

Abstract
Pulmonary intracellular infections, such as tuberculosis, anthrax, and tularemia, remain a significant challenge to conventionl antibiotic therapy. Ineffective antibiotic treatment of these infections can lead not only to undesired side effects, but also the emergence of antibiotic resistance. Aminoglycosides (e.g., streptomycin) have long been part of the therapeutic regiment for many pulmonary intracellular infections. Their bioavailability for intracellular bacterial pools, however, is limited by poor membrane permeability and rapid eliminaton. To address this challenge, polymer-augmented liposomes (PALs) were developed to provide improved cytosolic delivery of streptomycin to alveolar macrophages, an important host cell for intracellular pathogens. A multifunctional diblock copolymer was engineered to functionalize PALs with carbohydrate-mediated targeting, pH-responsive drug release, and endosomal release activity with a single functinoal polymer that replaces the pegylated lipid component to simplify the liposome formulation. The pH-sensing functionality enabled PALs to provide enhanced release of streptomycin under endosomal pH condition (70% release in 6 hours) with limited release at physiological pH 7.4 (16%). The membrane-destabilizing activity connected to endosomal release was characterized in a hemolysis assay and PALs displayed a sharp pH profile across the endosomal pH development target range. The direct connection of this membrane-destalizing pH profile to model drug release was demonstrated in an established pyranine / p-xylene bispyridinium dibromide (DPX) fluorescent dequenching assay. PALs displayed similar sharply pH-responsive release, whereas PEGylated control liposomes did not, and similar profiles were then shown for streptomycin release. The mannose-targeting capability of the PALs was also demonstrated with 2.5 times higher internalization compared to non-targeted PEGylated liposomes. Finally, the streptomycin-loaded PALs was shown with a significantly improved intracellular antibacterial activity in a Francisella-macrophage co-culture model, compared with free streptomycin or streptomycin delivered by control PEGylated liposomes (13× and 16×, respectively). This study suggests the potential of PALs as a useful platform to deliver antibiotics for the treatment of intracellular macrophage infections.
Graphical abstract
A simplified liposome formulation with three functionalities to enhance antibiotic bioavailability to cytosolic bacteria: carbohydrate-mediated targeting; pH-responsive release; endosomal-releasing activity.
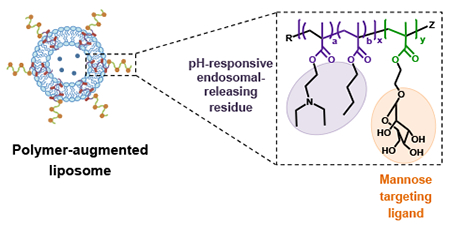
Introduction
Respiratory infections caused by intracellular bacteria, such as Mycobacterium tuberculosis, Bacillus anthracis, Francisella tularensis and Burkholderia pseudomallei, remain a major global health and biological threat1–4. For instance, inhaled Francisella tularensis can result in the most severe form of tularemia at even low infectious dose (estimated LD50 < 10 CFU)3,5. Without immediate antibiotic treatment, pulmonary tularemia can be fatal in 30% of cases3,5. The pathogenicity of intracellular bacteria is highly associated with their ability to seclude themselves and multiply within phagocytic cells, such as alveolar macrophages (AMs) in respiratory infections6–8. This intracellular localization protects the bacteria from the humoral and cellular immune responses, and limits the efficacy of conventional antibiotic treatments9–11.
Current treatment for F. tularensis infections relies on rigorous oral and IV antibiotic therapy with aminoglycoside, tetracycline, and/or fluoroquinolone drugs12. Aminoglycosides, including streptomycin and gentamicin, have been recommended as the agents of choice for the treatment of tularemia, due to their improved outcomes and lower relapse rates12. However, aminoglycosides suffer from suboptimal pharmacokinetics because of their rapid elimination rate (plasma half-life 2-3 hours)13. Additionally, owing to their hydrophilic nature, the efficacy of aminoglycosides is limited by their poor membrane permeability and slow cytosolic accumulation14. Due to those limitations, multiple daily high-dose injections are required for a prolonged period of time to achieve therapeutic efficacy, and this treatment regimen can have undesired side effects (e.g., nephrotoxicity and ototoxicity) and potentially induce drug resistance15,16. Consequently, drug delivery systems that can enhance cytosolic accumulation of aminoglycosides in the intracellular space might potentiate activity.
Liposomes have been previously utilized to improve antibiotic efficacy by enhancing cellular delivery and improving pharmacokinetics (PK) at the site of infections17–19. A liposomal ciprofloxacin (Lipoquin®, Aradigm Corp.) has shown improved antibacterial activity compared to free drug in F. tularensis Schu S4-infected mice20,21. Liposomal surfaces have also been modified with mannose to increase uptake by AMs through receptor-mediated endocytosis mediated by the macrophage mannose receptor22. This enhanced uptake improved PK properties in AMs compared to unmodified liposomes23. However, drugs released from those liposome formulations still rely on passive diffusion through vesicular or cellular membranes to reach cytosolic bacteria. This could limit the efficacy of aminoglycosides, as they preferentially accumulate in lysosomes following endocytosis10.
To achieve endosomal release, various approaches have been taken to provide drug carriers with membrane destabilizing and/or membrane fusion activities, such as fusogenic peptides and synthetic polymers24–26. Synthetic polymers with endosomal-destabilizing activity, particularly at a weakly acidic pH, are especially useful for cytoplasmic delivery27–29. When liposomes modified with these pH-sensitive polymers are taken up by cells through endocytosis, they can introduce entrapped cargos to the cytosol by destabilizing endo-lysosomal membranes. For example, a pH-sensitive fusogenic polymer, 3-methylglutarylated hyperbranched poly(glycidol), formulated with liposomes effectively introduced ovalbumin to the cytosol of dendritic cells30.
In this study, a multifunctional, diblock copolymer was developed to augment liposomes with macrophage-targeting and endosomal release functionality for the cytosolic delivery of hydrophilic aminoglycoside antibiotics against Francisella infection (Scheme 1a). This diblock copolymer was employed to functionalize liposomes to create the polymer-augmented liposome (PAL) system with the following advantageous properties: (1) surface mannose modification for targeted association and enhanced internalization by AMs; (2) pH-responsive streptomycin release from PALs; (3) membrane-destabilizing activity for enhancing streptomycin bioavailability to cytosolic bacteria; and (4) a simplified liposomal formulation where the polymer replaces the typical hydrophilic lipid (Scheme 1b). The efficacy against intracellular infections was characterized in an intracellular Francisella-macrophage co-culture model. The enhanced antibacterial activity of streptomycin delivered by PALs suggests that this carrier design could provide important benefits for intracellular delivery of drugs with poor membrane permeability.
Scheme 1.
