxamide (BTA) motif and investigate their regulation effect on the self-association behavior of the BTA derivatives. BTA derivatives decorated with aliphatic spacers covalently linked to tetra(ethylene glycol) are known to stack into supramolecular polymers of micrometers in length and a few nanometers in diameter in aqueous solution.48−50 Interestingly, simply replacing the tetra(ethylene glycol) with a carboxylic acid in one of the three solubilizing chains allows for the preparation of new supramolecular structures assembled from BTA derivatives, including ribbons, membranes, and hollow tubes of many micrometers in length. The formation of different structures results from the system’s tendency to maximize the hydrophobic interactions between the aliphatic spacers, which subsequently provide a local environment for the pairwise interaction of carboxyl groups from the neighboring BTAs to form hydrogen bonds. The different morphology of the supramolecular structures can be modulated by varying the length of the aliphatic spacer or, for specific BTA derivatives, simply by changing the solution conditions, such as temperature. The dynamic stability and molecular interactions of BTAs in different supramolecular structures are distinct from each other, as indicated by the spectroscopic experiments and hydrogen–deuterium exchange mass spectrometry.
Results and Discussion
Molecular Design of Asymmetric, Acid-Functionalized BTAs
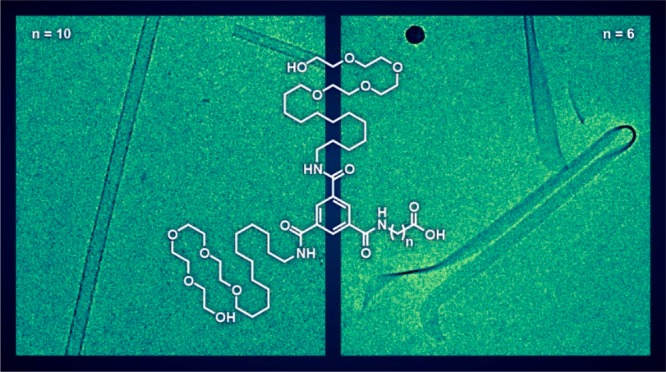
For this study, a series of four BTAs were designed and prepared; each containing a single carboxylic acid (Figure 1A, Scheme S1). All BTAs contain two identical arms consisting of 12-methylene units connected to a tetra(ethylene glycol). As previously reported, the methylene segment provides hydrophobic shielding for 3-fold hydrogen bonding in the BTA core while the tetra(ethylene glycol) provides water-compatibility.48 BTAs 1, 2, and 3 contain a third arm consisting of an alkyl spacer of 1, 6, or 10 methylene units, respectively, terminated with a carboxylic acid. This architecture, with the carboxyl groups at the end of a hydrophobic spacer, was chosen to increase the likelihood of acid–acid interactions in a hydrophobic environment when assembled in water. As a control, the third arm of BTA 4 contains a carboxylic acid at the end of the tetra(ethylene glycol). Placing the carboxylic acid at the periphery of a water-compatibilizing oligo(ethylene glycol) should result in typical one-dimensional supramolecular polymers decorated with carboxylic acids. Details on the synthesis, purification, and molecular characterization can be found in the SI.
Figure 1.

(A) Chemical structure of BTA derivatives 1–4. (B) Cartoon representation of an acid functionalized BTA. (C) Hypothesized packing morphology of the acid functionalized BTAs 2 and 3.
Aqueous Self-Assembly of BTAs
Solutions of BTA 1–4 were prepared in ultrapure water by subjecting the mixtures to successive cycles of heating and cooling. No pH adjustments were made to the solutions. BTA 1 proved to be insoluble in water at concentrations ranging from 5 mM to 50 μM; the resulting visible aggregates were not analyzed further. BTAs 2 and 3 formed opaque solutions at concentrations ranging from 6 mM to 50 μM, which were stable at room temperature over the course of weeks, while BTA 4 formed clear colorless solutions. At high concentration (e.g., 0.60 wt %), solutions of BTA 4 were noticeably viscous; this is presumably due to the presence of chain entanglements between the supramolecular polymers assembled from BTA 4 and is in accordance with previous reports on oligo(ethylene glycol) decorated BTAs. However, solutions of BTA 2 and 3 at the same concentration were considerably less viscous and appeared as free-flowing opaque liquids, suggesting the absence of significant chain entanglement from the assembled supramolecular structures. By comparing the appearance and viscosity of the solutions, a major structural difference between the assemblies of BTA 2 and 3 and BTA 4 is evident. Interestingly, a difference in the acidity of the carboxylic acids was observed between BTA 2 and 3 and BTA 4. The pH of 100 μM solutions of BTAs 2 and 3 was slightly basic (pH 7.1–7.4), whereas the pH of a 100 μM BTA 4 solution was slightly acidic (pH 6.0–6.5).
