

Abstract
Endolysosomes can be damaged by diverse materials. Terminally damaged compartments are degraded by lysophagy, but pathways that repair salvageable organelles are poorly understood. Here we found that the Endosomal Sorting Complex Required for Transport (ESCRT) machinery, known to mediate budding and fission on endolysosomes, also plays an essential role in their repair. ESCRTs were rapidly recruited to acutely injured endolysosomes via a pathway requiring calcium and ESCRT-activating factors that was independent of lysophagy. We used live cell imaging to demonstrate that ESCRTs responded to small perforations in endolysosomal membranes and enabled compartments to recover from limited damage. Silica crystals that disrupted endolysosomes also triggered ESCRT recruitment. ESCRTs thus provide a defense against endolysosomal damage likely to be relevant in physiological and pathological contexts.
One Sentence Summary:
Damaged endosomes and lysosomes rapidly recruit ESCRT machinery to promote membrane repair.
Endosomes and lysosomes together comprise the endolysosomal network that functions to sort, recycle, and degrade a wide range of substances from within and outside the cell. Endolysosomes are vulnerable to damage from diverse material such as incoming pathogens that seek to access the cytoplasm, molecules that intercalate into or otherwise destabilize the lipid bilayer, or particulate matter such as crystals that can puncture the membrane. Failure to promptly repair or sequester disrupted compartments can have deleterious consequences including in extreme cases cell death.
Damaged endolysosomes can be isolated from the rest of the cell and degraded by a form of selective autophagy known as lysophagy. Lysophagy is promoted by recruitment of cytoplasmic proteins including galectins and glycoprotein-specific ubiquitin ligases to abnormally exposed lumenal glycans on the afflicted compartment (1–6). Whether damaged endolysosomes can avoid autophagic degradation and instead be fully repaired is less clear but has been suspected (7–11). How this might work and how disrupted compartments would be triaged between these opposing fates is not known.
Membrane repair is known to occur elsewhere in the cell, particularly at the plasma membrane where coordinated responses counter mechanical and other disruptions (12, 13), but also at the nuclear envelope (14). In both of these contexts, recent studies highlight important roles played by ESCRT (Endosomal Sorting Complex Required for Transport) machinery (15–19). ESCRT proteins are organized into several modular complexes designated ESCRT-0, -I, -II, and -III, in addition to the ATPase VPS4 and associated factors, that together help power such processes as intralumenal vesicle formation, viral budding, and cytokinetic abscission, in addition to membrane repair (20). All of these processes share the involvement of ESCRT-III proteins (including CHMPs 1–7 and IST1), which form filaments around membrane orifices that are thought to drive constriction and typically promote membrane fission (21, 22).
ESCRTs respond to endolysosomal damage
To investigate whether ESCRT machinery might participate in repairing damaged endolysosomes, we first asked whether ESCRT components are recruited to these compartments following selective membrane damage. The lysosomotropic compound LLOME (L-leucyl-L-leucine O-methyl ester) is commonly used to rupture endolysosomes (3, 23, 24). LLOME rapidly permeates cellular membranes and accumulates in the lumen of acidified organelles, where it is condensed into a membranolytic polymer by the lysosomal enzyme cathepsin C (9, 23). We confirmed LLOME-induced damage in HeLa cells by noting concentration of cytoplasmic GAL3 (a galectin frequently used to monitor damage (24)) on punctate structures (Fig. 1A). Immunostaining LLOME-treated cells for the ESCRT-III component CHMP4A revealed that this typically cytoplasmic protein also accumulated on intensely stained puncta throughout the cell (Fig. 1A). These CHMP4A-containing structures did not completely correspond to those marked by GAL3, but they did overlap with the known CHMP4A-interacting protein ALIX (Fig. 1B).
Fig. 1. Damaged endolysosomes recruit ESCRT machinery.
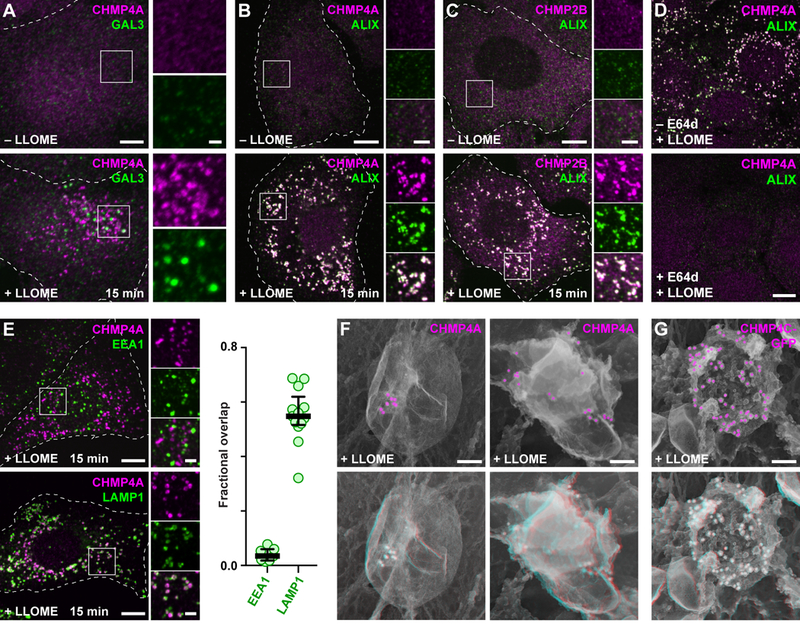
(A) HeLa cells treated with or without LLOME were costained for CHMP4A and GAL3. (B and C) U2OS cells treated as above were costained for ALIX and the indicated ESCRT-III proteins. (D) U2OS cells preincubated for 5 min in the absence or presence of the cathepsin inhibitor E64d were exposed to LLOME and costained for CHMP4A and ALIX. (E) LLOME-treated U2OS cells were stained for CHMP4A and EEA1 or LAMP1. Fractional overlap between CHMP4A puncta and labeled compartments is shown at right (n = 7 cells for EEA1, 13 cells for LAMP1). (F) U2OS cells or (G) HeLa cells producing CHMP4C-GFP were treated with LLOME and immunolabeled as indicated before processing for deep-etch electron microscopy. Top panels depict two-dimensional views with pseudocolored immunogold; bottom panels show corresponding anaglyphs, to be viewed with dual color glasses. In all fluorescence micrographs, representative cells are shown outlined by dashed white lines; boxed areas are magnified at right; and coincidence of green and magenta appears white. Scale bars equal 10 µm (A to E; 2 µm in magnified views); 100 nm (F and G).
Multiple ESCRT-III proteins, including CHMP2B (Fig. 1C) as well as CHMP1A, CHMP1B, CHMP3, CHMP4B, CHMP5, and IST1 (fig. S1) were co-recruited along with CHMP4A to LLOME-triggered puncta in U2OS cells, as were additional ESCRT-III interactors including VPS4A and VTA1 (fig. S1). The extensive accumulation and coincidence of this large number of ESCRT-III proteins suggests that ESCRT mobilization constitutes a coherent response to LLOME. We detected similar LLOME-dependent redistribution of ESCRT proteins in several different cell types, including breast adenocarcinoma and glioblastoma cells as well as macrophage-like THP-1 cells commonly used in studies of pathogen uptake and endolysosomal escape (fig. S2), and in HeLa cells expressing fluorescently-tagged ESCRT-III proteins (fig. S3).
We next confirmed that ESCRT recruitment correlated with LLOME-induced membrane damage. The effects of LLOME on ESCRTs were abolished by preincubating cells with the cathepsin inhibitor E64d (Fig. 1D), which prevents LLOME processing by cathepsin C and consequent membrane disruption (9). Accordingly, ESCRT-enriched structures induced by LLOME corresponded to late endosomes and lysosomes, coinciding well with LAMP1 but not with the early endosome protein EEA1 (Fig. 1E). ESCRT proteins localized to the limiting membrane of damaged endolysosomes (fig. S4). Closer inspection of ESCRT-labeled compartments by deep-etch electron microscopy confirmed that ESCRTs accumulated on vesicular structures (Fig. 1F, fig. S5). ESCRTs were seen to cluster in small domains on compartments exhibiting low immunolabel density, suggesting a role at discrete regions of the organelle membrane.
We next explored how ESCRT machinery might be targeted to damaged endolysosomes, using CHMP4A as a reporter of ESCRT recruitment. The ESCRT-I protein TSG101 is well known to nucleate ESCRT assembly in various contexts (20). TSG101 co-accumulated with CHMP4A on endolysosomes after LLOME-induced damage (fig. S6), and depleting cells of TSG101 delayed, but did not abolish, CHMP4A recruitment (fig. S7). TSG101 interacts with, and in certain contexts cooperates with ALIX to recruit ESCRT-III machinery (25, 26). Although ALIX co-localized with CHMP4A and other ESCRT components on endolysosomes after damage (Fig. 1B and fig. S1), CHMP4A accumulation was unperturbed in LLOME-treated cells depleted of ALIX alone (fig. S7) despite efficient protein reduction (fig. S8). However, depleting both TSG101 and ALIX suppressed the response of CHMP4A to LLOME (Fig. 2A, fig. S7 & S8). Other ESCRT-III proteins were similarly attenuated, although some residual signal was observed potentially attributable to differential antibody sensitivity and/or incomplete knockdown (fig. S9). Because TSG101 is known to be important for ESCRT-mediated endosomal sorting, we wanted to be sure that the reduction in ESCRT recruitment was not caused by impaired LLOME processing. GAL3 accumulation in response to LLOME was, however, unchanged in cells co-depleted of TSG101 and ALIX (Fig. 2B), confirming comparable membrane damage.
