

Significance
Understanding how pathogens cause disease is vital. Some bacteria start an infection with only a small number of cells in the initial inoculum, whereas others cause significant symptoms only if the initial dose is of the order of tens of thousands of cells or more. It has been hypothesized that these differences are explained by the distance at which the species’ pathogenetic mechanisms influence the host. Empirical studies have shown a statistical link between the infective dose and the scale of pathogenetic mechanisms, but a theoretical basis for this phenomenon is lacking. We show how this effect arises using a mechanistic model of pathogenesis and describe a within-host framework for investigating how different pathogenetic mechanisms affect the development of infectious diseases.
Keywords: infective dose, pathogenesis, spatial model, pathogen, parasite
Abstract
The initial amount of pathogens required to start an infection within a susceptible host is called the infective dose and is known to vary to a large extent between different pathogen species. We investigate the hypothesis that the differences in infective doses are explained by the mode of action in the underlying mechanism of pathogenesis: Pathogens with locally acting mechanisms tend to have smaller infective doses than pathogens with distantly acting mechanisms. While empirical evidence tends to support the hypothesis, a formal theoretical explanation has been lacking. We give simple analytical models to gain insight into this phenomenon and also investigate a stochastic, spatially explicit, mechanistic within-host model for toxin-dependent bacterial infections. The model shows that pathogens secreting locally acting toxins have smaller infective doses than pathogens secreting diffusive toxins, as hypothesized. While local pathogenetic mechanisms require smaller infective doses, pathogens with distantly acting toxins tend to spread faster and may cause more damage to the host. The proposed model can serve as a basis for the spatially explicit analysis of various virulence factors also in the context of other problems in infection dynamics.
The dose of pathogens needed to start an infection in an individual host varies between different pathogen species. The minimum amount is usually called the infective dose, although smaller doses are not guaranteed safe (1). The variation in infective doses is especially large between different bacterial pathogens (2, 3). Pathogens also vary in their pathogenetic mechanisms, that is, the ways in which they evade the immune defenses, use the nutrient-rich environment within the host, and eventually cause disease (4–8). A rough distinction can be made between pathogens that exert their effects locally, for example via membrane contact with (a certain target on) the host cells, and pathogens that produce diffusible toxins which may have their target at a distance from the invading pathogen (2–5). Microbial pathogens are well represented in both categories.
Schmid-Hempel and Frank (2) proposed that the differences in the infective dose among pathogen species are explained by their mechanism of pathogenesis. Namely, locally acting pathogenetic mechanisms are linked to smaller infective doses than mechanisms that depend on diffusible toxins, which may act at a distance from their source. Indeed, many pathogens, such as Shigella, that exert their harmful effect by contact to host cells or by entering host cells are highly infectious, requiring only tens or hundreds of bacteria to cause disease (9). Conversely, many toxin-producing bacterial pathogens have infective doses ranging from (e.g., Bacillus anthracis) to cells (e.g., Vibrio cholerae) (10, 11).
However, insight into the underlying reasons for the observed variation is lacking (7, 12). While the dose–response hypothesis of Schmid-Hempel and Frank (2) held against statistical testing for 43 human pathogens in a study by Leggett et al. (3), so far there has been no theoretical model to elucidate why the mode of action produces variation in the infective dose. In this work, we present mathematical models that explain this phenomenon. Furthermore, Schmid-Hempel and Frank (2) also hypothesized that pathogens with distantly acting pathogenetic mechanisms are more virulent in the sense that they cause more damage to the host; but Leggett et al. (3) found no support for this relationship. We also address this hypothesis.
Studying the mechanism of pathogenesis in relation to the infective dose and damage to the host requires accounting for the interactions between the invading pathogens and the immune effectors of the host, which can be extremely complex (8). The pathogen–immune system interaction has been modeled in both simplistic (13, 14) and detailed (15, 16) settings. However, prior models rarely take into account the spatial aspects of pathogenesis explicitly (but see, e.g., ref. 17). Indeed, while the importance of spatial effects has been widely recognized in ecology and evolutionary biology, much of the work in spatial epidemiological models has focused on between-host interactions (18–21); the spatial aspects of within-host interactions have received less attention, although spatial interactions of microbial communities have been investigated (22–25).
Microbes affect their environments in various ways via diffusible metabolites (6, 7) and use a wide array of strategies for defending themselves against the host immune system (4–8). Many bacteria, such as Yersinia pestis and Helicobacter pylori, secrete toxins which target the host’s immune system to suppress or modulate it (5). With this in mind, we focus on bacterial pathogens with toxins that inhibit the immune response of the host (2, 6, 7).
