

Abstract
Positron emission tomography (PET) has revealed key insights into the pathophysiology of movement disorders. This paper will focus on how PET investigations of pathophysiology are particularly relevant to Parkinson disease (PD), a neurodegenerative condition usually starting later in life marked by a varying combination of motor and non-motor deficits. Various molecular imaging modalities help to determine what changes in brain herald the onset of pathology; can these changes be used to identify presymptomatic individuals who may be appropriate for to-be-developed treatments that may forestall onset of symptoms or slow disease progression; can PET act as a biomarker of disease progression; can molecular imaging help enrich homogenous cohorts for clinical studies; and what other pathophysiologic mechanisms relate to non-motor manifestations. PET methods include measurements of regional cerebral glucose metabolism and blood flow, selected receptors, specific neurotransmitter systems, post synaptic signal transducers, and abnormal protein deposition. We will review each of these methodologies and how they are relevant to important clinical issues pertaining to PD.
Introduction
Positron emission tomography (PET) permits measurements of regional brain blood flow and metabolism, various brain receptors, neurotransmitter systems, post-synaptic signal transduction and abnormal protein deposition. Each of these measures can reveal various insights into the pathophysiology of the clinical manifestations of movement disorders including Parkinson disease (PD) and other related parkinsonian syndromes. This review will focus on PD since each of these PET imaging modalities have been used to address several key issues: what changes in brain herald the onset of pathology; can these changes be used to identify presymptomatic people who may be appropriate for to-be-developed treatments that may forestall onset of symptoms or slow disease progression; can PET act as a biomarker of disease progression; can molecular imaging help enrich homogenous cohorts for clinical studies; and what other pathophysiologic mechanisms relate to non-motor manifestations.
Some clinical and pathology clues provide some insights into these issues. PD begins commonly once a person hits the 60’s. However, the risk increases with age and yet as many as up to 5.8% may begin before age 401. Pathologically, deposition of misfolded α-synuclein in intracytoplasmic inclusions called Lewy bodies or in neuronal processes called Lewy neurites begins in pigmented nuclei in caudal brainstem (with or without olfactory tubercle involvement) and then progresses rostrally2. Once the pathologic process reaches the dopaminergic neurons of the substantia nigra in the midbrain, striatal dopamine loss occurs. However, the loss of nigrostriatal dopaminergic neurons must cross a threshold to produce motor dysfunction related to PD. This review will address how imaging can provide clues as to when the pathophysiologic process begins and how this relates to clinical manifestations.
Such studies have direct relevance for identifying when the pathophysiologic process begins in the brain, potentially to identify pre-symptomatic patients for possible treatment with to-be-developed therapies that may forestall onset of symptoms or slow disease progression. Many PET methods have been applied to such studies, and we critically review the relevant literature. Along the same lines, those people with selected co-morbid conditions may be at greater risk for development of PD. For example, those with rapid eye movement (REM) sleep behavior disorder (RBD) may have as much as a 50–60% risk of developing PD3. Applying PET to a higher risk group of people could be particularly revealing.
PET also may provide a measure of disease progression. A variety of PET methods have been used for this in clinical studies to determine efficacy of interventions to slow disease progression. These techniques have been fraught with challenges, with frequent application of PET methods prior to adequate validation of the technique. Of course, interpretation of such studies depends in large part on how accurately one can diagnose PD. Clinically, this can be challenging since many of the clinical features of PD overlap with other degenerative disorders such as multisystems atrophy (MSA) and progressive supranuclear palsy (PSP) as well as non-degenerative conditions (drug-induced parkinsonism).Many studies have tried to use PET or single photon emission computed tomography (SPECT) to distinguish these various conditions. This has clinical implications for potential response to treatment and research implications for selection of cohorts for clinical trials. We will address how well molecular imaging can help in this endeavor.
Finally, molecular imaging has begun to reveal new insights into the pathophysiology associated with non-motor manifestations of PD. These non-motor manifestations can be quite varied from autonomic deficits affecting multiple systems to cognitive and psychiatric dysfunction. Cognitive dysfunction is quite pervasive in PD with about 10% progressing to dementia every year; and ultimately affecting more than 80% of those who survive for two decades or more4,5. Cortical synucleinopathy with Lewy pathology accompanies this cognitive impairment. Thus PET imaging of α-synuclein could be highly informative and may identify cortical progression prior to symptom onset. Development of such a radiopharmaceutical is currently underway but one is not yet available. Other imaging approaches will be discussed.
Motor Manifestations of PD
Onset of PD
Several key questions revolve around the onset of brain pathologic changes that underlie PD. The initial studies in humans suggested that striatal dopamine deficiency must reach 70% to produce motor dysfunction6,7. These studies used postmortem measures and extrapolated clinical onset from retrospective review of patients’ charts6,7. Furthermore, the focus included striatum (caudate and putamen) rather than the putamen which preferentially affects motor function8,9. Postmortem studies in people with incidental Lewy pathology and no history of parkinsonism revealed about 50% to 70% striatal dopamine reduction8. However, these postmortem studies may overestimate dopamine loss as post-mortem interval may reduce subsequent striatal dopamine measurements10. Thus, an in vivo technique to measure the integrity of nigrostriatal dopaminergic neurons could help address these limitations.
The most direct PET based measure to address this question would use a radiotracer specific for α-synuclein, however, such a PET radiotracer is yet to be developed. Alternatively, multiple investigators have used PET measures of nigrostriatal neurons targeting presynaptic sites in the striatum. The integrity of these presynaptic terminals has been evaluated primarily with three classes of radiotracers: 6-[18F]fluorodopa (FD; reflects aromatic amino acid decarboxylase (AADC) that converts levodopa to dopamine and its uptake and storage in presynaptic terminals), [11C]dihydrotetrabenazine (DTBZ; reflects vesicular monoamine transporter type 2 (VMAT2) in the membranes of presynaptic vesicles that store dopamine in preparation for synaptic release), and 2-beta-[11C]carbomethoxy-3-beta-4-fluorophenyltropane (CFT; reflects membranous dopamine transporter (DAT) specific for dopaminergic neurons important for reuptake of synaptic dopamine back into the presynaptic neuron). Multiple different DAT radiotracers including radiopharmaceuticals for SPECT (mostly tropane derivatives) have been used. CFT is a commonly used PET radiotracer. We will critically evaluate the results of some key prior PET studies using these radiotracers and discuss their potential roles from a clinical and research perspective.
The potential clinical utility of PET to identify early changes in PD was promising as images revealed decreased striatal accumulation in people with PD11–14. Studies with the other classes of radiotracers targeting presynaptic nigrostriatal neurons also reported reduced striatal uptake in either people with PD or in animal models of parkinsonism15–18. This also set the stage for trying to determine when pathophysiologic changes begin in the brain with PD. All three radiotracer types gave similar answers - linear back extrapolation of rate of change in early PD suggested that onset of pathologic changes began less than seven years before diagnosis19.
Another entirely different approach yielded very similar result. Using PET scans of [18F]fluorodeoxyglucose to obtain regional measures related to glucose metabolism, a covariance pattern was found in a principal components analysis (PCA) of the regional metabolism. This pattern of covarying regions has been called the Parkinson disease related pattern (PDRP), and a single subject score can be calculated to indicate how strongly this covariance pattern exists in an individual. The strength of this single subject score correlated with severity of motor ratings of PD, thereby providing face validity for the approach. Back extrapolating the strength of these single subject scores would predict that the abnormal covariance pattern began about five years prior, close to the same range as found with the presynaptic markers20. This same group also noted abnormal PDRP expression in the ipsilateral presymptomatic hemisphere in hemiparkinsonian patients and concluded that the PDRP pattern expression becomes abnormal about two years before the onset of motor symptoms suggesting its potential as a prodromal marker of PD21. PDRP expression also was elevated in people with idiopathic REM sleep behavior disorder (RBD) at levels comparable to the early PD22,23. Progression to PD could be predicted by the baseline PDRP expression pattern24. Thus FDG PET can aid in identifying presymptomatic individuals especially in an at-risk population.
The limitation of all of these methods is the assumption of a linear degeneration of either the nigrostriatal pathway, a subsequent disturbed covariance pattern of brain metabolism and a linear change in motor severity. Subsequent studies have shown the limitations of these assumptions. In fact, applying a multivariable, nonlinear approach to presynaptic markers based upon longitudinal scans in people with PD produced an estimate of onset of striatal abnormality about 17 years before diagnosis25.
It remains to be determined whether the first pathologic changes in the brain that indicate onset of PD involve a widespread network dysfunction, that could in fact reflect early deficits in caudal brainstem pigmented nuclei that project to many cortical areas or is the initial deposition of abnormal α-synuclein. If the latter is true, then measurements of abnormal uptake of α-synuclein radiotracer may shed light on this remaining question.
Identification of presymptomatic PD patients
Identification of subclinical pathologic defects in the nigrostriatal pathway is critically important for research and potentially for clinical treatment. The importance for clinical treatment will be relevant once we actually have a treatment that can forestall disease onset in a population at risk or even retard the tempo of progression after onset. Hence an objective PET imaging marker that can detect the earliest pathophysiologic changes has substantial value. Molecular imaging measures of presynaptic nigrostriatal markers may be helpful in this regard.