Fig. 2. Calcium as well as TSG101 and ALIX are required for ESCRT assembly on damaged endolysosomes.
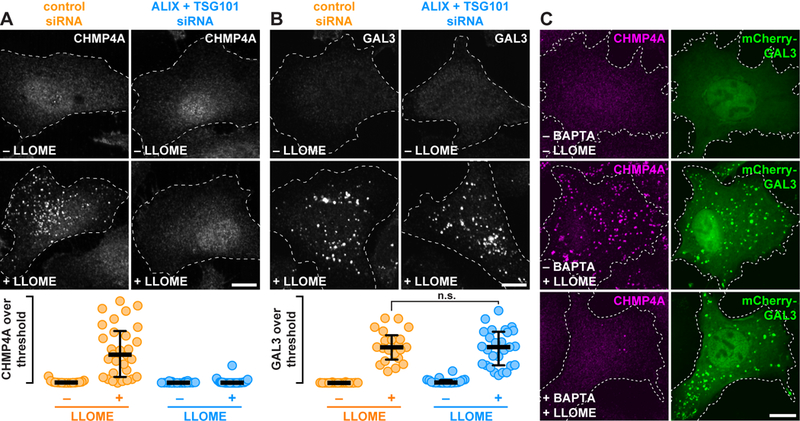
(A and B) HeLa cells transfected with the indicated siRNAs were treated with or without LLOME and stained for CHMP4A (A) or GAL3 (B). The proportion of each stain that exceeded a starting threshold is quantified (in the order shown: n = 20, 33, 21, and 33 cells in A; and 20, 21, 20, and 27 cells in B; n.s. denotes p = 0.6319 by Student’s two-tailed unpaired t-test). Horizontal bars mark the median; whiskers span the interquartile range. (C) U2OS cells producing mCherry-GAL3 were preincubated for 1 h with or without BAPTA-AM, then treated with DMSO or LLOME and stained for CHMP4A. Scale bars equal 10 µm.
TSG101 and ALIX direct ESCRT assembly to membranes in response to pathway-specific signals. Calcium is an established trigger of cellular responses to membrane damage at the plasma membrane (13), and is required to recruit ESCRTs to plasma membrane wounds (15, 16). Because endolysosomes are calcium-storing organelles and rupturing them elevates intracellular calcium (27, 28), we wondered whether ESCRT recruitment to damaged endolysosomes might similarly be regulated by calcium. Preloading cells with the calcium chelator BAPTA-AM prevented accumulation of ESCRTs on LLOME-disrupted endolysosomes without impairing organelle damage, demonstrated again by GAL3 accumulation (Fig. 2C). Supporting a role for calcium, ESCRT machinery coaccumulated on damaged endolysosomes with ALG-2 (fig. S10), a protein previously implicated in calcium-dependent ESCRT assembly at plasma membrane wounds (16, 29).
ESCRT response is separable from lysophagy
Calcium-triggered responses are typically rapid, prompting us to inspect the timing of ESCRT recruitment to damaged compartments. In U2OS and HeLa cells treated with LLOME for varying durations, ESCRT components were detectable on LAMP1-containing organelles within a minute (Fig. 3A), and reached maximal accumulation shortly thereafter (Fig. 3, B to D). In contrast, GAL3-reactive structures were largely undetectable at these early times, instead appearing steadily over a prolonged period (Fig. 3, B and C) as expected (3, 4). The delayed accumulation of GAL3 was accompanied by increasing levels of ubiquitin-conjugated material (Fig. 3D and fig. S11) and the ubiquitin-dependent disaggregase VCP/p97 (fig. S12), consistent with known roles for ubiquitination and associated proteins in lysophagy (2, 5, 30). Early ESCRT recruitment and delayed accumulation of GAL3 and ubiquitinated material were also apparent in macrophage-like THP-1 cells (fig. S13), suggesting that the ESCRT response is distinct from lysophagy.
Fig. 3. ESCRT machinery is rapidly recruited to LLOME-damaged endolysosomes and is independent of lysophagy.
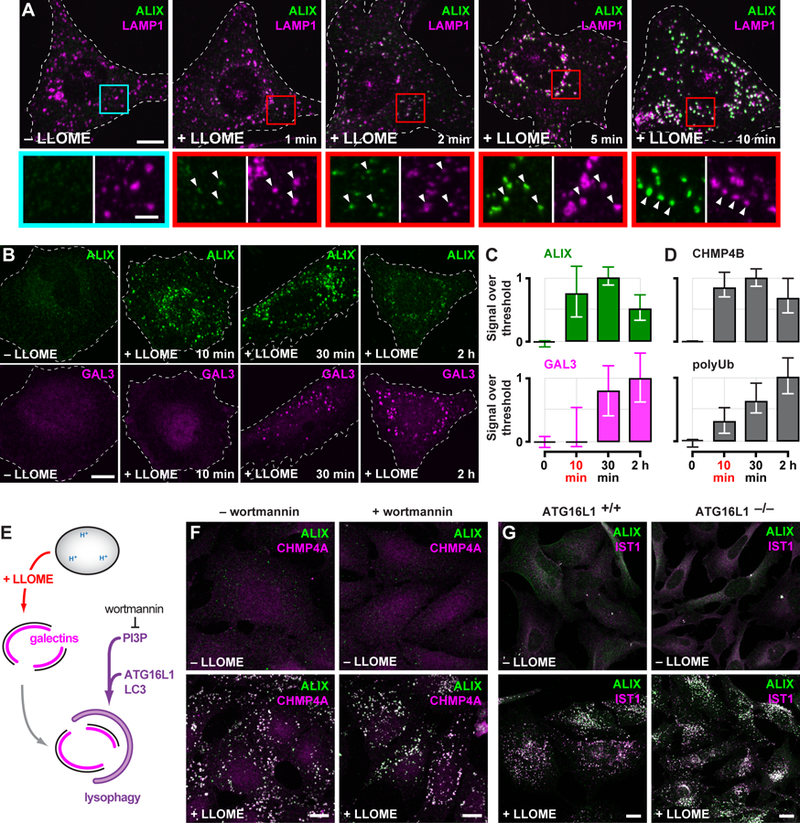
(A) U2OS cells exposed to LLOME for the indicated times were costained for ALIX and LAMP1. Boxed areas are magnified below; arrows highlight LLOME-induced ESCRT puncta. (B) HeLa cells treated with LLOME for the indicated times were costained for ALIX and GAL3. The proportion of each stain that exceeded a starting threshold is plotted in (C). Shown are the median and interquartile range (in chronological order: n = 3, 8, 10 and 8 cells). (D) Quantitation of CHMP4B or ubiquitinated material (polyUb) (see fig. S11) as in (C) (in chronological order: n = 10, 11, 15, and 16 cells for CHMP4B; n = 19, 18, 25 and 26 cells for polyUb). (E) Schematic depicting lysophagy and the role of factors targeted in adjacent experiments. (F) U2OS cells preincubated with or without wortmannin were treated with LLOME and costained for ALIX and CHMP4A. (G) HeLa cells expressing endogenous ATG16L1 (ATG16L1+/+) or lacking ATG16L1 as a result of CRISPR-mediated gene deletion (ATG16L1−/−), were treated with or without LLOME for 10 min and costained for ALIX and the ESCRT-III protein IST1. Scale bars equal 10 µm (2 µm in magnified views).
ESCRTs, however, have long been suspected to play a poorly defined role during autophagosome maturation (31), prompting us to further examine the relationship between lysophagy and ESCRT recruitment. Lysophagy involves engulfment of the damaged organelle by an autophagosome, whose formation requires phosphatidylinositol 3-phosphate (PI3P) (Fig. 3E). The ESCRT response to LLOME was unaffected by treating cells with wortmannin (Fig. 3F), an inhibitor of PI3P synthesis, despite clear physiological changes associated with loss of PI3P elsewhere in the cell (fig. S14). Organelle engulfment also requires conjugation of LC3 to the expanding autophagosome membrane (3) by the autophagy elongation complex (Fig. 3E), of which ATG16L1 is an essential component (32). Indeed, HeLa cells lacking ATG16L1 (33) failed to accumulate GFP-tagged LC3B in association with LLOME-damaged endolysosomes (fig. S15). In spite of this, LLOME-induced ESCRT redistribution was unperturbed (Fig. 3G). GFP-LC3B association with LLOME-damaged organelles was also unaffected in cells depleted of TSG101 and ALIX (fig. S16). The presence of ESCRT machinery on LLOME-damaged endolysosomes could thus be uncoupled from autophagosome formation.
ESCRTs preferentially sense small ruptures in the endolysosomal membrane
We next examined why ESCRTs respond differently than factors involved in lysophagy to LLOME-induced damage. LLOME forms hydrophobic adducts that destabilize the lipid bilayer, causing damage that ranges from small perturbations permeable to protons and small molecules (9) to larger ruptures that allow exchange of higher molecular weight material (24). Lysophagy of ruptured endolysosomes is directed in part by an influx of galectins, which bind to injury-exposed lumenal β-galactosides (1, 2) and would thus require sufficiently large membrane disruptions to access the compartmental interior. ESCRTs, in contrast, would not be similarly constrained because they assemble on the cytoplasmic surface of organellar membranes.