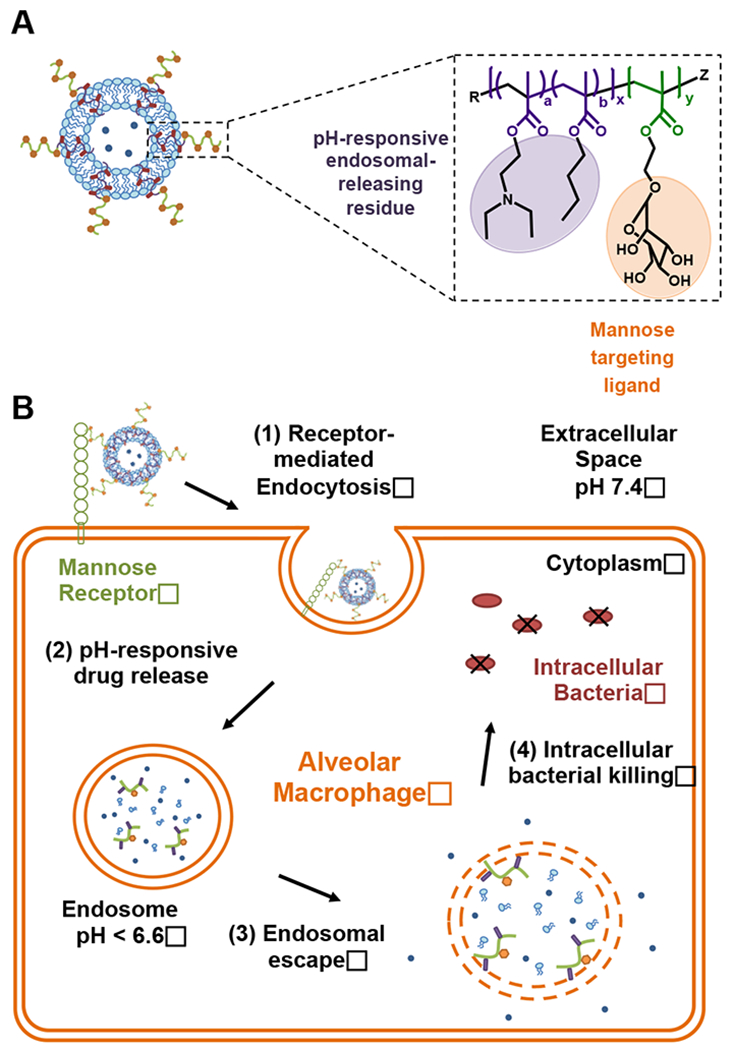
(A) Schematic of the formulation and functionalities of polymer-augmented liposomes (PALs). PAL was formulated through the hydrophobic effect between the lipid bilayer and hydrophobic block (purple) of the amphiphilic diblock copolymer; (B) Envisioned pathway for the cellular uptake of PALs and subsequent intracellular release of the cargo in alveolar macrophages. (Note: illustrations are not to scale.)
Experimental
Materials
Materials were purchased from Sigma Aldrich (St Louis, MO) unless otherwise specified. 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) were purchased from Avanti Polar Lipids (Alabaster, AL). Rhodamine B-labeled dihexadecanoyl-sn-glycero-3-phosphoethanolamine (DHPE-Rhodamine) was acquired from Thermo-Fisher Scientific (Pittsburgh, PA). Diethylaminoethyl methacrylate (DEAEMA) and butyl methacrylate (BMA) were distilled prior to use. 4-Cyano-4-(ethylsulfanylthiocarbonyl) sulfanylpentanoic acid (ECT) and mannose functionalized with ethyl methacrylate (ManEMA) were synthesized and fully characterized as described previously31,32. Spectra/Por regenerated cellulose dialysis membranes were purchased from Spectrum Laboratories (Houston, TX). Sephadex G-25 prepacked PD10 columns were obtained from GE Healthcare Life Sciences (Pittsburg, PA).
Reversible addition–fragmentation chain-transfer (RAFT) polymerization and characterization of poly[(DEAEMA-co-BMA)-b-ManEMA]
Please see supplementary information.
Formulation and characterization of streptomycin-loaded PALs
PALs were prepared using an ethanol injection method described previously with some modifications33. Briefly, DSPC (23.7 mg), cholesterol (7.8 mg), and poly[(DEAEMA-co-BMA)-b-ManEMA] (7.9 mg) were dissolved in ethanol (0.25 mL) to uniformly mix the lipids with polymer in a single phase. The lipid/polymer mixture was added into phosphate buffer saline (PBS, 3.15 mL, 10 mM, pH 7.4) containing streptomycin (100 mg/mL) under magnetic stirring to form streptomycin-loaded PALs. The PAL suspension was then sonicated for 15 min and extruded at 60 °C through the following polycarbonate filters: 800, 400, 200, and 100 nm (Whatman, GE Healthcare, Pittsburgh, PA USA) to obtain uniform PALs suspension. Ethanol and unencapsulated streptomycin were removed by dialysis against PBS using Spectra/Por dialysis tubing (MWCO 3kDa). Non-pH-responsive PEGylated control liposomes with similar formulation as Lipo-Dox® (DSPC: cholesterol: DSPC-PEG= 6:4:0.5 molar ratio) were also prepared to serve as a control34. For fluorescence labeled liposomes, fluorescent lipids, rhodamine DHPE, were incorporated in PALs and PEGylated liposomes at 0.5 mol% total lipids via the same preparation procedures. Co-formulation of the diblock copolymer with liposomes was characterized using high performance size exclusion chromatography (HPSEC); procedures were described in detail in Supplementary Information.
The hydrodynamic diameter of PALs and PEGylated liposomes were determined using a Malvern Instruments Zetasizer Nano series instrument equipped with a 22 mW He-Ne laser operating at 632.8 nm. To compare the hydrodynamic diameters of PALs and PEGylated liposomes in the pH range of the endosomal pathway (7.4, 7.0, 6.6, 6.2, and 5.8), both liposome formulations were dispersed in sodium phosphate buffer (100 mM, pH 7.4-6.2, supplemented with 150 mM NaCl) or sodium acetate buffer (100 mM, pH 5.8, supplemented with 150 mM NaCl). Mean diameters were reported as the intensity-average ± standard deviation from three or more independently prepared formulations.
Quantification of streptomycin encapsulation
Encapsulated streptomycin concentration in PALs was determined using a fluorescamine assay, which has been used to quantify the concentration of peptides, proteins, and aminoglycosides35,36. Fluorescamine is a non-fluorescent reagent that reacts readily under mild conditions with amines to form stable, highly fluorescent compounds37. The encapsulated streptomycin was first released from PALs by lysing the PAL suspension (0.015 mL, 5.6 mg/mL lipids) with 1% Triton X-100 (0.135 mL). The released streptomycin was then reacted with fluorescamine (3 mg/mL in dimethyl sulfoxide, 0.05 mL) for 15 min at room temperature. Fluorescence (λex=390 nm, λem=470 nm) was measured using a plate reader (Tecan M1000). Known concentrations of streptomycin solution were also reacted with fluorescamine solution to obtain a calibration curve.
Red blood cell hemolysis assay
The membrane destabilizing activity of the diblock copolymer and the streptomycin-loaded PALs was measured using a red blood cell hemolysis assay as previously described38,39. Polymer/PAL suspensions were incubated in triplicate wells of a 96-well plate (V-shaped bottom) for 1 h at 37 °C in the presence of human red blood cells at various polymer concentrations (10, 20, 40, 80 μg/mL) in 100 mM sodium phosphate buffers (pH 7.4, 7.0, 6.6, and 6.2, supplemented with 150 mM NaCl) or 100 mM sodium acetate buffer (pH 5.8, supplemented with 150 mM NaCl) intended to mimic the acidifying pH gradient in the lysosomal pathway. Polymer concentrations in PALs were estimated based on the amount of polymer added during PAL preparation procedures. To remove the unlysed cells, the plate was centrifuged at 1500 rpm for 5 min, and the supernatant was transferred to another 96-well plate (flat-bottom) for absorbance measurement. The percent hemolysis was determined by measuring the amount of released hemoglobin in the supernatant (λabs=541 nm) and normalized to a 100% hemolysis control (1% Triton X-100).
pH-dependent fluorescence dequenching assay
To study the pH-dependent release characteristics of PALs, a fluorescent dye-quencher pair, pyranine (35 mM) and p-xylene bispyridinium dibromide (DPX, 50 mM), was encapsulated in the aqueous compartment of PALs using the method described above. Untrapped dye was removed by gel filtration using PD-10 column twice. Subsequently, the PALs encapsulating pyranine and DPX were diluted 20 times in sodium phosphate buffers (pH 7.4, 7.0, 6.6, and 6.2) or sodium acetate buffer (pH 5.8 and 5.4) same as used for the hemolysis assay. Following a 3-h incubation in buffers at varying pH at 37 °C, the extent of contents release was quantified by measuring the fluorescence of pyranine (λex=413 nm, λem=512 nm) via a microplate reader (Tecan M1000). 100% release was achieved by adding 1% Triton X-100 to PAL suspension at pH 7.4 (final concentration 0.1%). Pyranine/DPX loaded PEGylated control liposomes were prepared similarly.
