

Abstract
Emerging evidence from various studies indicates that plasmid DNA (pDNA) is internalized by cells through an endocytosis-like process when it is used for electrotransfection. To provide morphological evidence of the process, we investigated ultrastructures in cells that were associated with the electrotransfected pDNA, using immunoelectron microscopy. The results demonstrate that four endocytic pathways are involved in the uptake of the pDNA, including caveolae- and clathrin-mediated endocytosis, macropinocytosis, and the clathrin- independent carrier/GPI-enriched early endosomal compartment (CLIC/GEEC) pathway. Among them, macropinocytosis is the most common pathway utilized by cells having various pDNA uptake capacities, and the CLIC/GEEC pathway is observed primarily in human umbilical vein endothelial cells. Quantitatively, the endocytic pathways are more active in easy-to-transfect cells than in hard-to-transfect ones. Taken together, our data provide ultrastructural evidence showing that endocytosis plays an important role in cellular uptake and intracellular transport of electrotransfected pDNA.
Keywords: electrotransfection, electroporation, intracellular trafficking, clathrin-mediated endocytosis, macropinocytosis
INTRODUCTION
Electrotransfection is a gene delivery method based on application of a controlled electric field to cells that allows cellular uptake of plasmid DNA (pDNA) both in vitro (Cervia, et al., 2017; Chang, et al., 2014; Mao, et al., 2017; Neumann, et al., 1982; Wu & Yuan, 2011), and in vivo (Heller & Heller, 2006; Heller & Heller, 2015; Henshaw, et al., 2008; Henshaw, et al., 2011; Mir, et al., 1999). The interest in electrotransfection has led to improvement of the technology. However, mechanisms by which pDNA molecules interact with cell membranes, pass through crowded cytoplasm, and finally reach the nucleus remain to be elucidated. The lack of understanding of the mechanisms has hindered its translation to clinical settings.
It is well known that few naked pDNA molecules can be internalized by cells under physiological conditions. When cells are treated with strong but short electric pulses, significant cellular uptake of pDNA will occur if it is present in the pulsing buffer. If pDNA molecules are added into the buffer after the application of electric pulses, they do not enter the cells (Escoffre, et al., 2009; Henshaw & Yuan, 2008). Several mechanisms have been proposed to explain pDNA transport across the plasma membrane. One of them considers the transport through transient pores created by pulsed electric fields (de Gennes, 1999; Neumann, et al., 1982). The modes of transport include diffusion (Michel, et al., 1988; Xie & Tsong, 1993), electrophoresis (Klenchin, et al., 1991; Mir, et al., 1999; Sukharev, et al., 1992), and electroosmosis (Hristova, et al., 1997). However, the pores have yet to be characterized directly because they are small and have a lifetime on the order of 10 msec (Neu & Krassowska, 1999; Zaharoff, et al., 2008).
Recently, evidence from various studies indicates that naked pDNA may not be able to enter cells via the transient pores (Chernomordik, et al., 1990; Hristova, et al., 1997; Spassova, etal., 1994). They are internalized by cells through endocytosis (Cervia, et al., 2017; Cervia & Yuan, 2018; Chang, et al., 2014; Rosazza, et al., 2016; Wu & Yuan, 2011), which is similar to mechanisms of internalization of most viral and non-viral gene delivery vectors (El-Sayed & Harashima, 2013; Mercer, et al., 2010). In an in vitro study, Golzio et al. directly visualized the early events of electric field-mediated pDNA uptake (Golzio, et al., 2002). They observed that electric pulses could facilitate pDNA molecules to form complexes with the plasma membrane on the side of cells facing the cathode, which were clearly visible under a light microscope, and that the complexes could not be detached from the membrane when the electric field was reversed. Additionally, pDNA in the complexes was resistant to exogenous DNase treatment (Eynard, et al., 1997), but could be removed by trypsinization of cells (Wu & Yuan, 2011), indicating that pDNA was inserted into surface structures of the plasma membrane or encapsulated by membranous structures. The complex formation was required for successful electrotransfection (Golzio, et al., 2002). To understand how the complexes are internalized by cells, we have investigated the roles that endocytosis plays in electrotransfection. We showed that both pDNA uptake and electrotransfection efficiency (eTE) could be significantly reduced by treatment of cells for a short period (10 to 30 min) with cold medium (4°C) or pharmacological inhibitors of endocytosis either before or after electric pulse application (Cervia, et al., 2017; Chang, et al., 2014; Mao, et al., 2017; Wu & Yuan, 2011). The same results were observed if specific genes related to clathrin-mediated endocytosis, i.e., clathrin heavy chain and dynamin II, were knocked down in cells treated with short interfering RNA (siRNA) (Chang, et al., 2014; Wu & Yuan, 2011). However, knock down of caveolin 1 had no effects on eTE (Chang, et al., 2014). Furthermore, we observed that pDNA uptake and eTE could be altered through manipulation of Rac1-dependent macropinocytosis (Mao, et al., 2017). In a recent study, Rosazza et al. showed that pDNA uptake involved all classical endocytic pathways, including caveolae-mediated endocytosis, clathrin-mediated endocytosis, and macropinocytosis (Rosazza, et al., 2016), although our data show that the caveolae-mediated endocytosis may not lead to successful gene delivery.
It has been generally accepted that there are no universal experimental protocols that are optimal for transfection of all types of cells because electrotransfection depends on a large number of factors. Some of them are known in the literature. They are related to (i) properties of cells, such as cell size, shape, membrane structures, and surface charge (Agarwal, et al., 2007), (ii) parameters of the electric field, such as electric field strength and pulse number, shape, duration, and frequency (Kotnik, et al., 2003), and (iii) cell microenvironment in tissues or pulsing buffers. However, how intracellular factors affect electrotransfection are still poorly understood. One of the challenges in studying intracellular transport is to visualize electrotransfected pDNA in different compartments of cells. In previous studies, the visualization often relied on light microscopy techniques that are currently limited by spatial resolutions in detecting locations of pDNA in cells at the ultrastructural level. To address this issue, we examined time-dependent pDNA distributions in cells, and quantitatively determined percentages of pDNA associated with different endocytic compartments using transmission electron microscopy (TEM).
MATERIALS AND METHODS
Cell culture
COS7 and 4T1 cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA), and HCT116 and HT29 cells were cultured in McCoy’s 5A medium, both containing 10% fetal bovine serum (FBS) (HyClone, Logan, UT), with penicillin/streptomycin (Invitrogen). Human umbilical vein endothelial cells (HUVEC) were cultured in EGM-2 medium (Lonza) with FBS, and experiments were conducted with cells in early passages (up to passage seven).
Plasmid DNA preparation
A 4.7 kbp plasmid (pEGFP-N1, Clontech, Palo Alto, CA), encoding enhanced green fluorescent protein (GFP), was prepared from transformed E. coli cells by using the Qiagen Plasmid Maxi Prep Kit (Qiagen, Valencia, CA). The pDNA was covalently labeled with digoxin at the ratio of 5:1 μg DNA to μl labeling reagent) (Mirus Corp., Madison, WI), according to the manufacturer’s protocol.
Electrotransfection
Electrotransfection was performed in a special chamber constructed on an 8-well chamber slide (EMD Millipore, CA). Two parallel scotch conductive tapes (VWR, PA) were used as electrodes that were separated by 5 to 6 mm on the bottom surface of each well. Approximately 5000 cells were seeded in each well and cultured overnight at 37°C in 5% CO2. Then, cells were rinsed once with Dulbecco’s Phosphate-Buffered Saline (DPBS), incubated on ice for 10 min in HeBS buffer (20 mM HEPES, 137 mM NaCl, 5 mM KCl, 6 mM dextrose, and 0.7 mM Na2HPO4, pH 7.05), containing digoxin-labeled (dig-pDNA) (60 μg/ml), or unlabeled pDNA at the same concentration, and treated with one of the following electric pulse sequences (BTX ECM 830 Square Wave Electroporation System, Harvard Apparatus, Holliston, MA). Four different sequences were used, which were a) 5 pulses at 400 V/cm, 5 msec duration, and 1 Hz frequency for HCT116 and COS7 cells; b) 4 pulses at 550 V/cm, 5 msec duration, and 1 Hz frequency for HT29 cells; c) 5 pulses at 500 V/cm, 5 msec duration, and 1 Hz frequency for 4T1 cells; and d) 1 pulse at 625 V/cm and 20 msec duration for HUVEC cells. The choices of the sequences were based on optimal conditions published in the literature (Chang, et al., 2014; Jordan, et al., 2008; Mao, et al., 2017; Wu & Yuan, 2011). After the application of electric pulses, cells were allowed to recover at 37°C for 10 min. For samples that needed to be processed at later time points, 200 μl fresh medium at 37°C was added to the pulsing buffer, and the cells were transferred to the incubator.
