

Abstract
Electronic effects in the transmetalation of an aryl group from gold to boron were investigated by NMR spectroscopy. The transmetalation reaction is more facile for increasingly electrophilic boron reagents and is in equilibrium under certain conditions. Observed tetracoordinate boronate compounds suggest a two-step, associative transmetalation reaction mechanism in which the organogold complex first delivers a nucleophilic phenyl group to the empty p orbital of boron. For certain substrates, this tetracoordinate intermediate decomposes to the tricordinate, final transmetallation product, and in others this tricordinate species remains in equilibrium with a tetracoordinate anionic boron compound. Experimental and theoretical investigations into the extension of this transmetalation reaction from a mechanistic step in our previously reported intramolecular gold-catalyzed addition of boron–oxygen σ bonds across alkynes to an intermolecular variant are discussed.2014 Elsevier Ltd. All rights reserved.
Keywords: transmetalation, boron, oxygen, gold, alkoxyboration
Graphical Abstract
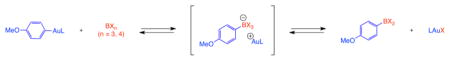
1. Introduction
Boronic acid derivatives are widely used building blocks in diversity-oriented synthesis.1 These organoboron reagents are often prepared through the addition of B–X σ bonds to C–C multiple bonds.2–17 Recently we reported the first alkoxyboration reaction, the formal addition of B–O σ bonds to C–C multiple bonds (Scheme 1).18 This intramolecular addition allowed for the synthesis of O-heterocyclic boronic acid derivatives. Central to our mechanistic hypothesis was a transmetalation reaction of neutral organogold intermediates (such as 3) with boron electrophiles (such as 4).18 This method, as the first report of transmetalation from organogold(I) to boron, showcases a complimentary and potentially diverse strategy for the formation of organoboron compounds by employing this transmetallation in a catalytic method for organic synthesis. The reverse transmetalation from organoboron to gold(I), in contrast, has been studied, especially stoichiometrically, wherein Gray has performed several founding studies.20–24 Thus, knowledge of the favored position (thermodynamics) and accessibility (kinetics) of this reaction in both directions would be helpful for further reaction design. After our initial publication, Stephan and Hashmi disclosed a stoichiometric organogold-to-boron transmetlation reaction,19 bolstering the range of systems for such transmetalation chemistry.
Scheme 1.

Previously published successful gold-catalyzed alkoxyboration reaction, showing proposed key organogold-to-boron transmetalation step. This transmetalation step motivated the stoichiometric transmetalation studies in this report.
We herein report an NMR spectroscopy study of the effect of electronic parameters on the organogold-to-boron transmetalation reaction. These electronic effects influence the accessibility of the transmetalation step. Further, transmetalation equilibria are established with certain substrates. We also disclose a computational inquiry regarding the role of electronic effects in our previously published successful intramolecular akoxyboration reaction. We anticipate that information gained through these studies will facilitate additional application of this step including the potential development of an intermolecular oxyboration reaction variant.
2. Results and Discussion
2.1. Electronic effects in the equilibrating organogold-to-boron transmetalation reaction
Arylgold complex 6 was selected as an appropriate model for proposed alkoxyboration catalytic intermediate 3. Complex 6 was treated with a range of boron reagents as shown in Table 1. The table is qualitatively arranged from the least electrophilic B reagent (entry 1) to the most electrophilic (entry 6).25 After addition of reagents the presence of transmetalation products was evaluated by 1H, 11B, and 19F spectroscopy. Particularly useful in the analysis of these reaction mixtures were diagnostic chemical shifts26 observable by 11B NMR spectroscopy: δ 33 ~ 31 ppm for boronic esters (RB[OR]2, such as 5), δ 23 ~ 18 ppm for boric esters (B[OR]3, such as 1), δ 10 ~ 0 ppm for mixed anionic borates or boronates ([BRx(OR)4-x]−).
Table 1.
Electronic effects on the stoichiometric aryl transmetalation from Au to B including presence of detectable equilibrium.

|
Ratios determined by 1H, 11B, and/or 19F NMR spectroscopy.
Indicates a reversible, thermal redistribution of products was observable by variable temperature NMR spectroscopy.