In vitro streptomycin release kinetics
The release kinetics of streptomycin from PALs was investigated in two pH conditions, pH 5.8 (endosomal) and pH 7.4 (physiologic), using acetate and phosphate buffered saline, respectively. The prepared streptomycin-loaded PALs were divided into two 2.5 ml aliquots and transferred to two dialysis tubes with a MWCO of 3.5 kDa. The two dialysis tubes were immersed into 30 mL of pH 5.8 and 7.4 buffer at 37 °C, respectively. At desired time intervals, 1 mL of release media was taken out and replenished with an equal volume of fresh media. The amount of streptomycin released was determined using fluorescamine assay as described above. Calibration curves were obtained with streptomycin in corresponding buffer solutions at pH 5.8 and 7.4. 100% streptomycin release was measured by lysing PALs with 1% Triton X-100 in PBS.
Cell uptake study
Cellular uptake of rhodamine-labeled PALs was investigated using flow cytometry and visualized via fluorescence microscopy. To induce higher expression of mannose receptors, RAW 264.7 cells were treated with 20 ng/mL IL-4 for 24 h40. RAW 264.7 cells with and without IL-4 pretreatment were suspended at a density of 1 × 106 cells/ml and blocked with growth media containing 10% FBS for 30 min at 37°C. Cells were subsequently treated with rhodamine-labeled PALs or PEGylated control liposomes at a total lipid concentration of 400 μg/ml for 20 min, 1 hr, and 3 hr at 37°C. The cells were then washed and resuspended in cold PBS containing 0.01% FBS to remove extracellular liposomes. The uptake level was presented as the intensity of rhodamine measured via a PE filter on a BD LSRII flow cytometer (BD Biosciences, San Jose, CA), and analyzed using FlowJo software (Tree Star, Inc.). For microscopy, cells were treated as above for 1 hr but instead fixed with 10% formaldehyde solution, and nuclei were counterstained with DAPI (Molecular probe). Random fields were observed at 600× or 100× magnification using a Nikon Ti-E live-cell fluorescence microscope.
Bacteria-macrophage co-culture therapeutic study
RAW 264.7 cells were seeded (500,000 cells/mL, 250 μL/well) into a 48 well plate with antibiotic free DMEM with 10% FBS, and incubated at 37°C with 5% CO2. After 18 hours, cells were infected with F. novicida U112 at early log phase of growth (OD600=0.2) at a multiplicity of infection of 50, and then incubated for 1 hour. Subsequently, growth media was replaced with fresh DMEM containing 10% FBS and 250 μg/mL kanamycin to eliminate extracellular bacteria not internalized by the cells; cells were then incubated for another hour. Growth media was then replaced with fresh, unsupplemented media containing varying concentrations of free streptomycin (0-115 μg/mL), streptomycin-loaded PEGylated control liposomes (equivalent to 0-115 μg/mL streptomycin), or streptomycin-loaded PALs (equivalent to 0-115 μg/mL streptomycin). Cells were incubated for another 22 hours (24 hours post-infection). After incubation, cells were washed three times with 1× PBS and lysed with 100 μL of PBS containing 0.1% (v/v) Triton X-100. Lysates were serially diluted and plated onto triplicate TSB agar plates and incubated for 24 hours and individual bacterial colonies were counted. Data represented as CFU/well vs. streptomycin dose.
Statistics
Numerical data were represented as mean ± SD. Statistical analysis was performed by Student’s paired t test. (**) denotes a P-value of < 0.005. All samples were performed in triplicate.
Results and discussion
Macrophages are key regulators of the inflammatory process and represent an attractive therapeutic target due to their many important roles in combating infection and maintaining homeostasis in the body41. Uncontrolled inflammatory response of macrophages becomes pathogenic resulting in significant levels of non-specific tissue damage, resulting in inflammatory and autoimmune disease42. Additionally, facultative or obligate intracellular parasites often use macrophages as a safe reservoir to protect them from host immune response and antibiotic treatment43. For example, prior research has shown that AMs are major reservoirs of F. tularensis during early-stage infection44, and that nanoparticles enhancing antibiotic delivery to macrophages led to better efficacy then free drug45. To eliminate intracellular pathogens within AMs, the intracellular antibiotic concentration must be higher than the minimum inhibitory concentration (MIC)11. Especially for concentration-dependent antibiotics, including aminoglycosides and fluoroquinolones, the rate and extent of their bactericidal activity has been shown to associate with the ratio of the maximum concentration to MIC (Cmax/MIC ≥ 10) in serum or target tissues46. Consequently, efficient cytosolic delivery of concentrated antibiotics to AMs is required to eradicate intracellular F. tularensis, and therefore may potentially minimize therapeutic failure as well as relapse rate. Due to its clinical use and poor membrane permeability, streptomycin was used as a model aminoglycoside in the present study to demonstrate the PAL system.
Synthesis of multifunctional diblock copolymer
The diblock copolymer, poly[(DEAEMA-co-BMA)-b-ManEMA], was prepared via RAFT polymerization according to Scheme 247. RAFT allows the three desired functionalities (targeting, pH-sensitivity, endosome releasing activity) to be integrated into this simple, well-defined diblock copolymer construct. poly(DEAEMA-co-BMA) was first prepared by copolymerizing DEAEMA and BMA with a comonomer feed ratio of 60 mol% DEAEMA and 40 mol% BMA, which has been shown to exhibit sharp pH-dependent response at pH 6.6 and strong endosomal-releasing properties48. The resultant polymer was used as a macro chain transfer agent (macroCTA) to prepare the poly[(DEAEMA-co-BMA)-b-ManEMA] diblock copolymer. The diblock copolymer was successfully synthesized under controlled conditions with consistent size and composition as determined by GPC and 1H-NMR spectroscopy. The obtained diblock copolymer has narrow and symmetric molecular weight distributions that cleanly move to lower elution volumes (Fig. 1a). Copolymer composition was confirmed by analysis of resonances associated with each of the comonomers (Fig. 1b). The feed ratios, monomer compositions, molecular weight (Mn), and dispersity (Đ) for macroCTA and the diblock copolymer are given in Fig 1c.
Scheme 2.