Pre-embedding immunostaining of pDNA for electron microscopy
Cell monolayers were collected at 10 or 40 min post electrotransfection and prepared for immunoelectron microscopy study by following the protocols reported in the literature (Pathak, et al., 1988; Sesack, et al., 2006; van den Pol, 1986). In brief, cell monolayers were fixed with a freshly prepared solution containing 4% paraformaldehyde and 0.2% glutaraldehyde prepared with 0.1 M phosphate buffer (PB) (3.1 g/L NaH2PO4-H2O and 10.9 g/L Na2HPO4 (anhydrous), pH 7.4) at room temperature for 20 min. The fixed cells were washed with fresh 0.1 M PB buffer, and incubated with 1% sodium borohydride (Sigma) for 10 min. Thereafter, they were washed 4 times for 5 min each time with 0.1 M PB buffer, and incubated for 30 min at room temperature in a blocking buffer containing 1% bovine serum albumin (BSA), 3% normal goat serum and 0.04% triton-100 in PBS (Sigma), followed by overnight incubation at 4°C in either the blocking buffer in the control group, or a mouse anti-digoxin antibody solution (1:1000 dilution with the blocking buffer) (Sigma) in the experimental group. The next day, the cells were rinsed 10 times for 3 min each time in PBS, incubated in either the blocking buffer (i.e., the control group), or a biotinylated goat anti-mouse antibody solution (1:400 dilution with the blocking buffer) (Vector Laboratories) for 1 h at room temperature. The samples were rinsed with PBS, and then exposed to an avidin-biotin-peroxidase complex (ABC, Vector Laboratories) for 1 h. After being briefly rinsed with PBS, the cells were incubated with a solution containing 0.05% 3,3ʹ-diaminobenzidine tetrahydrochloride (DAB, Sigma) and 0.003% hydrogen peroxide in 0.03 M PB buffer. After 5 min, the enzymatic reaction was stopped by rinsing the samples with 0.1 M PB. The pDNA in electron micrographs was identified as the darkest electron dense signals (EDS). To improve visual contrast of the EDS or confirm that the EDS were indeed originated from specific staining of pDNA, the reaction time in a few samples was extended to 8–20 min. Based on the experimental design described above, there were three different control groups. Compared to the experimental group, cells in control groups were either stained with the primary antibody only, the secondary antibody only, or not stained with any antibodies.
Sample preparation for electron microscopy
Immunostained samples
These samples were prepared for electron microscopy by following a protocol in the literature (Sesack, et al., 2006). After immunostaining as described above, cells in both experimental and control groups were treated with 2% osmium tetroxide in H2O for 30 min, and dehydrated in a graded series of acetone at increasing concentrations of 30%, 50%, 70%, 95% and 3 times in 100% for 20 min at each step. The cells were then infiltrated with graded mixtures of 100% acetone and EMBED 812 resin with different volume ratios (3:1, 1:1, and 1:3) (Electron Microscopy Science), followed by 3 changes of 100% epoxy resin for 30 min at each infiltration step. The samples were baked in epoxy resin at 60°C for at least 12 hours. After polymerization, specimens were heated with boiling water to make the resin pliable, and separated from the electrode chamber slide using a razor blade. The slabs were examined with an inverted light microscope, and an area with high cell density was cut out, re-mounted on Epon blocks, and ultrathin sectioned on a Reichert-Jung Ultracut E ultramicrotome (Leica, Buffalo Grove, IL) in a direction that was approximately parallel to the interface between cells and the chamber slide. The thickness of each section was approximately 80 nm. Sections were collected on 200 mesh copper grids covered with carbon-stabilized Formvar membranes. The conventional post-staining step using uranium and lead was purposefully omitted to keep cellular contrast low and thus, enhance visualization of the immune complexes. All grids were examined in a TEM (Philips EM CM 12, now FEI/Thermo Fisher) at 80 kV without post staining with customary uranium or lead to decrease contrast and enhance visibility of the immunolabels.
Non-immunostained samples
These samples were prepared with specific steps of staining (post- staining with uranyl acetate and lead citrate) to enhance the contrast of membranous structures in cells. They were used to study effects of electrotransfection on invagination of the plasma membrane. The details are as follows. COS7 cells were pulsed in the electrotransfection buffer with non-labeled pDNA. At 10 min and 40 min post application of electric pulses, the cells were harvested and prepared for electron microscopy study following the experimental procedures that were similar to those described above, except for a few minor modifications to improve visual contrast of membranous structures in cells. In brief, cells were fixed at room temperature for 20 min with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M PB buffer. After the cells were rinsed three times with 0.1 M PB buffer, they were treated with 1% osmium tetroxide in 0.1 M PB for 15 min, followed by the sequential dehydration with acetone at increasing concentrations. Then, they were embedded and ultrathin sectioned as described above. Unlike the immunostained sections, these were post stained on drops of 2% uranyl acetate for 15 min, rinsed and drained. Then, they were placed on drops of 0.5% lead citrate for 5–10 min in a chamber containing pellets of NaOH to trap CO2 and prevent PbCO3 precipitation. After several rinses in double-distilled water, and wicking with filter paper, they were dried in air.
Analysis of electron micrographs
TEM images with different resolutions were acquired. Images at high magnifications were used to show morphological details of ultrastructures in cells, and those at a low magnification (8.4 nm per pixel) were used for both morphological and quantitative analyses of the EDS (or pDNA) in cells. One challenge in the study was that even at the low magnification, the images contained only a few structures associated with the pDNA, which were inefficient for quantitative analysis. Thus, we pooled electron microscopy results from two independent experiments. From different regions of electron micrographs, we arbitrarily selected 40 cells in each group to manually count the numbers of different subcellular structures associated with the pDNA. To reduce false positive data, we neglected dark granular structures whose darkness was lighter than or similar to that of the nucleus, and those that were smaller than 3×3 pixels. As a result, we discounted electron dense aggregates smaller than 25 nm, which is smaller than the size of pDNA (~100 nm). For the non-immunostained samples, we arbitrarily selected 10 cells from different regions of electron micrographs in each group to manually count the numbers of clathrin- and caveolin-coated pits or vesicles in cells treated with or without electric pulses in the buffer containing non-labeled pDNA.
Statistical analysis
We counted the numbers of specific vesicles in thin sections of cells. To reduce the possibility of double counting the same vesicle appeared in adjacent sections, only 1 out of 5 sections were collected for image analysis. On the other hand, the numbers of the vesicles observed in the thin sections depend on the size of vesicles, which in turn might depend on experimental conditions. To this end, we only compared the numbers of vesicles under the same experimental conditions or with similar sizes.
The relative numbers of vesicles are reported in the paper because we only counted vesicles in thin sections whose total volume were less than that of cells. The experimental data were analyzed statistically using the bootstrap method. Briefly, the raw data in a given group were randomly resampled to generate 1000 bootstrapped data sets. For each data set, we calculated its average value. The mean and standard deviation of the average values were used, respectively, as the estimates of the mean and the standard error of the mean of the original data in the given group. Bootstrapped t-test was used to determine if samples in two different groups came from the same population. The hypothesis was rejected if the p-value was < 0.05. All statistically analyses were performed using a MATLAB® program created in-house.
