

Abstract
Autophagosome/amphisome-lysosome fusion is a highly regulated process at the protein, lipid and biochemical level. Each primary component of fusion, such as the core SNAREs, HOPS complex or physical positioning by microtubule-associated dynein motors, are regulated at multiple points to ensure optimum conditions for autophagic flux to proceed. With the complexity of the membrane fusion system, it is not difficult to imagine how autophagic flux defect-related disorders, such as Huntingtin’s disease, non-familial Alzheimer’s disease and Vici syndrome, develop. Each membrane fusion step is regulated at the protein, lipid and ion level. This review aims to discuss the recent developments toward understanding the regulation of autophagosome, amphisome, and lysosome fusion requirements for successful autophagic flux.
Keywords: Autophagy, autophagosome-lysosome fusion, STX17, SNAP29, HOPS complex, RAB7
From Phagophore to Autolysosome
Macroautophagy, hereafter referred to as autophagy, is a homeostatic process of degradation in the cell that clears protein aggregates, creates nutrients in times of deprivation and turns over damaged organelles. The process begins with a phagophore, or crescent-shaped double membrane [1,2], that is wrapped around targeted cargo until it is fully enveloped and the double membrane is closed to form an autophagosome [3]. Fusion of the autophagosome with an endosome or late endosome generates an amphisome. Amphisomes then fuse with lysosomes, forming an autolysosome, where contents can be recycled by lysosomal enzymes [3,4]. Each stage of this process, from the initiation of the phagophore to the degradation of the cargo in the autolysosome, is extensively regulated. Recently, a number of studies have identified new regulation of the fusion steps between the autophagosome/amphisome and the lysosome.
Prior to the last 5 years of research, the field understood that autophagic vesicle progression from autophagosome to autolysosome occurred, but discovery of the SNAREs coordinating the fusion in human and mouse cells propelled the field forward. It was known that HOPS complex components existed and played a role in fusion, but the mechanism had not been elucidated. Certain phosphoinositides, microtubule conditions and associated proteins related to autophagosome-lysosome fusion had been discovered, but not yet positioned in the larger picture. With an ever-increasing tool box of techniques to study this fusion step, it has become apparent that autophagosome-lysosome fusion requires a concert of proteins, lipids and ions to function optimally, all of which are discussed within. This review aims to discuss some of the more recent developments in autophagosome-lysosome fusion, to serve as a starting point for future medical research into the role of autophagy, and autophagic flux, in human disease.
STX17, SNAP29 and VAMP8
Syntaxin 17 (STX17), synaptosome associated protein, 29, (SNAP29) and vesicle associated membrane protein 8 (VAMP8) are the core three soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, discovered in 2012 in human cells and in 2013 in D. melanogaster [5,6]. These SNAREs are responsible for the physical fusion of the two opposing membranes of the lysosome and autophagosome/amphisome [6]. See Box 1 for a description of SNARE driven membrane fusion. These interactions require N-ethylmaleimide-sensitive factor (NSF) attachment protein, alpha (NAPA)-mediated SNARE priming (see glossary). NAPA is an adaptor protein for the enzyme NSF, which together initiate ATP hydrolysis and structural changes required for SNARE function [7]. Small amounts of NAPA are sufficient for partial rescue of the completion of the autophagy pathway by degradation of the autophagic cargo, also known as autophagic flux [7].
Box 1: How SNARE proteins mediate membrane fusion.
SNARE-domain containing proteins fall into broadly defining classifications depending on whether the SNARE protein contains a key glutamine (Q-SNAREs) or arginine (R-SNAREs) residue in the center of the SNARE motif. SNARE domains are structurally coiled-coil domains designed to interact with each other via SNARE zippering [90]. Q- and R-SNAREs function as v- or t-SNAREs based on whether they are associated with the incoming Vesicle membrane (typically the R-SNAREs) or with the Target compartment (Typically the Q-SNAREs). Four SNARE motifs are required for a SNARE bundle, sometimes referred to as a SNAREpin, 1 R-SNARE and 3 Q-SNAREs, subdivided as Qa, Qb, and Qc SNAREs based on peptide sequence. These motifs can be divided into 3 or 4 proteins, as the SNAP25-protein family members carry 2 Q-SNARE domains that provide Qbc SNARE domains for the interaction. Other involved protein families include the vesicle associated membrane proteins, or VAMP, family which is composed of R-SNAREs and the Syntaxin protein family which is composed of Q-SNAREs [91,92].