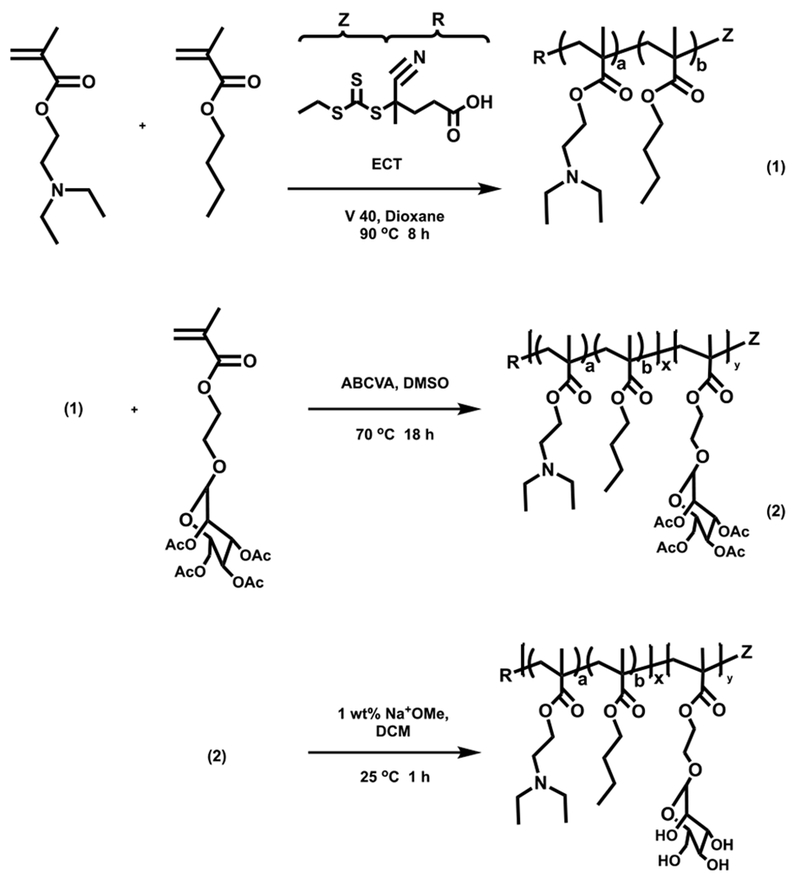
RAFT polymerization of poly([DEAEMA-co-BMA]-b-ManEMA). DEAEMA, diethylaminoethyl methacrylate; BMA, butyl methacrylate; ManEMA, mannose functionalized with ethyl methacrylate.
Fig. 1.
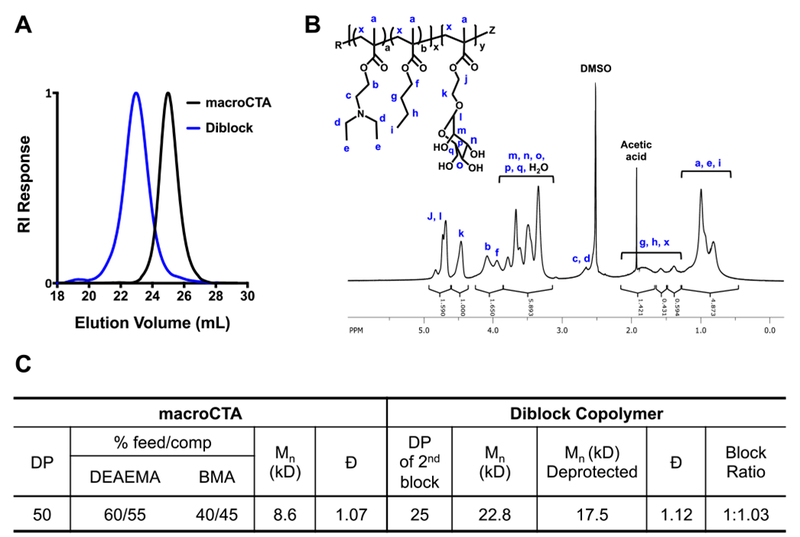
SEC-GPC and 1H-NMR characterization of macroCTA and diblock copolymer. (a) Representative SEC chromatogram showing the narrow and symmetric molecular weight distribution of poly(DEAEMA-co-BMA) macroCTA and of poly[(DEAEMA-co-BMA)-b-mannose ethyl methacrylate] (diblock); (b) 1H-NMR of the diblock copolymer in d-DMSO with assignment of the resonances associated with the respective comonomer residues; (c) Chemical composition and molecular weight of the diblock copolymer employed to formulate the polymer-augmented liposomes. Experimental Mn, Đ, and block ratios were determined by GPC analysis. The block ratios were determined as the ratio of the Mn of the second block to the first block. RI, refractive index; DP, degree of polymerization; DEAEMA, diethylaminoethyl methacrylate; BMA, butyl methacrylate; macroCTA, macro chain transfer agent; Đ, dispersity.
Preparation and characterization of polymer-augmented liposomes (PALs)
Two design principles were exploited to avoid potential side effects associated with the PALs. First, the PAL formulation was based upon lipid components (i.e., DSPC and cholesterol) that were previously approved by the US FDA for liposomal doxorubicin (Lipo-Dox®)34. Second, regarding the polymer clearance and safety properties, the molecular weight of the diblock copolymer was designed to be smaller than the renal clearance threshold (~ 50 kDa, ~ 6 nm)49, and all monomers were designed with ester junctions at the carbon backbone to degrade down to the minimal backbone segment (< 10 kDa).
Prior studies have shown that a variety of hydrophilic polymers with hydrophobic anchoring moieties can be co-formulated with liposomes50,51. The PAL was designed to incorporate poly[(DEAEMA-co-BMA)-b-ManEMA] with DSPC/cholesterol liposomes through the hydrophobic effect between the lipid bilayer and the hydrophobic polymer block, poly(DEAEMA-co-BMA). The incorporation of the diblock copolymer with the liposomal membrane was examined using high performance size exclusion chromatography (HPSEC)52. Thiocarbonyl RAFT agents exhibit absorption at 300-310 nm, which was used to assess co-formulation (Fig. S1a†). A rhodamine DHPE detectable at 580 nm was used to follow the lipid components. As shown in Fig. S1b†, co-elution of fluorescent lipids (fluorescence signal, 580 nm) and polymers (absorption signal, 310 nm) after injecting fluorescently-labeled PALs indicates that poly[(DEAEMA-co-BMA)-b-ManEMA] was associated with the liposomes, rather than forming micelles. This lack of micelle formation was confirmed by the monomodal size distribution of the PALs (Fig. 2a).
Fig. 2. Hydrodynamic size measurement as a function of pH.
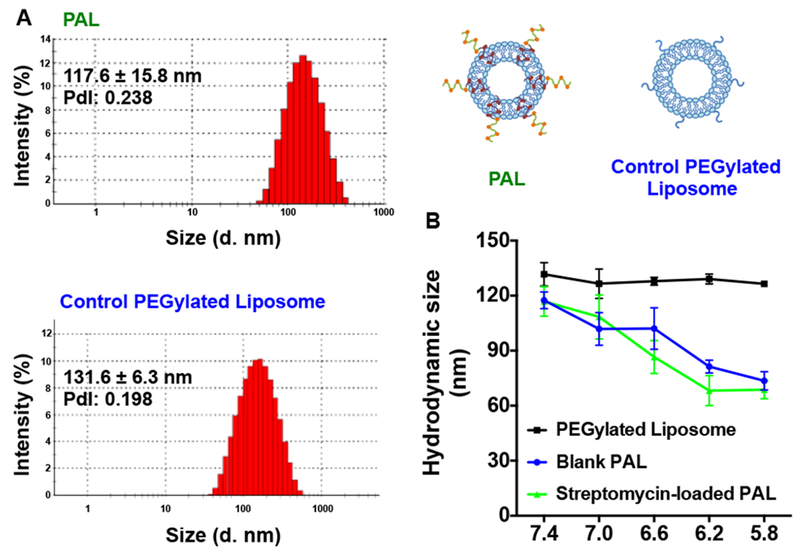
(A) The hydrodynamic distribution of streptomycin-loaded polymer-augmented liposomes (PAL) and control PEGylated liposomes in pH 7.4 phosphate buffer; (B) Blank or streptomycin-loaded PALs and non-pH-responsive PEGylated control liposomes (streptomycin-loaded) incubated in buffers ranging from pH 7.4 to 5.8. PdI: Polydispersity (Mean ± SD, n=3).