The motor symptoms of PD usually develop following the loss of a substantial proportion of putaminal dopamine terminal function whereas a lesser degree of nigrostriatal loss may not cause any clinical deficits. Thus, loss of nigrostriatal neurons begins before illness develops and presents a window of opportunity for presymptomatic identification of early neuronal damage in people with PD. Early studies provide support for this notion. An FD PET study showed abnormal uptake in four MPTP (a neurotoxin that selective destroys dopaminergic neurons) exposed individuals even before the onset of clinical parkinsonism12. Another FD PET study of 17 asymptomatic monozygotic and dizygotic twins of patients with PD reported significantly reduced putaminal FD uptake (83–87% of normal) but no significant difference was evident in the caudate26. Interestingly, one of the monozygotic twins in this study developed PD symptoms within two years highlighting the potential of FD PET to detect nigrostriatal deficits in presymptomatic individuals. But this type of study must be interpreted with caution. This subject had a postural tremor at the time of the PET, which strictly speaking can be considered to be a forme fruste initial manifestation of PD, thus this may have been a symptomatic patient rather than one without any manifestations26.
The data from animal models are somewhat clearer. MPTP treated cynmologous monkeys demonstrated significant reduction of striatal FD uptake even in the absence of clinical signs27. This was further corroborated by another group where sequential MPTP treated monkeys at 3–6 week interval demonstrated significant reduction of uptake of radiolabeled β-[11C]DOPA and a DAT radioligand [123I]PE2I even before the emergence of tremor. The tremor coincided with the reduction in [123I]PE2I binding to about 15% and β-[11C]DOPA uptake to about 34% of pretreatment levels28. In a separate, large scale longitudinal study using all the three presynaptic dopaminergic markers, only DTBZ but not FD or another DAT marker [11C]d-threo-methylphenidate (MP) was noted to be significantly reduced in PD patients with younger disease onset and estimates based on extrapolated calculations (without actual data points) suggested the ability of DTBZ, MP and FD to identify presynaptic dopaminergic deficits about 17, 13 and 6 years respectively prior to actual onset of motor symptoms19,29,30 while some others have projected disparate estimates ranging up to 50 years based on linear calculations31. Reduced DAT binding (using MP) was noted to be one of the earliest imaging markers in asymptomatic carriers of the autosomal dominant LRRK2 (leucine-rich repeat kinase 2) mutation. The reduced DAT binding was observed in the setting of normal FD PET, but the onset of clinical symptoms coincided with abnormal FD uptake32. A recent longitudinal study showed 14 out of 21 (67%) hyposmic individuals with an abnormal (uptake <65% of predicted) DAT SPECT converted to PD at 4 years compared to 2 of 22 hyposmic individuals with indeterminate and 3 of 109 with a negative DAT SPECT scan who went on to develop PD33. Thus molecular imaging can be more sensitive than clinical findings and this could be of particular value in an at-risk population, although caution must be exercised as the line of demarcation between normal and abnormal findings can be somewhat murky especially with minimal defects in the nigrostriatal pathway or in individuals with only subtle clinical deficits. The clinical utility of presymptomatic identification remains limited until development of a treatment which could forestall the disease manifestation.
On the other hand, such presymptomatic identification has research implications. For example, potential identification of an “affected” individual could act as an endophenotype for genetics research. PET potentially could “convert” a normal to an “affected” for genetic investigations of pedigrees with familial parkinsonism. However, some patients with overt early PD manifestations including asymmetric resting tremor with bradykinesia and rigidity may have entirely normal FD PET and yet go on to develop typical PD manifestations34. Thus, a normal PET does not exclude PD and may misidentify a person with presymptomatic PD.
Measurements of PD progression
An objective measure of PD progression can be a critical part of assessment of a disease modifying therapy35. Many have suggested that PET or SPECT measures of striatal uptake of presynaptic radiotracers may provide such an objective measure. This assumes that progressive loss of nigrostriatal neurons reflects disease progression21. As noted, quantification of abnormal deposition of α-synuclein may be a more direct measure but that is not yet available.
Multiple clinical trials of PD used molecular imaging to measure effects of an intervention. However, these presynaptic nigrostriatal molecular imaging biomarkers did not match clinical measures of disease progression. The ELLDOPA study prospectively tested three different doses of levodopa and placebo in a large cohort of PD patients (n=361)36. Clinical progression determined by a standardized clinical rating scale of motor parkinsonism yielded a completely different result from a SPECT-based measure of striatal DAT. The clinical measure suggested that those treated with the highest dose of levodopa had reduced disease progression over nine months whereas the SPECT DAT suggested in a large subgroup that this group fared worse. The clinical measures were thus discordant with the imaging measures. Several other interventional studies also reported discordant results37–41. These discrepancies raise questions about the validity of the neuroimaging measures and how they had been validated42–44. The bottom line is that the striatal uptake of any of the three presynaptic tracers correlated well with striatal dopamine but not with nigral dopaminergic cell bodies45.
To address these issues, we performed a meticulous analysis of striatal uptake using the three PET tracers (FD, DTBZ, CFT) in MPTP treated non-human primates45,46. All animals had PET imaging at baseline and at 8 weeks after unilateral intracarotid infusion of varying doses of MPTP. We previously demonstrated that this approach produces a stable, chronic hemiparkinsonism by 3–4 weeks47. Animals had blinded video motor ratings, were euthanized after the second PET for striatal dopamine measurements and unbiased counts of tyrosine hydroxylase stained nigral cells (this identifies dopaminergic cells). The relevant finding for this part of the review is that parkinsonism developed once animals had 25–35% loss of either striatal dopamine or unbiased stereologic counts of dopaminergic cell bodies in substantia nigra. Striatal PET measures of all three radiotracers types correlated strongly with striatal dopamine and with severity of parkinsonism. However, striatal uptake of any of the tracers no longer correlated with parkinsonism or nigral dopaminergic cell counts once nigral cell loss exceeded 50%. These data suggest that nigrostriatal reserve is substantially less than previously thought, in other words, less nigrostriatal injury is required to produce parkinsonism, at least in nonhuman primates after unilateral MPTP-induced injury.
This means that motor parkinsonism correlated with nigral cell counts but not with striatal dopamine. Once nigral cell counts decreased to about 50% striatal dopamine were near zero, yet motor parkinsonism continued to progress as nigral cell counts further decreased. One could speculate that this relationship occurred only in the MPTP (toxin) induced injury. However, similar results were found in people with PD. Postmortem measures from humans that had PD revealed that nigrostriatal neurons appear to reach a nadir when a person had only mild to relatively moderate PD48. Similarly, PET measures of striatal uptake of a presynaptic radiotracer in a longitudinal study in PD demonstrated that posterior putaminal uptake of a DAT marker reached a nadir within five years of PD diagnosis49. Thus two studies in human PD confirm the findings in MPTP-treated nonhuman primates.
Thus surprisingly, striatal uptake of any of the presynaptic markers only correlates with motor progression during the early course of PD45. Interestingly, PET measures of midbrain uptake of either a DAT or VMAT2 radiotracer did correlate well with nigral dopaminergic cell counts and severity of parkinsonism throughout the full range of parkinsonism severity and has potential to act as a neuroimaging biomarker of nigral dopaminergic cell loss and severity of parkinsonism50.
These findings also likely explain many of the reported discrepancies in studies of PD progression. Most of the studies recruited unmedicated, newly diagnosed patients and then followed them in the study. During the course of the follow up, many may have reached the nadir in striatal measures—leading to striatal molecular imaging measures that no longer correlate with severity of parkinsonism and is likely fraught with substantial noise35,45.
FDG and principal components analysis have been proposed as a measure of disease progression. A longitudinal analysis spanning 4 years was carried out in 15 early PD patients within 2 years of diagnosis51. The patients underwent scanning with FDG as well as PET imaging with a presynaptic dopaminergic terminal marker [18F]-fluoropropyl βCIT (FP-CIT), a marker of presynaptic DAT at multiple time points including baseline, 24 and 48 months. UPDRS motor measures also were collected at each time point. Statistical parametric mapping (SPM) analysis revealed longitudinal increase in glucose utilization over time in subthalamic nucleus, globus pallidus interna, dorsal pons, primary motor cortex with concomitant decrease in the prefrontal and inferior parietal regions. This was associated with a significant increase in both PDRP and the PD related cognitive pattern (PDCP); the latter progressed at a much slower pace reaching abnormal values only at the 4 year mark. As had been noted in multiple prior cross sectional FDG PET studies in PD patients of varying motor severity52,53, the PDRP expression correlated well with UPDRS at each time point. The PDRP also correlated with the declining striatal FP-CIT uptake. The PDCP did not correlate with the UPDRS or the DAT binding and no cognitive measures were available for correlation. The trajectory of progression of the disease specific spatial covariance patterns and their behavioral correlations has not been assessed in longitudinal studies with a bigger population or over a longer time interval.