In this work, we develop three models of toxin-dependent pathogenesis: a nonspatial model, a spatial diffusion model, and a stochastic individual-based model. We consider microorganisms that have the ability to harm the host when spreading. However, our models do not make an explicit distinction between parasites (organisms that have adapted to live and feed on a host organism) and pathogens (microorganisms capable of causing damage to the host) in general. We model the following scenario: The initial dose of the pathogen enters to a small inoculation area, from where it can spread out to the available space within the host that we call the focal area. The pathogen reproduces by consuming the host’s tissue (nutrients) and thereby causes damage to the host. Once the immune system detects the pathogens, immune effectors attempt to eliminate them. We assume that the host had no prior exposure to the pathogen and limit our attention to the initial phases of the pathogenesis in which the host’s innate immunity reacts, but its acquired immune response has not yet developed. If the pathogen is cleared out quickly, then little damage is inflicted upon the host; if the pathogen manages to overcome the innate immune defenses and consumes most of the nutrients in the focal area, then the infection may proceed to further stages of pathogenesis and cause disease. In general, virulence is an elusive concept with a multitude of different definitions (2, 26–28). In the eco-evolutionary sense, it can refer to the pathogen-induced decrease in host fitness (28), but also simply to the relative capacity to inflict damage in the host (26, 27). We use the latter definition and quantify virulence as the amount of tissue consumed by the pathogen.
One of the key benefits of our models is that we can examine the influence of the different spatial scales in the toxin’s mode of action, from local (e.g., the pathogen transmits toxins to host cells on membrane contact) to distant action (the pathogen secretes diffusible systemic toxins), while keeping all other properties of both the host and the pathogen the same. Obviously, this would be difficult—if not impossible—to achieve in empirical work. Moreover, our individual-based stochastic model accounts for demographic stochasticity (29) causing random variation in the outcome of an infection. By recording the distribution of outcomes, we can estimate the risk of serious infection in different scenarios.
Our spatial models support the first hypothesis: Increasing the spatial scale of toxin diffusion increases the infective dose. Regarding the second hypothesis, the stochastic model exhibits a threshold phenomenon: Given a high enough initial dose, a pathogen with a diffusible toxin can spread faster and can eventually consume (marginally) more of the host tissue than a locally acting pathogen. We also investigate how the spatial aggregation of the initial inoculum influences the difference between locally and distantly acting pathogens.
Modeling Toxin-Dependent Pathogenesis
First, we start with a simple analytical model and extend it into a spatial diffusion model. These models show that the pathogen dynamics exhibit an Allee effect and that increasing dilution and diffusion of the toxin increases the infective dose. Next, we consider an individual-based simulation model which allows us to examine the effects of demographic stochasticity, incorporate explicit resource-consumer dynamics for the pathogen, and model the immune response more mechanistically.
Simple Analytical Models.
Suppose that the pathogen () follows logistic population dynamics in the absence of the immune system (due to nutrient-limited growth) with intrinsic growth rate and carrying capacity scaled to 1. Immune effectors () eliminate the pathogens at rate . To fight the immune system, the pathogens secrete toxin molecules at rate , which are removed from the host system at a constant rate . The toxin particles decapacitate the immune effectors at rate . When decapacitated, the immune effectors cannot eliminate any pathogen until they recover, which happens at rate . Finally, we assume that the total amount of immune effectors remains constant such that the amounts of active and decapacitated immune effectors are and , respectively.
Nonspatial model.
Assuming that the toxin and immune effectors reach a fast quasi-equilibrium (see SI Appendix, section A for details), the pathogen dynamics are given by
where and are dimensionless parameters. If , the pathogen grows even when the immune system is fully activated, and the pathogen can invade the system without the toxin. On the other hand, if and , where depends only on , the model exhibits an Allee effect. If the initial density of the pathogen is below the Allee threshold, the pathogen goes extinct; above the threshold, the pathogens collectively produce enough toxin to facilitate growth. Moreover, the Allee threshold increases with decreasing , i.e., with increasing the removal rate . Thus, the more the toxin dilutes or leaks out of the system, the higher initial dose the pathogen requires to spread; and if is higher than a critical value, is violated and the spread of the pathogen becomes impossible.
Diffusion model.
The above model can be extended into a spatial reaction–diffusion model. We consider the limiting cases of slow and fast toxin diffusion in 1D space to show (SI Appendix, section B) that (i) with slow diffusion, the spread or extinction of the pathogen is independent of the initial dose, assuming that the pathogen attains a traveling-wave solution, and (ii) with fast diffusion, the initial dose must exceed a threshold for the pathogen to invade the host. A highly diffusing toxin leaks to parts of the host where the pathogen is not yet present. A high initial dose is then needed to overcome the dilution effect found in the nonspatial model.