To study the pH-responsive properties of the PALs, the hydrodynamic sizes were evaluated by dynamic light scattering (DLS). Non-pH-responsive PEGylated liposomes were prepared as a control. PALs with and without streptomycin loading show similar hydrodynamic size transitions in response to the pH gradient (Fig. 2b). The hydrodynamic size of PALs gradually decreased with decreasing pH, in contrast to the control PEGylated liposomes maintained a constant size independent of pH. The destabilization of the PALs results from the protonation of DEAEMA at acidic pH, which increases the charge density of the hydrophobic segment, thus destabilizing the liposomal membranes48. It is postulated that with the higher charge density on DEAEMA, the poly(DEAEMA-co-BMA) block was no longer well embedded in the liposomal membrane at low pH, and that lipids were released from the disrupted bilayer structure, forming random aggregates with the polymer. Therefore, we speculate that the smaller hydrodynamic diameters measured by DLS at lower pH reflect the formation of the random aggregates driven by the hydrophobic effects between the lipid tails and the hydrophobic BMA in the diblock copolymer.
The capability of PALs to encapsulate streptomycin was evaluated by reacting amines on streptomycin with fluorescamine to form a fluorescent streptomycin analogue (Fig. S2a†), and quantified using a calibration curve (Fig. S2b†)37. The streptomycin loading concentrations in PALs and PEGylated liposomes were 0.77 ± 0.04 mg/mL and 0.65 ± 0.01 mg/mL. The loading of streptomycin was 12.06 ± 0.36% and 10.44 ± 0.12%, respectively (weight of encapsulated streptomycin/weight of loaded PALs or PEGylated liposomes).
In vitro pH-dependent release
The pH-dependent release from PALs was examined using a pyranine/p-xylene bispyridinium dibromide (DPX) fluorescent dequenching assay, widely used to investigate the stability or pH-dependent release properties of liposomes53,54. PALs co-loaded with both pyranine and DPX were diluted with buffers mimicking the endosomal acidification profile. The degree of release was normalized relative to a positive control, pyranine/DPX-loaded PALs lysed with 1% v/v Triton X-100 (Fig. 3a). As expected, no significant pyranine release was detected from PEGylated liposomes throughout the test pH range. In contrast, the release from PALs was nearly complete at pH 5.4 within three hours of incubation. Above pH 6.2, only background pyranine fluorescence was observed. This pH-dependent response correlates well with the pH gradient found in endosomal-lysosomal trafficking55, facilitating the preferential release of therapeutic cargo in the endosomal compartment.
Fig. 3. pH-dependent release characteristics.
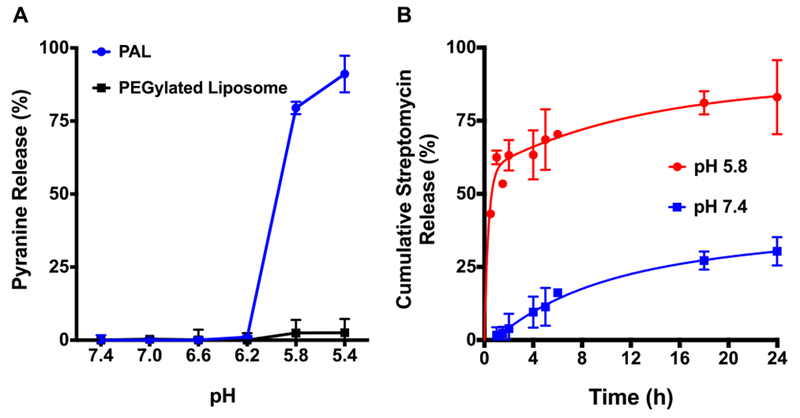
(A) Fluorescent dequenching assay. A fluorescent dye-quencher pair, pyranine and p-xylene bispyridinium dibromide, were encapsulated in polymer-augmented liposomes (PALs) and non-pH-responsive PEGylated liposomes. Pyranine release was evaluated in the pH range of the endosomal processing pathway (λex=413 nm, λem=512 nm); (B) Streptomycin release kinetics of PALs at pH 5.8 and 7.4. The release studies were carried out by exposing streptomycin-loaded PALs to physiological (pH 7.4) and acidic (pH 5.8) conditions at 37 °C. Percentage release was normalized relative to a positive control, 1% w/v Triton X-100 (Mean ± SD, n=3).
Streptomycin release kinetics were then evaluated in pH 5.8 and pH 7.4 buffers via a dynamic dialysis technique. Notably, the release profiles of PALs at pH 5.8 contained two phases, which included a relatively large burst effect in the initial stage (Fig. 3b, about 60% within 1 hour) followed by a slower release phase. We postulated that the burst release at pH 5.8 was due to the rapid transition of DEAEMA from hydrophobic to hydrophilic, thus disturbing the integrity of the lipid bilayer. The slower release phase may be attributed to the gradual detachment of streptomycin that attached to lipids or polymers. In contrast, at pH 7.4, streptomycin released from PALs was much slower but not negligible, which is postulated to result from a small fraction of surface bound drug in the liposome formulation. This controlled release nature of PALs has potential to deliver concentrated streptomycin to the intracellular infection site, and therefore to provide improved therapeutic efficacy.
pH-dependent membrane-destabilizing activity
The endosomal-releasing potential of synthetic polymers is frequently assessed using a red blood cell hemolysis assay due to a proven correlation between hemolytic efficiency and endosomal release56. The pH-responsive, membrane destabilizing activity of the free diblock copolymer and corresponding PALs were evaluated at several different pH values to mimic the endosomal acidification profile. No significant hemolytic activity was observed at pH 7.4 for polymer alone (Fig. 4a) or the PALs (Fig. 4b) at any of the concentrations evaluated. A significant increase in red blood cell hemolysis was observed upon reduction of the pH to 6.6. Also, as expected, hemolysis increased in a dose-dependent manner with increasing polymer concentration. Indeed the hemolysis profiles as a function of pH were very similar for both the polymer and PALs. In contrast, no significant hemolytic activity was present in the group treated with control PEGylated liposomes across pH range studied.
Fig. 4.
Hemolytic assay demonstrating pH-dependent membrane-destabilizing activity of (A) diblock copolymer alone as a function of pH at concentrations of 10, 20, 40, and 80 μg/mL and (B) polymer-augmented liposomes with same polymer concentration range in (A). Non-pH-responsive PEGylated liposomes were used as a negative control. Hemolytic activity was normalized relative to a positive control, 1% v/v Triton X-100, and the data represent a single experiment conducted in triplicate ± SD.