RESULTS
pDNA was observed in regions close to plasma membrane shortly after electrotransfection
At 10 min after pulse application, pDNA molecules were observed on only one side of cells (Fig. 1A through 1C, thin arrows). Some pDNA molecules formed aggregates near the membrane (Fig. 1A and 1B, thin arrows), which was consistent with previous observations by fluorescence microscopy (Chang, et al., 2014; Golzio, et al., 2002; Wu & Yuan, 2011; Zaharoff, et al., 2008). At a higher magnification, pDNA was observed as either thin layers within membranous structures or aggregates. The thin layer could be co-localized with membrane invaginations (Fig. 1D, thick arrow) or membranes of large intracellular vesicles (Fig. 1D, thin arrow). Some aggregates of pDNA were observed in the cytoplasm under the plasma membrane, and they were surrounded by vesicles of various sizes (50–500 nm) (Fig. 1E, thick arrow). Other aggregates were in contact with or were enwrapped in protrusions from cells (Fig. 1F, thick arrow). The structures shown in Figs. 1D and 1F suggested that within the first 10 min, pDNA could be internalized by cells via macropinocytosis.
Figure 1. Distribution of dig-pDNA in different cell lines at 10 min after electrotransfection.
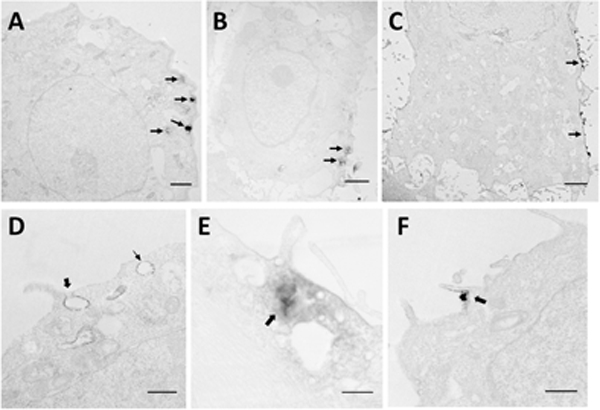
The digoxin labeled pDNA in cells was detected by immunostaining prior to TEM processing. All images in the figure have low contrast, because we purposefully chose not to post stain the samples with uranium and lead in order to improve visual contrast between the EDS from pDNA and subcellular structures. As a result, we could reduce false positive signals in image analysis. (A to C) These low magnification electron micrographs show unilateral distribution of the EDS (arrows), which indicates initial locations of electrotransfected pDNA in (A) COS7 cell, (B) HT29 cell, and (C) 4T1 cell. Bars = 2 μm. (D to F) The high magnification micrographs show associations of the EDS with the subcellular structures in (D) HCT116 cell, (E) HT29 cell, and (F) HCT116 cell. In D, the thick arrow indicates the formation of a macropinosome, and the thin arrow indicates an early endosome-like structure. Bar = 500 nm. In E, the arrow points to the EDS intermingled with vesicles of different sizes. Bar = 100 nm. In F, the thick arrow indicates a membrane protrusion around an electron dense particle. Bar = 500 nm.
pDNA molecules spread in cytoplasm at 40 min after electrotransfection
At 40 min post application of the electric pulses, pDNA was observed throughout the cytoplasm (Fig. 2A), rather than just near the plasma membrane observed at 10 min (Fig. 1A through 1C). Some aggregates even reached the perinuclear region. At a high magnification, we observed pDNA in the membrane of invaginated pits with a bulb-like shape (caveolae) (Figs. 2B through 2D). To confirm that the EDS indeed originated from immunostaining of pDNA, we prolonged the HRP reaction time from 5 min (Figs. 2B) to 8 min in some additional experiments (Figs. 2A, 2C, and 2D) before the electron microscopy examination. The prolonged reaction enhanced the contrast in regions of the dark samples, with little change in the darkness of structures in surrounding regions, indicating that the EDS in the subcellular structures were indeed due to the product from the reaction of HRP substrate (i.e., DAB). Thus, we could rely on the EDS to determine locations of pDNA.
Figure 2. Subcellular features associated with dig-pDNA at 40 min after electrotransfection.
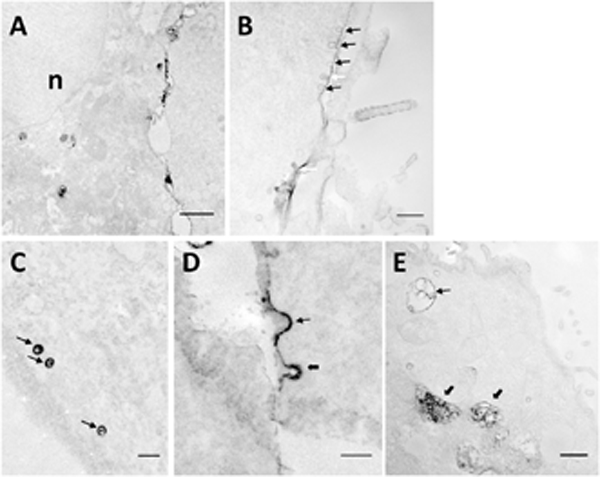
All images were acquired on COS7 cells. (A) The low magnification image shows the distribution of the EDS in the cell; n, nucleus. Bar = 1 μm. (B) A train of electron dense caveolae-like structures is visible. Black arrows indicate caveolae with membranes associated with the EDS. White arrows indicate caveolae partially filled with the EDS. Bar = 100 nm. (C) Typical caveolae-like structures are filled with the EDS indicated by arrows. Bar = 100 nm. (D) Two membrane invaginations are associated with the EDS. Thin and thick arrows indicate pits with smooth and rough membranes, respectively. Bar = 100 nm. (E) An early endosome-like structure and two late endosome/lysosome- like structures are associated with the EDS, as indicated by the thin and the thick arrows, respectively. Bar = 500 nm.
The pDNA molecules were either concentrated in the membrane (thin black arrows in Fig. 2B) or partially filled the pits (thin white arrow in Fig. 2B). Some of the pits aligned as a linear array (thin arrows in Fig. 2B), which is a characteristic of caveolin-coated pits (Parton & Simons, 2007; Stan, 2005). Some dark pits had a smooth membrane (thin arrow in Fig. 2D), suggesting that they were either uncoated or coated with caveolin (Bruns & Palade, 1968a; Bruns & Palade, 1968b; Palade & Bruns, 1968; Stan, 2005). Other pits had a rough membrane (thick arrow in Fig. 2D), which is a characteristic of clathrin-coated structures (Bretscher, et al., 1980; Krijnse Locker & Schmid, 2013). The studies described above were performed with the four stable cell lines, and the EDS associated with the pits were observed more frequently in COS7 cells than in other cells. Invaginated pits can bud off to form 50- to 100-nm vesicles in the cytoplasm. Most of the vesicles associated with electron dense structures had a smooth surface and were filled with the EDS (thin arrows in Fig. 2C) in all stable cell lines examined in the study. This was in contrast to the pits associated with the EDS, where both smooth and rough membranes could be observed (see Fig. 2D).
At 40 min post pulsing, pDNA molecules were also observed frequently in early endosomes (thin arrow in Fig. 2E) and late endosomes/lysosomes/multivesicular bodies (LLM) (thick arrow in Fig. 2E) in COS7 and HCT116 cells. These vesicles were either partially or completely filled with electron dense structures. In HT29 and 4T1 cells, relatively less EDS could be observed in LLM. Most of the EDS were associated with macropinosomes and early endosomes.