Stochastic Individual-Based Spatial Model.
The diffusion model captures key characteristics of within-host infection dynamics, but it is confined to traveling-wave solutions in 1D and considers only the limiting cases of slow and fast toxin diffusion; it neglects demographic stochasticity, which is important for initially small pathogen populations; and it oversimplifies the reaction of the immune system. To overcome these limitations, we constructed a more realistic spatiotemporal point process model to simulate the dynamics of toxin-dependent pathogen infection. The model is a continuous-time Markov process, where the state of the system at any time is given by the spatial locations (in continuous space) of every individual particle.
Elementary reactions.
In the individual-based model, the entity types are as follows: pathogens (); toxin (); tissue (); and immune effectors in seeking (), killing (), and decapacitated () states. The dynamics of the pathogen and toxin are given by the reactions
where the symbols above the arrows indicate the rates at which the reaction occurs and denotes that the reaction does not produce any new particles.
The immune response is typically not immediate but gradual, as the immune system needs time to react to a new threat. To model this, we assume a two-tier activation mechanism, where the active immune effectors can be in two different states: “seek” (initial stage of activation, ) and “kill” (second stage of activation, ). The toxin reacts with immune effectors in the initial stage of activation, sending them to an inactive or “decapacitated” state (). The response dynamics of the immune system are governed by the following reactions:
Spatial interactions.
Each individual particle is characterized by its location in the focal area and a mark denoting its type. The state of the system at time is given by the set of locations of each particle type . Reactions occur only if the particles are sufficiently close to each other, and when a new pathogen or toxin particle is produced, it is placed in the neighborhood of its parent. In general, a kernel describes how the locations of two particles influence the reaction rate. We used top-hat kernels, which assign a constant rate for points that are within distance from each other and zero otherwise (see Materials and Methods and SI Appendix, section C for further information).
Movement.
The tissue particles and the decapacitated immune effectors are immobile, and all other particles move by jump processes such that a particle of type at location moves to a small neighborhood of point at rate per unit area. The maximum distance of a single jump is given by the length-scale parameter of the top-hat kernel ; in other words, particles jump randomly to a point within radius . We assumed that jumps occur for each mobile particle at total rate 1, but the particles differ in the length of their jumps. We took and such that the seeking immune effectors move fast to locate the pathogens, and once they encounter pathogens, they “slow down” to eliminate them. To investigate local vs. distant action in pathogenesis, we varied the length-scale parameter of toxin movement; increasing yields more distantly acting mechanisms.
Results
The Experimental Setup.
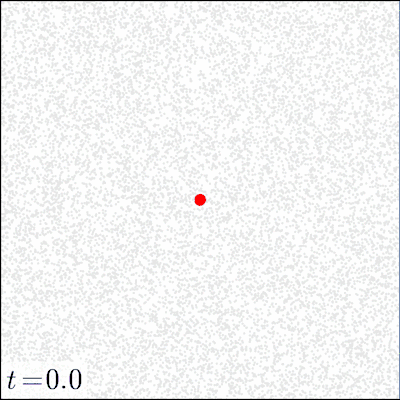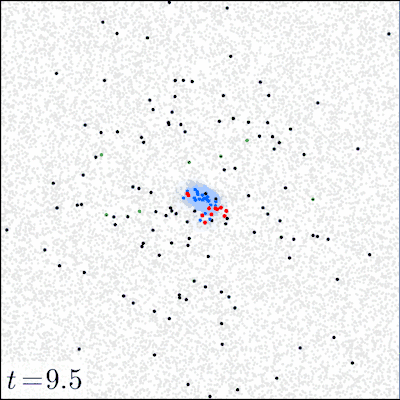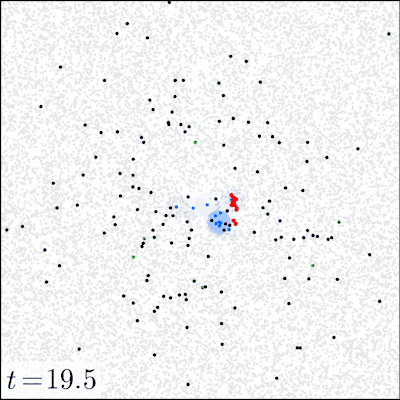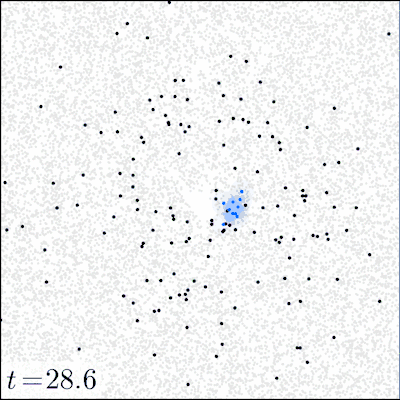
In our experiments, we varied (i) the initial dose of the pathogen, (ii) the mode of action (local vs. distant) of the pathogen via the toxin movement scale parameter , and (iii) the radius of the initial inoculation area. All other parameters were kept constant; SI Appendix, section D, Table S2 gives the parameter values used and a sensitivity analysis of the model. Before the inoculation, the focal area (a torus of size ) was occupied only by tissue particles and immune effectors in seek state. The dynamics of the model were simulated until either all pathogens were eliminated (by the immune system) or all of the tissue was consumed (by the pathogen). For each combination of the initial dose (21 different doses ranging from 1 to pathogens), inoculation area (radii 1, 4, 8, and 16), and toxin movement scale (1, 2, 4, 8, 16, or 32), we ran at least 2,000 simulation replicates for the first 20 doses and 1,000 replicates for the highest dose of pathogens. Fig. 1 illustrates how the model evolves over time.