Starting the evaluation with the most electron rich boron compounds, borate 7a and electron-rich boric esters 7b and 7c, indicated that these compounds made poor acceptors for the phenyl group from gold(I). No detectable quantities of aryl transfer products (9 or 10) were observed by NMR spectroscopy after prolonged heating (entries 1–3; 80 °C to 90 °C). These reaction mixtures were also examined inside the NMR spectrometer at their highest elevated temperatures; characterization at 80 °C to 90 °C indicated no detectible transmetalation products 9 or 10 at elevated temperature either. Thus, these transmetalation reactions to electron-rich boron acceptors are disfavored by thermodynamic or kinetic parameters or both, to the extent that elevated temperature does not perturb which compounds are present in the mixture by detectible quantities.
Substitution of two methoxy substituents at boron for the less electron-donating catechol ligand in compound 7d afforded the first indication of phenyl group transfer from gold: a small amount of arylated ion pair 8d, characterized by 11B NMR spectroscopy as a signal at δ = 15.2 ppm. Variable temperature NMR data measured from 25 °C to 80 °C characterized a reversible, thermal dependence on the product distribution, indicating that 8d was present in equilibrium with starting materials. This equilibrium was established in less than 1 h at ambient temperature after combination of the reagents. At ambient temperature, 7e, 8e, and 9e were observed in a ratio of 1 : 0.5 : 0.4, respectively. Heating to 80 °C in the NMR spectrometer afforded a greater equilibrium concentration of boronate 8e relative to the other boron-containing compounds; the same three components were then observed in a ratio of 1 : 0.9 : 0.5. Cooling of the mixture to the original ambient temperature regenerated the initial compound ratios (see Supplementary Data for variable temperature NMR spectroscopy data).
Similarly, the slightly more electrophilic boric ester 7e afforded a greater equilibrium concentration of the ion pair 8e, as well as providing the first access in the table to the neutral final transmetalation products 9e. Finally, the reaction of organogold complex 6 with the most electrophilic boron reagent studied,25 BF3·Et2O (entry 6), fully consumed 6 to rapidly provide a mixture of arylated ion pair 8f and neutral difluoroborane 9f in less than 20 min at ambient temperature. The resulting mixture subsequently underwent slow ambient-temperature conversion of ion pair 8f into neutral 9f through a reaction with a currently unidentified fluoride acceptor.
These data suggest that (1) the organogold-to-boron transmetalation is best represented electronically as the addition of a nucleophilic C–Au σ-bond to a boron electrophile, (2) tetracoordinate anionic boron complexes are present as on-path transmetalation intermediates or off-path components that can nevertheless funnel to products in some gold-to-boron transmetalation reactions, and (3) a mechanistic requirement to form the tetracoordinate anionic boron as an intermediate (the on path option) is consistent with the higher reactivity displayed by electrophilic boron reagents. These conclusions lend additional understanding to the mechanism of previously studied organoboron-to-gold transmetalation reactions20–24 according to the principle of microscopic reversibility, which dictates that if the organogold-to-boron transmetalation proceeds through a tetracoordinate boronate intermediate, the organoboron-to-gold transmetalation must also proceed through this type of intermediate.
The results of these studies can be interpolated for added understanding of the potential intermediates in our previously published alkoxyboration reaction. The proposed transmetalation partner from the alkoxyboration reaction, B-trifluoroacetoxycatecholborane (Scheme 1, 4), was not included in this study owing to difficulties encountered in its independent synthesis. The electrophilicity of 4, however, is likely greater than that of 7e since a phenyoxy group is a better donor than a trifluoroacetoxy group (as can be estimated by comparison of gas-phase and solution-phase acidities of the corresponding acids; aqueous pKa phenol = 10, pKa trifluoroacetic acid = 0.5).27 Therefore 4 is also anticipated to undergo facile transmetalation with arylgold reagents under catalytic reaction conditions.
2.2. DFT calculations examine electronic effect of boron transmetalation partner on overall alkoxyboration thermodynamics
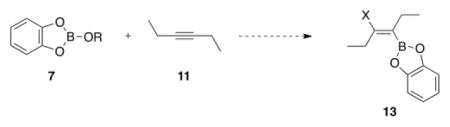
Having identified progressively more electrophilic boron reagents as progressively better transmetalation partners with organogold complexes, we next examined if the same electronic trend was present in the thermodynamics of the overall alkoxyboration reaction, which would effect its ability to be catalyzed (i.e., chemical reactions that are thermodynamically downhill when the full system is taken into account can be effectively catalyzed for useful conversions). For these studies we turned to density functional theory (DFT) calculations to provide insight due to the ability to DFT calculations to separate entropic and enthalpic contributions for detailed consideration.