We used live cell imaging to assess whether the nature of membrane damage dictates whether and when ESCRTs or galectins are recruited. Fluorescently tagged ESCRT-III proteins, in this case CHMP4B-GFP (34), responded to LLOME earlier than did mCherry-GAL3 (Fig. 4A) as observed for their endogenous counterparts (Fig. 3, B to D). The proteins also did not always appear together, with some compartments only recruiting CHMP4B-GFP, some only recruiting mCherry-GAL3, and others accumulating both (see arrows in Fig. 4A). Dually labelled compartments were typically first surrounded by ESCRTs and only later acquired mCherry-GAL3, occasionally through what appeared to be a single breach of the limiting membrane (Fig. 4B) consistent with restricted access of GAL3 to the endolysosomal interior.
Fig. 4. ESCRT machinery preferentially responds to small membrane disruptions.
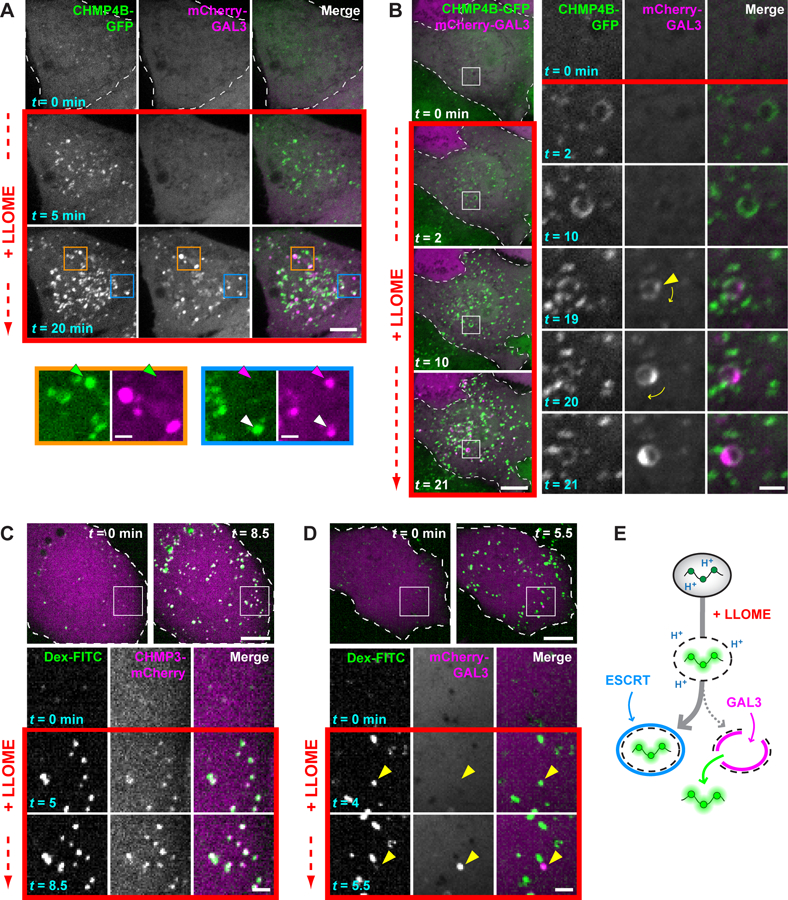
(A) HeLa cells producing CHMP4B-GFP were additionally transfected with a plasmid encoding mCherry-GAL3, and imaged live before and after adding LLOME. Colored arrows in magnified views of boxed areas designate structures containing only CHMP4B-GFP (green), only mCherry-GAL3 (magenta), or both (white). (B) Cells imaged as in (A). Boxed areas are magnified at right, with the leftmost two columns showing each signal in grayscale; these are merged and colored in the third column. Arrowhead marks the initial appearance of GAL3. Note that this compartment rotates over time, as denoted by the curved arrows. (C and D) U2OS cells were transfected with plasmids encoding CHMP3-mCherry (C) or mCherry-GAL3 (D) and loaded with 40-kDa pH-sensitive FITC-dextran chased into endolysosomes. Cells were imaged live before and after addition of LLOME. Boxed areas are magnified as in (B). Arrowheads in (D) indicate a dextran-laden compartment that rapidly acquires GAL3 after sudden loss of dextran. (E) Interpretive illustration of the different events seen in (C) and (D). In all panels, single planes of a representative cell are shown at the times indicated from each recording. Scale bars equal 10 µm (2 µm in magnified views).
We clarified this behavior by loading endolysosomes with high molecular weight dextrans conjugated to pH-sensitive FITC (fluorescein isothiocyanate), enabling us to distinguish perturbations permeable to protons from larger ruptures that allow dextran escape. Adding LLOME increased the fluorescence of dextran-loaded endolysosomes (Fig. 4C), indicating a dissipated pH gradient in an otherwise intact organelle. Most brightened dextran puncta were quickly surrounded by the ESCRT-III protein CHMP3-mCherry (Fig. 4C, fig. S17), whereas similar recruitment was not observed after raising lumenal pH with ammonium chloride (fig. S18). We did not see much mCherry-GAL3 on brightened dextran puncta during the same early time period (Fig. 4D). There were, however, occasional events in which sudden loss of dextran coincided with rapid concentration of mCherry-GAL3 (see arrows in Fig. 4D), linking the galectin response to substantial membrane perforation. Similarly abrupt GAL3 accumulation accompanies adenoviral escape from endosomes (35). ESCRTs thus responded to proton-permeable disruptions on the majority of afflicted compartments that GAL3 did not perceive (Fig. 4E). In support of this conclusion, treating cells with a low concentration of LLOME to limit damage redistributed ESCRTs onto LAMP1-reactive compartments, but only a few contained substantial GAL3 (fig. S19).
ESCRTs are transiently recruited following acute membrane damage
The preceding experiments were performed in the continuous presence of LLOME, and thus reported on cumulative ESCRT recruitment to perturbed compartments. However, ESCRT machinery operates transiently in most contexts that use its membrane remodeling activity (20). We thus sought a way to follow ESCRT dynamics during both the onset and resolution of endolysosomal injury. Briefly exposing cells to lysosomotropic rupturing agents induces transient membrane disruption (9, 11, 27, 36) and accordingly, cells treated with LLOME for less than a minute acquired ESCRT puncta shortly after adding drug but not at later times (fig. S20). Under these conditions of limited disruption, CHMP1B-GFP transiently appeared on dextran-labeled endolysosomes and then gradually dissociated over time (Fig. 5A). Similar dynamics were apparent with additional ESCRT-III components (fig. S21).
Fig. 5. Transient ESCRT recruitment accompanies and is important for recovery of endolysosomes from acute damage.
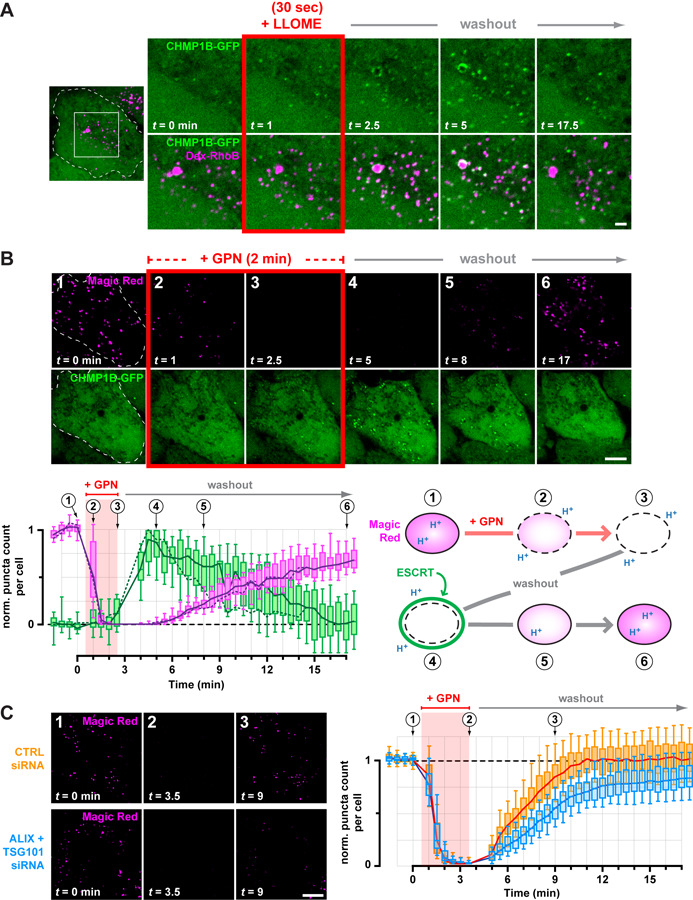
(A) HeLa cells producing CHMP1B-GFP were loaded with 40-kDa Rhodamine B dextran to label endolysosomes, and imaged live during and after a 30 sec exposure to LLOME. Shown are select frames from the resulting timelapse, corresponding to the boxed area of the outlined cell. Similar transient recruitment was observed using GPN (see fig. S23). (B) CHMP1B-GFP cells were incubated with Magic Red to label functional endolysosomes, and then imaged live at 30-sec intervals during and after a 2-min exposure to GPN. Magic Red was maintained at the same concentration throughout (see also Movie S1). The number of Magic Red or CHMP1B-GFP puncta detected at each time is plotted below (n = 17 cells), normalized to the initial number of puncta. Solid lines trace the median; boxes span the interquartile range; error bars designate the 10-th and 90-th percentiles. Dotted lines plot the number of puncta in the cell depicted in the time series, and the corresponding frames are numbered above the graph. An interpretive illustration of the observed events is shown at right. (C) U2OS cells transfected with the indicated siRNA were labeled with Magic Red and recorded live at 30-sec intervals during and after a 3-min pulse of GPN. Left panels depict a representative cell from each cohort, showing dissipation of Magic Red during the GPN pulse and a delay in Magic Red reacquisition in ESCRT-deficient cells. This behavior is plotted at right in terms of the number of Magic Red puncta at each time per cell from each cohort (n = 97 cells from 4 experiments). Scale bars equal 10 µm (2 µm in magnified views).