Fig. 1.

Snapshots of four simulation experiments at four different time points. The low dose was 200 and the high dose was 10,000 pathogens inoculated at time onto a circle of radius . Local action denotes a toxin movement scale of and global action refers to . The gray dots represent tissue particles, red points pathogens, green points toxin particles, blue points activated immune effectors that are consuming the pathogens, and black points immune effectors that have been decapacitated by a toxin particle.
We measured the total number of tissue particles consumed by the pathogen by the end of the simulation. Note that this also gives the total number of pathogens produced during the infection, as each consumed tissue particle yields one new pathogen individual in our model. We then analyzed the distribution of the outcomes and calculated dose–response relationships for the infective dose and tissue consumption.
Local vs. Distant Action.
Figs. 2 and 3A summarize the results of this experiment for the smallest inoculation area (). The experiment clearly demonstrated a strong effect of the initial dose and the mode of action on the infection process. With local action (toxin movement scale ), the amount of tissue consumed by the pathogen is high and not very sensitive to the initial dose. In contrast, with distant action (high ), there is a threshold effect. With low initial doses, most infections die out without consuming much of the tissue, and the average fraction of tissue consumed is low; with high initial dose, however, most infections spread such that the pathogen consumes most of the host tissue, indicating a severe infection. As the curves in Figs. 2 and 3A show, the expected amount of tissue consumed increases drastically when the initial dose exceeds a threshold. For distantly acting toxin (), the expected fraction of tissue consumed exceeds 0.5 only if there are several thousand pathogens in the initial inoculum.
Fig. 2.

(A–F) The dose–response relationships for different modes of action. The toxin movement scale quantifying the mode of action increases across the panels. The radius of the inoculation area is . The logarithm of the initial dose of the pathogen is on the horizontal axis, and the vertical axis shows the fraction of available tissue particles consumed by the pathogen during the course of the infection. The contour plots show the distribution of the stochastic outcomes; for a particular dose, darker areas indicate more typical outcomes. The lines give the average dose–response curve fitted to the Hill equation . At low doses, local action (A) leads to a higher tissue consumption on average than the more distantly acting mechanisms (B–F); at high doses, the situation is reversed. For locally acting mechanisms, almost all doses lead to a high response, whereas with distant mechanisms, only high doses lead consistently to a high response. The dotted lines show the dose for which at least 75% of tissue is consumed on average. Below this dose, most infections with distant mechanisms fail, whereas above this dose, most infections invade the host (compare shading).
Fig. 3.

(A–D) Effects of spatial aggregation on the dose response. Each panel shows the fraction of tissue particles consumed by the pathogen, averaged over all simulation replicates, as a function of the initial dose, for different toxin movement scales. The spatial aggregation of the initial dose decreases (the radius of the inoculation area increases) from left to right across A–D.
The strong difference between the local and distant mechanisms is also evident when we look at the dynamics of the system. Fig. 1 gives examples of four simulations that illustrate how the infection develops under two different initial doses and modes of action; in SI Appendix, Movies S1–S4 give animated versions of these scenarios. Fig. 1 shows that a 50-fold increase in the initial dose does not drastically change the qualitative behavior of the system with a locally acting toxin. However, for distant action, there are substantial differences in the progress of the infection; the immune system readily clears the pathogen in low-dose scenarios, whereas in high-dose scenarios, the pathogen spreads out.
Note that pathogens with local action do not always consume more tissue (and thus reproduce more) than pathogens with distant action. While at low initial doses a locally acting toxin clearly outperforms distant action, the trend reverses at high initial doses; with an initial dose of , all but the most distantly acting toxin provide on average better pathogen growth than the most locally acting toxin (Fig. 3A).
Effects of Spatial Aggregation.