Imaging Self-Assembled BTA Architectures in Water
PEGylated BTA assemblies readily sequester small hydrophobic molecules while retaining their self-assembled structures.48 This allows for visualization of BTA assemblies by staining with Nile Red, a solvatochromic dye, for examination by fluorescence microscopy (Figure S1) and fluorescence spectroscopy (Figure S2). Fluorescence microscopy of BTAs 2, 3, and 4 coassembled with Nile Red by annealing at high temperatures in solution reveal micrometer-sized assemblies. Although the resolution limits of this technique are unsuitable for the determination of precise morphologies, large micrometer-sized aggregates are readily observed for BTA 2 (Figure S1A, Video S1). Fluorescence microscopy of BTA 3 coassembled with Nile Red revealed high aspect ratio supramolecular structures that are several micrometers in length (Figure S1B, Video S2). As expected, BTA 4 coassembled with Nile Red yielded micrometer long thin fibers as previously reported for water-soluble BTAs of similar architecture (Figure S1C, Video S3). Cryogenic transmission electron microscopy (cryo-TEM) was employed to determine the self-assembled structure of BTAs 2, 3, and 4. To date, all previously reported PEGylated BTAs containing 11 to 13 methylene units in the aliphatic spacer have produced thin (∼5 nm) multimicrometer long fibers.48−50 As expected, this was also the case for BTA 4, which formed micrometers long fibers with a width of ∼5 nm (Video S3). In contrast, BTA 2 assembles into a diverse assortment of membranes and ribbons (Figure 2A–C), presumably due to the formation of bilayers stabilized by hydrophobically shielded acid–acid interactions. Cryo-TEM of aqueous solutions of BTA 3 reveals the formation of hollow tubes measuring ∼32 nm in width and ranging from hundreds of nanometers to several micrometers in length, with tube walls measuring ∼4 nm (Figure 2D–F). There also exist some ribbons and small membranes (Figure 2E). Furthermore, tubes in various states of assembly from membranes (Figure 2E,F) are found indicating that BTA 3 may share a similar packing morphology with the membrane forming BTA 2. As the tubular BTA 3 supramolecular structures are particularly interesting, further efforts were taken to characterize their morphology in solution.
Figure 2.

Distinct supramolecular structures assembled from 2 (A–C, c = 0.90 mM) and 3 (D–F, c = 0.95 mM), characterized by cryo-TEM. 100 nm scale bar for A, B, D, and F. 200 nm scale bar for C and E.
Structural Characterization by Small-Angle X-ray Scattering in Solution
The structure of the tubes in solution was further
analyzed by in situ small-angle X-ray scattering
(SAXS) using synchrotron X-rays at the ESRF (beamline BM29). Figure 3 shows the scattering
intensity profiles, I(q) as a function
of scattering vector q, collected from BTA 3 samples with concentrations of 0.75, 1.5, 3, and 6 mg/mL
(0.67, 1.35, 2.70, 5.39 mM), respectively, in water. The scattering
vector is defined as 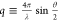 where θ is the scattering angle and
λ is the wavelength of the X-rays. Scattering patterns obtained
from the four samples are nearly identical, with the intensity difference
due to the sample concentration. The scattering profiles, I(q), are fitted by a combination of a
hollow cylinder model and a rectangular parallelepiped model to represent
the coexistence of the hollow tubes and the unclosed small membranes
in solution (please see the Supporting Information for the equations used in the model analysis). The simultaneous
fit of the scattering profiles in Figure 3 suggests that the tubular assemblies from
BTA 3 have a width of ∼36 nm, a shell thickness
of ∼5 nm, and an average length larger than 110 nm. The membranes
have a thickness of ∼5 nm, a width of at least ∼25
nm, and an average length of larger than 60 nm. The volume fraction
of the tubular supramolecular structures in the mixture of tubes and
membranes is ∼93% at a concentration of 0.75 mg/mL BTA 3. The volume fraction of membranes increases at higher concentrations
of BTA 3, reaching ∼41% at a concentration of
6 mg/mL. However, centrifugation steps applied before the SAXS measurements
in solution may have removed large supramolecular structures and altered
the proportion of the tube and membrane structures. In general, the
structural information obtained from the in situ SAXS
experiments agrees well with supramolecular structures observed by
cryo-TEM.
where θ is the scattering angle and
λ is the wavelength of the X-rays. Scattering patterns obtained
from the four samples are nearly identical, with the intensity difference
due to the sample concentration. The scattering profiles, I(q), are fitted by a combination of a
hollow cylinder model and a rectangular parallelepiped model to represent
the coexistence of the hollow tubes and the unclosed small membranes
in solution (please see the Supporting Information for the equations used in the model analysis). The simultaneous
fit of the scattering profiles in Figure 3 suggests that the tubular assemblies from
BTA 3 have a width of ∼36 nm, a shell thickness
of ∼5 nm, and an average length larger than 110 nm. The membranes
have a thickness of ∼5 nm, a width of at least ∼25
nm, and an average length of larger than 60 nm. The volume fraction
of the tubular supramolecular structures in the mixture of tubes and
membranes is ∼93% at a concentration of 0.75 mg/mL BTA 3. The volume fraction of membranes increases at higher concentrations
of BTA 3, reaching ∼41% at a concentration of
6 mg/mL. However, centrifugation steps applied before the SAXS measurements
in solution may have removed large supramolecular structures and altered
the proportion of the tube and membrane structures. In general, the
structural information obtained from the in situ SAXS
experiments agrees well with supramolecular structures observed by
cryo-TEM.
Figure 3.