GPN (glycyl-L-phenylalanine 2-naphthylamide) is an alternative rupturing agent that accumulates in endolysosomes like LLOME (11, 27). However, GPN is processed into metabolites thought to promote osmotic rupture (27, 37) potentially similar to damage caused by materials that normally transit the endolysosomal network. On initial examination, ESCRT recruitment in GPN-treated cells was less pronounced than seen with LLOME (fig. S22). However, similarly to LLOME, briefly treating cells with GPN caused ESCRT proteins to transiently accumulate on dextran-marked endolysosomes (fig. S23).
ESCRTs facilitate endolysosomal membrane repair
We next compared the timing of ESCRT recruitment with loss and recovery of endolysosomal function, using the Magic Red fluorogenic indicator of lysosomal protease activity (11). Magic Red fluorescence was lost after brief exposure to GPN as above, but returned after washing GPN away (Fig. 5B and movie S1). ESCRT puncta appeared transiently after Magic Red dissipation but before complete recovery (Fig. 5B, fig. S24, and movie S1). A similar relationship between transient ESCRT puncta and Magic Red was observed in cells treated briefly with LLOME (fig. S25) but not in mock-treated controls (fig. S26 and movie S2).
The temporal correlation between transient ESCRT recruitment and endolyososomal recovery suggested that ESCRTs might be involved in repairing damaged endolysosomal membranes, a hypothesis supported by known roles of ESCRT proteins in resealing wounds at the plasma membrane and nuclear envelope (15–17). We thus implemented the protocol just described to examine endolysosomal recovery in cells depleted of TSG101 and ALIX. ESCRT-deficient cells acquired Magic Red to a similar extent as controls, and lost Magic Red fluorescence with similar kinetics after adding GPN (Fig. 5C), indicating comparable pathway function and GPN processing. However, reacquisition of Magic Red fluorescence after removing GPN was significantly delayed in ESCRT-deficient cells. In addition, approximately 20% fewer Magic Red structures recovered than were present at the start of the assay, revealing a net loss of functional endolysosomes when the ESCRT response was impaired.
Recovery of endolysosomal activity after damage is arguably a complex process, in which restoration of membrane integrity is an early but a critical step. Endolysosomal pH is well known to be acutely sensitive to changes in membrane permeability (38). We thus adapted a ratiometric pH-reporting system as a readout of membrane integrity (Fig. 6A), loading endolysosomes with a mixture of 40–70 kDa dextrans conjugated to pH-sensitive FITC or pH-insensitive Rhodamine B fluorophores. FITC fluorescence was initially quenched in the acidic endolysosomal environment but transiently increased during brief exposure to LLOME, revealing transient pH dissipation consistent with membrane disruption and recovery. Compartments in cells depleted of TSG101 and ALIX responded to LLOME-induced damage with increased FITC fluorescence comparable to that in controls (Fig. 6A). By plotting the average compartmental fluorescence ratio from individual cells over time, however, we noted that many ESCRT-deficient cells recovered organellar pH significantly more slowly than controls (Fig. 6, B and C) (39). Delayed recovery in ESCRT-deficient cells was also apparent after GPN-induced rupture (Fig. 6, D and E). Under both conditions, a significant proportion of knockdown cells failed to recover by the end of the assay, suggesting a greater number of irreparably damaged compartments. Importantly, knockdown cells responded similarly to controls to pH modulation by the weak base ammonium chloride (Fig. 6, F and G). Thus, ESCRT activity is required in endolysosomal repair.
Fig. 6. Depleting ESCRT machinery impairs endolysosomal membrane repair.
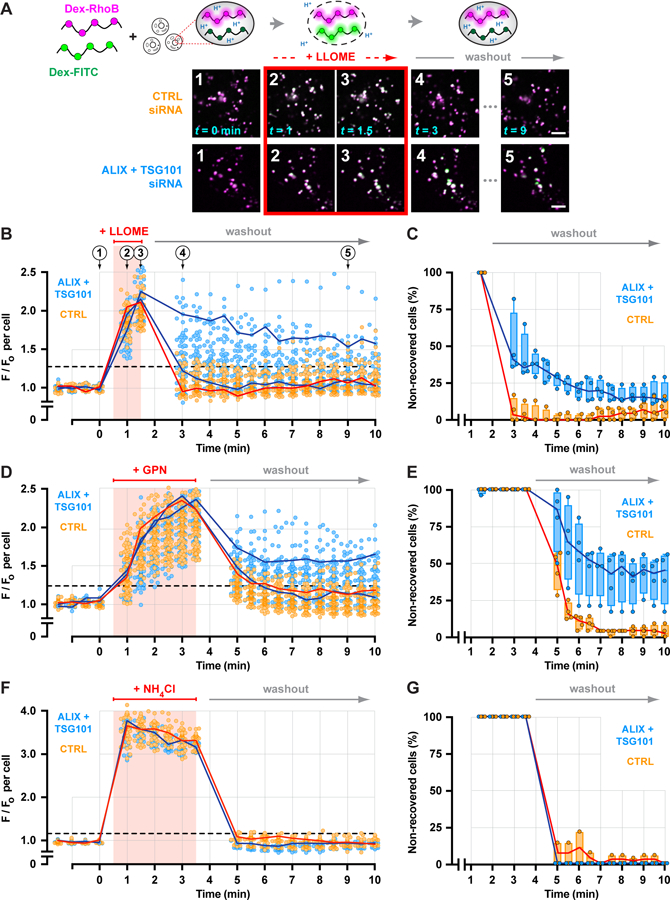
(A) A ratiometric pH-sensing assay was used to monitor endolysosomal integrity. U2OS cells transfected with the indicated siRNAs were loaded with 40–70 kDa dextrans conjugated to pH-sensitive FITC and pH-insensitive Rhodamine B fluorophores chased into endolysosomes. Loaded cells were imaged at 30-sec intervals during and after a 1-min exposure to LLOME. Fluorescence micrographs show a representative field of endolysosomes from each cohort, depicting the changes in FITC fluorescence associated with key steps in the assay. Scale bars equal 10 µm. (B) Changes in the median ratio of FITC-to-Rhodamine B fluorescence per cell at each time are plotted (n = 97 control and 85 knockdown cells from 4 experiments). Red line traces the response of a representative control cell. Blue lines designate two cells from the ESCRT-depleted cohort, one in which recovery was impaired and another in which recovery was similar to control. Horizontal dashed line specifies a recovery threshold derived from the control population (see Methods). In (C), the percentage of cells in each cohort that reached this threshold during recovery was counted, in each of 4 experiments. Lines connect the median; boxes and whiskers span the corresponding quartiles. (D-G) Using the same approach, the behavior of control and ESCRT-deficient cells was compared after a 3-min exposure to GPN (D and E; n = 99 control and 91 knockdown cells from 4 experiments) or to 10 mM ammonium chloride (F and G; n = 43 control and 29 knockdown cells from 4 experiments).
ESCRTs respond to membrane perforation by silica crystals
Previous studies have established that vesicular membranes can be damaged by particulate material such as crystals of silica, alum, and uric acid (10, 40, 41). In phagocytic cells, crystal-induced damage can lead to inflammasome activation that triggers secretion of pro-inflammatory cytokines and in dire cases activates cell death pathways. Although vesicles extensively damaged by silica are subject to lysophagy (3), silica-laden vacuoles have also been seen to undergo repeated cycles of small and resolvable membrane disruption (10).
To assess whether ESCRT machinery would respond to silica-induced damage, we examined ESCRT localization in macrophage-like THP-1 cells that had been fed silicon dioxide nanoparticles. Multiple ESCRT-III proteins were heavily enriched on vacuolar as well as smaller structures in a majority of silica-treated cells, whereas comparable structures were not observed in mock-treated controls (Fig. 7, A and B). ESCRTs colocalized on silica-damaged compartments and covered a large portion of their surface area (Fig. 7C and fig. S27). Similar ESCRT accumulation was apparent in U2OS cells exposed to silica nanoparticles (Fig. 7D). We additionally noted that ESCRT-positive vacuoles only sometimes contained GAL3 (Fig. 7D), supporting our conclusion that ESCRT machinery responds to sublytic membrane damage independently of galectins.
Fig. 7. ESCRTs are recruited to vacuolar compartments disrupted by silica nanocrystals.