The general mechanism of action of the SNAREs is to pull 2 opposing membranes together for fusion to occur. The fusion-permissive SNARE bundle is formed by sequential binding stages with non-fusogenic intermediates. Typically, the Q-SNAREs first complex together on the target membrane to form a receptor for the incoming R-SNARE laden vesicle. In the case of the autophagosome-lysosome core SNARE proteins fusion, this would indicate a binding of STX17 to SNAP29, which would be required before the addition of VAMP8’s SNARE domain could be “zippered” into the SNARE bundle [90]. Once the Qabc SNARE bundle is formed, it can bring the R-SNARE into the complex. Though this complex now has the required components, it is not yet fusogenic. Calcium ions are thought to be required for permissive fusion (see Box 3). Clamping factors, such as complexin, hold SNARE bundles ready for permissive fusion conditions and are regulated by the calcium-activated synaptotagmin family proteins. Upon Ca2+ availability, synaptotagmin can remove the fusion clamp from the complex and allow rapid membrane fusion to occur. Synaptotagmin family members have not been yet implicated for a role in autophagic membrane fusion, though a few have been reported in functions in related to the lysosome [90–94].
STX17 is a Qa-SNARE (see Box 1) that contains two transmembrane domains which are necessary for correct localization and a LC3-interacting region (LIR). LC3 is a cytosolic protein (LC3-I) that belongs to the ATG8 protein family including LC3 proteins and GABARAP proteins. LC3 is lipidated upon autophagic induction, which is a response to numerous stressors in the cell, such as starvation. Lipidated LC3 (LC3-II), LC3 conjugated to a phosphatidylethanolamine, is bound to membranes and localizes to autophagosomes. LC3 is traditionally used to measure autophagy induction by either western blot, separating non-lipidated LC3-I from lipidated LC3-II, or by immunofluorescence, to observe the relocalization of LC3 into small punctate structures or foci in the cytoplasm. Immunogold electron microscopy revealed that STX17 localizes specifically to the outer membrane of the completed autophagosome [6]. Colocalization studies using LC3, LAMP1 and LBPA showed that STX17 is required for autophagosome to endosome or lysosome fusion events [6]. The localization of STX17 is regulated by LAMP2, LC3/GABARAP proteins, and IRGM [6,8,9]. Autophagic flux, can be measured by a tandem fluorescent-tagged LC3 (tf-LC3) of either mCherry-GFP-LC3 or RFP-GFP-LC3 that measures the acidification state of the LC3+ vesicles (see Box 2) [10]. LAMP2 knockout mouse embryonic fibroblasts (MEFs) exhibit reduced autophagic flux, measured by tf-LC3, while expression of the LAMP2a isoform was able to reconstitute normal autophagy. Loss of LAMP2 disrupts the process due to a mislocalization of STX17 [8]. IRGM binds to STX17 via its dual transmembrane domains. The silencing of IRGM disrupts the normal colocalization of STX17 with LC3, demonstrating a targeting role for IRGM [9].
Box 2: Tandem-fluorescent tagged LC3 as a tool for fluorescent microscopy.
LC3 lipidation and subsequent localization to autophagosomes is considered a hallmark of autophagic induction. LC3 can insert in either side of the autophagosome during initiation. Over the course of the autophagosome maturation, the interior of the autophagosome becomes acidified, by endosome or lysosome fusion, and degradative, upon lysosome fusion. In the degradative step, LC3 on the interior of the autophagosome is degraded by lysosomally-delivered enzymes. Using 2 different fluorophores, an acid-stable RFP or mCherry, and an acid-labile GFP, different autophagosome conditions can be quantified using fluorescent microscopy (Figure I). With either fluorophore set up conjugated to LC3, RFP-GFP or mCherry-GFP, autophagosomes that have not been acidified will overlay as yellow puncta while either acidified amphisomes or autolysosomes will appear only as red puncta. These constructs, originally developed in [10] and available on Addgene, have been used in the field to measure the effect of factors of interest on autophagic flux. The red:yellow puncta in varying conditions can give a researcher a quantification of the process.