Selection of participants for clinical trials of PD
Since parkinsonian manifestations can overlap with other types of parkinsonism, a means to reliably identify those with underlying PD could potentially enhance the homogeneity of recruited participants for clinical trials of PD. Can molecular imaging help in this regard? Even if this works, the generalizability of the study will be limited. If a molecular imaging is used to screen participants in an interventional trial then the findings in the trial may only apply to those who are screened in the same manner. Thus, if an intervention demonstrates slowing of progression in this type of study, then application of the intervention may require similar molecular imaging screening.
Approaches to this differential diagnostic process can be considered at two levels: the ability to reliably differentiate PD from healthy individuals and at the second level to differentiate PD from atypical parkinsonism (APs that includes MSA, PSP, etc.). Multiple PET imaging studies with presynaptic terminal markers have reasonably distinguished PD from controls (Fig 1) but as noted some false negatives do occur13,54,55. Nevertheless, presynaptic terminal imaging markers have great value from a research perspective to help define a more homogeneous group with abnormal scans as inclusion criteria for enrollment, but in the same vein it would reduce the generalizability of the results of the study to only those who meet the scanning criteria.
Figure 1.

6-[18F]fluorodopa (FD) PET in a normal individual (A) and two PD patients with mild (B) and moderate (B) motor impairments showing asymmetric, gradient loss of putaminal FD uptake in PD patients compared to controls.
But the ability of PET with presynaptic dopaminergic terminal markers to distinguish PD from APs remains to be proven. The pattern of striatal FD uptake in PD lacks specificity; other conditions including MSA, post encephalitic parkinsonism, spinocerebellar ataxia type 2 can produce similar changes56. The striatal FD uptake in clinically diagnosed MSA was heterogeneous ranging from putaminal sparing identical to PD to a more uniform striatal involvement that has been described as the characteristic pattern of PSP57. One study initially suggested the ability of DAT SPECT to pick up a statistically significant reduction in DAT uptake in the caudate in PSP58. Another larger cross sectional DAT SPECT study reliably differentiated parkinsonian syndromes (57/61 had abnormal scans) from ET (none of 11 had abnormal DAT uptake) but failed to differentiate PD from APs 59,60. Similar limitations occur with DAT SPECT scans and this been reviewed recently35,61.
PET imaging could rationally influence clinical decision making process. PET-assisted diagnosis could help select appropriate candidates for deep stimulation surgery or help identification of implantation sites for deep brain stimulation (DBS). Whether this targeting strategy produces clinical benefit remains to be determined. It is also important to reiterate that optimal clinical management still heavily relies on meticulous clinical assessment and close follow-up. Hence PET imaging with presynaptic dopaminergic terminal imaging markers can reasonably distinguish PD (especially with moderate motor impairment) from controls but has not yet proven its worth in differentiating PD from atypical parkinsonisms or in their accurate differential diagnosis.
PCA based disease specific spatial covariance pattern of FDG may help differentiate AP from PD. An initial FDG PET analysis identified relative striatal and thalamic hypometabolism in 75% of AP cases when compared to PD62. Specific patterns of regional hypometabolism were also noted in MSA marked by reductions in putamen and caudate compared with disease duration/severity matched PD patients or controls63 while another separate study on an independent cohort found regional hypometabolism additionally in the cerebellum and brainstem in nine early stage clinically diagnosed MSA patients64. The autonomic symptoms correlated with brainstem glucose metabolism while the striatal FD uptake correlated with the motor symptoms64. Analysis of 41 clinically diagnosed probable or possible PSP subjects found relative reductions in glucose utilization quite diffusely in the frontal, parietal association, thalamus, caudate and cerebellum65. Cortico-thalamic metabolic asymmetry that was greater than control subjects or asymmetric PD patients were noted in FDG PET scans from five patients with corticobasal degeneration (CBD)66. Another study used statistical parametric imaging (SPM) tools to create disease related templates from FDG PET scans from a random selection of eight patients from each of PD, MSA, CBD and PSP patient groups. Application of this template either by visual assessment or a blinded SPM based computer assisted interpretation of individual FDG PET scans showed a diagnostic accuracy (compared to clinical diagnosis at 2 year follow-up) of 85.4% and 92.4% respectively67. While the numbers were not drastically different from prior clinico-neuropathologic correlations68,69, it is interesting to note that the computer assessment agreed with the clinical assessment for only 86.5% of healthy control subjects while it was about 90.9% for the visual inspection. Specific disease related spatial covariance patterns may exist for MSA70,71, PSP71 and CBD72 but their utility in accurate differential diagnosis is not entirely clear. In a recent study of 51 PD with 127 AP patients, the PDRP in nondemented PD patients while significantly different from controls was not significantly different compared to the MSA-P, PSP, or CBD subgroups. Considerable between groups overlap was also evident with MSA, PSP or CBD related patterns, although it is important to note that dopaminergic medications were not held prior to scans which could have eroded some of the group differences in this analysis73. Another group utilizing a distinct methodological approach of automated voxel-based multivariate supervised machine learning reported even lower diagnostic accuracy of specific AP subtypes, varying between 45–62%74. A more recent study utilizing the previously delineated metabolic patterns and a two level logistic classification algorithm reported the ability to distinguish PD from AP with 94% specificity and 96% PPV in the first step. The indeterminate cases (~19%) were not included in the second step of analysis, where the AP cases were further differentiated into either MSA or PSP. This further revealed 79% sensitivity, 90% specificity and 85% PPV for MSA while the sensitivity, specificity and PPV for PSP was reported at 100%, 94% and 94% respectively. 60% of the subjects had disease duration of less than 2 years and a greater rate of false negatives was noted for classifying AP patients with short duration of symptoms. Also, important to note that the PPVs reported here were more reflective of the study population with 37% AP patients, that was significantly higher than the general population23,75. Of note, the clinical diagnosis was selected as the gold standard and there was no neuropathologic confirmation of the inferences made with imaging in any of the abovementioned studies. As previously discussed the positive predictive value of a clinical diagnosis of the AP subtypes hovers around 80% and could be even worse early in the disease process, so drawing critical inferences based on the above results might be premature. FDG-PET could have a role in the differential diagnosis of AP subtypes, but further studies and preferably pathologic correlations are required at this stage.
Thus, overall, molecular imaging may help identify a more homogenous cohort for clinical trials but at the expense of loss of some of the generalizability of the study findings.
Non-motor manifestations
Measure of cognitive impairment
Several molecular imaging approaches have been used to investigate pathophysiology underlying cognitive impairment that commonly occurs in people with PD. In this section, we will review FDG based measures, then discuss the role of receptor measures and finally address proteinopathy based approaches. The value of such studies is to try to identify new targets of engagement for therapies aimed at addressing this poorly treated aspect of PD.
FDG PET:
PCA based approaches of FDG PET has been a major focus of multiple studies. The PDCP spatial covariance pattern in FDG PET images is characterized by relative metabolic reductions in frontal and parietal association areas with concomitant increases in cerebellar vermis and dentate and was noted even in cognitively normal PD patients76. The strength of the individual subject PDCP expression predicted memory performances, visuospatial function and executive functioning. Comparison of the PDCP pattern in cognitively intact PD versus single domain mild cognitive impairment (MCI) and multiple cognitive domains MCI revealed a stepwise increment in brainstem/cerebellar metabolism and concurrent decrements in the prefrontal and parietal metabolism with worsening cognition77. The PDCP pattern has some topographic resemblance to the frontoparietal control network. Levodopa mediated changes in verbal learning occurred only in a subset of PD patients with cognitive impairment; these patients had a higher baseline PDCP expression and the cognitive response correlated with the levodopa mediated reduction of PDCP expression. PDCP expression was not different in the non-responders or in the subset of patients with improvement in cognitive measures after placebo78. Analysis of FDG and FD PET in 106 PD subjects showed correlation of PDRP expression with FD uptake in caudate and putamen (and only putamen after controlling for PDCP expression). In contrast, PDCP expression correlated with the anterior striatum, but only with modest effect sizes suggesting the metabolic patterns did not only reflect underlying nigrostriatal denervation79. Another longitudinal FDG-PET study by a separate group utilizing a volume-of-interest and stereotactic surface projection (SSP) based analysis revealed hypometabolism in the visual association cortex, posterior cingulate cortices and to a lesser extent in the caudate that could predict subsequent dementia onset in cognitively intact PD patients80. Follow-up scans in a subset of these patients who progressed to dementia revealed interval reduction of metabolism in multiple cortical areas including the thalamus, posterior cingulate, occipital, parietal and frontal cortices compared to baseline with relatively preserved metabolism in the temporal cortex. A recent longitudinal study with 79 newly diagnosed PD patients and 20 controls reported hypometabolism in the occipital and inferior parietal lobes in the PD cohort; the parietotemporal and frontal metabolism correlated with memory based and attentional tasks; while the baseline parietal hypometabolism predicted the MMSE and MoCA at 18 months81. Prior cross-sectional and longitudinal SPECT studies revealed regional hypoperfusion in PD patients with dementia in those same regions as compared to PD patients with intact cognition82,83. Interestingly the cuneus has the most severe cholinergic denervation in demented PD patients and the reduced glucose metabolism could possibly reflect an ongoing cholinergic deafferentation80.