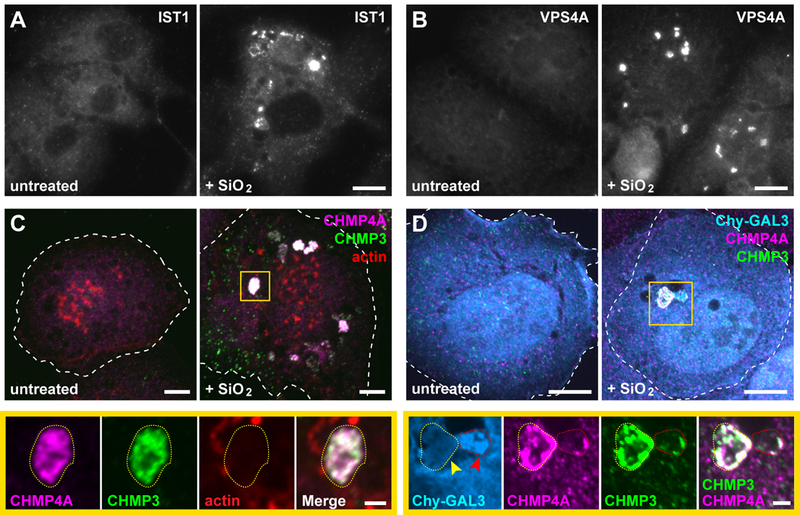
(A and B) PMA-differentiated THP-1 monocytes were treated with or without 50 µg/ml silicon dioxide (SiO2) nanoparticles for 3 h and stained for IST1 or VPS4A. Shown are widefield fluorescence images of representative cells from each cohort. (C) THP-1 cells treated as above were co-stained for the indicated ESCRT-III proteins along with rhodamine-conjugated phalloidin. Note that the prominent actin-rich puncta correspond to podosomes characteristic of this cell type. Shown are maximum intensity projections of representative cells. Magnified below are single planes corresponding to the boxed area, highlighting the type of compartment (outlined by yellow dotted lines) on which ESCRTs were predominantly observed in SiO2-fed cells (see fig. S27 for a corresponding three-dimensional rendering). (D) U2OS cells producing mCherry-GAL3 (Chy-GAL3) were fed silica nanoparticles for 1.5 h and costained for the indicated ESCRT-III proteins. Shown are maximum intensity projections and magnified single-plane views of the boxed area. Yellow arrow indicates a compartment (elsewhere encircled by a dotted yellow line) intensely stained for both ESCRT-III proteins but lacking Chy-GAL3; red arrow marks a compartment (elsewhere encircled by a dotted red line) that has accumulated Chy-GAL3 along with some ESCRT. Scale bars equal 10 µm (2 µm in magnified views).
Discussion
ESCRTs are best known – and named for – their involvement in endosomal trafficking, and are essential for generating vesicles that bud into multivesicular endosomes. Here we have shown that ESCRT machinery plays an additional role on endolysosomal organelles, responding to and promoting the repair of damaged or perforated membrane. ESCRT proteins readily accumulate on compartments disrupted by lysosomotropic peptides or internalized silica crystals, and are important for restoring compartmental pH and hydrolytic function in an experimental paradigm of acute and reversible membrane damage. Notably, ESCRTs are recruited to small disruptions in endolysosomes independently of factors involved in lysophagy such as galectins, which are preferentially engaged by more extensively damaged compartments. Together with earlier studies showing that ESCRTs respond to wounds at the plasma membrane (15–17) and nuclear envelope (18, 19), our study bolsters the idea that ESCRTs help to protect membrane integrity.
The need to protect endolysosomal integrity has broad implications for many situations, perhaps most critically in highly phagocytic cells that internalize and process substantial loads of potentially disruptive material. Much of what is currently understood about damage to endolysosomal compartments comes from studies of pathogen entry, which collectively reveal a wide variety of strategies for interacting with and modulating host endolysosomal membranes. Roles for ESCRT proteins in bacterial (42, 43) and viral (44–47) entry into cells have been reported, but never fully developed, because of challenges in reconciling them with trafficking functions classically associated with ESCRTs on endolysosomes. A role for ESCRTs in protecting endolysosomal integrity may provide an additional context for further understanding these connections.
Finally, central to this and previous studies of ESCRT function in membrane repair is the question of how this machinery acts to resolve membrane damage. Current thinking regarding ESCRT involvement in plasma membrane repair envisions that ESCRTs package wounded membrane into vesicles for release from the cell surface (15–17). We propose an alternative although not mutually exclusive possibility in which ESCRT-III proteins and the filaments they form may repair damaged membranes on endolysosomes, and potentially elsewhere, by directly resealing wounds. While clearly speculative, ESCRT-III filaments exhibit a propensity to assemble into spirals on highly curved membranes (21, 48) and can readily be envisioned to surround, constrict, and ultimately close an open hole.
Materials and Methods
Reagents and cell culture
All chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise indicated. Compounds were used at the following concentrations unless explicitly stated: 25 µM BAPTA-AM (no. 15551; Cayman Chemical, Ann Arbor, MI, USA); 200 µM E64d (no. 13533; Cayman Chemical); 200 µM GPN (no. sc-252858; Santa Cruz Biotechnology Dallas, TX, USA); 1 mM LLOME (no. L7393; Sigma-Aldrich); and 10 µM wortmannin (no. W1628; Sigma-Aldrich). Concentrated stock solutions of all compounds were prepared in dimethyl sulfoxide (DMSO) and stored at −80 ºC in single-use aliquots.
The mammalian expression vector encoding mCherry-GAL3 was kindly supplied by C. Weihl at Washington University in St. Louis (MO, USA) and has been described (27). The vector for p97-GFP was previously described (44). The plasmid encoding CHMP3-mCherry was constructed by cloning the human CHMP3 coding sequence into pmCherry-N1 (Clontech, Mountain View, CA, USA) using restriction enzymes XbaI and HindIII.
All cells were maintained at 37 ºC and supplemented with 5 % CO2. HeLa human cervical adenocarcinoma, U2OS human osteosarcoma, U-87 MG human glioblastoma, and MCF7 human breast adenocarcinoma cells originally from the American Type Culture Collection (ATCC; Manassas, VA, USA) were grown in Dulbecco’s modified Eagle’s medium (DMEM) (no. 11965–084; Gibco, Carlsbad, CA, USA) supplemented with 8 % v/v fetal bovine serum (FBS; Atlanta Biologicals, Flowery Branch, GA, USA). HeLa cells constitutively producing C-terminal GFP-tagged CHMP1B, CHMP4B, or CHMP4C were a generous gift from S. Simon at Rockefeller University (New York, NY, USA), and have been previously described (49). HeLa cells lacking ATG16L1 as a result of genetic deletion (clone G9), as previously described (33), were kindly provided by R. Xavier at the Broad Institute (Boston, MA, USA) and H. Virgin at Washington University; these were maintained in DMEM supplemented with 10 % v/v heat-inactivated FBS, 1 mM sodium pyruvate, and 10 mM HEPES (pH 7.4). The THP-1 (ATCC TIB-202) monocytic cell line was grown in suspension in Roswell Park Memorial Institute (RPMI) 1640 medium (R8758; Sigma-Aldrich) supplemented with 8 % v/v FBS, 1 mM sodium pyruvate, and 50 µM 2-mercaptoethanol. Prior to an experiment, 2–3 × 105 THP1 cells/ml were seeded over glass coverslips and differentiated into a macrophage-like state by incubating for 3–4 days in growth medium additionally supplemented with 0.4 µg/ml PMA (phorbol-12-myristate-13-acetate) (no. 524400; Calbiochem, San Diego, CA, USA).
Drug treatments for immunofluorescence
Cells were seeded over coverslips under the conditions appropriate for each cell line as described above. Growth media for all manipulations and drug dilutions were warmed to 37 ºC and equilibrated in 5 % CO2 before adding to cells. For experiments requiring continuous exposure to rupturing agents, coverslips were bathed in medium containing the appropriate agent, typically for 5–10 min unless otherwise indicated, then processed for immunofluorescence. For pulse-chase experiments, coverslips were immersed in medium containing the appropriate agent for the time indicated, rinsed in drug-free medium, and then incubated in excess drug-free medium as indicated before processing for immunofluorescence. For experiments assessing the effect of inhibitors or chelators on cellular responses to endolysosomal damage, cells were incubated in medium containing the inhibitor or chelator as specified, then switched to medium containing the inhibitor or chelator together with the indicated rupturing agent.
Crystal nanoparticle uptake
Silicon dioxide nanoparticles (no. tlrl-sio; InvivoGen, San Diego, CA, USA) were suspended in ultrapure water according to the manufacturer’s instructions and diluted in complete growth medium to 50–200 µg/ml. The resulting suspension was added to subconfluent cells on glass coverslips, and cells were incubated for the times indicated before being fixed and prepared for immunofluorescence.
RNA interference
Subconfluent cells were suspended by trypsinization and transfected using DharmaFECTTM 1 (Dharmacon, Lafayette, CO, USA) according to the manufacturer’s instructions. The resulting cell suspension was diluted in growth medium to a final concentration of 0.5–1 × 105 cells/ml, 25 nM total siRNA, and 0.12 % v/v transfection reagent, and dispensed into a well of a 6-well plate and incubated as described above. Medium was replaced after ca. 12 h. After 36–48 h, cells were suspended by trypsinization, reseeded at an appropriate density for subsequent experiments, and incubated for an additional 16–24 h. Where indicated, cells were additionally transfected with plasmid DNA, as described below, prior to reseeding. The siRNA sequences used were against human ALIX (5’ CCU GGA UAA UGA UGA AGG A), TSG101 (5’ CCU CCA GUC UUC UCU CGU C), or Firefly luciferase (5’ AUG UAU UGG CCU GUA UUA G) as a non-targeting control. Each siRNA was synthesized with dideoxythimidine 3’ overhangs by Dharmacon.