In order to investigate the thermodynamic basis for the alkoxyboration reaction in Scheme 1, a series of computational experiments were conducted using DFT to study the simple reaction of anti addition of catecholboric esters to 3-hexyne (Table 2). The data suggest that the alkoxyboration reaction is essentially neutral enthalpically for electron-rich boric esters (entries 1 and 2) and becomes more favorable for comparatively electron-poor boric esters (entries 3 and 4). This trend can readily be explained by the relative stabilization of the boron empty p orbital through resonance;30,31 the alkoxyboration reaction forms an electron-rich vinyl boronic acid derivative, affording an enthalpic gain associated with increased resonance stabilization of the p orbital when compared to electron-poor boric ester starting materials.
Table 2.
Calculated thermochemical dependence of an intermolecular alkoxyboration reaction on the electronic character of the boric ester reagent.a
| ||||
|---|---|---|---|---|
| Entry
|
OR
|
ΔH°calc (kcal/mol)
|
ΔS°calc (cal/[mol·K])
|
ΔG°calc (kcal/mol)
|
| 1 |
7g |
−0.5 | −43.9 | +12.6 |
| 2 |
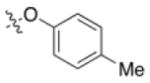 7e |
−0.2 | −40.3 | +11.8 |
| 3 |
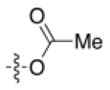 7h |
−5.3 | −34.6 | +5.0 |
| 4 |
 7i |
−8.2 | −34.1 | +2.0 |
Gas phase DFT calculations performed at the B3LYP/6-311G level of theory. Thermochemical parameters were calculated at 298.15 K and 1.000 atm.
These DFT calculations show that electron-poor boron reagents provide the highest thermodynamic driving force for the alkoxyboration reaction. Our experiments in Table 1 show that these same electron-poor boron reagents provide the most favorable transmetalation reactions with organogold complexes. Thus, the same boron reagents that are best matched for the transmetalation step in this reaction as are most thermodynamically favorable for the alkoxyboration reaction overall. This coincidence is not presupposed, given that the transmetalation step provides the catalytic pathway (kinetics) but the overall reaction thermodynamics are necessarily path independent. Such an overall oxyboration reaction could feature a transmetalation of a nucleophilic, catalytic organogold intermediate (e.g., 3, Scheme 1) either directly with electron-poor boron partners such as those shown in Table 2 or with a catalytic intermediate generated in situ from such reagents (e.g., 4, Scheme 1).
These DFT studies provide additional insight into the feasibility of catalyzing an overall intermolecular alkoxyboration reactions to C–C multiple bonds; the previously published reaction was demonstrated only in intramolecular examples. Each calculated intermolecular reaction in Table 2 is thermodynamically disfavored overall. The modest enthalpic gains in the calculated intermolecular variant are more than offset by the highly unfavorable entropic nature of an intermolecular reaction. Thus, any potential intermolecular alkoxyboration reaction would require a careful consideration of thermodynamic parameters.
3. Conclusion
A general intermolecular alkoxyboration faces significant challenges. The potential reactions examined through DFT calculations were found to be thermodynamically disfavored; substituent manipulations affected modest variations in ΔH, but these variations were insufficient to overcome the uniformly large negative ΔS. DFT calculations suggested the thermodynamic unfavorability of such a reaction would be minimized with electron-poor boric esters. However, these same boric esters are inherently less nucleophilic and therefore may suffer from an increased kinetic challenge, which could arise from a requirement for the boron substituent to act as a nucleophile in the proposed alkyne-attack mechanism. Overall, these calculations indicate that our previous report of a successful catalytic benzofuran-forming alkoxyboration reaction was enabled by the small enthalpic difference between the B–O σ bond of the starting materials and the B–C σ bond of the products, as well by as the comparatively low entropic cost of intramolecular reactivity. It is possible that an intermolecular alkyne alkoxyboration may yet be realized in more specialized systems. Such a reaction will likely involve an organogold-to-boron transmetalation reaction and thus an understanding of the electronic parameters effecting this potentially equilibrating process will be valuable.