Varying the size of the inoculation area () demonstrates that spatial aggregation has a strong effect; increasing the initial inoculation area leads to more tissue consumed on average for all toxin movement scales and all initial doses (naturally with the exception of the initial dose of a single pathogen; Fig. 3 A–D). The dose–response curves change such that the difference between locally and distantly acting toxins is diminished by spreading out the initial inoculum. It, however, remains true that for distantly acting toxins, most infections lead to little tissue consumed when the initial dose is below a threshold, whereas most infections spread well when the initial dose is above the threshold; the threshold shifts toward smaller initial doses with increasing (SI Appendix, Figs. S7–S9).
A large inoculation area implies less competition between the pathogens in the early phase of the infection, when demographic stochasticity critically affects the outcome. With a large inoculation area, the pathogens behave to some extent as if there were several independent inocula, and if one of these manages to spread, the infection takes hold. This benefits pathogens with both locally and distantly acting toxins. Pathogens with locally acting toxins, however, lose the benefit of high local toxin concentration. As a result, pathogens with distant action benefit more from decreasing spatial aggregation and thus get closer to pathogens with local action in Fig. 3.
Speed of Infection.
The speed at which the pathogen spreads in the host depends on both the initial dose and the mode of action (Fig. 4). In high-dose scenarios, pathogens using a distant mechanisms tend to spread more quickly than pathogens with a local mechanism, whereas the opposite holds in low-dose scenarios.
Fig. 4.

(A and B) Progression of the infection as a function of the initial dose and the mode of action (toxin movement scale). The plots show the mean time until 50% (A) and 90% (B) of the initial tissue are consumed (with inoculation radius ), averaged over replicates where the infection spreads as far.
Discussion
Schmid-Hempel and Frank (2) hypothesized that the variation in observed infective doses is explained by the pathogen’s mode of action, that is, whether the underlying mechanism of pathogenesis is locally acting or distantly acting. Leggett et al. (3) showed that empirical evidence supports the hypothesis, but the mechanism behind the phenomenon has not been shown previously. Our models demonstrate that the mode of action can give rise to the variation in infective doses: All else being equal, pathogens with locally acting toxins have smaller infective doses than pathogens with highly diffusive toxins. The empirical evidence in prior studies (2, 3) relies on data from various different pathogen species and strains with varying phenotypes and pathogenetic mechanisms, whereas our models show that the effect can emerge from varying the diffusibility of the toxin while keeping all other properties of the pathogen and the host the same. The analytical models show that when the toxin is highly diffusive, the initial pathogen population grows and establishes only if the initial dose is sufficiently high; with a low initial dose, the pathogen is eliminated by the initial immune response. In contrast, with low diffusion, the pathogen can grow also if the initial dose is small. In the individual-based simulation model we observe similar results: At low initial doses, pathogens with locally acting toxins inflict on average more damage (Figs. 2 and 3). Assuming that more damage in the early phase of the infection implies a higher chance to develop symptomatic disease, this yields that pathogens with locally acting mechanisms have lower infective doses than pathogens with highly diffusive toxins.
The way in which the toxin benefits the pathogen induces an Allee effect (30–32), because the toxin concentration has to be sufficiently high to protect the pathogen from the immune system. Toxin production is thus a cooperative defense mechanism (7, 33, 34) for the pathogen. All else being equal, a highly diffusible toxin spreads to a large area and has a less concentrated effect, thus not protecting a small initial inoculum of the pathogen effectively. The Allee effect yields a threshold for the initial dose that increases with the diffusibility of the toxin especially when the initial inoculum is aggregated (Fig. 3). For distant action the dose response exhibits a switch between the initial inoculum typically failing to typically spreading (Fig. 2 and SI Appendix, Figs S7–S9).
The benefit from a locally acting toxin is more or less immediate, but with distant action, the benefits are not realized until the pathogens manage to spread far enough. A small initial pathogen population may simply die out before reaching far, but the diffusible toxin may speed up the pathogen’s spread if the infection takes hold (Fig. 4). Our model predicts that the infective dose decreases when the pathogens are initially more spread out, and this is particularly so in case of distant action (Fig. 3 and SI Appendix, Figs S7–S9). Typically, the initial dose of the pathogen is clumped, but the degree of aggregation can vary depending on the route of infection (skin wound, digestion, inhalation, and so on). Pathogens that are initially scattered over a large area may invade the host easier, especially in case of distantly acting toxins.