Cholinergic system:
Measures of cholinergic neurons also may be particularly pertinent to non-motor features of PD. Initial PET studies using N-[11C]methyl-piperidin-4yl-propionate (PMP), an acetylcholine analogue and marker of acetylcholinesterase (AChE) activity showed significantly reduced cortical AChE activity in both PD with dementia (PDD) (−20.9%) and PD without dementia (−12.7%) compared to controls and the AChE activity correlated with attentional and executive functioning as well as working memory but not with severity or duration of motor symptoms84. Another PMP PET study reported similar findings additionally showing maximal reduction in medial occipital cortex (BA18) in PDD and dementia with Lewy bodies (DLB) patients; no group difference in AChE activity was noted between early or advanced PD patients or between DLB and PDD85. PET study utilizing PMP, DTBZ and VMAT2 compared PD patients with versus without falls; the PD fallers had significantly reduced cortical and thalamic AChE activity compared to nonfallers. But more interestingly, the thalamic AChE activity was significantly reduced only in the fallers but not the nonfallers compared to controls; this was also noted to be decreased in fallers even after adjusting for nigrostriatal dysfunction86. This highlights the importance of the cholinergic outputs from the PPN to the thalamus in regulating gait and postural stability. AChE activity also correlated with gait speed in PD patients without dementia and no significant difference in gait speed was noted with isolated nigrostriatal dysfunction in the absence of cholinergic denervation87. Another PET study using radiopharmaceuticalN-methyl-[11C]2-(4’-methylaminophenyl)-6-hydroxybenzothiazole also known as Pittsburgh compound B (PiB), DTBZ and PMP in 143 PD patients of which 20 had freezing of gait (FoG) found frequency of FoG was the highest (41.7%) in PD patients who had both increased amyloid burden (PiB+) as well as reduced AChE activity, the frequency was 30.4% if they were just PiB+ and 4.8% if they were neither PiB+ nor had any cholinergic denervation88. Even though the PD patients with FoG had significantly reduced DTBZ uptake, the role of extra nigral pathology in FoG was highlighted by this study. Studies with radiotracers targeting the postsynaptic muscarinic receptor showed increased muscarinic acetylcholine receptor (AChR) density in the frontal cortex of PD patients but without any behavioral correlates89,90. Postsynaptic nicotinic AChR density was also analyzed in PD patients with 2-[18F]fluoro-3-(2[S]-2-azetidinylmethoxy)-pyridine (2FA); this showed reduced uptake in multiple cortical and subcortical regions in PD patients compared to controls, and the binding correlated with both depressive and cognitive symptoms91.
PET studies using radiotracers targeting vesicular acetylcholine transporter (VAChT) is a substantial advance that enables qualitative and quantitative analysis of presynaptic cholinergic nerve terminals. The initial human PET study in healthy controls using (−)-5-[18F]-fluoroethoxybenzovesamicol (FEOBV) tracer, a vesamicol derivative that selectively binds VAChT showed highest binding in the striatum, followed by thalamus, vermis and cerebral cortex92. The original developers of this radioligand are currently applying this to a large longitudinal cohort of people with people but no studies in PD have been reported to date. We also characterized a VAChT tracer in nonhuman primates93,94 and studies in PD patients are currently ongoing. Thus cholinergic PET imaging has immense potential to delineate the pathophysiologic bases of two key causes of morbidity and mortality in PD patients that are refractory to levodopa, namely gait and cognition. With the advent of superior radiotracers that are able to better demonstrate cholinergic pathology in subcortical areas of interest especially cerebellar vermis, lot remains to be told in this area.
Proteinopathy:
PET assays with radiotracers targeting abnormal proteins have enhanced our understanding of the underlying pathophysiology of neurodegenerative disorders. The prodromal pathology or disease progression and response to therapy could be best assessed by a radiotracer specific for α-synuclein. This still continues to elude the field because of technical challenges of finding a lipophilic ligand with a low background signal that would effectively cross the blood brain barrier and then specifically bind to the intracellular oligomeric α-synuclein95. Radiolabeled benzoxazole compounds or phenothiazines lack specificity given their affinity for β-amyloid and/or tau and high signal to noise ratio precludes their use for invivo imaging96.
PiB PET amyloid imaging has enabled the in vivo measurement of fibrillary β-amyloid (Aβ)97. This has been particularly important in delineating the pathophysiology of cognitive impairment in Alzheimer disease (AD) where Aβ deposition precedes the abnormal deposition of tau and onset of dementia; in fact the increased uptake of PiB predicts an early onset of dementia98,99. The presence of amyloid pathology has long been reported in neuropathologic studies of PD especially in the presence of dementia but the prevalence of classical AD pathology or its role in dementia in PD was far from clear100–102. A PET study with PiB in 13 DLB, 12 PDD, 10 PD with normal cognition and 41 age matched controls showed significantly increased amyloid load in over 80% of subjects with DLB and in only two of the subjects with PDD, although there was no significant difference in the regional pattern of uptake. None of the nondemented PD subjects had an increased cortical PiB uptake103. Another study found a similar proportion of DLB and PDD subjects with increased PiB uptake (PiB+) but additionally noted reduced cerebrospinal fluid (CSF) Aβ42 level, lower MMSE and higher ApoE4 prevalence in PiB+ patients. Motor symptoms did not correlate with the PiB binding; this led them to contemplate the predominance of AD pathology in the subjects with increased PiB uptake104. But it was far from clear whether a positive PiB scan actually indicated an AD pathology and the true answer lay in the pathologic validation of the PET findings.
Initial data from a large scale ongoing longitudinal study at our center has helped clarify this. Analysis with only a limited number of subjects found an equal proportion of subjects in each of the groups based on cognitive status (PD with normal cognition, PD-MCI, PDD, DLB and age matched healthy controls) with elevated PiB binding potential (BP) for predefined cortical regions and mean cortical binding potential (MCBP). Elevated PiB BP was associated with worse global cognition but did not correlate with earlier onset or faster rate of progression of cognitive impairment105. Initial pathologic correlation from 3 individuals with PDD who had a PiB PET imaging within 15 months of death showed the two PiB+ PET scans had abundant diffuse Aβ plaques but only scant neuritic plaques and intermediate neurofibrillary tangle pathology and hence did not meet the NIA-Raegan criteria for the diagnosis of AD. All three patients had abundant cortical Lewy bodies (Braak PD stage 6), thus the PiB-PET scan in the third individual who had only rare diffuse Aβ plaques proved the specificity of PiB-PET for fibrillary Aβ amyloid and not α-synuclein (Fig 2). More importantly this study showed that PiB+ PET was not sufficient to indicate coexisting AD pathology in a PD patient106. In a subsequent autopsy series of 32 cases, it was clearly evident that classical AD pathology (with moderate tauopathy) was present in only about 3% of the PD cases. But further analysis showed substantial accumulation of Aβ in addition to cortical synucleinopathy predicted significantly shorter survival rates. Although there was a trend towards it, it did not necessarily predict an earlier onset of dementia107. Interestingly even though Aβ pathology was noted in 59% of the PD patients, only about 20% had PiB+ PET scans in this study. While the time lag between initial PiB PET and autopsy could contribute to this discrepancy, the specificity of the PiB tracer for labeling only the fibrillar Aβ seems to be the major determinant in this case108. A study with baseline PiB PET and longitudinal clinical measures showed the increased PiB uptake predicted rapid decline in cognition but did not alter the tempo of motor progression in PD patients109. A cross sectional PiB PET study of 40 patients of PD with MCI showed only 6 (15%) subjects with PiB binding at levels comparable to AD, but a significant correlation between cortical PiB binding and global composite cognitive function was noted110. This pattern of higher PiB+ studies in DLB compared to PDD, and the lower prevalence of PiB+ scans in PD-MCI as compared to non-PD related MCI was also evident in a recent systematic review of the literature111. The pooled prevalence of PiB+ studies was 0.68 in the DLB group, 0.34 in the PDD group and 0.05 in the PD-MCI group. Interestingly this was lower than the 25–30% range reported in prior studies of unaffected elderly subjects. Another study found higher cortical amyloid burden in the DLB group, comparable to AD patients, but amyloid deposition in the PDD group was low and comparable to the PD and normal controls. The amyloid deposition in the lateral parietal/precuneus and posterior cingulate regions in the DLB, PDD and PD groups correlated with visuospatial impairment112. A PCA analysis of PiB PET scans from AD patients and PD patients with cognitive impairment showed contrasting PiB binding pattern in the two groups possibly alluding to a different pathophysiologic role of Aβ deposition in cognitive impairment in PD113. CSF-Aβ was found to be lower in nondemented PD patients compared to controls and the PiB MCBP inversely correlated with the CSF-Aβ level. Interestingly, the CSF-α-synuclein level was also found to correlate with the CSF-Aβ level thus suggesting a pathophysiologic connection between their metabolisms in PD114. Thus a PiB PET scan in PD patients reflects fibrillary amyloid burden but this does not necessarily translate to a coexisting AD pathology; and this may predict a more rapid cognitive decline and possibly shorter survival.