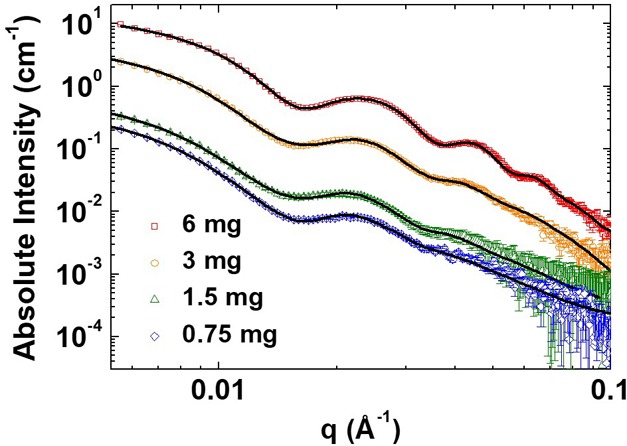
In situ small-angle X-ray scattering of BTA 3. Open symbols represent the experimental data where squares, circles, triangles, and diamonds represent 6, 3, 1.5, and 0.75 mg/mL BTA 3. The solid black lines stand for the best fit results.
Apparently, the formation of two-dimensional supramolecular polymers in BTA 2 and 3 rather than the typical 1D fibrous assemblies results from the collective effects of the now C2 symmetric subunits, a shorter aliphatic spacer in the arm with the carboxylic acid in comparison to that in the other arms, and the need to effectively suppress the ionization of carboxyl groups in the assemblies. While the structures may look very different, a tubular assembly is a two-dimensional polymer that possesses curvature in only one direction. Tube formation occurs when the gain of free energy from merging the edge of a surface lattice overcomes the excess free energy due to distortion. The emergence of some tubular structures in BTA 3, which only differs from BTA 2 by 4 methylene units in the spacer, indicates that the arrangement and strengths of interactions between the subunits are considerably different in the supramolecular polymers. Therefore, we first examine whether the 3-fold intermolecular hydrogen bonding between BTA core amides still contributes to the formation of supramolecular structures in BTAs 2 and 3, as in the 1D fibers assembled from conventional PEGylated BTAs with C3 symmetry.
Investigating BTA H-bonding interactions via FT-IR
The assemblies formed by BTAs 2 and 3 in solution were probed via FT-IR to determine the presence of intermolecular hydrogen bonds between aligned BTAs in the membranes and tubes. When molecularly dissolved in deuterated methanol, both BTA 2 (Figure 4A) and BTA 3 (Figure 4B) display strong amide I vibrations at 1649 cm–1.49 In deuterated water, both BTA 2 (Figure 4A) and BTA 3 (Figure 4B) amide I vibrations are shifted to 1627 and 1628 cm–1, respectively. The observed shift of the amide I vibration to lower wavelengths indicates intermolecular hydrogen bonding between BTA core amides. Due to the presence of intermolecular hydrogen bonding, it is likely that the stacking of BTA cores is retained in the acid modified BTAs 2 and 3, at least to some extent, as schematically shown in Figure 1C. However, near the amide I vibration for BTA 2 at 1628 cm–1, a pronounced shoulder that resembles the amide I in deuterated methanol is present, indicating that a substantial population of BTA 2 is not participating or is only temporarily participating in 3-fold hydrogen bonding with neighboring BTAs. These “defects” in the stacking of BTA 2 should give rise to weaker and liquid-like associations of BTA 2 in the two-dimensional membrane and explain why large distortion or strains were not observed under the cryo-TEM. In contrast, the amide I vibration of BTA 3 in deuterium oxide is shifted to 1627 cm–1, with no shoulder corresponding to molecularly dissolved BTAs. This suggests that with four additional methylene units in the aliphatic spacer, the gain of the free energy from closing a membrane into a tube and from the formation of a continuous network of intermolecularly hydrogen bonded BTA cores is able to compensate the distortion energy arising from the stacking of BTAs in one direction. Furthermore, BTA 3 displays a broadened carboxylic acid carbonyl vibration at 1728 cm–1 and a new vibration at 1655 cm–1. The vibration at 1655 cm–1 is consistent with carboxyl groups either in a hydrophobic environment51 or involved in an acid–acid dimer.52 This result supports our hypothesis that the pairwise interaction of carboxyl groups can be realized in a local hydrophobic environment shielded from the aqueous solution (Figure 1C). With the 4 additional methylene units in the aliphatic spacer and presumably more ordered packing, BTA 3 is also superior to BTA 2 in hydrophobically burying carboxyl groups. Furthermore, we expect that this subtle difference in packing morphology and ordering will lead to different solvent accessibility and distinct monomer exchange dynamics of the supramolecular polymers. For example, the 3-fold intermolecular hydrogen bonding present in BTA 3 tubular aggregates should be far less labile than those in BTA 2 membranes. Microscopy and FT-IR together provide a static understanding of the BTA 2 and 3 assemblies, which indicates that BTA cores are aligned via triple hydrogen bonds within the membrane and tubular assemblies. With this knowledge in hand, it is essential to probe how monomer exchange dynamics contribute to the formation of complex BTA architectures, such as membranes and tubes.
Figure 4.

FT-IR spectra of BTA 2 and 3 in solution. (A) FT-IR spectra of BTA 2 (c = 20 mg mL–1) in deuterated CD3OD (dashed lines) and D2O (solid lines). (B) FT-IR spectra of BTA 3 (c = 20 mg mL–1) in deuterated CD3OD (dashed lines) and D2O (solid lines).