The pDNA-positive pits were observed more frequently at 40 min than at 10 min post electrotransfection (see Figs. 1 and 2), indicating that more pDNA uptake at the later time point (i.e., 40 min) was mediated by clathrin- and caveolin-mediated endocytosis. The observation suggested that these endocytic pathways might be inhibited by electric pulses during the first 10 min. To further investigate this possibility, we divided COS7 cells into 4 groups. In the first two groups, the cells were treated with five electric pulses (400 V/cm, 5 msec, and 1 Hz) in the buffer with non-labeled pDNA. In the last two groups, the cells were incubated in the same buffer but without pulsing. After 10 min, the cells in one of the pulsed and one of the non-pulsed groups were fixed and sectioned for electron microscopy examination. Ten cells were randomly selected from each group for quantification of the total numbers of clathrin- and caveolin-coated pits and vesicles (see Methods section). The procedures were repeated at 40 min for the other two groups. The majority of the vesicles with the sizes of 50 to 100 nm were observed to be located within a short distance (less than 0.5 μm) from the plasma membrane. The number of pits was less than 10% of that for vesicles in each group. To demonstrate effects of electric pulsing on clathrin- and caveolin-mediated endocytosis, the total numbers of pits and vesicles normalized by the matched, non-pulsed controls are reported for each group in Fig. 3. The data indicate that treatment of cells with electric pulses had insignificant effects on the number of clathrin coated pits and vesicles but transiently decreased the number of caveolin-coated pits and vesicles (p < 0.05). At 40 min post pulsing, there was no significant difference in the number of either of these structures between pulsed cells and non-pulsed controls.
Figure 3. Effects of cell treatment with electric pulses on numbers of small vesicles.
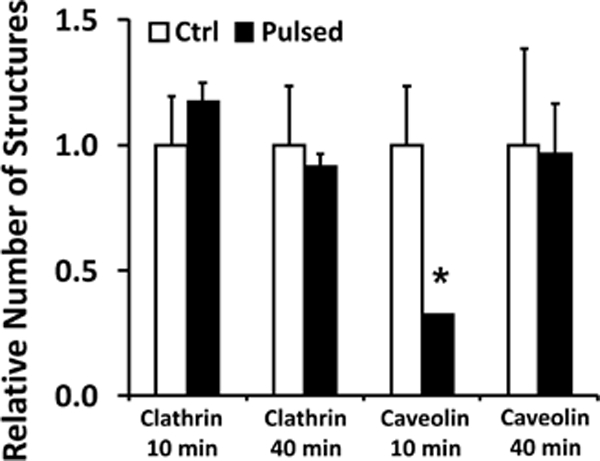
COS7 cells in the buffer containing non-labeled pDNA were treated with five electric pulses (400 V/cm, 5 msec, and 1 Hz). Cells in the control groups were prepared with the same procedures except that the electric pulses were not delivered. At 10 or 40 min post treatment, membranous structures in cells were post-stained with uranyl acetate and lead citrate to enhance their contrast under the electron microscope (see the Materials and Methods section), but no immunostaining was performed for these cells. Subcellular structures of pits and vesicles (50 – 100 nm in diameter) were grouped together. The structures with smooth and rough membranes were considered to be clathrin- and caveolin-coated, respectively. The total numbers of the structures were counted in 10 different cells (i.e., one section per cell); and the data in the pulsed groups were normalized by the corresponding controls. Bars and error bars represent the mean and the standard error of the mean, respectively. *p < 0.05, Ctrl vs Pulsed.
Endocytosis mediated pDNA uptake was more active in easy-to-transfect cells
The amount of pDNA in the cytoplasm varied among four stable cell lines after the cells were electrotransfected under similar conditions. Qualitatively, more pDNA was observed in COS7 and HCT116 cells, compared to HT29 and 4T1 cells. This observation was consistent with the efficiency of electrotransfection reported for these cell lines (Chang, et al., 2014; Melkonyan, et al., 1996; Rothnie, et al., 2011). To quantitatively compare the number of subcellular structures that were associated with pDNA (i.e., regions with EDS), we analyzed TEM images of two cell lines: COS7 and 4T1, which represented, respectively, easy- and hard-to-transfect cells. For each cell line, we randomly selected one region per cell, and examined 40 cells that were electrotransfected with dig-pDNA and 40 with non-labeled pDNA (control). To determine specific intracellular structures that were associated with pDNA, we developed a set of morphological criteria to divide pDNA positive structures into five groups. One is defined as surface complex (SC), which refers to membrane-pDNA complexes on the cell surface or within a short distance from the plasma membrane that could not be resolved by the TEM at the low magnification (see Fig. 1C). Each continuous complex was counted as one structure no matter how large it was. The SC includes pDNA-associated clathrin- and caveolin-coated pits and vesicles (Figs. 2B-2D), and membrane protrusion/ruffle with (Fig. 4A) or without closure (Fig. 1F). It also includes aggregates of pDNA surrounded by vesicles of various sizes (50–500 nm) in the vicinity of the plasma membrane (Fig. 1E). The second group includes pDNA complexes with early endosome-like compartments (EE) identified as large vesicles (up to 1 μm) whose lumen was electron-translucent and might contain a few smaller vesicles (Fig. 4B). The third group includes the pDNA complex with late endosome-, lysosome-, or multivesicular body-like compartments (LLM). They were lumped together and identified as vesicles with various sizes in TEM images whose lumens were filled with electron dense materials or contained numerous smaller vesicles (Figs. 4C and4D). The forth group includes pDNA in macropinosome-like compartments (ME), that contained large vesicles without internal smaller vesicles, although it could also be early endosomes at a very early stage. The last group is called the intracellular complex (IC), which is an aggregate of pDNA located away from the plasma membrane where the aggregate is surrounded by vesicles of various sizes (50–500 nm) without a membrane envelope (Fig. 4E). Although the criteria for the subcellular structure categorization are less accurate than molecular markers, they have been used routinely in previous studies to identify the intracellular structures (Huotari & Helenius, 2011; Mukherjee, et al., 1997; Zeigerer, et al.,2012).
Figure 4. Representative electron micrographs of pDNA-positive structures.
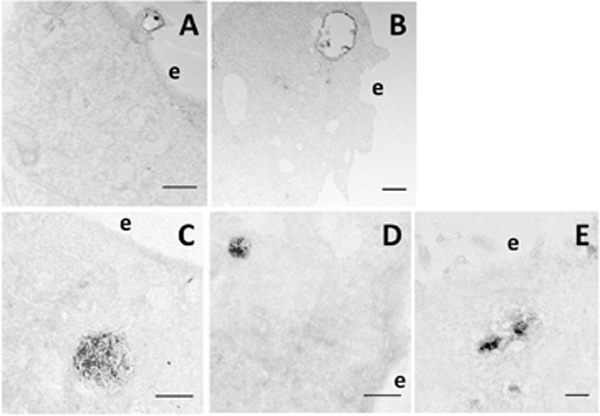
Morphological criteria were developed in the study for the identification of subcellular structures associated with pDNA. Some morphological features are shown in Figs. 1 and 2, respectively. Others are shown in this figure. (A) A membrane protrusion/ruffle with a closure is associated with the EDS in a COS7 cell, indicating that macropinocytosis is involved in pDNA uptake. Bar = 500 nm. (B) An early endosome-like structure with an electron-translucent lumen contains a few internal vesicles that are positive for the EDS in an HCT116 cell. Bar = 500 nm. (C) A large and (D) a relatively smaller late endosome/lysosome-like structures are positive for the EDS in COS7 cells. Bar = 1 μm. (E) A few electron dense aggregates can be seen in an HT29 cell; and each aggregate is surrounded by multiple vesicles. Bar = 200 nm. e, extracellular space.
We counted the total numbers of the pDNA-positive structures in each group, and normalized the data by that for COS7 at 10 min. The final results shown in Fig. 5 demonstrated that COS7 cells had more pDNA-positive structures than 4T1 cells (p < 0.05), and the numbers of these structures might decrease with time, presumably due to pDNA degradation in cells, but the decrease was statistically insignificant (p > 0.05). For cells pulsed with non-labeled pDNA, subcellular structures should not contain EDS although we did observe it in each group, presumably due to non-specific staining of the samples. Quantitatively, the false-positive signals in the non-labeled pDNA groups were significantly lower (p < 0.05) than those from the samples pulsed with dig-pDNA in both types of cells and at both time points (Fig. 5).
Figure 5. Relative numbers of subcellular structures associated with dig-pDNA.