SNAP29 is a promiscuous, non-lipid anchored Qbc-SNARE (see Box 1) that donates 2 coiled-coil domains to the forming SNARE bundle [11]. SNAP29 binds STX17 on the autophagosome and facilitates binding to VAMP8 [6]. SNAP29 knockout cells have showed significant accumulation of vesicles in D. melanogaster [12]. SNAP29-STX17 binding is inhibited by an O-GlcNAcylation on SNAP29 in nutrient-sufficient conditions. When O-GlcNAcylation levels decrease in starvation conditions, the result is a more stable STX17-SNAP29-VAMP8 complex [13]. The localization of SNAP29 is dependent on LAMP2, although this is hypothesized to be a secondary effect of the mislocalization of STX17 [8].
VAMP8 is the transmembrane R-SNARE (see Box 1) on the lysosome that binds STX17-SNAP29 [14]. VAMP8 has been shown to be important in pathogen-containing autophagic vacuoles, as knockdown of VAMP8 decreases the antimicrobial effects of the autophagy pathway [15]. VAMP8’s localization is not dependent on LAMP2 [8], but is dependent on the small GTPase RAB21 [16]. In starvation conditions, RAB21 requires activation by SBF2, a guanine nucleotide exchange factor for GTPases, to interact with VAMP8. However, neither RAB21 nor SBF2 affect lysosome quantity, acidification, or the endocytic degradation pathway [16].
The role of YKT6 as a newly described autophagosomal SNARE in autophagosome-lysosome fusion has recently been discovered in yeast, human cells and D. melanogaster [17–19]. It was shown in humans that STX17 depletion alone doesn’t inhibit fusion, but depletion of both STX17 and YKT6 simultaneously completely inhibits autophagosome-lysosome [17]. YKT6 was also found localized on the lysosome in starved fly larval cells [18]. Together, these suggest that YKT6 is a fourth SNARE required for autophagosome-lysosome fusion.
Other SNARE proteins have been reported to have a role in autophagosome/amphisome to lysosome fusion. Two groups have reported the SNARE VTI1B, whose interacting partners are STX6 and VAMP3, to have a role in lysosome fusion with either pathogen-containing autophagosomes or recycling endosomes [15,20]. Another study demonstrated that VTI1B localizes to late endosomes and lysosomes and plays no role in autophagic fusion [6]. These conflicting results may indicate differential regulation and binding partners depending on the type of autophagic stimulation. VAMP3 and VAMP7 have been shown to mediate fusion events between autophagosome to multivesicular bodies, for formation of amphisomes, and the fusion event for the formation of autolysosomes, respectively [21]. Recently, our group presented evidence that SNAP47, a protein with partial homology to SNAP29, has a role in autophagic flux [22]. SNAP47 has been shown to colocalize with ERGIC markers and facilitates the proper localization of VAMP4 and VAMP7 [23]. SNAP47’s role in autophagosome-lysosome fusion could be to coordinate a fusion that is either required within the autophagy pathway itself, or, to coordinate the localization of a required component of the pathway.