The observed pH-responsive hemolytic effects of PALs near pH 6.6 could be attributed to the composition of the hydrophobic block, poly(DEAEMA-co-BMA). Our group has shown that the transition pH could be tuned by varying the hydrophobic content (i.e. BMA) in a relevant diblock copolymer composed of poly(DEAEMA-co-BMA)48. With 40% BMA and 60% DEAEMA, at pH 6.6, the diblock copolymer has a sharp transition from micelles to unimers and from nonhemolytic to hemolytic activity48. Poly[(DEAEMA-co-BMA)-b-ManEMA] developed in this study has similar BMA to DEAEMA ratio, which could account for the observed pH-responsive transition at ~pH 6.6.
To evaluate if the endosomal-releasing activity of the PALs lead to cytoxicity, RAW 264.7 cells were incubated with the polymer alone and PALs separately for 24 h. The viability of RAW cells was evaluated using a MTS assay (Fig. S3†). Although the polymer alone shows notable toxicity (~75% viability at 0.05 mg/mL), formulating the polymer with liposomes significantly reduced the toxicity (~92% viability, at 0.05 mg/mL).
Taken together, those results indicate that poly[(DEAEMA-co-BMA)-b-ManEMA] augmented the liposomes with pH-responsive endosomal-releasing activity and the incorporation of the hydrophobic block into the liposomal membrane did not reduce the pH-selective, membrane destabilizing properties of the polymer. Additionally, the endosomal-releasing activity of PALs did not lead to notable toxicity. These results together with in vitro release results suggest that poly[(DEAEMA-co-BMA)-b-ManEMA] can enhance endosomal release to provide concentrated antibiotics to the cytoplasm where intracellular bacteria accumulate.
Mannose ligands enhance macrophage uptake of PALs
AMs highly express internalizing mannose-binding receptors (CD206)57,58 for recognizing carbohydrates on infectious agents such as bacteria, fungi, and viruses59. Carbohydrate-based ligands therefore represent a promising approach for targeting therapeutic agents to macrophages22,23. The pH-responsive polymer was thus designed to display multivalent mannose ligands by incorporating poly[(DEAEMA-co-BMA)-b-ManEMA] into liposomes. RAW 264.7 murine macrophage cells, expressing moderate levels of the CD206 mannose receptor31, were used to study the cellular uptake of rhodamine-labeled PALs. Rhodamine-labeled PEGylated liposomes without mannose modification were used as a negative control. Cells were respectively incubated with either rhodamine-labeled PALs or PEGylated liposomes (400 μg/mL) with similar fluorescent intensities (Fig. S4†). Uptake levels were evaluated using flow cytometry and fluorescence microscopy. The observed fluorescent intensity in the group treated with the PEGylated control liposomes was limited, which was likely due to the surface PEGylation (Fig. 5a), as PEG is well-known to inhibit uptake by phagocytic cells of the mononuclear phagocyte system54. Conversely, the PAL treatment group produced an increased fluorescent intensity as incubation progressed and revealed considerably greater cellular uptake, an essential feature to enhance the delivery of intracellular drug. The enhanced uptake in RAW 264.7 cells of PALs was also visualized by fluorescence microscopy (Fig. 5b & Fig. S5†)
Fig. 5. Mannose-receptor mediated uptake of rhodamine-labeled polymer-augmented liposomes by RAW 264.7 cells.
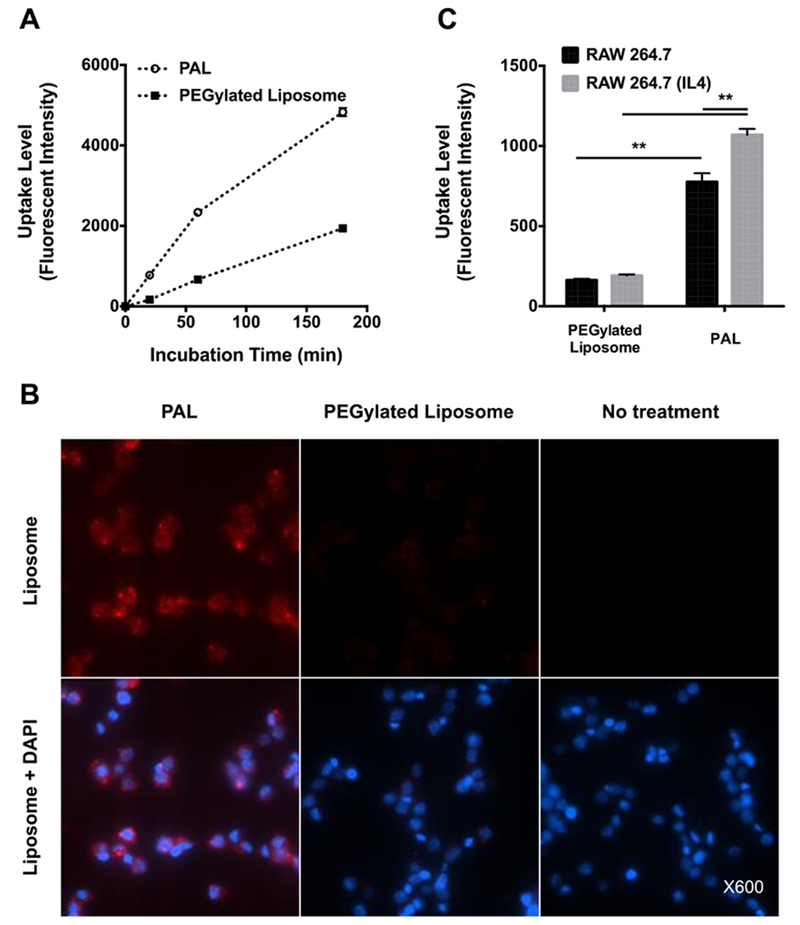
(A) Time course of RAW 264.7 cell uptake of mannose-functionalized polymer-augmented liposomes (PAL) and non-targeted PEGylated liposomes; (B) Representative fluorescence microscopy images of RAW 264.7 incubated with rhodamine-labeled PALs or PEGylated liposomes (400 μg/mL) for 1 h; (C) Liposome uptake level of RAW 264.7 cells with and without IL-4 stimulation (mannose receptor upregulation). Data were reported as mean fluorescence intensity ± SD (n=3, **p < 0.005).
To further demonstrate mannose receptor-mediated interactions between the PALs and RAW 264.7 cells, cells were treated with IL-4 to upregulate their expression of the mannose receptor40. Following 20 min incubation, the uptake of PALs was significantly higher in IL-4 treated cells than in non-treated cells (P < 0.005), indicating the involvement of mannose receptors in the effective PAL uptake by macrophages (Fig. 5c). As expected, PEGylated control liposomes show significantly lower uptake levels in RAW 264.7 cells than that of PALs, regardless of IL-4 pretreatment. Those results were in agreement with a prior study showing that the uptake mechanism of mannosylated liposomes was mannose-receptor mediated endocytosis in NR8383, a cell line expressing mannose receptors23. Additionally, our lab has previously demonstrated that mannose functionalized polymers show specific mannose receptor-mediated uptake by IL-4 treated RAW 264.7 macrophages31. Taken together, the uptake study results demonstrate that mannose modification significantly increased the internalization of PALs, likely via the mannose receptor-mediated endocytosis.
Bactericidal effect of streptomycin-loaded PALs
The intracellular bactericidal efficacy of streptomycin-loaded PALs was evaluated using a co-culture challenge assay with F. novicida infected RAW 264.7 cells. F. novicida is commonly used as a laboratory surrogate for F. tularensis, due to its low virulence for humans and high degree of genetic similarities with F. tularensis (~95%)55. Additionally, F. novicida exhibits the same evasion of phagolysosome fusion and phagosome escape within infected human-derived macrophages as F. tularensis56. Bactericidal activity was quantified by enumerating the colony forming units (CFU) of surviving intracellular bacteria after incubating streptomycin-loaded PAL, streptomycin-loaded PEGylated control liposomes or free streptomycin with infected RAW 264.7 cells for 22 hours. Non-drug containing blank PALs or PEGylated liposomes showed no inhibitory effect (Fig. 6a), similar to the no treatment control.