4. Experimental section
4.1.1. General
All chemicals were used as received from commercial sources unless otherwise noted. Anhydrous 3-hexyne was obtained by distillation over CaH2 under N2 atmosphere. Toluene-d8 was dried over CaH2, degassed using three freeze-pump-thaw cycles, and vacuum transferred prior to use. Commercially available boric esters were distilled over Na0 and stored under N2 atmosphere at −35 °C. All manipulations were conducted in a glovebox under nitrogen atmosphere or using standard Schlenk techniques unless otherwise specified. All proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded on a Bruker DRX-400 spectrometer, Bruker DRX-500 spectrometer outfitted with a cryoprobe, or a Bruker AVANCE-600 spectrometer. All boron nuclear magnetic resonance (11B NMR) spectra were recorded on a Bruker AVANCE-600 spectrometer. All chemical shifts (δ) are reported in parts per million (ppm) downfield of tetramethylsilane, and referenced to the residual protiated solvent peak (δ = 2.08 ppm for toluene-d8 or δ = 5.32 ppm for dichloromethane-d2 in 1H NMR spectroscopy experiments; δ = 20.43 ppm for toluene-d8 or δ = 53.84 ppm for dichloromethane-d2 in 13C NMR spectroscopy experiments). 11B NMR spectroscopy experiments are referenced to the absolute frequency of 0 ppm in the 1H dimension according to the Xi scale. NMR spectra for variable temperature transmetalation studies are located in the Supplementary Material.
4.2. Synthesis of transmetalation partners
4.2.1. IPrAu(4-OMePh) (6)
Aryl gold complex 6.1 was prepared from IPrAuCl according to a literature procedure32 in 63% yield. 1H NMR (d8-toluene, 400 MHz)™ 7.27 (d, J = 6.5 Hz, 2H), 7.20 (t, J = 6.7 Hz, 2H), 7.09–7.05 (m, 4H), 6.73 (d, J = 6.6 Hz, 2H), 6.40 (s, 2H), 3.34 (s, 3H), 2.68 (br m, 4H), 1.46 (d, J = 5.5 Hz, 12 H), 1.11 (d, J = 5.6 Hz, 12 H).
4.2.2. Tetrabutylammonium 1-hydroxy-4-methyl-2,6,7-trioxa-1-borabicyclo[2.2.2]octan-1-uide (7a)
Triol borate 7a was prepared using a procedured adapted from the synthesis of the corresponding Na salt.33 A 20 mL vial was charged with boric acid (180 mg, 2.9 mmol, 1.0 equiv) and water (1 mL). Tetra-n-butylammonium hydroxide (1.7 g of 44 wt % aq solution, 2.9 mmol, 1.0 equiv) and 1,1,1-tris(hydroxymethyl)ethane (350 mg, 2.9 mmol, 1.0 equiv) were added, and the reaction mixture was heated in a preheated 100 °C oil bath for 30 min. Volatiles were removed in vacuo, and 250 mg of the resulting white powder was purified by reverse-phase silica gel chromatography eluting using a gradient from 100% water to 100% MeCN. Volatiles were removed in vacuo at 30 °C and ca. 10 mTorr overnight to afford 50 mg 7a as a white powder (20% recovery). This salt exhibits a temperature- and solvent-dependent equilibrium with a dimer and higher order oligomers34 that obfuscates analysis by NMR spectroscopy.
4.2.3. 2-(p-Tolyloxy)benzo[d][1,3,2]dioxaborole (7e)
A flame-dried 100 mL Schlenk tube with a stirbar under a dynamic N2 atmosphere was charged with B-chlorocatecholborane (810 mg, 5.3 mmol, 1.0 equiv) and anhydrous DCM (7.5 mL). The reaction vessel was equipped with a 25 °C water bath to control any potential exotherm, and a solution of p-cresol (570 mg, 5.3 mmol, 1.0 equiv) in anhydrous DCM (7.5 mL) was added dropwise over 5 min. The resulting solution was stirred at 25 °C for 50 min before being concentrated in vacuo using Schlenk techniques. To the resulting oily residue was added anhydrous pentane (10 mL), and the solution was again concentrated to afford a viscous, pale yellow oil that solidified upon storage at −35 °C for 18 h. Boric ester 7e was obtained as a cream-colored solid in 81% yield (970 mg). 1H NMR (anhydrous CD2Cl2, 600 MHz): δ 7.20 (br d, J = 8.0 Hz, 2H), 7.17 (dd, J = 5.7, 3.4 Hz, 2H), 7.13 (br d, J = 7.3 Hz, 2H), 7.08 (dd, J = 5.8, 3.4 Hz, 2H), 2.36 (s, 3H). 11B NMR (anhydrous CD2Cl2, 193 MHz): δ 22.7 (br s). 13C NMR (anhydrous CD2Cl2, 125 MHz): δ 150.6, 148.1, 134.4, 130.5, 123.0, 119.6, 112.4, 20.8.