DNA transfections
Cells were suspended by trypsinization and transfected using Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The resulting cell suspension was diluted in growth medium to a final concentration of 0.5–1 × 105 cells/ml, 1 µg/ml total DNA, and 0.14 % v/v transfection reagent; dispensed into a culture vessel appropriate for the intended application; and incubated for 16–24 h before use.
Immunoblotting
Cell lysates were electrophoretically resolved under denaturing and reducing conditions according to the method of Laemmli, and transferred to Protran® nitrocellulose membranes (Amersham, Piscataway, NJ, USA) before immunoblotting with indicated antibodies against ALIX (mouse; no. 634502, Biolegend, San Diego, CA, USA), TSG101 (mouse; no. sc-7964, Santa Cruz), or α-tubulin (mouse; no. T-6199, Sigma-Aldrich), in Tris-buffered saline containing 0.01 % v/v Tween® 20 (Fisher Scientific, Waltham, MA, USA) and 3 % w/v nonfat milk solids (Carnation, Vevey, Switzerland). Immunoblots were developed by chemiluminescence and captured on a ChemiDocTM MP Imaging System (Bio-Rad Laboratories, Hercules, CA, USA).
Microscopy
The following imaging platforms were employed where designated. Settings appropriate for individual applications are specified in each corresponding section in the Methods.
Platform 1, widefield fluorescence:
Images were acquired via Metamorph Advanced software (64-bit, v. 7.7.5.0; Molecular Devices, Sunnyvale, CA, USA) on an Olympus (Tokyo, Japan) IX-81 microscope, using a 60× 1.42 NA PLAPON Apochromat oil-immersion objective, an X-Cite® 120Q excitation light source (Excelitas Technologies, Waltham MA, USA), an ORCA-Flash 2.8 scientific CMOS camera (Hamamatsu Photonics, Hamamatsu City, Japan), and the following Brightline® filter sets (Semrock, Rochester, NY, USA): long-pass blue emission (no. DAPI-11LP-A-000); single-band green emission (no. GFP-3035D-000); single-band red emission (no. mCherry-C-000); and single-band far-red emission (no. Cy5–4040C-000).
Platform 2, Olympus spinning disk confocal:
This configuration utilized the same microscope and objective as in Platform 1, with the exception that images were acquired via µManager (50) using a Yokogawa (Tokyo, Japan) CSU10 spinning disk confocal scanner; a two-line (488 and 561 nm) Sapphire laser module (Coherent, Santa Clara, CA, USA) equipped with an acousto-optic tunable filter (Gooch & Housego, Ilminster, UK); green emission (525 ± 25 nm) and red emission (605 ± 26 nm) ET series single-band bandpass filters (Chroma, Bellow Falls, VT, USA); and a Cascade® 512B camera (Photometrics, Tucson, AZ, USA) in charge multiplication mode. For all timelapse recordings, the Olympus IX3 Z-drift compensation module (IX3-ZDC2) was engaged to compensate for axial drift.
Platform 3, Nikon spinning disk confocal, single-camera mode:
Images were acquired on a Nikon (Tokyo, Japan) spinning disk confocal system, comprised of a Nikon Ti-E inverted microscope, 60× 1.40 NA and 100× 1.45 NA CFI Plan Apochromat Lambda oil-immersion objectives, and a 4-line (405, 488, 561, and 647 nm) laser module coupled to a Yokogawa CSU-X1 variable speed scanner through a filter wheel equipped with the following single-band bandpass filters (Chroma): blue emission ET455/50m (455 ± 25 nm); green emission ET525/36m (525 ± 18 nm); red emisson ET605/70m (605 ± 30 nm); and far-red emission ET700/75m (700 ± 37.5 nm). Images were acquired via NIS-Elements AR software (version 4.51; Nikon) on a Zyla 4.2-megapixel scientific CMOS camera (Andor, Belfast, UK). For all timelapse recordings, the Nikon Perfect Focus System (PFS) was used to compensate for axial drift. Where indicated, the camera was configured to use a 16-bit dual amplifier to improve the signal-to-noise ratio (SNR) yet preserve a sufficiently wide dynamic range to avoid pixel saturation. Binning was additionally employed to further improve the SNR as appropriate.
Platform 4, Nikon spinning disk confocal, dual-camera mode:
Images were acquired on the same system as Platform 3, with the following modifications. For ratiometric pH assessment, samples were simultaneously illuminated with 488 and 561 nm excitation light, and emitted fluorescence was simultaneously directed through an Andor TuCam dual camera adapter to a pair of Andor Zyla 4.2-megapixel sCMOS cameras, using a 580 nm BrightLine® dichroic beamsplitter (no. FF580-FDi01; Semrock) coupled to red emission (617 ± 36.5 nm; no. FF02–617/73) and green emission (514 ± 15 nm; no. FF01–514/30) BrightLine® single-band bandpass filters (Semrock). For all other experiments, samples were sequentially illuminated with 488 and then 561 nm excitation light, and only the green component of the 488 emission and the red component of the 561 emission (as determined by the above filter cut-offs), were retained.
Immunofluorescence
Cells grown over no. 1.5 round glass coverslips (Electron Microscopy Sciences, Hatfield, PA, USA) were fixed in 2.5–4 % w/v paraformaldehyde (Electron Microscopy Sciences) in PBS for 15 min at room temperature (RT), rinsed with PBS, and permeabilized either in 0.1 % v/v TritonTM X-100 (no. 28314; Pierce Biotechnology, Waltham, MA, USA) or, for all immunostains involving LAMP1, in 0.4 % w/v saponin (no. 84510; Sigma-Aldrich), in PBS for 10 min at RT. Cells were next rinsed in PBS and blocked for 30 min in 5 % v/v goat serum in PBS with or without 0.1 % w/v saponin, as appropriate. Alternatively, cells were fixed by immersing the coverslips in cold methanol on dry ice for 30 min, then air-dried for 5 min before blocking.
Cells were immunolabeled in blocking solution for 1 hr at RT with antibodies against the following proteins: ALG-2 at a 200-fold dilution (rabbit; no. 12303–1-AP, ProteinTech Group, Rosemont, IL, USA); ALIX at 5 µg/ml (mouse; no. 634502, Biolegend); CHMP1A at 1 µg/ml (mouse; no sc-271617, Santa Cruz); CHMP1B at 0.7 µg/ml (rabbit; no. 14639–1-AP, ProteinTech Group); CHMP2B at 5 µg/ml (rabbit; no. ab33174, Abcam, Cambridge, United Kingdom); CHMP3 at 1 µg/ml (mouse; no. sc-166361, Santa Cruz); CHMP4A at 1.3 µg/ml (rabbit; (51)); CHMP4B at a 200-fold dilution (rabbit; clone 485, a generous gift from A. Shiels at Washington University); CHMP5 at 0.8 µg/ml (rabbit; no. sc-67230, Santa Cruz); GAL3 at 1 µg/ml (rabbit; no. sc-20157, Santa Cruz) or at 1 µg/ml (mouse; no. sc-32790, Santa Cruz); IST1 at 0.4 µg/ml (rabbit; no. 19842–1-AP, ProteinTech); EEA1 at 0.5 µg/ml (mouse; no. 610456, BD Biosciences, Franklin Lakes, NJ, USA); LAMP1 at a 1000-fold dilution of ascites fluid (mouse; clone H4A3, the Developmental Studies Hybridoma Bank, Iowa City, IA, USA); TSG101 at 0.2 µg/ml (mouse; no. sc-7964, Santa Cruz); VPS4A at 5 µg/ml (mouse; no. SAB4200215, Sigma-Aldrich); or VTA1 at 3.7 µg/ml (rabbit; no. GTX107286, GeneTex, Irvine, CA, USA).
Following rinsing with PBS, goat secondary antibodies conjugated to Alexa Fluor® 488, 555, or 647 fluorescent dyes (Molecular Probes, Carlsbad, CA, USA) were diluted to 1 µg/ml in blocking solution together with 0.25 µg/ml of 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes), and added to immunolabeled cells for 30 min at RT. After rinsing with PBS, coverslips were mounted in a solution of 100 mM Tris (pH 8.5 at RT), 10.5 % w/v polyvinyl alcohol, and 21 % v/v glycerol, and allowed to cure for 24 h before imaging.
Images of fixed cells were acquired on the platform indicated in each figure legend. Where appropriate, multi-color images were computationally corrected for lateral and axial chromatic aberration using as a reference fluorescent microspheres (Invitrogen) imaged using identical parameters. Images to be compared were collected with identical acquisition settings, configured to maximize the dynamic range while avoiding saturation of relevant features.