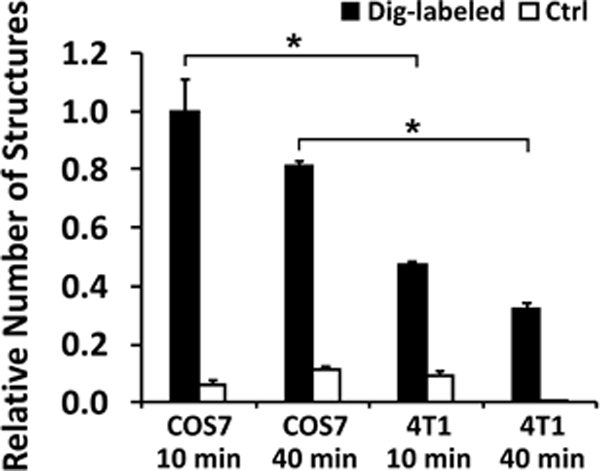
COS7 and 4T1 cells were collected at 10 or 40 min post electrotransfection, and examined under the electron microscope. At each time point, we arbitrarily selected electron micrographs from 40 different cells that were electrotransfected with dig-labeled pDNA, and 40 from the control group (Ctrl, where unlabeled pDNA was for electrotransfection). The selection was performed without prior knowledge of whether the selected cells contained the EDS. The total numbers of the subcellular structures associated with the pDNA in these cells were counted manually for each group. The results are reported as the total number of pDNA-positive structures per group normalized by the data of COS7 at 10 min. The data show that the numbers of pDNA- positive structures are higher in easy-to-transfect cells (COS7) than in hard-to- transfect cells (4T1). The numbers of false positive structures in the control groups were negligible, compared to the corresponding data in the experimental groups. Bars and error bars represent the mean and the standard error of the mean, respectively. *p < 0.05.
In addition to the total numbers of subcellular structures, we quantified distributions of pDNA among the specific structures defined above. By comparing the distributions at 10 and 40 min, respectively, we could show how pDNA was transported in cells. For COS7 cells, pDNA distribution at 10 min was observed to be: 49.0% in SC, 11.5% in ME, 10.0% in EE, 29.2% in IC, and none in LLM (Fig. 6A). At 40 min after pulsing, the percentage of pDNA dropped to 26.0% in SC (p < 0.05), but more than doubled (27.5%) in EE (p < 0.05). No pDNA was observed in LLM at 10 min, but the percentage was increased to 14.1% at 40 min (p < 0.05) (Fig. 6A). The shift in the distribution pattern was presumably due to inward transport of pDNA. In 4T1 cells, most pDNA molecules were associated with SC at both 10 and 40 min, and the percentage did not change significantly with time (68.0% at 10 min and 69.8% at 40 min) (Fig. 6B). For EE, ME, and LLM, the numbers of these structures with pDNA did not vary with time (p > 0.05) when comparing the data between the 10-min and 40-min groups (Fig. 6B). The only significant change was observed for the number of the IC structure with pDNA. It was decreased from 12.2% at 10 min to zero at 40 min (p < 0.05). These observations suggested that pDNA in 4T1 cells was trapped near the cell surface instead of being actively transported towards the nucleus. Interestingly, the fraction of ME-associated signals was comparable between easy- and hard-to-transfect cells (Fig. 6), suggesting that macropinocytosis was a common pathway involved in electrotransfection.
Figure 6. Percentages of different subcellular structures associated with dig-pDNA.
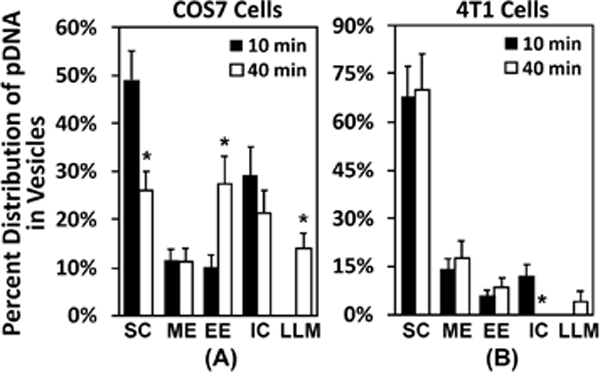
The raw data for this figure are the same as those for Fig. 5. Instead of reporting the total numbers, the figure shows the percentages of each subcellular structures that were associated with the EDS. The structures include surface complex (SC), macropinosome-like compartment (ME), early endosome-like compartment (EE), intracellular complex (IC), and late endosome-, lysosome-, or multivesicular body-like compartment (LLM). The details of the definition of each structure are provided in the results section. The data demonstrate that most pDNA molecules were located near the plasma membrane at 10 min. (A) In easy-to-transfect cells (COS7), a large fraction of pDNA was transferred to the inner vesicles at 40 min. (B) The transfer was minimal in hard-to-transfect cells (4T1). Bars and error bars represent the mean and the standard error of the mean, respectively. *p < 0.05, 10 min vs 40 min for each structure.
Endocytic pathways involved in pDNA uptake by HUVEC in electrotransfection
Endothelial cells (ECs) are also hard to transfect by many commercially available methods (Hunt, et al., 2010), but can be successfully transfected by using electric pulses. Additionally, transcytosis is not common in most cells, but is essential in ECs for systemic delivery of molecules to extravascular space in tissues with continuous capillaries (Oh, et al., 2007). Therefore, endocytic pathways involved in electrotransfection of ECs may differ from those in other cells. In the current study, cellular uptake of electrotransfected pDNA was investigated in HUVEC, one of the most popular models for ECs. Similar to that in the stable cell lines described above, pDNA uptake in HUVEC occurred at only one side of the cells, and formed aggregates near the plasma membrane shortly (10 min) after electric pulsing (Fig. 7A). At a higher magnification, we observed the electron dense aggregates surrounded by vesicles of various sizes (50–300 nm) (Fig. 7B), and clusters of caveolae-like structures associated with the EDS on the cell surface (Fig. 7C). Some of the electron dense aggregates on the cell surface were engulfed by the plasma membrane to form macropinosomes (Fig. 7D). At 40 min, several clusters of caveolae-like vesicles with the EDS could be observed in the cytoplasm that were far from the plasma membrane (Fig. 7E, arrow head versus arrow). The observations differed from those in the stable cell lines, where the EDS were infrequently associated with the caveolae-like structures at 10 min post electrotransfection, and most EDS associated with the caveolae-like structures were observed near the plasma membrane only at 40 min (see Fig. 2). Although the mechanisms are still unknown, the differences in the observations may be related to the high level of caveolin expressed in HUVEC that is critical for transcytosis of macromolecules (Oh, et al., 2007).
Figure 7. Subcellular structures associated with dig-pDNA in HUVEC and COS cells after electrotransfection.
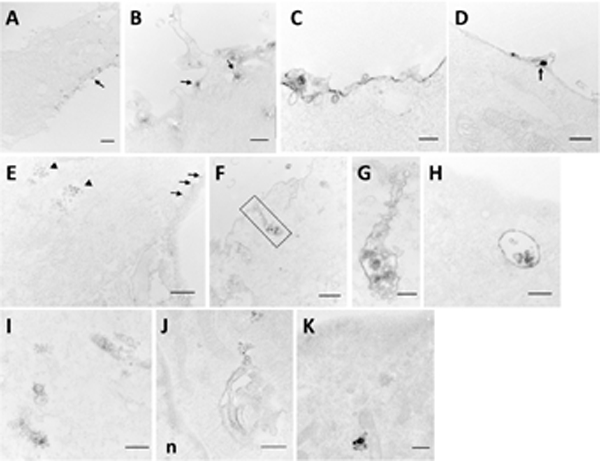
(A) A portion of HUVEC with unilateral distribution of the EDS is indicated by the arrow at 10 min after electrotransfection. Bar = 2 μm. (B) Electron dense aggregates near the plasma membrane are shown to be surrounded by vesicles of various sizes (50–300 nm), indicated by the arrows, at 10 min after electrotransfection. Bar = 500 nm. (C) A train of electron dense caveolae-like structures is seen in a HUVEC at 10 min after electrotransfection. Bar = 100 nm. (D) A macropinosome has formed to engulf an electron dense particle (arrow) in HUVEC at 20 min after electrotransfection. (E) A portion of a HUVEC contains clusters of intracellular caveolae-like structures (arrowheads), and membrane invaginations (arrows) with the EDS at 40 min after electrotransfection. Bar = 500 nm. (F) An elongated tubular structure from the plasma membrane of a HUVEC is seen to be associated with electron dense particles at 20 min after electrotransfection. Bar = 500 nm. The boxed area with a high magnification is shown in Panel (G). Bar = 100 nm. (H) An early endosome-like structure is present in a HUVEC at 40 min after electrotransfection. Bar = 500 nm. (I) Tubular vesicular structures associated with the EDS in the cytoplasm of a HUVEC at 20 min after electrotransfection. Bar = 100 nm. (J) A Golgi-like structure is associated with the EDS in caveolae rosette-like vesicles in a HUVEC at 40 min after electrotransfection; n, nucleus. Bar = 500 nm. (K) A tubulovesicular structure, similar to that observed in HUVEC, is associated with the EDS in a COS7 cell at 40 min after electrotransfection. Bar = 100 nm.