Fig. 6. Antibacterial activity of streptomycin-loaded polymer-augmented liposomes.
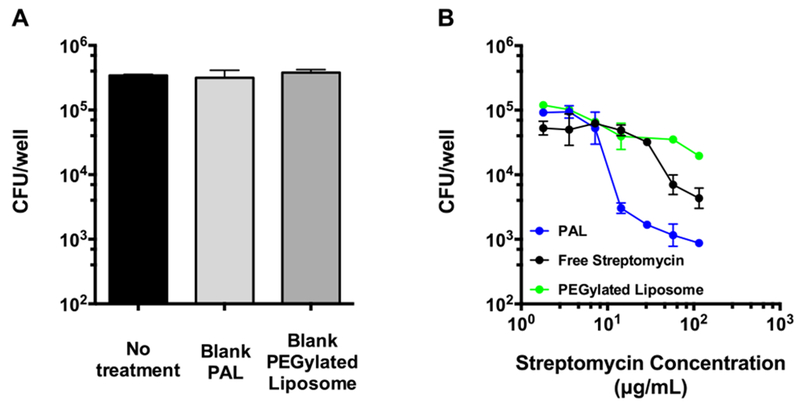
(A) RAW 264.7 cells were treated with blank polymer-augmented liposomes (PAL) or control PEGylated liposomes following an infection with F. novicida. Concentrations used here equaled to the maximum liposome concentration (highest streptomycin dose) used in Fig. 5b. (B) RAW 264.7 cells were treated with varying streptomycin concentrations (115 μg/mL to 1.8 μg/mL) of different formulations following an infection with F. novicida to determine the efficacy against intracellular bacteria. CFU: colony-forming unit. (Mean ± SD, n=3).
Streptomycin-loaded PALs showed concentration-dependent intracellular bacterial growth inhibition, and exhibited better inhibitory capability against intracellular bacteria than streptomycin-loaded PEGylated liposomes and free streptomycin (Fig. 6b). At ~15 μg/mL dosing, the PAL system demonstrated a 13-16 fold increased efficacy, compared to free streptomycin and PEGylated liposomes. The better efficacy of PALs than PEGylated liposomes may result from the fact that endosomal-releasing activity of PALs facilitates the cytosolic delivery of streptomycin, which would otherwise be mainly entrapped in acidic lysosomes due to the polycationic nature of streptomycin10. This observation was in accordance with a previous study showing that gentamicin-loaded fusogenic liposomes had better efficacy against intracellular bacteria than non-fusogenic liposomes18. It is worth noting that PALs did not show significant cytotoxicity in RAW 264.7 cells within the dose range (Fig. S3b†). Those findings revealed the superior inhibition of intracellular bacterial growth by streptomycin-loaded PALs, which can account for its efficient intracellular drug release in infected RAW 264.7 cells. Additional studies are needed to elucidate the individual contribution of the targeting and pH-responsive functionalities to the superior activity of PALs.
Building upon the findings of the current study, future design criteria for antibiotic carriers against intracellular infections may include differential release mechanisms that respond to the microenvironemnt of the intracellular infection setting (e.g., bacterial enzymes60), or respond to physical stimuli (e.g, ultrasound) to minimize off-target release61. Additionally, antibiotic carriers that can provide sustained release at the site of infection are postulated to improve drug retention, and therefore potentially reduce dosing frequency62.
Conclusions
This study demonstrates a utility of a multifunctional diblock copolymer, poly[(DEAEMA-co-BMA)-b-ManEMA], to functionalize liposomes for intracellular antibiotic delivery. Liposomes formulated with this polymer (PALs) enhanced the cellular uptake level through mannose receptor-mediated endocytosis, as confirmed in macrophage uptake study. Additionally, the PALs show endosomal pH-induced enhanced drug release and endosomal-releasing activity. Streptomycin loaded in PALs shows better bactericidal efficacy than free drug and non-pH responsive and non-targeted PEGylated control liposomes in a representative bacteria-macrophage co-culture model. Taken together, the PAL system developed here provides a new delivery platform for effective cytosolic antibiotic delivery against Francisella and potentially other intracellular bacterial infections.
Supplementary Material
Acknowledgements
This work was funded by the Defense Threat Reduction Agency (Grant #HDTRA1-13-1-0047). F.Y.S. is an international student research fellow of the Howard Hughes Medical Institute. J.C. and A.M.K. were graduate research fellows of the National Science Foundation.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details including polymer synthesis, GPC, NMR, high performance size exclusion chromatography, and cell viability assay. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Zaman K, Journal of Health, Population, and Nutrition, 2010, 28, 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goel AK, World Journal of Clinical Cases: WJCC, 2015, 3, 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis J, Oyston PCF, Green M and Titball RW, Clin. Microbiol. Rev, 2002, 15, 631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perumal Samy R, Stiles BG, Sethi G and Lim LHK, PLoS Neglected Trop. Di.s, 2017, 11, e0004738. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Oyston PCF, Sjostedt A and Titball RW, Nat. Rev. Microbiol, 2004, 2, 967–978. [DOI] [PubMed] [Google Scholar]
- 6.Ray K, Marteyn B, Sansonetti PJ and Tang CM, Nat. Rev. Microbiol, 2009, 7, 333–340. [DOI] [PubMed] [Google Scholar]
- 7.Flannagan RS, Cosio G and Grinstein S, Nat. Rev. Microbiol, 2009, 7, 355–366. [DOI] [PubMed] [Google Scholar]
- 8.Fink A, Hassan MA, Okan NA, Sheffer M, Camejo A, Saeij JPJ and Kasper DL, mBio, 2016, 7, e02243–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufmann SH, Annu. Rev. Immunol, 1993, 11, 129–163. [DOI] [PubMed] [Google Scholar]
- 10.Carryn S, Chanteux H, Seral C, Mingeot-Leclercq M-P, Van Bambeke F and Tulkens PM, Infectious Disease Clinics of North America, 2003, 17, 615–634. [DOI] [PubMed] [Google Scholar]
- 11.Van Bambeke F, Barcia-Macay M, Lemaire S and Tulkens PM, Curr. Opin. Drug Discovery Dev, 2006, 9, 218–230. [PubMed] [Google Scholar]
- 12.Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Tonat K and for the Working Group on Civilian Biodefense, JAMA, 2001, 285, 2763–2773. [DOI] [PubMed] [Google Scholar]
- 13.Pechere J-C and Dugal R, Clin. Pharmacokinet, 1979, 4, 170–199. [DOI] [PubMed] [Google Scholar]
- 14.Tulkens PM, Scandinavian Journal of Infectious Diseases. Supplementum, 1990, 74, 209–217. [PubMed] [Google Scholar]
- 15.de Jager P and van Altena R, Int. J. Tuberc. Lung Dis, 2002, 6, 622–627. [PubMed] [Google Scholar]
- 16.Jernberg C, Lofmark S, Edlund C and Jansson JK, Microbiology, 2010, 156, 3216–3223. [DOI] [PubMed] [Google Scholar]
- 17.Wong JP, Yang H, Blasetti KL, Schnell G, Conley J and Schofield LN, J. Controlled Release, 2003, 92, 265–273. [DOI] [PubMed] [Google Scholar]