HOPS and other membrane tethers in autophagosome-lysosome fusion
Although SNARE proteins drive the fusion of membranes, the SNARE proteins alone do not provide efficient fusion and require other factors. Two major tether complexes exist for vesicle fusion within the cell, the homotypic fusion and protein sorting complex (HOPS) or class C core vacuole/endosome tethering complex (CORVET). CORVET has specificity for membranes that contain the RAB5 GTPase. HOPS is an evolutionarily conserved membrane tethering complex for membranes containing the RAB7 GTPase and has been shown to be required for efficient autophagosome-lysosome fusion [24–26]. RAB5 is the predominant GTPase on early autophagic and endosomal membranes, but is later replaced by RAB7 [27,28]. RAB5 has been shown dispensable for autophagosome-lysosome fusion [29]. This switch of GTPases from RAB5 to RAB7 is required for autophagosome-lysosome fusion [20]. The exchange may be coordinated by the localization of a GEF complex for RAB7, Mon1-CCZ1, which localizes to the autophagosome by binding to ATG8 in yeast and ATG8a in D. melanogaster [19,29]. The GEF complex then activates RAB7 on the autophagosome [19]. HOPS is a 6-membered complex made up of 4 core subunit proteins: VPS11, VPS16, VPS18, and VPS33A, and 2 supplementary proteins: VPS39 and VPS41 [30].
The complex interacts directly with STX17 to coordinate autophagic flux [31], and is required for proper core SNARE bundle assembly [32]. UVRAG increases autophagosome maturation by recruitment of the HOPS core complex to autophagosomes by increasing the RAB7 localization to autophagosomes [33]. BLOC-one-related complex (BORC) is an 8-membered complex that recruits ARL8, a small GTPase, and HOPS to lysosomal membranes to facilitate interactions with LC3 and STX17 that are required for fusion. Without BORC, fewer LC3+ vesicles acidify, as measured by tf-LC3, and interactions between VAMP8 and SNAP29 to STX17 decrease [34]. Other tethers related to this fusion event include ATG14 and BIRC6. ATG14 homooligomers interact with STX17 and SNAP29 on closed autophagosomes, to act as a tethering component of the autophagosome-lysosome fusion [35]. BIRC6, also known as BRUCE, is an E2/E3 ubiquitin conjugating enzyme localized to the lysosome that interacts with GABARAP proteins and STX17 to increase autophagosome-lysosome fusion [36].
Non-tethering Factors Associated with Autophagosome-Lysosome Fusion
Autophagosome-lysosome membrane fusion utilizes several other required proteins. The ATG8 proteins are a family of 6 proteins in mammals that encompass LC3A, LC3B, LC3C, GABARAP, GABARAPL1 and GABARAPL2. Surprisingly, when all 6 proteins are knocked out, STX17+ puncta are still able to be detected. These STX17+ vesicles no longer fuse with LAMP1+ vesicles in starvation conditions, indicating a role in fusion for those proteins [37]. STX17+ vesicles, described as “autophagosome-like structures,” are able to fuse with lysosomes under starvation conditions in ATG conjugation-deficient cells [38]. These authors suggest this discovery helps resolve why many autophagy-related protein knockout mice are embryonically lethal, whereas elimination of the ATG8 family proteins or their lipidation process enzymes are able to survive for a brief period after birth [38]. More recently, another group used GABARAP CRISPR knockout cells to show a role exists specifically for the GABARAP proteins in autophagic flux [39]. One role for GABARAP is in phosphoinositide kinase recruitment to autophagosomes, as described below in “Lipids in Autophagosome-Lysosome fusion.”`
As briefly mentioned previously, RAB7 colocalizes with autophagosome markers and is required for autophagic flux [27–29]. The conversion of RAB5+ membranes, from early in autophagy or the endocytic cycle, to RAB7+ membranes is important for normal maturation of vesicles to autolysosomes [27]. RAB7-GTP presence on the autophagosome, which can be induced by rapamycin treatment, is required for autophagosome-lysosome fusion [27]. Treatment of cells with Vacuolin-1 inhibits RAB7− mediated fusion of the autophagosome with the lysosome by activating RAB5a [40]. Regulation of RAB7 localization by UVRAG increases the colocalization of RAB7 with LC3+ vesicles [33]. RAB7’s membrane localization is itself regulated by prenylation. It was recently discovered that N6-isopentenyladenosine (iPA), which is a cellular product of the isoprenoid synthesis pathway, induces autophagy through PRKAA1 signaling and inhibition of the MTOR pathway and inhibits autophagic flux by decreasing the prenylation of RAB7 [41]. EPG5 uses RAB7 to localize to late endosomes, amphisomes (upon autophagy induction) and lysosomes. EPG5 binds STX17-SNAP29 to enhance the STX17-SNAP29-VAMP8 complex formation, determined by increased interactions via co-immunoprecipitation. Interestingly, silencing of EPG5 changes the SNAP protein binding partner preference for STX17 from SNAP29 to SNAP25 in HeLa cells [28], which could suggest a role in endosome-autophagosome fusion. Other RABs reported to influence autophagic flux include RAB2 in D. melanogaster [42,43], and RAB33B [44]. RAB33B is regulated by TBC1D25, previously known as OATL1. TBC1D25 is a GTPase activating protein (commonly known as a GAP) for RAB33B, which interacts with LC3 and GABARAPs and colocalizes with LC3+ puncta. Overexpression of TBC1D25 pushes the equilibrium of the GTPase RAB33B from a GTP-bound to GDP-bound protein and inhibits autophagic flux [44]. Based on the model established for RAB7, the GDP-bound form of related GTPases should be inhibitory for fusion [27].