In addition to the subcellular structures described above, we observed that pDNA could be internalized by a long tubular structure with an elongated, narrow neck connected to the plasma membrane (Fig. 7F and 7G). The tubular structure was surrounded by numerous small vesicles, which was morphologically similar to those in the clathrin-independent carriers/GPI- enriched early endosomal compartments (CLIC/GEEC) pathway (Doherty & McMahon, 2009), and also resembled the tubular invagination with caveolae observed in the sinus endothelial cells in the rat spleen (Uehara & Miyoshi, 1999).
Similar to the pDNA distribution in stable cell lines (Fig. 2), pDNA molecules were observed in a few early endosomes (Fig. 7H). But most pDNA molecules were associated with tubulovesicular structures in HUVEC cells (Fig. 7I). Numerous small vesicles (about 50 nm in diameter) were seen budding off the structure, forming a rosette or grape-like polymorphous structure. Finally, we observed pDNA in a Golgi-like structure with opposing rosette-like structures in HUVEC (Fig. 7J), but not in the stable cell lines used in the study, implying that pDNA-positive vesicles could fuse with the ER-Golgi network in HUVEC.
In addition to HUVEC, we detected some tubular vesicle-like structures with the EDS in COS7 cells although at low abundance (Fig. 7K), after a long reaction time (20 min) for DAB to increase the contrast between pDNA and subcellular structures. The observation indicated that the CLIC/GEEC pathway might also be involved at a lower frequency in the uptake of electrotransfected pDNA in non-endothelial cells.
DISCUSSION
Different appearances of vesicular ultrastructures were observed to be associated with electrotransfected pDNA at or near the plasma membrane. These structures have also been reported in previous studies unrelated to electrotransfection (Hansen & Nichols, 2009). The structural data indicated that electrotransfected pDNA could be internalized by cells through multiple pathways, including caveolae-mediated and clathrin-mediated endocytosis, macropinocytosis, and the CLIC/GEEC pathway (Doherty & McMahon, 2009). Compared to the easy-to-transfect cells (e.g., COS7), the total number of vesicular structures with positive EDS was low in the hard-to-transfect cells (e.g., 4T1), and less pDNA was actively transported away from the cell surface, suggesting a correlation between the ease of electrotransfection and the activity of endocytosis among different cells. Macropinocytosis was the most common pathway shared by all types of cells, and it occurred at both earlier (10 min) and later (40 min) time points, whereas caveolae-mediated endocytosis was involved in pDNA uptake mainly at the later time point in the stable cell lines. In the HUVEC, endocytic pathways with tubulovesicular structures played a major role in the uptake of electrotransfected pDNA. In all cells, few pDNA molecules were observed in the cytosol with no association with surrounding vesicles. Together, the data described above demonstrate that endocytosis is a key mechanism for cellular uptake and intracellular trafficking of electrotransfected pDNA.
Distribution of electrotransfected pDNA near the cell surface
Previous studies have stated that cellular uptake of electrotransfected pDNA requires formation of complexes between the pDNA and the plasma membrane (Golzio, et al., 2002; Wu & Yuan, 2011). The statement is based on observations with fluorescence microscopy, where fluorescently labeled pDNA accumulates on the surface of cells facing the cathode with continuous or punctate patterns right after application of electric pulses; and there are no pDNA molecules in the cytoplasm. However, these observations are limited by the spatial resolution of light microscopes that cannot reveal structural details of the complexes. In the current study, we observed four different ultrastructures that might be considered as the complexes described above: a) the electron dense membrane, which could be a portion of the plasma membrane or membrane invagination (Figs. 2B,2D,7C), b) the membrane of vesicles near the cell surface (Figs. 1D, 2C, 7C), c) the electron dense aggregates engulfed by membrane protrusions (Figs. 1F, 4A, 7D), and d) the electron dense aggregates surrounded by vesicles of various sizes (Figs. 1E, 7B). The ultrastructures demonstrate that the complexes observed with light microscopy are non-specific and can be structurally different. The difference may explain why cellular uptake and intracellular trafficking of pDNA are mediated simultaneously by multiple endocytic pathways (Chang, et al., 2014; Mao, et al., 2017; Rosazza, et al., 2016; Wu & Yuan, 2011). Understanding mechanisms of the ultrastructure formation will be important for development of new strategies to improve gene delivery in cells.
Roles of caveolae-dependent pathway in uptake of electrotransfected pDNA
Caveolae are small Ω-shaped invaginations of the plasma membrane primarily composed of cholesterol and sphingolipids. They are present in many cell types and have been implicated in a variety of cellular processes including endocytosis. In the current study, caveolae containing electrotransfected pDNA were observed as either Ω-shaped invaginations of the plasma membrane, fully invaginated caveolae or grape-like clusters. Since pDNA (~ 100 nm) can be larger than the size of caveolae, which are about 50–100 nm in diameter, the former was unlikely to be engulfed by the latter. We observed that only a portion of the membrane of invaginations or vesicles was covered with the EDS if the enzymatic reaction with the substrate DAB was short (i.e., 5 min). When the reaction time was increased to 8–20 min, the EDS from the membrane became significantly stronger. And some membrane buds were filled with the EDS (Fig 2C, thin arrows), indicating that the product from the DAB reaction could accumulate in the caveolae-like vesicles. These observations suggested that caveolae could mediate the uptake of electrotransfected pDNA, and that the pDNA formed a flat complex with the membrane before being internalized by caveolae. In fact, a number of larger pathogens such as FimH-expressing E. coli (about 2 μm) (Shin, et al., 2000), E Coli K1 (Sukumaran, et al., 2002), and Pseudomonas aeruginosa (Zaas, et al., 2005) have been suggested to make use of caveolae as endocytic vehicles to avoid degradation in lysosomes. The involvement of caveolae-mediated endocytosis in cellular uptake of electrotransfected pDNA was also in agreement with the data from a recent study using fluorescence microscopy (Rosazza, et al., 2016).
Roles of clathrin-dependent pathway in uptake of electrotransfected pDNA
Clathrin-mediated endocytosis has been shown to be involved in electrotransfection (Chang, et al., 2014; Rosazza, et al., 2016; Wu & Yuan, 2011). In the current study, we observed EDS in clathrin-coated pits at 40 min after the application of electric pulses (Fig. 2). However, we rarely detected EDS associated with clathrin-coated vesicles (CCV) in all cell lines at any time points after electrotransfection. The discrepancy was not due to the lack of the CCV in cells because their number was comparable to that of caveolae-like vesicles (Fig. 3), but likely due to the uncoating event of the CCV, as it is well known that clathrin can quickly dissociate from the vesicles after CCV bud off from the plasma membrane (Jiang, et al., 2000; Prasad, et al., 1993; Rothnie, et al., 2011). The lifetime of CCV is on the order of a few minutes (Boulant, et al., 2011; Massol, et al., 2006; Tan, et al., 2015). Therefore, it is possible for electrotransfected pDNA to be internalized via the clathrin-mediated endocytosis while not being detected in the CCV. Although the number of CCV containing EDS may decrease quickly with time, regular CCV are formed constantly in cells. As a result, it was possible for the number of total CCV shown in Fig. 3, which depended on the difference in the rates between uncoating and formation of CCV, to be significantly higher than the number of CCV containing EDS.