Probing BTA Dynamics by Hydrogen–Deuterium Exchange Mass Spectrometry
We recently introduced hydrogen–deuterium exchange mass spectrometry (HDX-MS) for synthetic aqueous supramolecular polymers to investigate monomer exchange dynamics.50 The advantage of this technique is that it does not require labeling of monomers with a probe, such as a fluorophore or spin label, which can alter monomer exchange behavior. Specifically, HDX-MS analysis of 1D water-soluble BTA supramolecular polymers revealed structural diversity within fibers, consisting of fast and slow exchanging domains corresponding to disordered and ordered segments of the supramolecular polymer, respectively. Concentrated solutions of BTA 2 and 3 (500 μM) were diluted by a factor of 100 into deuterium oxide and the exchange of all labile hydrogen atoms (−OH, −COOH, and −NH) to deuterium was monitored in time via ESI-MS (Figure 5A). H/D exchange occurs instantaneously when labile hydrogen atoms are not involved in hydrogen bonding and are solvent exposed, for example, when water penetrates the aggregate or when a BTA monomer exits the assembly. Strong hydrogen bonding or hydrophobic shielding, in contrast, slows down H/D exchange. Within minutes after the dilution step, all of the labile hydrogen atoms of BTA 2 were exchanged, indicating that the monomers within the assembly interact very weakly with one another or that all of the labile hydrogen atoms are continuously solvent exposed (Figure S5). This result is in agreement with the more “disordered”, liquid-like packing of BTA 2 in the two-dimensional membrane structures as determined by FT-IR. The H/D exchange profile is drastically different for BTA 3 (Figure 5B), which exchanges far slower than BTA 2. Three minutes after dilution, ∼82% BTA 3 remains as the 3 times deuterated species (BTA 3-3D). Upon dilution, exchange occurs at the solvent exposed alcohols and at the carboxylic acids. H/D exchange of the carboxylic acids to the deuterated form is likely due to rapid proton transfer in the network of hydrogen bonded carboxyl groups. The three remaining hydrogens of BTA 3-3D likely correspond to the BTA amides, which are deeply shielded from water due to hydrophobic collapse of the alkyl spacers. Furthermore, the initial ∼18% “burst” H/D exchange of BTA 3 to the fully deuterated species is likely due to the heterogeneity of the BTA 3 assemblies, with immediate exchange occurring in the non-tube or -membrane structures present in solution. H/D exchange of BTA 3-3D to the fully deuterated species was monitored for an additional 100 h at which point ∼50% of the BTA 3-3D remained. Comparing the H/D exchange behavior of BTA 2 and 3 indicates that the differences in macroscopic morphology are due to an additional increase in hydrophobicity provided by four additional methylene units, which results in a considerably different in-plane interaction and molecular ordering in the two-dimensional supramolecular polymers. In self-assembled systems, directing pathway complexity and dynamics at the molecular scale are critical to obtaining desired mesoscale morphologies. As revealed by H/D exchange experiments, monomer exchange or interaction with water can be drastically increased via subjecting solutions of BTA 3 to elevated temperatures. After a 100-fold dilution of BTA 3 into deuterium oxide, the solution was incubated at 40 °C for 1 h at which point ∼10% of BTA 3-3D remained and ∼90% of the BTA 3 was fully deuterated (Figure 5B). The same HDX-MS experiment conducted at 60 °C resulted in a complete exchange to the fully deuterated species within 1 h. At this point, it is necessary to investigate polymorphism in self-assembled acid functionalized BTAs in conditions where monomer exchange dynamics are accelerated.
Figure 5.

(A) Chemical structures of BTA 3 and its deuterated analogues as shown in a reaction scheme depicting the H/D exchange experiment. (B) Kinetic exchange experiment revealing the conversion of BTA 3-3D into BTA 3-6D at 20 and 40 °C. (C) ESI-MS spectra of the BTA 3 double sodium adducts showing the isotopic distribution at 3 min and 96 h after D2O dilution at 20 °C.
Temperature-Induced Polymorphism of BTA 3
The success in generating different morphologies by designing BTAs with varying strengths of hydrophobic interaction in the assembly encouraged us to explore the polymorphism from the same monomers. BTA 3 is the obvious candidate as its supramolecular polymers at room temperature show different stages of membrane and tube formation. Temperature-dependent UV–vis experiments revealed a thermal transition through an isosbestic point at 198 nm upon heating of BTA 3 in aqueous solution (Figure 6A). From 5 to 25 °C, BTA 3 exhibits a UV–vis profile with a maximum at 194 nm and shoulders at 210 and 226 nm. Upon heating to 45 °C, the maximum at 194 nm disappears, and the shoulders become more pronounced resulting in a maximum at 210 nm with a shoulder remaining at 226 nm. A continued increase in intensity of the maximum and the shoulder is observed upon heating to 65 °C. This transition is reversible upon cooling and can be reproduced with the same sample at least several times. Interestingly, at high temperatures the UV–vis profile of BTA 3 closely resembles the UV–vis profile of the fiber forming BTA with three PEGylated arms in water at 20 °C previously reported by our group.48 This may indicate that at high temperatures the BTA 3 chromophores are aligned in similar fashion as helically stacked BTAs in aqueous 1D assemblies; the tubes consist of helically winding strands. This suggests that although the stacking order between successive BTA 3 molecules improves at the higher temperature, the overall packing of the strands may become more flexible and more accessible to solvent exchange, as we will discuss later. Together, the UV–vis and HDX-MS data indicate that BTA 3 undergoes thermal reordering via a dynamic process. Temperature dependent UV–vis and HDX-MS experiments did not reveal the same phenomenon for BTA 2.