- 18.Lutwyche P, Cordeiro C, Wiseman DJ, St-Louis M, Uh M, Hope MJ, Webb MS and Finlay BB, Antimicrob. Agents Chemother, 1998, 42, 2511–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vyas SP, Kannan ME, Jain S, Mishra V and Singh P, Int. J. Pharm, 2004, 269, 37–49. [DOI] [PubMed] [Google Scholar]
- 20.Cipolla D, Blanchard J and Gonda I, Pharmaceutics, 2016, 8, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamblin KA, Armstrong SJ, Barnes KB, Davies C, Wong JP, Blanchard JD, Harding SV, Simpson AJH and Atkins HS, Antimicrob. Agents Chemother, 2014, 58, 3053–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wijagkanalan W, Kawakami S, Takenaga M, Igarashi R, Yamashita F and Hashida M, J. Controlled Release, 2008, 125, 121–130. [DOI] [PubMed] [Google Scholar]
- 23.Chono S, Tanino T, Seki T and Morimoto K, J. Controlled Release, 2008, 127, 50–58. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi S, Nakase I, Kawabata N, Yu H-H, Pujals S, Imanishi M, Giralt E and Futaki S, Bioconjugate Chem, 2009, 20, 953–959. [DOI] [PubMed] [Google Scholar]
- 25.Yen J, Zhang Y, Gabrielson NP, Yin L, Guan L, Chaudhury I, Lu H, Wang F and Cheng J, Biomater. Sci, 2013, 1, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song W-J, Du J-Z, Sun T-M, Zhang P-Z and Wang J, Small, 2010, 6, 239–246. [DOI] [PubMed] [Google Scholar]
- 27.Lane DD, Su FY, Chiu DY, Srinivasan S, Wilson JT, Ratner DM, Stayton PS and Convertine AJ, Polym. Chem, 2015, 6, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raisin S, Morille M, Bony C, Noel D, Devoisselle J-M and Belamie E, Biomater. Sci, 2017, 5, 1910–1921. [DOI] [PubMed] [Google Scholar]
- 29.Keller S, Wilson JT, Patilea GI, Kern HB, Convertine AJ and Stayton PS, J. Controlled Release, 2014, 30, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshizaki Y, Yuba E, Sakaguchi N, Koiwai K, Harada A and Kono K, Biomaterials, 2014, 35, 8186–8196. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Son H-N, Hill JJ, Srinivasan S, Su F-Y, Stayton PS, Convertine AJ and Ratner DM, Nanomedicine, 2016, 12, 2031–2041. [DOI] [PubMed] [Google Scholar]
- 32.Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS and Stayton PS, J. Controlled Release, 2009, 133, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stano P, Bufali S, Pisano C, Bucci F, Barbarino M, Santaniello M, Carminati P and Luisi PL, J. Liposome Res, 2004, 14, 87–109. [DOI] [PubMed] [Google Scholar]
- 34.Chang H-I and Yeh M-K, Int. J. Nanomed, 2012, 7, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen AC and Mayer RT, Journal of Chromatography A, 1981, 207, 445–448. [Google Scholar]
- 36.Tekkeli SEK, Onal A and Sagirli AO, Luminescence, 2014, 29, 87–91. [DOI] [PubMed] [Google Scholar]
- 37.Udenfriend S, Stein S, Böhlen P, Dairman W, Leimgruber W and Weigele M, Science, 1972, 178, 871–872. [DOI] [PubMed] [Google Scholar]
- 38.Chen C, Zheng P, Cao Z, Ma Y, Li J, Qian H, Tao W and Yang X, Biomater. Sci, 2016, 4, 412–417. [DOI] [PubMed] [Google Scholar]
- 39.Murthy N, Robichaud JR, Tirrell DA, Stayton PS and Hoffman AS, J. Controlled Release, 1999, 61, 137–143. [DOI] [PubMed] [Google Scholar]
- 40.Stein M, Keshav S, Harris N and Gordon S, J. Exp. Med, 1992, 176, 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh A, Talekar M, Raikar A and Amiji M, J. Controlled Release, 2014, 190, 515–530. [DOI] [PubMed] [Google Scholar]
- 42.Laskin DL, Sunil VR, Gardner CR and Laskin JD, Annual review of pharmacology and toxicology, 2011, 51, 267–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Donowitz GR, Clin. Infect. Dis, 1994, 19, 926–930. [DOI] [PubMed] [Google Scholar]
- 44.Roberts LM, Tuladhar S, Steele SP, Riebe KJ, Chen C-J, Cumming RI, Seay S, Frothingham R, Sempowski GD, Kawula TH and Frelinger JA, Infect. Immun, 2014, 82, 2504–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Z, Clemens DL, Lee B-Y, Dillon BJ, Horwitz MA and Zink JI, ACS Nano, 2015, 9, 10778–10789. [DOI] [PubMed] [Google Scholar]
- 46.Rodvold KA, Pharmacotherapy, 2001, 21, 319S–330S. [DOI] [PubMed] [Google Scholar]
- 47.Moad G, Rizzardo E and Thang SH, Aust. J. Chem, 2005, 58, 379–410. [Google Scholar]
- 48.Manganiello MJ, Cheng C, Convertine AJ, Bryers JD and Stayton PS, Biomaterials, 2012, 33, 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Longmire M, Choyke PL and Kobayashi H, Nanomedicine, 2008, 3, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ringsdorf H, Venzmer J and Winnik FM, Angew. Chem. Int. Ed. Engl, 1991, 30, 315–318. [Google Scholar]
- 51.Kitano H, Akatsuka Y and Ise N, Macromolecules, 1991, 24, 42–46. [Google Scholar]
- 52.Grabielle-Madelmont C, Lesieur S and Ollivon M, Size-Exclusion Chromatography, 2003, 56, 189–217. [DOI] [PubMed] [Google Scholar]
- 53.Bertrand N, Simard P and Leroux J-C, Methods Mol. Biol, 2010, 605, 545–558. [DOI] [PubMed] [Google Scholar]
- 54.Di Marzio L, Esposito S, Rinaldi F, Marianecci C and Carafa M, Colloids Surf., B, 2014, 104, 200–206. [DOI] [PubMed] [Google Scholar]
- 55.Ohkuma S and Poole B, Proc. Natl. Acad. Sci. U.S.A, 1978, 75, 3327–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Plank C, Oberhauser B, Mechtler K, Koch C and Wagner E, J. Biol. Chem, 1994, 269, 12918–12924. [PubMed] [Google Scholar]
- 57.Ezekowitz RAB, Williams DJ, Koziel H, Armstrong MYK, Warner A, Richards FF and Rose RM, Nature, 1991, 351, 155–158. [DOI] [PubMed] [Google Scholar]
- 58.Hussell T and Bell TJ, Nat. Rev. Immunol, 2014, 14, 81–93. [DOI] [PubMed] [Google Scholar]
- 59.Azad AK, Rajaram MVS and Schlesinger LS, Journal of cytology & molecular biology, 2014, 1, 1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Komnatnyy VV, Chiang W-C, Tolker-Nielsen T, Givskov M and Nielsen TE, Angew. Chem. Int. Ed. Engl, 2014, 53, 439–441. [DOI] [PubMed] [Google Scholar]
- 61.Noble ML, Mourad PD and Ratner BD, Biomater. Sci, 2014, 2, 839–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mountziaris PM, Shah SR, Lam J, Bennett GN and Mikos AG, Biomater. Sci, 2016, 4, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.