4.3. General procedure for organgold-to-boron transmetalation study
All stoichiometric transmetalation reactions were set up in a N2-filled glovebox. A solution of boric ester 7 (17 μmol, 1.0 equiv) in anhydrous d8-toluene (0.5 mL) was added to a dram vial containing organogold complex 6 (12 mg, 17 μmol). The resulting solution was mixed well and then transferred to a J. Young NMR tube for observation. For reactions conducted at elevated temperatures, the NMR tube was heated in a preheated oil bath as indicated. Particularly useful in the analysis of the resulting mixtures were diagnostic chemical shifts observable by 11B NMR spectroscopy: δ 33 ~ 31 ppm for boronic esters such as 9e, δ 23 ~ 18 ppm for boric esters such as 7c, δ 10 ~ 0 ppm for mixed borates or boronates such as 8e and 8f.26
4.4. Density functional theory (DFT) calculations
Thermochemical data for compounds from Table 2 were obtained for optimized geometries of each molecule. Structures were first modeled using Avogadro35 1.1.1 and optimized using UFF molecular mechanics. Each structure was then further optimized in the gas phase using Gaussian 0936 using density functional theory (DFT) calculations using the B3LYP functional37,38 and the 6-311G basis set.39 Thermochemical data were calculated at the same level of theory at a temperature of 298.15 K and a pressure of 1.000 atm.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (1R01GM098512-01). We thank the Alexander von Humboldt Foundation for a fellowship to S.A.B and Prof. David L. Van Vranken and Prof. Christopher D. Vanderwal (University of California-Irvine) for their helpful comments on a draft of this manuscript. J.J.H. also thanks the UC-Regents for a dissertation fellowship.
Footnotes
References and notes
- 1.Hall DG. Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine, and Materials. 2. Wiley-VCH; Weinheim, Germany: 2011. [Google Scholar]
- 2.Hurd DT. J Am Chem Soc. 1948;70:2053–2055. [Google Scholar]
- 3.Brown HC. Tetrahedron. 1961;12:117–138. [Google Scholar]
- 4.Männig D, Nöth H. Angew Chem, Int Ed Engl. 1985;24:878–879. [Google Scholar]
- 5.Carrol A-M, O’Sullivan TP, Guiry P. J Adv Synth Catal. 2005;347:609–631. [Google Scholar]
- 6.Miyaura N. Hydroboration, Diboration, and Stannylboration. In: Togni A, Grützmacher H, editors. Catalytic Heterofunctionalization. Wiley-VCH; Weinheim: 2001. pp. 1–46. [Google Scholar]
- 7.Suginome M, Yamamoto A, Murakami M. J Am Chem Soc. 2003;125:6358–6359. doi: 10.1021/ja0349195. [DOI] [PubMed] [Google Scholar]
- 8.Suginome M, Shirakura M, Yamamoto A. J Am Chem Soc. 2009;128:14438–14439. doi: 10.1021/ja064970j. [DOI] [PubMed] [Google Scholar]
- 9.Suginome M. Chem Rec. 2010;10:348–358. doi: 10.1002/tcr.201000029. [DOI] [PubMed] [Google Scholar]
- 10.Suginome M, Nakamura H, Ito Y. Chem Commun. 1996:2777–2778. [Google Scholar]
- 11.Suginome M, Nakamura H, Ito Y. Angew Chem, Int Ed Engl. 1997;36:2516–2518. [Google Scholar]
- 12.Onozawa S, Hatanaka Y, Sakakura T, Shimada S, Tanaka M. Organometallics. 1996;15:5450–5452. [Google Scholar]
- 13.Ishiyama T, Nishijima K, Miyaura N, Suzuki A. J Am Chem Soc. 1993;115:7219–7225. [Google Scholar]