Figure 2.
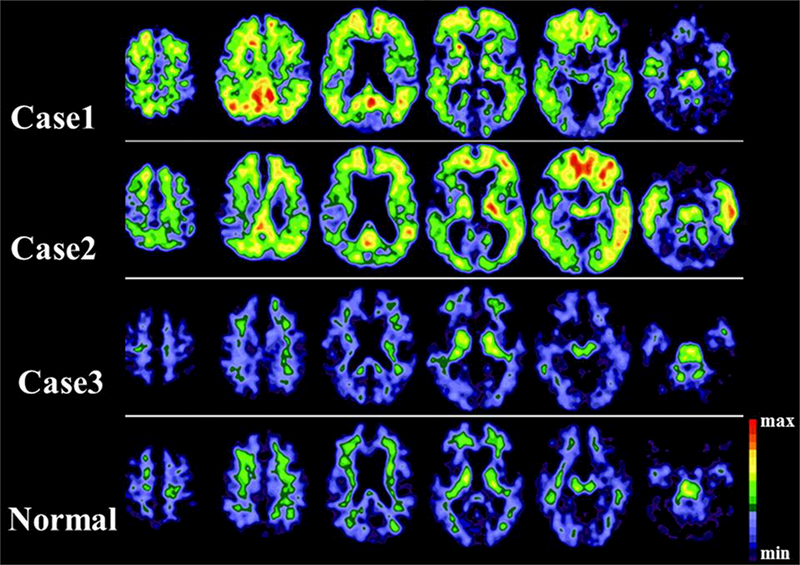
[11C]-PiB PET images from three PD patients with dementia (Case 1–3) and a normal individual. Case 1 and Case 2 both have increased PiB signal in multiple cortical areas signifying diffuse amyloid burden, while Case 3 had only minimal cortical PiB signal like the normal individual. The signal in the white matter areas is likely secondary to non-specific PiB binding. (Image adapted from Burack et al.106)
Tau depositions reflecting neurofibrillary tangle (NFT) pathology have been detected with [18F]-AV-1451 (AV) radiotracer in non-PD patients with MCI115. A recent PET study with 26 PD patients (nine had MCI) and 25 controls reported no significant difference between the PD-MCI and PD patients with normal cognition, in keeping with our prior pathologic analysis116. Another cross sectional study of AV-PET and PiB PET in 7 DLB, 8 PD with cognitive impairment and 9 PD patients with normal cognition reported somewhat contrasting findings. AV cortical uptake was significantly elevated in DLB patients compared to controls particularly in the inferior temporal gyrus and precuneus and this correlated with cognitive measures117. Quite interestingly, the PiB uptake did not correlate with AV PET measures. Also, 17 out of the 21 patients with Lewy body disease had a low amyloid burden (distribution volume ratio <1.15) but 4 of these 17 patients had elevated AV uptake in spite of a low amyloid burden. This was in stark contrast to our prior pathologic findings107. The nascent data with AV PET needs to be interpreted with caution given the important lesson from our experience with PiB that further validation especially pathologic correlation could be the key missing piece; also binding specificity of the AV tracer in an environment with PD pathology may be different from AD. So this remains to be sorted out.
Dopamine receptors:
Dopamine receptors belong to two broad categories: the D1—like family (D1, D5; usually presynaptic; stimulates adenylyl cyclase and generates cAMP), and D2—like family (D2, D3 and D4; inhibits adenylyl cyclase and decrease cAMP levels; D2 receptors can be both pre or postsynaptic). The striatum harbors an equal density of D1 and D2 receptors118. No pathophysiologic correlates of striatal D1-like receptor density were found in PD patients90,119 perhaps due to the low selectivity of the radiotracers120. The D2-like ligands can either be antagonists of high affinity that bind irreversibly to the D2-like receptor, or of low affinity like ([11C]raclopride, (RAC)). RAC because of its low affinity to the D2-like receptor can be subject to competition by endogenous striatal dopamine concentration and striatal changes in RAC binding potential can reflect dynamic changes in the synaptic concentration of dopamine121. Multiple studies have highlighted the role of RAC PET in dissecting the pathophysiology of levodopa induced dyskinesia90,122 and in predicting onset of motor fluctuations in PD patients123. But more recent RAC PET studies have shed light on the pathophysiology of certain non-motor manifestations of PD. A RAC PET study showed significantly reduced dopamine release in the dorsal caudate but not in the anterior cingulate cortex of PD patients compared to controls during the performance of an executive task implicating the association of nigrostriatal dopaminergic deficit with executive dysfunction in PD124; but detection of changes in anterior cingulate could be challenging. Another RAC PET study revealed greater synaptic release of dopamine in the ventral striatum of PD patients with pathological gambling both during gambling and during performance of a control task as compared to PD patients without pathological gambling125. Significant reduction in the extrastriatal D2 receptor density was noted in the hypothalamus of PD patients compared to controls126.
Few studies have explored the role of D3 receptors, in large part due to inadequate specific radiotracers. A modestly selective D3 radioligand [11C]-PHNO found low levels of hypothalamic D3 receptors that correlated with excessive daytime sleepiness in PD patients127.
Serotonergic receptors:
PET studies with [11C]DASB, a marker of serotonin transporter have highlighted the pathophysiologic role of serotonergic dysfunction in multiple non-motor symptoms in PD including depression128, fatigue129, visual hallucinations and sleep problems90,130. An inverse relationship between cortical serotonergic innervation and the amyloid burden in PD patients has also been reported; the relationship in the striatum is less clear131,132.
Phosphodiesterases:
Phosphodiesterases (PDE) are intracellular enzymes that catalyze hydrolysis of the second messengers cAMP and cGMP; PDE10A is almost exclusively found in the axons of striatal medium spiny neurons and plays an important role in the dopaminergic signaling cascade133. A PET study using [11C]-IMA107, a PDE10A specific radioligand showed lower expression of PDE10A in caudate, putamen and globus pallidus and this correlated with disease duration and motor severity134. A more recent study with [11C]rolipam, a marker of PDE4 showed reduced expression of PDE4 in multiple subregions of the striato-thalamo-cortical circuit including caudate, thalamus, hypothalamus, dorsolateral prefrontal cortex, medial frontal cortex and supplementary motor area in PD patients compared to controls that correlated with deficits of spatial working memory135.
Conclusion
In summary, multiple molecular imaging methods have provided exciting new critical insights into the pathophysiology of PD. These methods can help detect the onset of brain pathology, identify presymptomatic people destined to develop PD; provide a measure of disease progression - at least during the early milder stages of PD; can help to enrich cohorts of PD for clinical trials and investigate non-motor aspects of PD. However, the approaches used for these studies have important limitations that, in some cases, may restrict application or interpretation of findings, yet careful investigations can lead to significant advances in our understanding of PD.