Roles of CLIC/GEEC in uptake of electrotransfected pDNA in HUVEC
To show unique endocytic pathways involved in electrotransfection that might be different from those in the stable cell lines, we examined distribution of pDNA in HUVEC. Shortly after electrotransfection, some long, cell surface-connected tubular invaginations and vesicles were observed to be associated with the EDS in HUVEC. Similar structures were also observed in the cytoplasm. To our knowledge, this data provides the first morphological evidence that the CLIC/GEEC pathway could be involved in the uptake of electrotransfected pDNA in any cells. This pathway is known to be independent of clathrin, caveolin, and dynamin. It functionally relies on actin cytoskeleton and membrane cholesterol (Kumari & Mayor, 2008; Lundmark, et al., 2008; Sabharanjak, et al., 2002). The ultrastructural morphology of nascent CLIC/GEEC at the plasma membrane is polymorphous tubular membrane invagination and vesicles (Romer, et al., 2007). This endocytic pathway has been shown to have high-capacity and be constitutively active (Nonnenmacher & Weber, 2011). It can lead to a vast uptake of fluid and rapid turnover of the plasma membrane (Howes, et al., 2010). For example, the entire membrane of fibroblasts can be internalized via this pathway in approximately 15 min (Howes, et al., 2010). Due to its high capacity in membrane internalization, a large amount of membrane-bound pDNA may be internalized within a short period. Unlike the observations in stable cell lines, most EDS in intracellular compartments of HUVEC were associated with tubular vesicles. Some vesicles with the EDS were observed to be connected to the Golgi apparatus (Fig. 7J). The observations in the current study were similar to findings in the study of adeno-associated virus 2 (AAV2) internalization (Nonnenmacher & Weber, 2011). AAV2 has shown great promise as a gene transfer vector. It uses the CLIC/GEEC pathway as one of the major entry routes for infection. The internalized AAV2 appears to bypass the classical endosomal systems to reach the Golgi apparatus. Only CLIC/GEEC-mediated entry of AAV2 can lead to efficient transduction, suggesting that the transduction requires trafficking of the virus particles to the Golgi apparatus.
CONCLUSIONS
The data in the study provide the ultrastructural evidence that endocytosis plays an essential role in cellular uptake and intracellular transport of electrotransfected pDNA. Although the intracellular trafficking alone may not be able to predict the eTE, which also depends on nuclear entry of pDNA and efficiency of transgene expression, results from the study can be used to determine mechanisms of the electrotransfection and guide development of novel strategies to improve this non-viral gene delivery technique.
ACKNOWLEDGMENT
This research was supported by grants from National Institutes of Health (GM098520) and National Science Foundation (CBET-1264186).
References
- Agarwal A, Zudans I, Weber EA, Olofsson J, Orwar O & Weber SG (2007). Effect of cell size and shape on single-cell electroporation. Anal Chem 79(10), 35893596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BoulanT S, Kural C, Zeeh JC, Ubelmann F & Kirchhausen T (2011). Actin dynamics counteract membrane tension during clathrin-mediated endocytosis. Nat Cell Biol 13(9), 1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher MS, Thomson JN & Pearse BM (1980). Coated pits act as molecular filters. Proc Natl Acad Sci U S A 77(7), 4156–4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RR & Palade GE (1968a). Studies on blood capillaries. I. General organization of blood capillaries in muscle. J Cell Biol 37(2), 244–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RR & Palade GE (1968b). Studies on blood capillaries. II. Transport of ferritin molecules across the wall of muscle capillaries. J Cell Biol 37(2), 277–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervia LD, Chang CC, Wang L & Yuan F (2017). Distinct effects of endosomal escape and inhibition of endosomal trafficking on gene delivery via electrotransfection. PLoS One 12(2), e0171699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervia LD & Yuan F (2018). Current Progress in Electrotransfection as a Nonviral Method for Gene Delivery. Mol Pharm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Wu M & Yuan F (2014). Role of specific endocytic pathways in electrotransfection of cells. Mol Ther Methods Clin Dev 1, 14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik LV, Sokolov AV & Budker VG (1990). Electrostimulated uptake of DNA by liposomes. Biochim Biophys Acta 1024(1), 179–183. [DOI] [PubMed] [Google Scholar]
- DE Gennes PG (1999). Passive entry of a DNA molecule into a small pore. Proc Natl Acad Sci U S A 96(13), 7262–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty GJ & McMahon HT (2009). Mechanisms of endocytosis. Annu Rev Biochem 78, 857–902. [DOI] [PubMed] [Google Scholar]
- El-Sayed A & Harashima H (2013). Endocytosis of gene delivery vectors: from clathrin- dependent to lipid raft-mediated endocytosis. Mol Ther 21(6), 1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoffre JM, Portet T, Wasungu L, Teissie J, Dean D & Rols MP (2009). What is (still not) known of the mechanism by which electroporation mediates gene transfer and expression in cells and tissues. Mol Biotechnol 41(3), 286–295. [DOI] [PubMed] [Google Scholar]
- Eynard N, Rols MP, Ganeva V, Galutzov B, SabrI N & Teissie J (1997). Electrotransformation pathways of procaryotic and eucaryotic cells: recent developments. Bioelectrochemistry and Bioenergetics 44(1), 103–110. [Google Scholar]
- Golzio M, Teissie J & Rols MP (2002). Direct visualization at the single-cell level of electrically mediated gene delivery. Proc Natl Acad Sci U S A 99(3), 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG & Nichols BJ (2009). Molecular mechanisms of clathrin-independent endocytosis. J Cell Sci 122(Pt 11), 1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller LC & Heller R (2006). In vivo electroporation for gene therapy. Hum Gene Ther 17(9), 890–897. [DOI] [PubMed] [Google Scholar]
- Heller R & Heller LC (2015). Gene electrotransfer clinical trials. Adv Genet 89, 235–262. [DOI] [PubMed] [Google Scholar]
- Henshaw J, Mossop B & Yuan F (2008). Relaxin treatment of solid tumors: effects on electric field-mediated gene delivery. Mol Cancer Ther 7(8), 2566–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshaw J, Mossop B & Yuan F (2011). Enhancement of electric field-mediated gene delivery through pretreatment of tumors with a hyperosmotic mannitol solution. Cancer Gene Ther 18(1), 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshaw JW & Yuan F (2008). Field distribution and DNA transport in solid tumors during electric field-mediated gene delivery. J Pharm Sci 97(2), 691–711. [DOI] [PubMed] [Google Scholar]
- Howes MT, Kirkham M, Riches J, Cortese K, Walser PJ, Simpson F, Hill MM, Jones A, Lundmark R, Lindsay MR, Hernandez-Deviez DJ, Hadzic G, McCluskey A, Bashir R, Liu L, Pilch P, McMahon H, Robinson PJ, Hancock JF, Mayor S & Parton RG (2010). Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J Cell Biol 190(4), 675–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova NI, Tsoneva I & Neumann E (1997). Sphingosine-mediated electroporative DNA transfer through lipid bilayers. FEBS Lett 415(1), 81–86. [DOI] [PubMed] [Google Scholar]
- Hunt MA, Currie MJ, Robinson BA & Dachs GU (2010). Optimizing transfection of primary human umbilical vein endothelial cells using commercially available chemical transfection reagents. J Biomol Tech 21(2), 66–72. [PMC free article] [PubMed] [Google Scholar]
- Huotari J & Helenius A (2011). Endosome maturation. EMBO J 30(17), 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Gao B, Prasad K, Greene LE & EisenBerg E (2000). Hsc70 chaperones clathrin and primes it to interact with vesicle membranes. J Biol Chem 275(12), 84398447. [DOI] [PubMed] [Google Scholar]
- Jordan ET, Collins M, Terefe J, Ugozzoli L & Rubio T (2008). Optimizing electroporation conditions in primary and other difficult-to-transfect cells. J Biomol Tech 19(5), 328–334. [PMC free article] [PubMed] [Google Scholar]