UVRAG, mentioned above as a protein that helps recruit RAB7, and therefore the HOPS complex, to the autophagosome, also forms a complex with beclin 1 (BECN1) and phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), which is a PI(3)P kinase. UVRAG is regulated through phosphorylation by the MTORC1 complex, a negative regulator of autophagic signaling [45]. During nutrient-sufficient conditions, phosphorylated UVRAG associates with RUBCN, a protein that negatively regulates autophagy at multiple stages [46,47]. This interaction decreases the amount of UVRAG that associates with the HOPS complex and therefore, decreases autophagic flux, while inhibition of MTORC1’s phosphorylation on UVRAG restores flux [45]. RUBCNL, previously known as Pacer, is a poorly-characterized protein which colocalizes with LC3 and increases the interaction of UVRAG to LC3. Competition for binding to UVRAG by RUBCNL dissociates RUBCN in vitro, and in vivo, creating a UVRAG-BECN1-PIK3C3. This complex allows autophagosome site-specific phosphorylation activity to create phosphoinositide-3-phosphate, which benefits autophagosome-lysosome fusion [48].
Other proteins thought to promote autophagosome-lysosome fusion include Presenilin 1 (PSEN1), PLEKHM1, Mahogunin (MGRN1), HSPB8, thioredoxin-interacting protein (TXNIP), and transglutaminase 2 (TGM2). PSEN1 is a gamma secretase known to contain mutations found in inherited Alzheimer’s disease patients [49]. Mutant forms of PSEN1 unable to be phosphorylated on S367 increase the number of incompletely fused autophagosome-lysosome pairs in mouse brains, visualized by electron microscopy. Upon phosphorylation at S367, PSEN1 interacts with Annexin A2 (ANXA2), a calcium regulated membrane protein of the Annexin family, which is primarily responsible for crosslinking the plasma membrane to the cytoskeleton. This interaction allows ANXA2 to bind to VAMP8. The ANXA2-VAMP8 interaction increases VAMP8-STX17 interactions, which increases autophagic flux [49]. PLEKHM1 is an adaptor protein that regulates fusion by interaction with HOPS and the ATG8 family proteins to coordinate tethering for both the autophagosome-lysosome and the endosome-lysosome fusion pathway [50]. Knockdown of PLEKHM1 has recently been shown to inhibit the autophagosome-lysosome, as well as the endosome-lysosome, fusion pathway [51]. MGRN1, a cytosolic E3 ubiquitin ligase associated with deficient lysosomal-storage neurodegenerative disorders, is required for flux, as a knockdown of the protein increases the size and number of LC3+ vesicles, but decreases autophagosome-lysosome fusion events. MGRN1 acts by ubiquitinating TSG101, an ESCRT-I complex member, and ubiquitinated TSG101 can rescue blocked autophagic flux either by its overexpression or monoubiquitination [52]. HSPB8 functions in a complex with the nucleotide exchange factor BAG3, and together recognize misfolded proteins to initiate misfolded protein clearance through autophagy [53]. In neuroblastoma cells treated with high glucose (33mM), a treatment used in some cell types to induce signaling that leads to autophagy, knockdown of HSPB8 blocks acidification of the autophagosome while ectopic expression causes a decrease in SQSTM1 levels, indicating an increase in autophagic flux. While a specific mechanism of action for HSPB8 is yet unknown, a link to autophagic flux is clear [54]. TXNIP is a thio-oxidoreductase that functions to regulate metabolic state, as well as ER stress. TXNIP also inhibits flux, according to measurement of total LC3 and SQSTM1 in high glucose-induced autophagic conditions [55]. TGM2 has also been reported as a positive regulator for autophagic flux, but the mechanism of action is not yet known [56]. In summary, there are many protein factors that play a regulatory part, both positively and negatively, in autophagosome-lysosome fusion.