Acknowledgments
We would like to thank our funding sources, including NIH/NINDS/NIA (NS075321; NS097437; NS103957, NS41509), the American Academy of Neurology Clinical Research Training Fellowship in Parkinson disease funded by American Brain Foundation and AbbVie, the American Parkinson Disease Association (APDA) Advanced Research Center at Washington University, the St. Louis Chapter of the APDA, the Jo Oertli Fund, the Washington University ICTS, and the Barnes Jewish Hospital Foundation (Elliot Stein Family Fund & Parkinson disease research Fund).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marttila RJ, Rinne UK. Epidemiology of Parkinson’s disease in Finland. Acta neurologica Scandinavica. February 1976;53(2):81–102. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell and tissue research. October 2004;318(1):121–134. [DOI] [PubMed] [Google Scholar]
- 3.Iranzo A, Fernandez-Arcos A, Tolosa E, et al. Neurodegenerative disorder risk in idiopathic REM sleep behavior disorder: study in 174 patients. PloS one. 2014;9(2):e89741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aarsland D, Kurz MW. The epidemiology of dementia associated with Parkinson’s disease. Brain pathology May 2010;20(3):633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Callaghan C, Lewis SJG. Cognition in Parkinson’s Disease. International review of neurobiology. 2017;133:557–583. [DOI] [PubMed] [Google Scholar]
- 6.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. Journal of the neurological sciences. December 1973;20(4):415–455. [DOI] [PubMed] [Google Scholar]
- 7.Riederer P, Wuketich S. Time course of nigrostriatal degeneration in parkinson’s disease. A detailed study of influential factors in human brain amine analysis. J Neural Transm. 1976;38(3–4):277–301. [DOI] [PubMed] [Google Scholar]
- 8.Scherman D, Desnos C, Darchen F, Pollak P, Javoy-Agid F, Agid Y. Striatal dopamine deficiency in Parkinson’s disease: role of aging. Annals of neurology. October 1989;26(4):551–557. [DOI] [PubMed] [Google Scholar]
- 9.Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Progress in neurobiology. November 1996;50(4):381–425. [DOI] [PubMed] [Google Scholar]
- 10.Carlsson A, Winblad B. Influence of age and time interval between death and autopsy on dopamine and 3-methoxytyramine levels in human basal ganglia. J Neural Transm. 1976;38(3–4):271–276. [DOI] [PubMed] [Google Scholar]
- 11.Eidelberg D, Moeller JR, Dhawan V, et al. The metabolic anatomy of Parkinson’s disease: complementary [18F]fluorodeoxyglucose and [18F]fluorodopa positron emission tomographic studies. Movement disorders : official journal of the Movement Disorder Society. 1990;5(3):203–213. [DOI] [PubMed] [Google Scholar]
- 12.Calne DB, Langston JW, Martin WR, et al. Positron emission tomography after MPTP: observations relating to the cause of Parkinson’s disease. Nature. September 19–25 1985;317(6034):246–248. [DOI] [PubMed] [Google Scholar]
- 13.Leenders KL, Salmon EP, Tyrrell P, et al. The nigrostriatal dopaminergic system assessed in vivo by positron emission tomography in healthy volunteer subjects and patients with Parkinson’s disease. Archives of neurology. December 1990;47(12):1290–1298. [DOI] [PubMed] [Google Scholar]
- 14.Hoshi H, Kuwabara H, Leger G, Cumming P, Guttman M, Gjedde A. 6-[18F]fluoro-L-dopa metabolism in living human brain: a comparison of six analytical methods. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. January 1993;13(1):57–69. [DOI] [PubMed] [Google Scholar]
- 15.De La Fuente-Fernandez R, Lim AS, Sossi V, et al. Age and severity of nigrostriatal damage at onset of Parkinson’s disease. Synapse. February 2003;47(2):152–158. [DOI] [PubMed] [Google Scholar]
- 16.Huang WS, Ma KH, Chou YH, Chen CY, Liu RS, Liu JC. 99mTc-TRODAT-1 SPECT in healthy and 6-OHDA lesioned parkinsonian monkeys: comparison with 18F-FDOPA PET. Nuclear medicine communications. January 2003;24(1):77–83. [DOI] [PubMed] [Google Scholar]
- 17.Ribeiro MJ, Vidailhet M, Loc’h C, et al. Dopaminergic function and dopamine transporter binding assessed with positron emission tomography in Parkinson disease. Archives of neurology. April 2002;59(4):580–586. [DOI] [PubMed] [Google Scholar]
- 18.Kazumata K, Dhawan V, Chaly T, et al. Dopamine transporter imaging with fluorine-18-FPCIT and PET. J Nucl Med. September 1998;39(9):1521–1530. [PubMed] [Google Scholar]
- 19.Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson’s disease with [18F]dopa PET. Journal of neurology, neurosurgery, and psychiatry. March 1998;64(3):314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moeller JR, Eidelberg D. Divergent expression of regional metabolic topographies in Parkinson’s disease and normal ageing. Brain : a journal of neurology. December 1997;120 (Pt 12):2197–2206. [DOI] [PubMed] [Google Scholar]
- 21.Tang CC, Poston KL, Dhawan V, Eidelberg D. Abnormalities in metabolic network activity precede the onset of motor symptoms in Parkinson’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. January 20 2010;30(3):1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu P, Yu H, Peng S, et al. Consistent abnormalities in metabolic network activity in idiopathic rapid eye movement sleep behaviour disorder. Brain : a journal of neurology. December 2014;137(Pt 12):3122–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meles SK, Teune LK, de Jong BM, Dierckx RA, Leenders KL. Metabolic Imaging in Parkinson Disease. J Nucl Med. January 2017;58(1):23–28. [DOI] [PubMed] [Google Scholar]
- 24.Holtbernd F, Gagnon JF, Postuma RB, et al. Abnormal metabolic network activity in REM sleep behavior disorder. Neurology. February 18 2014;82(7):620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuramoto L, Cragg J, Nandhagopal R, et al. The nature of progression in Parkinson’s disease: an application of non-linear, multivariate, longitudinal random effects modelling. PloSone. 2013;8(10):e76595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burn DJ, Mark MH, Playford ED, et al. Parkinson’s disease in twins studied with 18F-dopa and positron emission tomography. Neurology. October 1992;42(10):1894–1900. [DOI] [PubMed] [Google Scholar]
- 27.Guttman M, Yong VW, Kim SU, et al. Asymptomatic striatal dopamine depletion: PET scans in unilateral MPTP monkeys. Synapse. 1988;2(5):469–473. [DOI] [PubMed] [Google Scholar]
- 28.Nagai Y, Obayashi S, Ando K, et al. Progressive changes of pre- and post-synaptic dopaminergic biomarkers in conscious MPTP-treated cynomolgus monkeys measured by positron emission tomography. Synapse. October 2007;61(10):809–819. [DOI] [PubMed] [Google Scholar]
- 29.Stoessl AJ. Developments in neuroimaging: positron emission tomography. Parkinsonism & related disorders. January 2014;20 Suppl 1:S180–183. [DOI] [PubMed] [Google Scholar]
- 30.de la Fuente-Fernandez R, Schulzer M, Kuramoto L, et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Annals of neurology. May 2011;69(5):803–810. [DOI] [PubMed] [Google Scholar]
- 31.Vingerhoets FJ, Snow BJ, Lee CS, Schulzer M, Mak E, Calne DB. Longitudinal fluorodopa positron emission tomographic studies of the evolution of idiopathic parkinsonism. Annals of neurology. November 1994;36(5):759–764. [DOI] [PubMed] [Google Scholar]
- 32.Nandhagopal R, Mak E, Schulzer M, et al. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology. November 25 2008;71(22):1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jennings D, Siderowf A, Stern M, et al. Conversion to Parkinson Disease in the PARS Hyposmic and Dopamine Transporter-Deficit Prodromal Cohort. JAMA neurology. August 1 2017;74(8):933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Racette BA, Good L, Antenor JA, et al. [18F]FDOPA PET as an endophenotype for Parkinson’s Disease linkage studies. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. April 5 2006;141B(3):245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perlmutter JS, Norris SA. Neuroimaging biomarkers for Parkinson disease: facts and fantasy. Annals of neurology. December 2014;76(6):769–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fahn S, Oakes D, Shoulson I, et al. Levodopa and the progression of Parkinson’s disease. The New England journal of medicine. December 9 2004;351(24):2498–2508. [DOI] [PubMed] [Google Scholar]
- 37.Whone AL, Watts RL, Stoessl AJ, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Annals of neurology. July 2003;54(1):93–101. [DOI] [PubMed] [Google Scholar]
- 38.Ravina B, Eidelberg D, Ahlskog JE, et al. The role of radiotracer imaging in Parkinson disease. Neurology. January 25 2005;64(2):208–215. [DOI] [PubMed] [Google Scholar]
- 39.Lang AE, Gill S, Patel NK, et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Annals of neurology. March 2006;59(3):459–466. [DOI] [PubMed] [Google Scholar]
- 40.Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. The New England journal of medicine. March 8 2001;344(10):710–719. [DOI] [PubMed] [Google Scholar]
- 41.Dorsey ER, Holloway RG, Ravina BM. Biomarkers in Parkinson’s disease. Expert review of neurotherapeutics. June 2006;6(6):823–831. [DOI] [PubMed] [Google Scholar]
- 42.Pate BD, Kawamata T, Yamada T, et al. Correlation of striatal fluorodopa uptake in the MPTP monkey with dopaminergic indices. Annals of neurology. September 1993;34(3):331–338. [DOI] [PubMed] [Google Scholar]
- 43.Snow BJ, Tooyama I, McGeer EG, et al. Human positron emission tomographic [18F]fluorodopa studies correlate with dopamine cell counts and levels. Annals of neurology. September 1993;34(3):324–330. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez-Fischer D, Blessmann G, Trosowski C, et al. Quantitative [(123)I]FP-CIT pinhole SPECT imaging predicts striatal dopamine levels, but not number of nigral neurons in different mouse models of Parkinson’s disease. NeuroImage. October 15 2007;38(1):5–12. [DOI] [PubMed] [Google Scholar]