Figure 6.
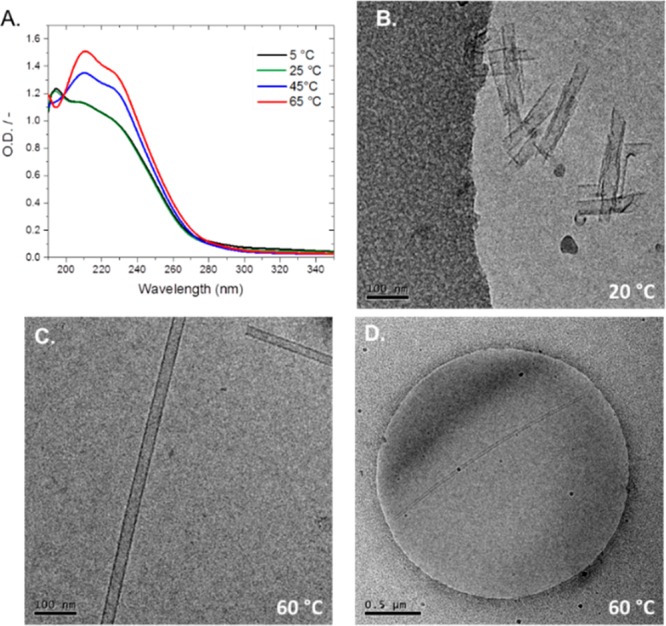
Polymorphism of supramolecular assemblies induced by the change of temperature. (A) UV–vis (c = 50 μM) of BTA 3 in water as a function of temperature. (B) Cryo-TEM of a centrifuged sample of BTA 3 in water at 20 °C, showing short nanotubes with ill-formed tube walls (c = 0.90 mM). (C) Cryo-TEM of elongated BTA 3 nanotubes in water after heating to 60 °C, showing well-formed nanotubes (100 nm scale bar, c = 0.90 mM). (D) Cryo-TEM of BTA 3 in water after heating to 60 °C, showing a several micrometer long nanotube (0.5 μm scale bar, c = 0.90 mM).
Striking structural changes in the BTA 3 aggregates were further observed by temperature-dependent experiments using cryo-TEM. Simply by centrifugation of a concentrated solution of BTA 3 (0.6 wt %) at room temperature, we can isolate short tubes measuring several hundred nanometers in length from the supernatant (Figure 6B). These BTA 3 assemblies measure ∼33 nm in width and several hundred nanometers in length with wall widths of ∼4 nm. A number of these nanotubes are noticeably ill-formed and contain curved, incompletely closed tubes. After heating to 60 °C and immediate imaging by cryo-TEM, we observe a dramatic elongation of the hollow nanotubes to micrometers in length with a tube width of ∼31 nm and a wall width of ∼4 nm (Figure 6C,D). The increase in degree of polymerization of the tubular polymers should be facilitated by the free energy gain from the stronger hydrophobic interactions at higher temperature. Interestingly, all of the hollow nanotubes tubes formed at 60 °C appeared perfectly straight, with almost defect-free tube walls under the resolution of the microscope and absence of any membrane-like structures. A straight tube is formed only when the distortion is distributed uniformly across the supramolecular structure. Although a straight nanotube gives the impression of high rigidity, this phenomenon is typically due to the effect of geometric stiffness. The in-plane flexibility of the 2D lattice may indeed be greater than that at 20 °C, resulting from the weaker interactions between the BTA strands in the tubes at high temperature, as indicated by the HDX-MS experiments.
The temperature-induced structural transitions were then examined on an in-house SAXS instrument. Figure 7A shows the scattering profiles collected from BTA 3 samples with concentration of ∼6 mg/mL upon heating from 20 to 70 °C. At each set temperature, the sample was given at least 30 min to equilibrate. As shown in Figure 7A, only slight changes occurred in the SAXS profiles upon increasing the temperature from 20 to 40 °C. In contrast, the scattering profile changes markedly upon heating the sample from 40 to 50 °C, after which the profiles largely remain the same at 60 and 70 °C. The changes are completely reversible, albeit with some hysteresis, as evidenced by the scattering profiles collected from cooling the sample from 70 °C back to 20 °C. The scattering profiles at temperatures below and above 50 °C can, again, be fitted with a combination of a hollow tube and a membrane model. The model analysis suggests that in the tubular structures, the thickness of wall changes slightly from ∼5 nm to ∼4 nm when the temperature increases to 50 °C, while the width of the tubes first increases to ∼38 nm at 40 °C and then decreases to ∼32 nm at 50 °C. The volume fraction (Figure 7B) of tubular structures in solution increases from ∼53% to ∼95% upon heating the sample from 40 to 50 °C, presumably due to transformation of the small membranes into tubes. It is still unclear whether the packing morphology of BTA 3 in the supramolecular structures is also changed. Nevertheless, the in situ SAXS provides structural information that confirms the subtle temperature-induced changes in the molecular interactions and organization of BTA 3 in the supramolecular structures.
Figure 7.

(A) SAXS patterns of BTA 3 (c = 6 mg/mL) in water as a function of temperature. The open symbols represent the experimental data where squares, circles, triangles, diamonds, hexagons, and stars represent 20, 30, 40, 50, 60, and 70 °C and the solid line represents the best fit results. (B) Volume fraction (closed square) and core radius (closed circle) of BTA 3 as a function of temperature. The dashed lines are point-to-point guidelines.