Electron microscopy
Cells were grown to near confluence on polylysine-coated 12-mm no. 1 coverslips (BioCoat, no. 354085; Discovery Labware, Bedford, MA, USA), then incubated with LLOME for 10–15 min. Coverslips were next briefly rinsed in 0.3× KHMgE (at 1×: 30 mM Hepes pH 7.2 at RT, 70 mM KCl, 5 mM MgCl2, and 3 mM EGTA), unroofed by brief ultrasonication, and immediately immersed in KHMgE containing 2 % w/v paraformaldehyde for ca. 1 hr. Antibody staining was performed as previously described (52) using 18-nm gold particles conjugated to goat anti-rabbit or anti-mouse (Jackson Immunoresearch, West Grove, PA, USA) antibodies. The area of coverslip with the highest yield of plasma membranes was identified by phase contrast microscopy and trimmed with a diamond knife to ca. 3 × 3 mm. Replicas were prepared as previously described (52) and viewed on a JEM-1400 transmission electron microscope (JEOL, Tokyo, Japan) at two different tilt angles (+/− 5°). Images were captured using an XR111 camera (Advanced Microscopy Techniques, Woburn, MA, USA). Digital image pairs were made into anaglyphs as described (52).
Sample preparation for timelapse recordings
The following procedure was used to prepare cells for imaging in all timelapse experiments; modifications and additional information specific to any given application are described in the corresponding sections below. Cells from a subconfluent culture were seeded in a 4-chamber, no. 1.5 glass-bottom dish (Cellvis, Mountain View, CA, USA) and cultured as appropriate for the intended experiment. Prior to imaging, medium was replaced with warmed imaging solution (composed of 20 mM HEPES pH 7.4 at RT, 140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM D-glucose, and 5 % v/v FBS), and the dish immediately transferred to a Tokai Hit (Fujinomiya City, Japan) INU series stage-top incubator preheated to 37 ºC. The dish was allowed to equilibrate for 20 min before initiating acquisition. Imaging solution intended for all manipulations and drug dilutions was warmed to 37 ºC in a separate vessel placed within the stage-top incubator together with the cells, in order to prevent slight differences in temperature and atmospheric conditions from influencing the cellular response.
Timelapse recordings of fluorescently-tagged proteins
U2OS cells transfected with plasmids encoding the indicated mCherry-tagged proteins, or HeLa cells constitutively producing the indicated GFP-tagged proteins, were seeded at 5–1 × 104 cells per chamber of a 4-well dish and cultured for 16–24 h. Where indicated, cells were additionally transfected with plasmids encoding mCherry-tagged proteins prior to seeding, or loaded with FITC-conjugated or Rhodamine B-conjugated dextran (see section on ratiometric pH sensing below) before being prepared for imaging. Single-color recordings were acquired at 30-sec intervals on 20–30 fields of cells using Platform 2. Dual-color recordings were acquired at 30-sec intervals using Platform 4, with both cameras operating at a readout rate of 200 MHz and utilizing a 16-bit dual amplifier with binning. Acquisition was paused for 30 sec after the first four timepoints to allow cells to be bathed in imaging solution containing rupturing agent or vehicle alone, and subsequently continued without interruption.
Timelapse recordings using Magic Red
Stock solutions of Magic Red (no. 938; ImmunoChemistry Technologies, Bloomington, MN, USA) were prepared in DMSO according to the manufacturer’s instructions, and stored at −80 ºC in single-use aliquots. A fresh aliquot was thawed for each experiment prior to use. For imaging GFP-tagged ESCRT-III proteins in the presence of Magic Red, appropriate HeLa cells were cultured in 4-well dishes and prepared for imaging. Magic Red was diluted 2000-fold in excess imaging solution, and equilibrated in an additional vessel on the stage-top incubator together with the cells and regular imaging solution. Prior to each experiment, 0.5 ml of this labeling solution was added to cells. Compartments labeled with Magic Red began to be discernible by 2 min, at which time 20–30 cells with ca. 10 or more bright puncta were selected for imaging; and by 10 min, the intensity of Magic Red-labeled compartments had stabilized and acquisition was initiated. Acquisition was paused for 30 sec after the first four timepoints and the cells bathed in imaging solution containing Magic Red together with the rupturing agent or vehicle. Acquisition was then continued for the duration specified; paused again for 1 min as the cells were washed with 1 ml of fresh imaging solution and re-immersed in additional imaging solution containing only the Magic Red reagent; and subsequently continued without further interruption. Recordings were acquired at 30-sec intervals using Platform 4.
For imaging Magic Red in cells depleted of TSG101 and ALIX, U2OS cells were seeded at no more than 5 × 104 cells per chamber of a 4-well dish and cultured for 16–24 h before labeling with Magic Red and imaging as above. Fields were imaged consecutively, for 100 msec each, at 30-sec intervals on Platform 3, using 561 nm excitation and the 100× 1.45 NA objective, and the camera configured to operate at 200 MHz with the 16-bit dual amplifier and 4×4 binning.
Ratiometric pH sensing
U2OS cells were seeded at no more than 5 × 104 cells per chamber of a 4-well glass-bottom dish and allowed to adhere for ca. 12 h. Medium was then replaced with fresh growth medium containing 0.5 mg/ml fluorescein isothiocynate (FITC)-conjugated 40 kDa dextran (no. D-1844, Molecular Probes) and 0.2 mg/ml Rhodamine B-conjugated 70 kDa dextran (no. D-1841, Molecular Probes). After 6 h, cells were washed in excess medium and incubated for a further 6 h in dextran-free medium to allow the dextrans to accumulate in late endosomes and lysosomes, before being prepared for imaging.
For each experiment, 20–30 fields were selected that consisted of a single cell containing ≥ 10 compartments with bright Rhodamine B fluorescence and dim FITC fluorescence. Fields were imaged consecutively, for 100 msec each, at 30-sec intervals on Platform 4 using the 100× 1.45 NA objective. Both cameras were configured to operate with a readout rate of 200 MHz using a 16-bit dual amplifier and 4×4 binning. After the first four timepoints, the recording was paused for 30 sec as cells were bathed in imaging solution containing an appropriate concentration of rupturing agent or other compound. The recording was continued for the duration indicated, paused again for 1 min as the cells were washed once with 1 ml of imaging solution and then bathed in 0.5 ml of imaging solution, and subsequently continued without interruption.
Image analysis
All analyses were performed on the original, unmodified image data using ImageJ (53) with the aid of custom-written scripts that are available on request. For timelapse recordings, all fields in which cells detached or otherwise moved out of the focal plane at any time during imaging were omitted from analysis.
In Fig. 1D, fractional overlap between the ESCRT and endosomal marker immunostains was calculated as follows. Images were first automatically thresholded to generate regions of interest corresponding to the compartments that stained either for the ESCRT component or for each respective marker. Next, additional regions of interest were generated that corresponded to the intersection of the ESCRT compartments and those identified by each respective marker. Finally, the total pixel area of the intersecting set was calculated, and divided by the total pixel area occupied by the ESCRT compartments in each image.
In Fig. 2 (A and B) and Fig. 3 (C and D), the proportion of each indicated immunostain that exceeded a starting threshold was calculated as follows. Only images of individual cells, and in which the majority of the cellular area fit within the imaging field, were considered for analysis. The outlines of individual cells and the corresponding nuclei were manually traced to further limit the analysis to the pixels within the cytoplasmic area of each cell. A starting intensity threshold was first derived by extracting and pooling pixel intensity values from all images of untreated cells, and then calculating the corresponding 99-th percentile for that cohort. Next, the proportion of total signal that exceeded this threshold (i.e., denoted as signal over threshold) was calculated for each image, and normalized to the median signal over threshold for the timepoint cohort with the maximal intensity.
In Fig. 5B, the number of Magic Red and ESCRT puncta was calculated as follows. For each channel, a mean background value was manually calculated by averaging, across several fields, the pixel intensity in an extracellular area at least 100 × 100 pixels wide. This value was then subtracted from all pixels in the respective channel at every timepoint for each field. Background-corrected images were next linearly contrast-stretched to normalize the pixel intensity distribution: for each field, lower and upper intensity bounds were calculated that allowed 0.1 % pixel saturation at the initial timepoint; the remaining timepoints were then adjusted to this range. Finally, puncta were identified in terms of local intensity maxima above an empirically-determined threshold. (The same threshold was applied to all recordings of all samples analyzed within each experiment.) For Magic Red, the number of puncta identified at each timepoint was normalized to the initial number of puncta, calculated as the average of the first four timepoints. For fluorescent ESCRT-III proteins, the number of puncta identified at each timepoint was normalized to the initial puncta count as described above, as well as to the maximum number of puncta identified within the respective field at any timepoint during the recording.
In Fig. 5C, the number of Magic Red puncta was determined as follows. Images of Magic Red fluorescence were background-subtracted as above, and further corrected for local intensity variations using the Rolling Ball Background Subtraction algorithm in ImageJ. Compartments containing Magic Red were then identified by calculating local intensity maxima above a predetermined threshold as above. For every cell, the number of compartments thus identified at each timepoint was normalized to the average number of compartments for that cell during the initial four timepoints.