Schmid-Hempel and Frank (2) also suggested that pathogens with distantly acting mechanisms, and thus high infective doses, tend to be more virulent. While this may at first sound tautological, since a higher dose of a certain pathogen can readily be expected to increase the severity of the infection, this need not be so across different pathogenic species. In our stochastic model, the amount of harm to the host (3, 27) can be identified with the amount of host tissue consumed. Therefore, we can examine the second hypothesis of Schmid-Hempel and Frank (2) in this sense. We observed that the expected amount of tissue consumed as a function of the initial dose increases strongly for distantly acting pathogens; at high initial doses, it (marginally) surpasses the locally acting pathogens (Fig. 3). We also observed that once the initial dose passes a threshold, distantly acting pathogens spread faster than those with local action (Fig. 4). Inflicting more damage and, in particular, spreading faster may hinder adequate host defenses (such as the development of the specific immune response) before the infection spreads beyond the focal area (e.g., before a skin infection becomes systemic). This can lead to more harm, so that the second hypothesis of Schmid-Hempel and Frank (2) is in this way supported by our model. Note, however, that the empirical study of Leggett et al. (3) found no support for the second hypothesis.
More work is needed to understand how the mode of action influences the epidemiology of pathogens by, e.g., developing models that link within-host dynamics to between-host dynamics (35). Our models do not consider the life history traits, ecology, or evolution of the pathogen species and thus cannot answer the question, Why do pathogens exhibit such vastly different strategies of local vs. distant action (7, 12)? Indeed, a locally acting mechanism may at first seem to be more beneficial to the pathogen, since the gains from the toxin are immediate and the infective dose can be small. This can even be seen as a “stealth attack strategy” (5), as localized mechanisms may lower the chances of the immune system detecting the pathogen. Our model suggests that while pathogens with distantly acting toxins have higher infective doses, they can spread faster than locally acting pathogens with the same initial dose given that the dose is sufficiently high.
In general, locally acting toxins can be seen to resemble nonshareable private goods, whereas diffusible toxins are shareable public goods. Indeed, bacteria produce various kinds of public goods, that is, beneficial diffusible factors and metabolites, into their surrounding environment (6). There is evidence that habitat structure may drive the selection between the use of private and public goods (24, 36, 37). Moreover, many pathogenetic bacteria with distantly acting toxins are environmentally transmitted (e.g., V. cholerae), opportunistic or facultative (e.g., Staphylococcus aureus), or coincidentally pathogenic (e.g., Clostridium tetani) and therefore can be subject to different selective forces than obligate parasites. This suggests that some species with distantly acting toxins may be in general less adapted to an obligate parasitic lifestyle.
Our model treats the host immune system–pathogen interactions in a simplistic way; we exclude many known bacterial defenses (5, 7, 8) and ignore the vast complexity of the immune system. Nevertheless, our model captures many general properties of an immune response where the immune system gradually identifies and eliminates the pathogens. The way immune effectors act in our model best resembles the role of macrophages in the innate immune response. Despite the simplifications, we observe that the growth of the initial inoculum strongly depends on its size when the toxin acts distantly, i.e., when the pathogen depends on a public good. The importance of intra- and interspecific cooperation in overcoming the immune system has been postulated in several experimental (7, 38) and modeling studies (39–41). Our results indicate that pathogens with distant action depend more on cooperative effort in infection formation, but locally acting pathogens may cause severe infections starting from a few individuals.
Understanding the underlying mechanisms of pathogenesis and host–parasite interactions has been identified as one of the key issues in evolutionary ecology and immunology (5, 7), which can potentially help in developing novel therapeutic agents and combat increasing antibiotic resistance. Our work shows that techniques from spatial ecology can illuminate the within-host dynamics of pathogens with different pathogenetic mechanisms.
Materials and Methods
The focal area, i.e., the spatial domain , was a torus of size . The initial state of the system at time consisted of tissue particles and immune effectors in seek state, whose distribution followed complete spatial randomness with densities and per unit area, respectively. The initial inoculum of the pathogen was spatially aggregated, and a total of pathogens were randomly distributed within the inoculation area, in a disk of radius . All other particle types were absent at .
For the spatial reactions and movement, we used top-hat kernels, which assign the value for points that are within distance from each other and 0 otherwise; here is the total rate, i.e., for the reactions and 1 for the movement of all mobile particles (tissue and decapacitated immune effectors are immobile).
Specifically, a pathogen at location consumes a tissue particle at location at the rate given by the consumption kernel . Once a pathogen consumes a tissue particle, it immediately produces a new pathogen, whose location is determined by the pathogen movement kernel . The immune effectors in kill state eliminate pathogens in their vicinity according to the kernel , such that a pathogen at location is killed at rate . To counteract the immune system, the pathogens secrete toxins according to the kernel , such that the rate at which toxin particles are secreted to the vicinity of point is per unit area. The toxin is inactivated and disappears at rate . A toxin particle at decapacitates an immune effector in seek state at at rate . The toxin particle is consumed when it decapacitates an immune effector. An immune effector in seek state at transitions into the kill state at rate , where is the activation kernel. The immune effectors in kill state revert to the seek state at the per-capita rate and decapacitated immune effectors recover back to the seek state at rate . Note that if there are many pathogens nearby, an immune effector in the seek state transitions to the kill state at a high rate.