Together, the presented data displays how modulating dynamic behavior of small molecules in water via subtle changes to molecular structure, that is, addition or subtraction of methylene or water-soluble oligo(ethylene glycol) units or introducing environmental stimuli, such as heat, can result in drastically different self-assembly structures via similar molecular arrangements. Furthermore, monomer exchange dynamics are critical to the self-assembled structures obtained. From the evidence collected, it is clear that the acid functionalized BTA 2 and 3 share a common base membrane morphology, likely a bilayer. Tube formation is driven by an increase in stability of the membrane structure, as evidenced by the slow exchange dynamics of the BTA at room temperature, and the higher energetic cost for exposing the edges of the membranes to water. Interestingly, heating BTA 3, and thus increasing exchange dynamics at the molecular level, results in apparent ordering at the mesoscale, likely due to the alleviation of some strain energy in the assemblies. A complete molecular packing model for this system is still unclear, specifically with regards to the location of the carboxylic acid in the supramolecular assembly. Furthermore, the solvation state of the oligo(ethylene glycol)s at elevated temperatures may play a role in the formation of well-defined BTA 3 nanotubes.53 However, we do not believe that a temperature-dependent desolvation of the oligo(ethylene glycol) units or lower critical solution temperature effects dominate the temperature-dependent behavior due to the increased monomer exchange dynamics observed by HDX-MS experiments coupled with no apparent change in solution stability of the aggregates at elevated temperature. Herein, the formation of acid–acid dimers between BTA fibers has been proposed as the lateral supramolecular interaction responsible for bridging 1D BTA supramolecular polymers into bilayers to form membranes and tubes. However, another likely scenario for bridging fibers into bilayers to form membranes and tubes are acid–amide H-bonding interactions.54−56 Further investigation of the precise molecular organization of these molecules in water, for example, by SANS and fiber diffraction, and the morphological response to pH are subjects of future studies.
Conclusion
In summary, we have introduced acid functionalized BTAs, which assemble into membrane and tube-like structures in water. In this solvent, small molecule self-assembly is dominated by hydrophobic forces and order can be induced via the introduction of directional supramolecular interactions, such as H-bonding and π–π interactions. Pairwise interactions of carboxyl groups are a popular supramolecular motif in organic solvent and are present in the regulation of biological structures; however this motif is quite rare in synthetic aqueous supramolecular assemblies, where carboxyl groups are employed as water-solubilizing moieties. By sufficiently shielding carboxyl groups in a hydrophobic environment in the acid functionalized BTAs, we have introduced an additional directional supramolecular interaction, which together with the 3-fold intermolecular H-bonding of BTA core amides results in the generation of two- and three-dimensional objects in water. Furthermore, introducing carboxylic acids into BTA assemblies opens new possibilities for the regulation of complex structures from simple building blocks that can be reversibly modulated via external conditions such as temperature, pH, and the addition of salts.
Intriguingly, BTA 3, where the carboxylic acid is positioned at the periphery of the 10-methylene hydrophobic domain, displays reversible temperature-dependent polymorphism in which the tubular assemblies are elongated into well-ordered and micrometers long hollow tubes at elevated temperatures. The nature of this apparent ordering at the mesoscopic scale can be attributed to an increase in dynamic behavior at the molecular scale. At room temperature the dynamic exchange of monomeric units in the aggregates is slowed down significantly, and then both sheets and tubes are formed. The results presented indicate that for synthetic structures and their assembled aggregates, it is important to control the subtle trade-off between well-defined structures, with high precision and yields, and the dynamic nature of the assemblies. As the latter can be tuned by temperature, it is proposed that in molecular engineering of self-assembled architectures, next to optimizing the molecular structure and solvent conditions, temperature must be optimized as well. This is not as simple as it may seem. Optimization of temperature, as a design parameter in self-assembled architecture, presents the practical challenge of characterizing the self-assembled aggregate across a broad range of temperatures and times. However, as in organic and polymer synthesis when creating new covalent bonds, the noncovalent synthesis of objects, in high yields with well-defined architectures, requires that all parameters of the pathway control must be optimized; otherwise, uncontrolled polymorphism will be an obstacle for future progress.
Acknowledgments
Jolanda Spiering is acknowledged for fruitful discussions. We acknowledge financial support from the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035). Y.L. acknowledges financial support from the US National Science Foundation (DMR-1150742 and the program of “Research Opportunities in Europe for NSF CAREER Awardees”). I.K.V. is grateful for financial support from The Netherlands Organization for Scientific research (NWO VIDI grant 723.014.006, ECHO-STIP 717.013.005).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b07697.
The authors declare no competing financial interest.