- Klenchin VA, Sukharev SI, Serov SM, Chernomordik LV & Chizmadzhev Yu A (1991). Electrically induced DNA uptake by cells is a fast process involving DNA electrophoresis. Biophys J 60(4), 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotnik T, Pucihar G, Rebersek M, Miklavcic D & Mir LM (2003). Role of pulse shape in cell membrane electropermeabilization. Biochim Biophys Acta 1614(2), 193–200. [DOI] [PubMed] [Google Scholar]
- KRIJNSE Locker J & Schmid SL(2013)._Integrated electron microscopy: suger-duper resolution. PLoS Biol 11(8), e1001639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari S & Mayor S (2008). ARF1 is directly involved in dynamin-independent endocytosis. Nat Cell Biol 10(1), 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark R, Doherty GJ, Howes MT, Cortese K, Vallis Y, Parton RG & McMahon HT (2008). The GTPase-activating protein GRAF1 regulates the CLIC/GEEC endocytic pathway. Curr Biol 18(22), 1802–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao M, Wang L, Chang CC, Rothenberg KE, Huang J, Wang Y, Hoffman BD, LIton PB & Yuan F (2017). Involvement of a Raci-Dependent Macropinocytosis Pathway in Plasmid DNA Delivery by Electrotransfection. Mol Ther 25(3), 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massol RH, Boll W, Griffin AM & Kirchhausen T (2006). A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating. Proc Natl Acad Sci U S A 103(27), 10265–10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELKONYAN H, Sorg C & Klempt M (1996). Electroporation efficiency in mammalian cells is increased by dimethyl sulfoxide (DMSO). Nucleic Acids Res 24(21), 4356–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J, Schelhaas M & Helenius A (2010). Virus entry by endocytosis. Annu Rev Biochem 79, 803–833. [DOI] [PubMed] [Google Scholar]
- Michel MR, Elgizoli M, Koblet H & Kempf C (1988). Diffusion loading conditions determine recovery of protein synthesis in electroporated P3X63Ag8 cells. Experientia 44(3), 199–203. [DOI] [PubMed] [Google Scholar]
- Mir LM, Bureau MF, Gehl J, Rangara R, Rouy D, Caillaud JM, Delaere P, Branellec D, Schwartz B & Scherman D (1999). High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc Natl Acad Sci U S A 96(8), 4262–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Ghosh RN & Maxfield FR (1997). Endocytosis. Physiol Rev 77(3), 759–803. [DOI] [PubMed] [Google Scholar]
- Neu JC & Krassowska W (1999). Asymptotic model of electroporation. Physical Review E 59(3), 3471–3482. [Google Scholar]
- Neumann E, Schaefer-Ridder M, Wang Y & Hofschneider PH (1982). Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J 1(7), 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOnnenmacher M & WEBER T (2011). Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway. Cell Host Microbe 10(6), 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh P, Borgstrom P, Witkiewicz H, Li Y, Borgstrom BJ, Chrastina A, Iwata K, ZInn KR, Baldwin R, Testa JE & Schnitzer JE (2007). Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat Biotechnol 25(3), 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade GE & Bruns RR (1968). Structural modulations of plasmalemmal vesicles. J Cell Biol 37(3), 633–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG & Simons K (2007). The multiple faces of caveolae. Nat Rev Mol Cell Biol 8(3), 185–194. [DOI] [PubMed] [Google Scholar]
- Pathak RK, Merkle RK, Cummings RD, Goldstein JL, Brown MS & Anderson RG (1988). Immunocytochemical localization of mutant low density lipoprotein receptors that fail to reach the Golgi complex. J Cell Biol 106(6), 1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad K, Barouch W, Greene L & Eisenberg E (1993). A protein cofactor is required for uncoating of clathrin baskets by uncoating ATPase. J Biol Chem 268(32), 23758–23761. [PubMed] [Google Scholar]
- Romer W, Berland L, Chambon V, Gaus K, Windschiegl B, Tenza D, Aly MR, Fraisier V, Florent JC, PERRAIS D, Lamaze C, Raposo G, Steinem C, Sens P Bassereau P & Johannes L (2007). Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 450(7170), 670–675. [DOI] [PubMed] [Google Scholar]
- Rosazza C, Deschout H, Buntz a ., Braeckmans K, Rols MP & Zumbusch A. (2016). Endocytosis and Endosomal Trafficking of DNA After Gene Electrotransfer In Vitro. Mol Ther Nucleic Acids 5, e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothnie a. , Clarke AR, Kuzmic P, Cameron A & Smith CJ (2011). A sequential mechanism for clathrin cage disassembly by 70-kDa heat-shock cognate protein (Hsc70) and auxilin. Proc Natl Acad Sci U S A 108(17), 6927–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharanjak S, Sharma P, Parton RG & Mayor S (2002). GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell 2(4), 411–423. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Miner LH & Omelchenko N (2006). Preembedding Immunoelectron Microscopy: Applications for Studies of the Nervous System In Neuroanatomical Tract-Tracing 3, Zaborszky L, Wouterlood FG and Lanciego JL (Eds.), New York, NY: Springer. [Google Scholar]
- Shin JS, Gao Z & Abraham SN (2000). Involvement of cellular caveolae in bacterial entry into mast cells. Science 289(5480), 785–788. [DOI] [PubMed] [Google Scholar]
- Spassova M, Tsoneva I, Petrov AG, Petkova JI & Neumann E (1994). Dip patch clamp currents suggest electrodiffusive transport of the polyelectrolyte DNA through lipid bilayers. Biophys Chem 52(3), 267–274. [DOI] [PubMed] [Google Scholar]
- STan RV (2005). Structure of caveolae. Biochim Biophys Acta 1746(3), 334–348. [DOI] [PubMed] [Google Scholar]
- Sukharev SI, Klenchin VA, Serov SM, Chernomordik LV & Chizmadzhev Yu A (1992). Electroporation and electrophoretic DNA transfer into cells. The effect of DNA interaction with electropores. Biophys J 63(5), 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran SK, Quon MJ & Prasadarao NV (2002). Escherichia coli K1 internalization via caveolae requires caveolin-1 and protein kinase Calpha interaction in human brain microvascular endothelial cells. J Biol Chem 277(52), 50716–50724. [DOI] [PubMed] [Google Scholar]
- Tan X, Heureaux J & Liu AP (2015). Cell spreading area regulates clathrin-coated pit dynamics on micropatterned substrate. Integr Biol (Camb) 7(9), 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UEhara K & Miyoshi M (1999). Tubular invaginations with caveolae and coated pits in the sinus endothelial cells of the rat spleen. Histochem Cell Biol 112(5), 351–358. [DOI] [PubMed] [Google Scholar]
- van den Pol AN (1986). Tyrosine hydroxylase immunoreactive neurons throughout the hypothalamus receive glutamate decarboxylase immunoreactive synapses: a double pre-embedding immunocytochemical study with particulate silver and HRP. J Neurosci 6(3), 877–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M & Yuan F (2011). Membrane binding of plasmid DNA and endocytic pathways are involved in electrotransfection of mammalian cells. PLoS One 6(6), e20923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIe TD & TSONG TY (1993). Study of mechanisms of electric field-induced DNA transfection. V. Effects of DNA topology on surface binding, cell uptake, expression, and integration into host chromosomes of DNA in the mammalian cell. Biophys J 65(4), 1684–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaas DW, Duncan MJ, Li G, Wright JR & Abraham SN (2005). Pseudomonas invasion of type I pneumocytes is dependent on the expression and phosphorylation of caveolin-2. J Biol Chem 280(6), 4864–4872. [DOI] [PubMed] [Google Scholar]
- Zaharoff DA, Henshaw JW, Mossop B & Yuan F (2008). Mechanistic analysis of electroporation-induced cellular uptake of macromolecules. Exp Biol Med (Maywood) 233(1), 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeigerer A, Gilleron J, Bogorad RL, Marsico G, Nonaka H, Seifert S, Epstein- Barash H, Kuchimanchi S, Peng CG, Ruda VM, Del Conte-ZerIal P, Hengstler JG, Kalaidzidis Y, Koteliansky V & Zerial M (2012). Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 485(7399), 465–470. [DOI] [PubMed] [Google Scholar]