The simulations were based on a Gillespie-style algorithm (42) adapted to spatial point processes. Each simulation replicate was run until either all pathogens or all tissue particles disappeared. We recorded the particle locations of each particle type every time units. The source code for the implementation is available on Zenodo (DOI 10.5281/zenodo.1421750). See SI Appendix, sections C—F for further details.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the anonymous reviewers for their helpful comments and Otso Ovaskainen and Panu Somervuo for many discussions regarding the simulation framework. The authors acknowledge CSC—IT Center for Science, Finland, for computational resources. This project received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Grant 754411 (to J.R.) and from the Academy of Finland through the Center of Excellence in Analysis and Dynamics Research (E.K.). J.R. and J.V.A. were also supported by the Academy of Finland Grants 1273253 and 267541.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The source code for the implementation is available on Zenodo (DOI 10.5281/zenodo.1421750).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1721061115/-/DCSupplemental.
References
- 1.Buchanan RL, Smith JL, Long W. Microbial risk assessment: Dose-response relations and risk characterization. Int J Food Microbiol. 2000;58:159–172. doi: 10.1016/s0168-1605(00)00270-1. [DOI] [PubMed] [Google Scholar]
- 2.Schmid-Hempel P, Frank SA. Pathogenesis, virulence, and infective dose. PLoS Pathog. 2007;3:e147. doi: 10.1371/journal.ppat.0030147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leggett HC, Cornwallis CK, West SA. Mechanisms of pathogenesis, infective dose and virulence in human parasites. PLoS Pathog. 2012;8:e1002512. doi: 10.1371/journal.ppat.1002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donnenberg MS. Pathogenic strategies of enteric bacteria. Nature. 2000;406:768–774. doi: 10.1038/35021212. [DOI] [PubMed] [Google Scholar]
- 5.Scott Merrell D, Falkow S. Frontal and stealth attack strategies in microbial pathogenesis. Nature. 2004;430:250–256. doi: 10.1038/nature02760. [DOI] [PubMed] [Google Scholar]
- 6.West SA, Diggle SP, Buckling A, Gardner A, Griffin AS. The social lives of microbes. Annu Rev Ecol Evol Syst. 2007;38:53–77. [Google Scholar]
- 7.Schmid-Hempel P. Parasite immune evasion: A momentous molecular war. Trends Ecol Evol. 2008;23:318–326. doi: 10.1016/j.tree.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Schmid-Hempel P. Evolutionary Parasitology: The Integrated Study of Infections, Immunology, Ecology, and Genetics. Oxford Univ Press; Oxford: 2011. [Google Scholar]
- 9.Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: Controlling host signaling, invasion and death by type III secretion. Clin Microbiol Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.WHO 2008. Anthrax in Humans and Animals, 4th Ed (WHO, Geneva)
- 11.Kothary MH, Babu US. Infective dose of foodborne pathogens in volunteers: A review. J Food Saf. 2001;21:49–68. [Google Scholar]
- 12.Cressler CE, McLeod DV, Rozins C, van den Hoogen J, Day T. The adaptive evolution of virulence: A review of theoretical predictions and empirical tests. Parasitology. 2016;143:915–930. doi: 10.1017/S003118201500092X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alizon S, Baalen M. Acute or chronic? Within-host models with immune dynamics, infection outcome, and parasite evolution. Am Nat. 2008;172:E244–E256. doi: 10.1086/592404. [DOI] [PubMed] [Google Scholar]
- 14.Pujol JM, Eisenberg JE, Haas CN, Koopman JS. The effect of ongoing exposure dynamics in dose response relationships. PLoS Comput Biol. 2009;5:e1000399. doi: 10.1371/journal.pcbi.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perelson AS. Modelling viral and immune system dynamics. Nat Rev Immunol. 2002;2:28–36. doi: 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- 16.Hancioglu B, Swigon D, Clermont G. A dynamical model of human immune response to influenza A virus infection. J Theor Biol. 2007;246:70–86. doi: 10.1016/j.jtbi.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 17.Grant AJ, et al. Modelling within-host spatiotemporal dynamics of invasive bacterial disease. PLoS Biol. 2008;6:e74. doi: 10.1371/journal.pbio.0060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satō K, Matsuda H, Sasaki A. Pathogen invasion and host extinction in lattice structured populations. J Math Biol. 1994;32:251–268. doi: 10.1007/BF00163881. [DOI] [PubMed] [Google Scholar]