Supplementary Material
References
- Aida T.; Meijer E. W.; Stupp S. I. Science 2012, 335, 813–817. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber M. J.; Appel E. A.; Meijer E. W.; Langer R. Nat. Mater. 2016, 15, 13–26. 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]
- Yan X. H.; Zhu P. L.; Li J. B. Chem. Soc. Rev. 2010, 39, 1877–1890. 10.1039/b915765b. [DOI] [PubMed] [Google Scholar]
- Hendricks M. P.; Sato K.; Palmer L. C.; Stupp S. I. Acc. Chem. Res. 2017, 50, 2440–2448. 10.1021/acs.accounts.7b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terech P.; de Geyer A.; Struth B.; Talmon Y. Adv. Mater. 2002, 14, 495–498. . [DOI] [Google Scholar]
- Madenci D.; Egelhaaf S. U. Curr. Opin. Colloid Interface Sci. 2010, 15, 109–115. 10.1016/j.cocis.2009.11.010. [DOI] [Google Scholar]
- Görl D.; Zhang X.; Würthner F. Angew. Chem., Int. Ed. 2012, 51, 6328–6348. 10.1002/anie.201108690. [DOI] [PubMed] [Google Scholar]
- Ryu J. H.; Hong D. J.; Lee M. Chem. Commun. 2008, 1043–1054. 10.1039/B713737K. [DOI] [PubMed] [Google Scholar]
- Brunsveld L.; Lohmeijer B. G. G.; Vekemans J. A. J. M.; Meijer E. W. Chem. Commun. 2000, 0, 2305–2306. 10.1039/b007502g. [DOI] [Google Scholar]
- Petkau-Milroy K.; Sonntag M. H.; van Onzen A. H. A. M.; Brunsveld L. J. Am. Chem. Soc. 2012, 134, 8086–8089. 10.1021/ja3029075. [DOI] [PubMed] [Google Scholar]
- Fuhrhop J. H.; Helfrich W. Chem. Rev. 1993, 93, 1565–1582. 10.1021/cr00020a008. [DOI] [Google Scholar]
- Schnur J. M. Science 1993, 262, 1669–1676. 10.1126/science.262.5140.1669. [DOI] [PubMed] [Google Scholar]
- Aggeli A.; Nyrkova I. A.; Bell M.; Harding R.; Carrick L.; McLeish T. C. B.; Semenov A. N.; Boden N. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 11857–11862. 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunitake T. Angew. Chem., Int. Ed. Engl. 1992, 31, 709–726. 10.1002/anie.199207091. [DOI] [Google Scholar]
- Capito R. M.; Azevedo H. S.; Velichko Y. S.; Mata A.; Stupp S. I. Science 2008, 319, 1812–1816. 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- Shimizu T.; Masuda M.; Minamikawa H. Chem. Rev. 2005, 105, 1401–1443. 10.1021/cr030072j. [DOI] [PubMed] [Google Scholar]
- Dankers P. Y. W.; Meijer E. W. Bull. Chem. Soc. Jpn. 2007, 80, 2047–2073. 10.1246/bcsj.80.2047. [DOI] [Google Scholar]
- Jung J. P.; Nagaraj A. K.; Fox E. K.; Rudra J. S.; Devgun J. M.; Collier J. H. Biomaterials 2009, 30, 2400–2410. 10.1016/j.biomaterials.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M.; Langer R.; Jia X. Q. J. Biomater. Sci., Polym. Ed. 2007, 18, 241–268. 10.1163/156856207779996931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson J. B.; Stupp S. I. Chem. Commun. 2011, 47, 7962–7964. 10.1039/c1cc12570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Rehm S.; Safont-Sempere M. M.; Würthner Nat. Chem. 2009, 1, 623–629. 10.1038/nchem.368. [DOI] [PubMed] [Google Scholar]
- Liang G.; Ren H.; Rao J. Nat. Chem. 2010, 2, 54–60. 10.1038/nchem.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyborna Y.; Vybornyi M.; Häner R. Chem. Commun. 2017, 53, 5179–5181. 10.1039/C7CC00886D. [DOI] [PubMed] [Google Scholar]
- Engelen W.; Wijnands S. P. W.; Merkx M. J. Am. Chem. Soc. 2018, 140, 9758–9767. 10.1021/jacs.8b06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korevaar P. A.; George S. J.; Markvoort A. J.; Smulders M. M. J.; Hilbers P. A. J.; Schenning A.; De Greef T. F. A.; Meijer E. W. Nature 2012, 481, 492–496. 10.1038/nature10720. [DOI] [PubMed] [Google Scholar]
- Tompa P.; Fuxreiter M. Trends Biochem. Sci. 2008, 33, 2–8. 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Oosawa F.Thermodynamics of the Polymerization of Protein; Academic Press: London, U.K., 1975. [Google Scholar]
- Gibbons I. R. J. Cell Biol. 1981, 91, 107s–124s. 10.1083/jcb.91.3.107s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspar D. L. D.; Cohen C.; Longley W. J. Mol. Biol. 1969, 41, 87–107. 10.1016/0022-2836(69)90128-4. [DOI] [PubMed] [Google Scholar]
- Namba K.; Stubbs G. Science 1986, 231, 1401–1406. 10.1126/science.3952490. [DOI] [PubMed] [Google Scholar]
- Oosawa F. J. Polym. Sci. 1957, 26, 29–45. 10.1002/pol.1957.1202611204. [DOI] [Google Scholar]