Lipids in Autophagosome-Lysosome fusion
Cholesterol regulates autophagic flux through localization to the autophagosome. This localization to the autophagosome is regulated by MYO1C, a class I myosin that has been previously implicated in the transport of lipid rafts from storage compartments within the cell to the plasma membrane. Knockdown of MYO1C decreases autophagic flux. This knockdown also affects the intracellular distribution of cholesterol, as visualized by filipin staining. It has been suggested that the defect in fusion is caused by the loss of cholesterol trafficking to autophagosomes [57].
One study looked at the effect cholesterol plays at the autophagosome. Oxysterol binding protein like 1A (OSBPL1A), previously known as ORP1L, is a RAB7 target that stabilizes RAB7-GTP on the membrane and interacts with cholesterol via its ORD domain. This domain is a core lipid binding domain found in oxysterol binding proteins and, for OSBPL1A, is required for autophagic flux. The cholesterol-ORD interaction facilitates the recruitment of PLEKHM1 and HOPS to RAB7+ membranes. In the absence of cholesterol, another domain of OSBPL1A, FFAT (named for having 2 phenylalanine residues in an acidic tract), interacts with the VAPA protein on the ER. VAPA is a vesicle associated membrane protein found on many organelles within the cell, as well as the plasma membrane [58]. This VAPA-FFAT interaction creates contact sites from the ER to the autophagosome, and this is hypothesized to inhibit the targeting of membrane tethers necessary for fusion, demonstrating the importance of cholesterol in autophagosome-lysosome fusion. The OSBPL1A-RAB7 oligomer can also bind to RILP, a lysosomal protein that interacts with dynactin for retrograde transport of lysosomes. This interaction can regulate the positioning of autophagosomes to the perinuclear region and regulate the acquisition of HOPS complex tethering [58].
Specific phosphorylated phosphoinositides are required for autophagic flux. While PI(3)P is an important lipid early in autophagy [59], PI(3,5)P2 is important for autophagic flux and colocalization of SQSTM1 and LAMP2 in mice [60]. Another study shows a correct balance of PI(3,5)P2:PI(3)P is required on the autophagosome for autophagic flux. INPP5E is a phosphatase that converts PI(3,5)P2 back into PI(3)P, and is required for fusion to occur. Absence of INPP5E causes decreased colocalization of LC3 and LAMP1 and decreased levels of activated cortactin on the lysosomes. Cortactin acts to stabilize the lysosome on actin filaments for fusion to autophagosomes/amphisomes to occur [61]. Together, these two studies suggest there is an ideal middle ground for the ratio of phospholipids at the stage of autophagosome-lysosomal flux.
PI(4)P and PI4K2A have been described to play a role in autophagosome-lysosome fusion. PI(4)P lipids have been found on LC3+ autophagosomes and are phosphorylated by one of four isoforms of phosphoinositide-4 kinases [62]. PI4K2A is a phosphoinositide kinase isoform that has been associated with a role in autophagy. PI4K2A localizes to membranes due to a post-translational palmitoylation, which is required for this kinase’s localization to autophagosomes [63]. The localization of the kinase to autophagosomes can be induced by starvation, and depleted or kinase-dead PI4K2A yields larger autophagosomes with less PI(4)P and decreased autophagosome acidification [62]. The presence of GABARAPs on autophagosomes directs association of PI4K2A with these membranes [62]. This newly described GABARAP-PI4K2A-PI(4)P relationship adds another level of understanding and grounds for future questions for the role of lipids in autophagic flux.