- 19.Tilman D, Kareiva P, editors. Spatial Ecoogy: The Role of Space in Population Dynamics and Interspecific Interactions. Princeton Univ Press; Princeton: 1997. [Google Scholar]
- 20.Lion S, van Baalen M. Self-structuring in spatial evolutionary ecology. Ecol Lett. 2008;11:277–295. doi: 10.1111/j.1461-0248.2007.01132.x. [DOI] [PubMed] [Google Scholar]
- 21.Riley S, Eames K, Isham V, Mollison D, Trapman P. Five challenges for spatial epidemic models. Epidemics. 2015;10:68–71. doi: 10.1016/j.epidem.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durrett R, Levin S. Allelopathy in spatially distributed populations. J Theor Biol. 1997;185:165–171. doi: 10.1006/jtbi.1996.0292. [DOI] [PubMed] [Google Scholar]
- 23.Czárán T, Hoekstra RF. A spatial model of the evolution of quorum sensing regulating bacteriocin production. Behav Ecol. 2007;18:866–873. [Google Scholar]
- 24.Allen B, Gore J, Nowak MA. Spatial dilemmas of diffusible public goods. Elife. 2013;2:e01169. doi: 10.7554/eLife.01169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bewick S, et al. Invasion speeds in microbial systems with toxin production and quorum sensing. J Theor Biol. 2017;420:290–303. doi: 10.1016/j.jtbi.2017.01.034. [DOI] [PubMed] [Google Scholar]
- 26.Casadevall A, Pirofski L-A. Host-pathogen interactions: Redefining the basic concepts of virulence and pathogenicity. Infect Immun. 1999;67:3703–3713. doi: 10.1128/iai.67.8.3703-3713.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Casadevall A, Pirofski L-A. The damage-response framework of microbial pathogenesis. Nat Rev Microb. 2003;1:17–24. doi: 10.1038/nrmicro732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Méthot P-O, Alizon S. What is a pathogen? Toward a process view of host-parasite interactions. Virulence. 2014;5:775–785. doi: 10.4161/21505594.2014.960726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lande R. Risks of population extinction from demographic and environmental stochasticity and random catastrophes. Am Nat. 1993;142:911–927. doi: 10.1086/285580. [DOI] [PubMed] [Google Scholar]
- 30.Allee WC, Bowen ES. Studies in animal aggregations: Mass protection against colloidal silver among goldfishes. J Exp Zool. 1932;61:185–207. [Google Scholar]
- 31.Dennis B. Allee effects: Population growth, critical density, and the chance of extinction. Nat Res Mod. 1989;3:481–538. [Google Scholar]
- 32.Stephens PA, Sutherland WJ, Freckleton RP. What is the Allee effect? Oikos. 1999;87:185–190. [Google Scholar]
- 33.Allee WC, et al. Principles of Animal Ecology. W B Saunders Company; Philadelphia: 1949. [Google Scholar]
- 34.Courchamp F, Clutton-Brock T, Grenfell B. Inverse density dependence and the Allee effect. Trends Ecol Evol. 1999;14:405–410. doi: 10.1016/s0169-5347(99)01683-3. [DOI] [PubMed] [Google Scholar]
- 35.Mideo N, Alizon S, Day T. Linking within- and between-host dynamics in the evolutionary epidemiology of infectious diseases. Trends Ecol Evol. 2008;23:511–517. doi: 10.1016/j.tree.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 36.Driscoll WW, Pepper JW. Theory for the evolution of diffusible external goods. Evolution. 2010;64:2682–2687. doi: 10.1111/j.1558-5646.2010.01002.x. [DOI] [PubMed] [Google Scholar]
- 37.Kmämerli R, Schiessl KT, Waldvogel T, McNeill K, Ackermann M. Habitat structure and the evolution of diffusible siderophores in bacteria. Ecol Lett. 2014;17:1536–1544. doi: 10.1111/ele.12371. [DOI] [PubMed] [Google Scholar]
- 38.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Regoes RR, Ebert D, Bonhoeffer S. Dose-dependent infection rates of parasites produce the Allee effect in epidemiology. Proc R Soc Lond B Biol Sci. 2002;269:271–279. doi: 10.1098/rspb.2001.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ackermann M, et al. Self-destructive cooperation mediated by phenotypic noise. Nature. 2008;454:987–990. doi: 10.1038/nature07067. [DOI] [PubMed] [Google Scholar]
- 41.Anttila J, et al. A mechanistic underpinning for sigmoid dose-dependent infection. Oikos. 2016;126:910–916. [Google Scholar]
- 42.Gillespie DT. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81:2340–2361. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.