- Caspar D. L. D.Assembly and Stability of the Tobacco Mosaic Virus Particle. In Advances in Protein Chemistry; Anfinsen C. B., Anson M. L., Edsall J. T., Eds.; Academic Press: 1964; Vol. 18, pp 37–121. [DOI] [PubMed] [Google Scholar]
- Sachse C.; Chen J. Z.; Coureux P.-D.; Stroupe M. E.; Fändrich M.; Grigorieff N. J. Mol. Biol. 2007, 371, 812–835. 10.1016/j.jmb.2007.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flocco M. M.; Mowbray S. L. J. Mol. Biol. 1995, 254, 96–105. 10.1006/jmbi.1995.0602. [DOI] [PubMed] [Google Scholar]
- Etter M. C. Acc. Chem. Res. 1990, 23, 120–126. 10.1021/ar00172a005. [DOI] [Google Scholar]
- Lackinger M.; Heckl W. M. Langmuir 2009, 25, 11307–11321. 10.1021/la900785f. [DOI] [PubMed] [Google Scholar]
- Gdaniec M.; Jankowski W.; Milewska M. J.; Połoñski Angew. Chem., Int. Ed. 2003, 42, 3903–3906. 10.1002/anie.200351432. [DOI] [PubMed] [Google Scholar]
- Ma X.; Tian H. Acc. Chem. Res. 2014, 47, 1971–1981. 10.1021/ar500033n. [DOI] [PubMed] [Google Scholar]
- Yan X. Z.; Wang F.; Zheng B.; Huang F. H. Chem. Soc. Rev. 2012, 41, 6042–6065. 10.1039/c2cs35091b. [DOI] [PubMed] [Google Scholar]
- Zhou S. L.; Matsumoto S.; Tian H. D.; Yamane H.; Ojida A.; Kiyonaka S.; Hamachi I. Chem. - Eur. J. 2005, 11, 1130–1136. 10.1002/chem.200400677. [DOI] [PubMed] [Google Scholar]
- Frisch H.; Nie Y.; Raunser S.; Besenius P. Chem. - Eur. J. 2015, 21, 3304–3309. 10.1002/chem.201406281. [DOI] [PubMed] [Google Scholar]
- Dimitrov I.; Trzebicka B.; Muller A. H. E.; Dworak A.; Tsvetanov C. B. Prog. Polym. Sci. 2007, 32, 1275–1343. 10.1016/j.progpolymsci.2007.07.001. [DOI] [Google Scholar]
- Pochan D. J.; Schneider J. P.; Kretsinger J.; Ozbas B.; Rajagopal K.; Haines L. J. Am. Chem. Soc. 2003, 125, 11802–11803. 10.1021/ja0353154. [DOI] [PubMed] [Google Scholar]
- Kim H.-J.; Lee J.-H.; Lee M. Angew. Chem. 2005, 117, 5960–5964. 10.1002/ange.200501270. [DOI] [Google Scholar]
- Alemán García M. A.; Magdalena Estirado E.; Milroy L. G.; Brunsveld L. Angew. Chem., Int. Ed. 2018, 57, 4976–4980. 10.1002/anie.201800148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidhar Y.; Weissman H.; Wolf S. G.; Gulino A.; Rybtchinski B. Chem. - Eur. J. 2011, 17, 6068–6075. 10.1002/chem.201003419. [DOI] [PubMed] [Google Scholar]
- Tena-Solsona M.; Rieß B.; Grötsch R. K.; Löhrer F. C.; Wanzke C.; Käsdorf B.; Bausch A. R.; Müller-Buschbaum P.; Lieleg O.; Boekhoven J. Nat. Commun. 2017, 8, 15895. 10.1038/ncomms15895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenders C. M. A.; Albertazzi L.; Mes T.; Koenigs M. M. E.; Palmans A. R. A.; Meijer E. W. Chem. Commun. 2013, 49, 1963–1965. 10.1039/c3cc38949a. [DOI] [PubMed] [Google Scholar]
- Leenders C. M. A.; Baker M. B.; Pijpers I. A. B.; Lafleur R. P. M.; Albertazzi L.; Palmans A. R. A.; Meijer E. W. Soft Matter 2016, 12, 2887–2893. 10.1039/C5SM02843D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X.; Lafleur R. P. M.; Leenders C. M. A.; Schoenmakers S. M. C.; Matsumoto N. M.; Baker M. B.; Van Dongen J. L. J.; Palmans A. R. A.; Meijer E. W. Nat. Commun. 2017, 8, 15420. 10.1038/ncomms15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susi H.; Zell T.; Timasheff S. N. Arch. Biochem. Biophys. 1959, 85, 437–443. 10.1016/0003-9861(59)90508-9. [DOI] [PubMed] [Google Scholar]
- Génin F.; Quilès F.; Burneau A. Phys. Chem. Chem. Phys. 2001, 3, 932–942. 10.1039/b008897h. [DOI] [Google Scholar]
- Kemper B.; Zengerling L.; Spitzer D.; Otter R.; Bauer T.; Besenius P. J. Am. Chem. Soc. 2018, 140, 534–537. 10.1021/jacs.7b08189. [DOI] [PubMed] [Google Scholar]
- Phadke A.; Zhang C.; Arman B.; Hsu C.-C.; Mashelkar R. A.; Lele A. K.; Tauber M. J.; Arya G.; Varghese S. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 4383–4388. 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes A. D.; Hawes C. S.; Ward E. N.; Haffner B.; Mobius M. E.; Byrne K.; Schmitt W.; Pal R.; Gunnlaugsson T. CrystEngComm 2017, 19, 1427–1438. 10.1039/C7CE00206H. [DOI] [Google Scholar]
- Menger F. M.; Caran K. L. J. Am. Chem. Soc. 2000, 122, 11679–11691. 10.1021/ja0016811. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.