Cytoskeleton and Autophagosome-Lysosome Fusion
Cytoskeletal cell components, microtubules and actin filaments, are protein polymers that are also considered to have a role in autophagosome-lysosome fusion. Microtubules support production of autophagosomes and while intact microtubules are not strictly required for autophagosome fusion to lysosomes, destabilizing microtubules causes a decrease in the percentage of fusion events observed [64]. Acetylation of microtubules, a reversible modification which promotes anterograde traffic, enhances autophagosome-lysosome fusion [65–67]. This was shown by differentially destabilizing either acetylated or non-acetylated microtubules. Vinblastine destabilizes all microtubules, whereas nocodazole destabilizes primarily non-acetylated microtubules. Vinblastine-treated cells develop an accumulation of LC3+ puncta that do not acidify, and this is not observed in nocodazole-treated cells [66].
Microtubules are used by molecular motors to move cargo about the cell and dynein-mediated retrograde transport of autophagosomes increases autolysosome formation [68]. Dynein is acquired by the retroactively moving vesicles through fusion with late endosomes, while blocking the endosome-autophagosome fusion abrogates retrograde autophagic vesicle movement [69]. Dynactin 1 (DCTN1) a key protein for dynein-mediated transport of vesicles, mediates the colocalization of LC3+ and LAMP1+ vesicles. DCTN1 protein levels are upregulated by TAR DNA binding protein (TARDBP) and silencing of TARDBP inhibits autophagosome-lysosome fusion. The action of TARDBP on DCTN1 supports DCTN1’s role in retrograde transport of autophagic vesicles for autophagosome-lysosome fusion [70]. TARDBP also regulates the translocation of TFEB to the nucleus by modulating mRNA expression for an MTORC1 component, RPTOR. Inhibition of this translocation, such as by iron, inhibits autophagic flux [71]. Tubulin polymerization promoting protein (TPPP) helps tubulin form microtubules and also stabilizes microtubule bundles for increased integrity. TPPP inhibits autophagosome-lysosome fusion, shown by tf-LC3 microscopy [72]. These results could suggest microtubule size or number must be balanced appropriately for successful completion of the autophagy pathway.
In addition to MYO1C, which was mentioned above as a molecule that traffics cholesterol to the autophagosome-lysosome fusion membranes, another myosin family motor, MYO6, has been implicated for a role in autophagosome-lysosome fusion. MYO6 colocalizes with LC3, is required for clearance of both Huntingtin protein aggregates and SQSTM1 puncta, and has autophagic protein binding partners: T6BP, NDP52 and optineurin (OPTN). The endosomal protein TOM1 interacts with MYO6, and loss of TOM1 inhibits autophagosome-lysosome fusion. These data suggest that MYO6 is involved with tethering TOM1+ endosomes to autophagosomes to promote amphisome formation [73].
Actin filaments also play a role in promoting autophagosome-lysosome fusion. As previously mentioned, the percentage of PI(3)P and PI(3,5)P2 regulates cortactin’s presence on the lysosome. Cortactin’s role in autophagosome-lysosome fusion was discovered because the role of F-actin in fusion had been previously shown. F-actin was first suggested to be important for basal autophagy because mice unable to recruit cortactin, due to a tubulin specific deacetylase HDAC6 knockout, have an autophagic flux defect [74]. HDAC6 is required to clear ubiquitinated and Huntingtin protein aggregates, as well as regulate lysosome positioning near the microtubule organizing centers [74,75]. HDAC6 recruits cortactin to organize F-actin, and this is required for fusion. It is interesting to note that under concurrent glucose, amino acid and serum starvation, cortactin and HDAC6 are not required for autophagic induction [74]. Taken together, these data suggest that actin filaments appear to promote autophagosome-lysosome fusion, but may not be required in all autophagic signaling conditions.