- 45.Karimi M, Tian L, Brown CA, et al. Validation of nigrostriatal positron emission tomography measures: critical limits. Annals of neurology. March 2013;73(3):390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tabbal SD, Tian L, Karimi M, Brown CA, Loftin SK, Perlmutter JS. Low nigrostriatal reserve for motor parkinsonism in nonhuman primates. Experimental neurology. October 2012;237(2):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tabbal SD, Mink JW, Antenor JA, Carl JL, Moerlein SM, Perlmutter JS. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys associated with low striatal dopamine. Neuroscience. September 1 2006;141(3):1281–1287. [DOI] [PubMed] [Google Scholar]
- 48.Kordower JH, Olanow CW, Dodiya HB, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain : a journal of neurology. August 2013;136(Pt 8):2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nandhagopal R, Kuramoto L, Schulzer M, et al. Longitudinal evolution of compensatory changes in striatal dopamine processing in Parkinson’s disease. Brain : a journal of neurology. November 2011;134(Pt 11):3290–3298. [DOI] [PubMed] [Google Scholar]
- 50.Brown CA, Karimi MK, Tian L, et al. Validation of midbrain positron emission tomography measures for nigrostriatal neurons in macaques. Annals of neurology. October 2013;74(4):602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang C, Tang C, Feigin A, et al. Changes in network activity with the progression of Parkinson’s disease. Brain : a journal of neurology. July 2007;130(Pt 7):1834–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eidelberg D, Moeller JR, Ishikawa T, et al. Assessment of disease severity in parkinsonism with fluorine-18-fluorodeoxyglucose and PET. J Nucl Med. March 1995;36(3):378–383. [PubMed] [Google Scholar]
- 53.Asanuma K, Tang C, Ma Y, et al. Network modulation in the treatment of Parkinson’s disease. Brain : a journal of neurology. October 2006;129(Pt 10):2667–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Frost JJ, Rosier AJ, Reich SG, et al. Positron emission tomographic imaging of the dopamine transporter with 11C-WIN 35,428 reveals marked declines in mild Parkinson’s disease. Annals of neurology. September 1993;34(3):423–431. [DOI] [PubMed] [Google Scholar]
- 55.Ishikawa T, Dhawan V, Chaly T, et al. Clinical significance of striatal DOPA decarboxylase activity in Parkinson’s disease. J Nucl Med. February 1996;37(2):216–222. [PubMed] [Google Scholar]
- 56.Au WL, Adams JR, Troiano AR, Stoessl AJ. Parkinson’s disease: in vivo assessment of disease progression using positron emission tomography. Brain research. Molecular brain research. March 24 2005;134(1):24–33. [DOI] [PubMed] [Google Scholar]
- 57.Brooks DJ, Ibanez V, Sawle GV, et al. Differing patterns of striatal 18F-dopa uptake in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Annals of neurology. October 1990;28(4):547–555. [DOI] [PubMed] [Google Scholar]
- 58.Messa C, Volonte MA, Fazio F, et al. Differential distribution of striatal [123I]beta-CIT in Parkinson’s disease and progressive supranuclear palsy, evaluated with single-photon emission tomography. European journal of nuclear medicine. September 1998;25(9):1270–1276. [DOI] [PubMed] [Google Scholar]
- 59.Brooks DJ. Molecular imaging of dopamine transporters. Ageing research reviews. September 2016;30:114–121. [DOI] [PubMed] [Google Scholar]
- 60.Plotkin M, Amthauer H, Klaffke S, et al. Combined 123I-FP-CIT and 123I-IBZM SPECT for the diagnosis of parkinsonian syndromes: study on 72 patients. Journal of neural transmission. May 2005;112(5):677–692. [DOI] [PubMed] [Google Scholar]
- 61.Perlmutter JS, Eidelberg D. To scan or not to scan: DaT is the question. Neurology. March 6 2012;78(10):688–689. [DOI] [PubMed] [Google Scholar]
- 62.Antonini A, Kazumata K, Feigin A, et al. Differential diagnosis of parkinsonism with [18F]fluorodeoxyglucose and PET. Movement disorders : official journal of the Movement Disorder Society. March 1998;13(2):268–274. [DOI] [PubMed] [Google Scholar]
- 63.Eidelberg D, Takikawa S, Moeller JR, et al. Striatal hypometabolism distinguishes striatonigral degeneration from Parkinson’s disease. Annals of neurology. May 1993;33(5):518–527. [DOI] [PubMed] [Google Scholar]
- 64.Taniwaki T, Nakagawa M, Yamada T, et al. Cerebral metabolic changes in early multiple system atrophy: a PET study. Journal of the neurological sciences. August 15 2002;200(1–2):79–84. [DOI] [PubMed] [Google Scholar]
- 65.Blin J, Baron JC, Dubois B, et al. Positron emission tomography study in progressive supranuclear palsy. Brain hypometabolic pattern and clinicometabolic correlations. Archives of neurology. July 1990;47(7):747–752. [DOI] [PubMed] [Google Scholar]
- 66.Eidelberg D, Dhawan V, Moeller JR, et al. The metabolic landscape of cortico-basal ganglionic degeneration: regional asymmetries studied with positron emission tomography. Journal of neurology, neurosurgery, and psychiatry. October 1991;54(10):856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eckert T, Barnes A, Dhawan V, et al. FDG PET in the differential diagnosis of parkinsonian disorders. NeuroImage. July 1 2005;26(3):912–921. [DOI] [PubMed] [Google Scholar]
- 68.Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology. October 23 2001;57(8):1497–1499. [DOI] [PubMed] [Google Scholar]
- 69.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain : a journal of neurology. April 2002;125(Pt 4):861–870. [DOI] [PubMed] [Google Scholar]
- 70.Poston KL, Tang CC, Eckert T, et al. Network correlates of disease severity in multiple system atrophy. Neurology. April 17 2012;78(16):1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eckert T, Tang C, Ma Y, et al. Abnormal metabolic networks in atypical parkinsonism. Movement disorders : official journal of the Movement Disorder Society. April 15 2008;23(5):727–733. [DOI] [PubMed] [Google Scholar]
- 72.Niethammer M, Tang CC, Feigin A, et al. A disease-specific metabolic brain network associated with corticobasal degeneration. Brain : a journal of neurology. November 2014;137(Pt 11):3036–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ko JH, Lee CS, Eidelberg D. Metabolic network expression in parkinsonism: Clinical and dopaminergic correlations. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. February 2017;37(2):683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garraux G, Phillips C, Schrouff J, et al. Multiclass classification of FDG PET scans for the distinction between Parkinson’s disease and atypical parkinsonian syndromes. NeuroImage. Clinical 2013;2:883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tripathi M, Tang CC, Feigin A, et al. Automated Differential Diagnosis of Early Parkinsonism Using Metabolic Brain Networks: A Validation Study. J Nucl Med. January 2016;57(1):60–66. [DOI] [PubMed] [Google Scholar]
- 76.Huang C, Mattis P, Tang C, Perrine K, Carbon M, Eidelberg D. Metabolic brain networks associated with cognitive function in Parkinson’s disease. NeuroImage. January 15 2007;34(2):714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang C, Mattis P, Perrine K, Brown N, Dhawan V, Eidelberg D. Metabolic abnormalities associated with mild cognitive impairment in Parkinson disease. Neurology. April 15 2008;70(16 Pt 2):1470–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mattis PJ, Tang CC, Ma Y, Dhawan V, Eidelberg D. Network correlates of the cognitive response to levodopa in Parkinson disease. Neurology. August 30 2011;77(9):858–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holtbernd F, Ma Y, Peng S, et al. Dopaminergic correlates of metabolic network activity in Parkinson’s disease. Human brain mapping. September 2015;36(9):3575–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bohnen NI, Koeppe RA, Minoshima S, et al. Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J Nucl Med. June 2011;52(6):848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Firbank MJ, Yarnall AJ, Lawson RA, et al. Cerebral glucose metabolism and cognition in newly diagnosed Parkinson’s disease: ICICLE-PD study. Journal of neurology, neurosurgery, and psychiatry. April 2017;88(4):310–316. [DOI] [PubMed] [Google Scholar]
- 82.Firbank MJ, Burn DJ, McKeith IG, O’Brien JT. Longitudinal study of cerebral blood flow SPECT in Parkinson’s disease with dementia, and dementia with Lewy bodies. International journal of geriatric psychiatry. August 2005;20(8):776–782. [DOI] [PubMed] [Google Scholar]
- 83.Firbank MJ, Colloby SJ, Burn DJ, McKeith IG, O’Brien JT. Regional cerebral blood flow in Parkinson’s disease with and without dementia. NeuroImage. October 2003;20(2):1309–1319. [DOI] [PubMed] [Google Scholar]
- 84.Bohnen NI, Kaufer DI, Hendrickson R, et al. Cognitive correlates of cortical cholinergic denervation in Parkinson’s disease and parkinsonian dementia. Journal of neurology. February 2006;253(2):242–247. [DOI] [PubMed] [Google Scholar]
- 85.Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. July 28 2009;73(4):273–278. [DOI] [PubMed] [Google Scholar]
- 86.Bohnen NI, Muller ML, Koeppe RA, et al. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology. November 17 2009;73(20):1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bohnen NI, Frey KA, Studenski S, et al. Gait speed in Parkinson disease correlates with cholinergic degeneration. Neurology. October 29 2013;81(18):1611–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bohnen NI, Frey KA, Studenski S, et al. Extra-nigral pathological conditions are common in Parkinson’s disease with freezing of gait: an in vivo positron emission tomography study. Movement disorders : official journal of the Movement Disorder Society. August 2014;29(9): 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Asahina M, Shinotoh H, Hirayama K, et al. Hypersensitivity of cortical muscarinic receptors in Parkinson’s disease demonstrated by PET. Acta neurologica Scandinavica. June 1995;91(6):437–443. [DOI] [PubMed] [Google Scholar]