- 14.Ishiyama T, Matsuda N, Miyaura N, Suzuki A. J Am Chem Soc. 1993;115:11018–11019. [Google Scholar]
- 15.Marder TB, Norman NC. Top Catal. 1998;5:63–73. [Google Scholar]
- 16.Lappert MF, Prokai B. J Orgmet Chem. 1964;1:384–400. [Google Scholar]
- 17.Hara S, Dojo H, Takinami S, Suzuki A. Tetrahedron Lett. 1983;24:731–734. [Google Scholar]
- 18.Hirner JJ, Faizi DJ, Blum SA. J Am Chem Soc. 2014;136:4740–4745. doi: 10.1021/ja500463p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansmann MM, Rominger F, Boone MP, Stephan DW, Hashmi ASK. Organometallics. 2014;33:4461–4470. [Google Scholar]
- 20.Sladek A, Hofreiter S, Paul M, Schmidbaur H. J Orgmet Chem. 1995;501:47–51. [Google Scholar]
- 21.Forward JM, Fackler JP, Jr, Staples R. J Organometallics. 1995;14:4194–4198. [Google Scholar]
- 22.Partyka DV, Zeller M, Hunter AD, Gray TG. Angew Chem Int Ed. 2006;45:8188–8191. doi: 10.1002/anie.200603350. [DOI] [PubMed] [Google Scholar]
- 23.Partyka DV, Zeller M, Hunter AD, Gray TG. Inorg Chem. 2012;51:8394–8401. doi: 10.1021/ic3009464. [DOI] [PubMed] [Google Scholar]
- 24.Lenker HK, Gray TG, Stockland RA., Jr Dalton Trans. 2012;41:13274–13276. doi: 10.1039/c2dt32129g. [DOI] [PubMed] [Google Scholar]
- 25.Beckett MA, Strickland GC, Holland JR, Varma KS. Polymer. 1996;37:4629–4631. [Google Scholar]
- 26.Nöth H, Wrackmeyer B. Nuclear Magnetic Resonance Spectroscopy of Boron Compounds. In: Diehl P, Fluck E, Kosfeld R, editors. NMR: Basic Principles and Progress. Vol. 14 Springer-Verlag; Berlin: 1978. [Google Scholar]
- 27.a) Oonishi Y, Gómez-Suárez A, Martin AR, Nolan SP. Angew Chem Int Ed. 2013;52:9767–9771. doi: 10.1002/anie.201304182. [DOI] [PubMed] [Google Scholar]; b) Ansyln Eric V, Cougherty Dennis A. Modern Physical Organic Chemistry. University Science Books; Sausalito, California: 2006. pp. 278–281. [Google Scholar]
- 28.Richard ME, Fraccica DV, Garcia KJ, Miller EJ, Ciccarelli RM, Holahan EC, Resh VL, Shah A, Findeis PM, Stockland RA., Jr Beilstein J Org Chem. 2013;9:2002–2008. doi: 10.3762/bjoc.9.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.This 11B NMR signal (here in neat, protiated 3-hexyne) is suggestive of a boronate intermediate but does not match the spectral data from the stoichiometric transmetalation experiments, which were conducted at a higher concentration in d8-toluene.
- 30.Good CD, Ritter DM. J Am Chem Soc. 1962;84:1162–1166. [Google Scholar]
- 31.Schulman JM, Disch RL. Organometallics. 1989;8:733–737. [Google Scholar]
- 32.Dupuy S, Crawford L, Bühl M, Slawin AMZ, Nolan SP. Adv Synth Catal. 2012;354:2380–2386. [Google Scholar]
- 33.Taylor MJ, Grigg JA, Rickard CEF. Polyhedron. 1992;11:889–892. [Google Scholar]
- 34.Taylor MJ, Grigg JA, Laban IH. Polyhedron. 1996;15:3261–3270. [Google Scholar]
- 35.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchinson GR. J Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian; Wallingford, CT: 2009. [Google Scholar]
- 37.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 38.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 39.Krishnan R, Binkley JS, Seeger R, Pople JA. J Chem Phys. 1980;72:650–654. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.