In Fig. 6, relative changes in endolysosomal pH were extracted from timelapse recordings of fluorescently-labeled dextrans. A mean background value was first manually calculated for each channel by averaging, across several fields, the pixel intensity in an area at least 100 × 100 pixels devoid of dextran puncta. This value was then subtracted from all pixels in the respective channel at every timepoint for each field. Dextran-loaded compartments were next computationally identified as follows, using the channel corresponding to the pH-insensitive fluorescence of the Rhodamine B-conjugated dextran. At each timepoint, the background-subtracted Rhodamine B image was first linearly contrast-stretched by allowing 0.1 % pixel saturation to normalize the pixel intensity distribution, and then thresholded according to a predetermined value that preserved the majority of visible dextran-loaded compartments. (The same threshold was applied to all recordings of all samples analyzed within each experiment.) The brightest pixels within the image were next identified by calculating local intensity maxima, and every pixel thus identified was symmetrically enlarged by one pixel in each direction to yield a square region 3×3 pixels wide that encompassed the majority of the corresponding compartment. The total intensity within this region was measured from the original background-subtracted image for each channel, and reported as the ratio of the FITC intensity to the Rhodamine B intensity for that compartment at that timepoint. For whole cell-based measurements, the median ratio for the respective field was calculated from the ratios of all compartments identified within that field at that timepoint, and normalized to the average ratio of the first four timepoints for that cell. The data were then filtered by first calculating the 5th and 95th percentiles of all control cells at the timepoint immediately preceding washout, and then excluding from further analysis any cell from either sample whose ratio value at this timepoint was less than the above 5th percentile or greater than the above 95th percentile. In addition, any cell whose ratio at any timepoint after washout exceeded the above 95th percentile was also rejected. The extent of recovery was then assessed as follows. A recovery threshold was first derived from the control population, by pooling all whole cell values from the control cohort throughout the duration of washout and calculating the corresponding 95th percentile. For each sample, the number of cells was then counted that exceeded this threshold at every timepoint after washout, and reported as a percentage of all cells analyzed.
Statistics
Statistical significance was calculated using Graphpad Prism 7.0c (La Jolla, CA, USA).
Image processing for publication
All digital images, including fluorescence and electron micrographs, immunoblots, and timelapse recordings, were linearly contrast stretched in ImageJ to display relevant features. All frames within a movie, and all images and movies intended to be compared, were processed identically. For images of pH-sensitive fluorophores, background fluorescence was first subtracted as described above in the section on timelapse recordings using Magic Red, before contrast stretching. In fig. S27, three-dimensional projections were rendered from spinning-disk confocal Z-stacks in NIS-Elements software using new engine in alpha blending mode. Data were plotted using Graphpad Prism 7.0c. Figures were assembled for publication using Adobe Illustrator CS4 (San Jose, CA, USA).
Supplementary Material
Acknowledgments:
We thank C. Weihl and J. Philips for insightful discussions, reagents, and sharing unpublished data; K. Blumer, D. Kast, and D. Piston for helpful discussions; R. Roth for making deep-etch EM replicas; A. Ustione for microscopy assistance; A. Clippinger and members of the Hanson laboratory for helpful discussions, reagents, microscopy assistance, and contributions to experimental design. Funding: This work was supported by R01 GM122434 from the National Institutes of Health (to P.I.H.). Imaging was performed in part through use of the Washington University Center for Cellular Imaging, partially supported by the Children’s Discovery Institute at Washington University (CDI-CORE-2015-505).
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
References and Notes:
- 1.Paz I et al. , Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 12, 530 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F, Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maejima I et al. , Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32, 2336 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hung YH, Chen LM, Yang JY, Yang WY, Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun 4, 2111 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Chauhan S et al. , TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev Cell 39, 13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida Y et al. , Ubiquitination of exposed glycoproteins by SCFFBXO27 directs damaged lysosomes for autophagy. Proc Natl Acad Sci U S A 114, 8574 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis MJ, Gregorka B, Gestwicki JE, Swanson JA, Inducible renitence limits Listeria monocytogenes escape from vacuoles in macrophages. J Immunol 189, 4488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreibich S et al. , Autophagy Proteins Promote Repair of Endosomal Membranes Damaged by the Salmonella Type Three Secretion System 1. Cell Host Microbe 18, 527 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Repnik U et al. , L-leucyl-L-leucine methyl ester does not release cysteine cathepsins to the cytosol but inactivates them in transiently permeabilized lysosomes. J Cell Sci 130, 3124 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Joshi GN, Goetjen AM, Knecht DA, Silica particles cause NADPH oxidase-independent ROS generation and transient phagolysosomal leakage. Mol Biol Cell 26, 3150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bright NA, Davis LJ, Luzio JP, Endolysosomes Are the Principal Intracellular Sites of Acid Hydrolase Activity. Curr Biol 26, 2233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews NW, Almeida PE, Corrotte M, Damage control: cellular mechanisms of plasma membrane repair. Trends Cell Biol 24, 734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper ST, McNeil PL, Membrane Repair: Mechanisms and Pathophysiology. Physiol Rev 95, 1205 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isermann P, Lammerding J, Consequences of a tight squeeze: Nuclear envelope rupture and repair. Nucleus 8, 268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jimenez AJ et al. , ESCRT machinery is required for plasma membrane repair. Science 343, 1247136 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Scheffer LL et al. , Mechanism of Ca²⁺-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun 5, 5646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong YN et al. , ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denais CM et al. , Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raab M et al. , ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Hurley JH, ESCRTs are everywhere. EMBO J 34, 2398 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiaruttini N, Roux A, Dynamic and elastic shape transitions in curved ESCRT-III filaments. Curr Opin Cell Biol 47, 126 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Schöneberg J, Lee IH, Iwasa JH, Hurley JH, Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol 18, 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiele DL, Lipsky PE, Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A 87, 83 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aits S et al. , Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 11, 1408 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christ L et al. , ALIX and ESCRT-I/II function as parallel ESCRT-III recruiters in cytokinetic abscission. J Cell Biol 212, 499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang S et al. , ESCRT-III activation by parallel action of ESCRT-I/II and ESCRT-0/Bro1 during MVB biogenesis. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haller T, Dietl P, Deetjen P, Völkl H, The lysosomal compartment as intracellular calcium store in MDCK cells: a possible involvement in InsP3-mediated Ca2+ release. Cell Calcium 19, 157 (1996). [DOI] [PubMed] [Google Scholar]
- 28.Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S, Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci 126, 60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maki M, Takahara T, Shibata H, Multifaceted Roles of ALG-2 in Ca(2+)-Regulated Membrane Trafficking. Int J Mol Sci 17, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papadopoulos C et al. , VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J 36, 135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefebvre C, Legouis R, Culetto E, ESCRT and autophagies: Endosomal functions and beyond. Semin Cell Dev Biol (2017). [DOI] [PubMed] [Google Scholar]
- 32.Ktistakis NT, Tooze SA, Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol 26, 624 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Begun J et al. , Integrated Genomics of Crohn’s Disease Risk Variant Identifies a Role for CLEC12A in Antibacterial Autophagy. Cell Rep 11, 1905 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. We used several fluorescently tagged ESCRT-III proteins interchangeably throughout this study and note that their recruitment to damaged endolysosomes was indistinguishable within the spatial and kinetic resolution of our assays.
- 35.Maier O, Marvin SA, Wodrich H, Campbell EM, Wiethoff CM, Spatiotemporal dynamics of adenovirus membrane rupture and endosomal escape. J Virol 86, 10821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinberg BE et al. , A cation counterflux supports lysosomal acidification. J Cell Biol 189, 1171 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jadot M, Colmant C, Wattiaux-De Coninck S, Wattiaux R, Intralysosomal hydrolysis of glycyl-L-phenylalanine 2-naphthylamide. Biochem J 219, 965 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishida Y, Nayak S, Mindell JA, Grabe M, A model of lysosomal pH regulation. J Gen Physiol 141, 705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. The range of recovery defects observed could be influenced, among others, by incomplete protein depletion through RNAi as noted in Fig. S9; by the stochastic nature of membrane damage induced by the lysosomotropic compounds used here; or by additional undefined repair machinery.
- 40.Hornung V et al. , Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9, 847 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mulay SR, Anders HJ, Crystal nephropathies: mechanisms of crystal-induced kidney injury. Nat Rev Nephrol 13, 226 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Cheng LW et al. , Use of RNA interference in Drosophila S2 cells to identify host pathways controlling compartmentalization of an intracellular pathogen. Proc Natl Acad Sci U S A 102, 13646 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Philips JA, Porto MC, Wang H, Rubin EJ, Perrimon N, ESCRT factors restrict mycobacterial growth. Proc Natl Acad Sci U S A 105, 3070 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S, Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog 7, e1002232 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva-Ayala D et al. , Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc Natl Acad Sci U S A 110, 10270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shtanko O, Nikitina RA, Altuntas CZ, Chepurnov AA, Davey RA, Crimean-Congo hemorrhagic fever virus entry into host cells occurs through the multivesicular body and requires ESCRT regulators. PLoS Pathog 10, e1004390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar B et al. , ESCRT-I Protein Tsg101 Plays a Role in the Post-macropinocytic Trafficking and Infection of Endothelial Cells by Kaposi’s Sarcoma-Associated Herpesvirus. PLoS Pathog 12, e1005960 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanson PI, Roth R, Lin Y, Heuser JE, Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Biol 180, 389 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jouvenet N, Zhadina M, Bieniasz PD, Simon SM, Dynamics of ESCRT protein recruitment during retroviral assembly. Nat Cell Biol 13, 394 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edelstein AD et al. , Advanced methods of microscope control using μManager software. J Biol Methods 1, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin Y et al. , Interaction of the mammalian endosomal sorting complex required for transport (ESCRT) III protein hSnf7–1 with itself, membranes, and the AAA+ ATPase SKD1. J Biol Chem 280, (2005). [DOI] [PubMed] [Google Scholar]
- 52.Cashikar AG et al. , Structure of cellular ESCRT-III spirals and their relationship to HIV budding. Elife 3, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider CA, Rasband WS, Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.