- 90.Politis M, Pagano G, Niccolini F. Imaging in Parkinson’s Disease. International review of neurobiology. 2017;132:233–274. [DOI] [PubMed] [Google Scholar]
- 91.Meyer PM, Strecker K, Kendziorra K, et al. Reduced alpha4beta2*-nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease. Archives of general psychiatry. August 2009;66(8):866–877. [DOI] [PubMed] [Google Scholar]
- 92.Petrou M, Frey KA, Kilbourn MR, et al. In vivo imaging of human cholinergic nerve terminals with (−)-5-(18)F-fluoroethoxybenzovesamicol: biodistribution, dosimetry, and tracer kinetic analyses. J Nucl Med. March 2014;55(3):396–404. [DOI] [PubMed] [Google Scholar]
- 93.Yue X, Bognar C, Zhang X, et al. Automated production of [(1)(8)F]VAT suitable for clinical PET study of vesicular acetylcholine transporter. Applied radiation and isotopes : including data, instrumentation and methods for use in agriculture, industry and medicine. January 2016;107:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tu Z, Zhang X, Jin H, et al. Synthesis and biological characterization of a promising F-18 PET tracer for vesicular acetylcholine transporter. Bioorganic & medicinal chemistry. August 1 2015;23(15):4699–4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brooks DJ, Tambasco N. Imaging synucleinopathies. Movement disorders : official journal of the Movement Disorder Society. June 2016;31(6):814–829. [DOI] [PubMed] [Google Scholar]
- 96.Bagchi DP, Yu L, Perlmutter JS, et al. Binding of the radioligand SIL23 to alpha-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PloS one. 2013;8(2):e55031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of neurology. March 2004;55(3):306–319. [DOI] [PubMed] [Google Scholar]
- 98.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. August 8 2006;67(3):446–452. [DOI] [PubMed] [Google Scholar]
- 99.Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Archives of neurology. December 2009;66(12):1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Archives of neurology. February 1993;50(2):140–148. [DOI] [PubMed] [Google Scholar]
- 101.Jellinger KA. Prevalence of Alzheimer lesions in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. October 2003;18(10):1207–1208. [DOI] [PubMed] [Google Scholar]
- 102.Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology. May 23 2000;54(10):1916–1921. [DOI] [PubMed] [Google Scholar]
- 103.Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. Journal of neurology, neurosurgery, and psychiatry. December 2008;79(12):1331–1338. [DOI] [PubMed] [Google Scholar]
- 104.Maetzler W, Liepelt I, Reimold M, et al. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiology of disease. April 2009;34(1):107–112. [DOI] [PubMed] [Google Scholar]
- 105.Foster ER, Campbell MC, Burack MA, et al. Amyloid imaging of Lewy body-associated disorders. Movement disorders : official journal of the Movement Disorder Society. November 15 2010;25(15):2516–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Burack MA, Hartlein J, Flores HP, Taylor-Reinwald L, Perlmutter JS, Cairns NJ. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology. January 5 2010;74(1):77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kotzbauer PT, Cairns NJ, Campbell MC, et al. Pathologic accumulation of alpha-synuclein and Abeta in Parkinson disease patients with dementia. Archives of neurology. October 2012;69(10):1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cairns NJ, Ikonomovic MD, Benzinger T, et al. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Archives of neurology. December 2009;66(12):1557–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gomperts SN, Locascio JJ, Rentz D, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology. January 1 2013;80(1):85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Petrou M, Bohnen NI, Muller ML, Koeppe RA, Albin RL, Frey KA. Abeta-amyloid deposition in patients with Parkinson disease at risk for development of dementia. Neurology. September 11 2012;79(11):1161–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Petrou M, Dwamena BA, Foerster BR, et al. Amyloid deposition in Parkinson’s disease and cognitive impairment: a systematic review. Movement disorders : official journal of the Movement Disorder Society. June 2015;30(7):928–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology. September 16 2008;71(12):903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Campbell MC, Markham J, Flores H, et al. Principal component analysis of PiB distribution in Parkinson and Alzheimer diseases. Neurology. August 6 2013;81(6):520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buddhala C, Campbell MC, Perlmutter JS, Kotzbauer PT. Correlation between decreased CSF alpha-synuclein and Abeta(1)(−)(4)(2) in Parkinson disease. Neurobiology of aging. January 2015;36(1):476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain : a journal of neurology. May 2016;139(Pt 5):1539–1550. [DOI] [PubMed] [Google Scholar]
- 116.Hansen AK, Damholdt MF, Fedorova TD, et al. In Vivo cortical tau in Parkinson’s disease using 18F-AV-1451 positron emission tomography. Movement disorders : official journal of the Movement Disorder Society. June 2017;32(6):922–927. [DOI] [PubMed] [Google Scholar]
- 117.Gomperts SN, Locascio JJ, Makaretz SJ, et al. Tau Positron Emission Tomographic Imaging in the Lewy Body Diseases. JAMA neurology. November 1 2016;73(11):1334–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brooks DJ. Functional imaging in relation to parkinsonian syndromes. Journal of the neurological sciences. March 1993;115(1):1–17. [DOI] [PubMed] [Google Scholar]
- 119.Turjanski N, Lees AJ, Brooks DJ. In vivo studies on striatal dopamine D1 and D2 site binding in L-dopa-treated Parkinson’s disease patients with and without dyskinesias. Neurology. September 1997;49(3):717–723. [DOI] [PubMed] [Google Scholar]
- 120.Ekelund J, Slifstein M, Narendran R, et al. In vivo DA D(1) receptor selectivity of NNC 112 and SCH 23390. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. May-Jun 2007;9(3):117–125. [DOI] [PubMed] [Google Scholar]
- 121.Stoessl AJ, Martin WW, McKeown MJ, Sossi V. Advances in imaging in Parkinson’s disease. The Lancet. Neurology November 2011;10(11):987–1001. [DOI] [PubMed] [Google Scholar]
- 122.de la Fuente-Fernandez R, Sossi V, Huang Z, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain : a journal of neurology. December 2004;127(Pt 12):2747–2754. [DOI] [PubMed] [Google Scholar]
- 123.de la Fuente-Fernandez R, Lu JQ, Sossi V, et al. Biochemical variations in the synaptic level of dopamine precede motor fluctuations in Parkinson’s disease: PET evidence of increased dopamine turnover. Annals of neurology. March 2001;49(3):298–303. [DOI] [PubMed] [Google Scholar]
- 124.Sawamoto N, Piccini P, Hotton G, Pavese N, Thielemans K, Brooks DJ. Cognitive deficits and striato-frontal dopamine release in Parkinson’s disease. Brain : a journal of neurology. May 2008;131(Pt 5):1294–1302. [DOI] [PubMed] [Google Scholar]
- 125.Steeves TD, Miyasaki J, Zurowski M, et al. Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a [11C] raclopride PET study. Brain : a journal of neurology. May 2009;132(Pt 5):1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Politis M, Piccini P, Pavese N, Koh SB, Brooks DJ. Evidence of dopamine dysfunction in the hypothalamus of patients with Parkinson’s disease: an in vivo 11C-raclopride PET study. Experimental neurology. November 2008;214(1): 112–116. [DOI] [PubMed] [Google Scholar]
- 127.Pagano G, Molloy S, Bain PG, et al. Sleep problems and hypothalamic dopamine D3 receptor availability in Parkinson disease. Neurology. December 6 2016;87(23):2451–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Politis M, Wu K, Loane C, et al. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology. November 23 2010;75(21):1920–1927. [DOI] [PubMed] [Google Scholar]
- 129.Pavese N, Metta V, Bose SK, Chaudhuri KR, Brooks DJ. Fatigue in Parkinson’s disease is linked to striatal and limbic serotonergic dysfunction. Brain : a journal of neurology. November 2010;133(11):3434–3443. [DOI] [PubMed] [Google Scholar]
- 130.Politis M, Piccini P. In vivo imaging of the integration and function of nigral grafts in clinical trials. Progress in brain research. 2012;200:199–220. [DOI] [PubMed] [Google Scholar]
- 131.Kotagal V, Bohnen NI, Muller ML, Koeppe RA, Frey KA, Albin RL. Cerebral amyloid deposition and serotoninergic innervation in Parkinson disease. Archives of neurology. December 2012;69(12):1628–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kotagal V, Spino C, Bohnen NI, Koeppe RA, Albin RL. Serotonin, beta-amyloid, and cognition in Parkinson disease. Annals of neurology. April 17 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nishi A, Kuroiwa M, Miller DB, et al. Distinct roles of PDE4 and PDE10A in the regulation of cAMP/PKA signaling in the striatum. The Journal of neuroscience : the official journal of the Society for Neuroscience. October 15 2008;28(42):10460–10471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Niccolini F, Foltynie T, Reis Marques T, et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain : a journal of neurology. October 2015;138(Pt 10):3003–3015. [DOI] [PubMed] [Google Scholar]
- 135.Niccolini F, Wilson H, Pagano G, et al. Loss of phosphodiesterase 4 in Parkinson disease: Relevance to cognitive deficits. Neurology. August 8 2017;89(6):586–593. [DOI] [PubMed] [Google Scholar]