Ion regulation and Autophagosome-Lysosome Fusion
Calcium ions are potent regulators of autophagic flux and are required for the assembly of the fusion SNARE complex. Inhibitors of calcium release from the endoplasmic reticulum (ER), such as thapsigargin, inhibit autophagic flux. This suggests that Ca2+ is required for autophagosome-lysosome fusion [76]. Bafilomycin A1, an inhibitor of V-ATPase-mediated acidification in the cell, has a separate but related role as a sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor and therefore blocks autophagosome-lysosome fusion [77]. The inhibition of SERCA can lead to high levels of cytosolic calcium, which then inhibits autophagosome-lysosome fusion, rather than the inhibition of lysosomal acidification as previously thought [78]. Heavy metal ions, such as ethylmercury and cadmium, have been shown to disrupt autophagic flux. This is triggered by an increase of reactive oxygen species in the cell, which increases ER stress. This, in turn, dysregulates the calcium ion control on the cytosol, causing a calcium ion overload that inhibits autophagosome-lysosome fusion [79,80]. Specific lipid molecule accumulations, such as oleic and palmitate fatty acids from high-fat diet fed rats also create a block of autophagosome-lysosome fusion that stems from ER stress [81,82]; and may feed into this ER stress response/calcium dysfunction pathway.
The ER is not the only source of the calcium ions required for autophagosome-lysosome fusion as calcium is also held in lysosomes [83]. CACNA1A, a calcium-selective voltage-gated channel (see Box 3) found on lysosomes and the plasma membrane, has been demonstrated as being required for autophagosome-lysosome fusion, as knockouts of this channel decrease the colocalization of LC3+ to LAMP1+ puncta. To differentiate which population of CACNA1A channels were responsible for fusion, a non-cell permeable and a general cell-permeable calcium channel blocker was used to exclude the CACNA1A channels that are localized to the plasma membrane. This suggests that the population of CACNA1A on the lysosomes is required for autophagosome-lysosome fusion [84].
Box 3: Voltage-gated channels implicated in autophagosome-lysosome fusion.
Levels of ions, even at a local level, are responsible for the function of several classes of proteins in the cell. Ions are required for structural assembly of proteins, activation or inhibition signals of protein, and can change the rate of exocytosis (which is particularly studied in the case of neurotransmitter exocytosis from neurons). Voltage-gated ion pumps, such as CACNA1A, open and close the channel pore based on signals from surrounding potentials [95]. Specifically, as a voltage-gated channel, CACNA1A requires a change in membrane potential to open for localized efflux of Ca2+ ions, which increases membrane fusion [95]. There has not been a strict connection in the autophagy field as to which ion pumps can regulate a local membrane potential during fusion of the autophagosome to the lysosome. There are, however, two related channels that have been implicated as being regulated by MTOR (a negative regulator kinase for autophagy) in a nutrient-sensitive manner which could be involved in the fusion.
Two pore channel 1 and 2 (TPC1 and TPC2) are Na+-selective, voltage-gated channels on the lysosome that are PI(3,5)P2-activated and are inhibited by MTOR binding, until amino acid or ATP deprivation causes MTOR dissociation. Once active, the channels pump out Na+ ions from the lysosome to the local cytosol, creating a negatively charged lumen; this facilitates an influx of H+ pumping by v-ATPases, and therefore, lysosomal acidification, for cargo degradation and nutrient generation [96–98]. While not formally linked in the literature, these results suggest that the localized calcium efflux required for fusion may have additional regulation from the lipid composition and autophagic induction signals that regulate MTOR inhibition.
Concluding Remarks and Future Directions
Autophagy is a tightly regulated cytosolic process that is required for homeostatic cell maintenance, including organelle turnover and protein aggregate clearance, as well as nutrient generation upon induction of starvation signals. Here we have outlined three major regulatory components: proteins, including SNAREs and the HOPS complex; lipids, including cholesterol and specific phospholipids; and ions, such as calcium. A schematic of the web of regulations on these membrane fusion events has been summarized in Figure 1, Key Figure. This is a highly dynamic field of inquiry, and while many of these studies are rather recent, many questions remain and have been outlined in the Outstanding Questions.
Figure 1, Key Figure: A model of autophagosome-lysosome fusion requirements.
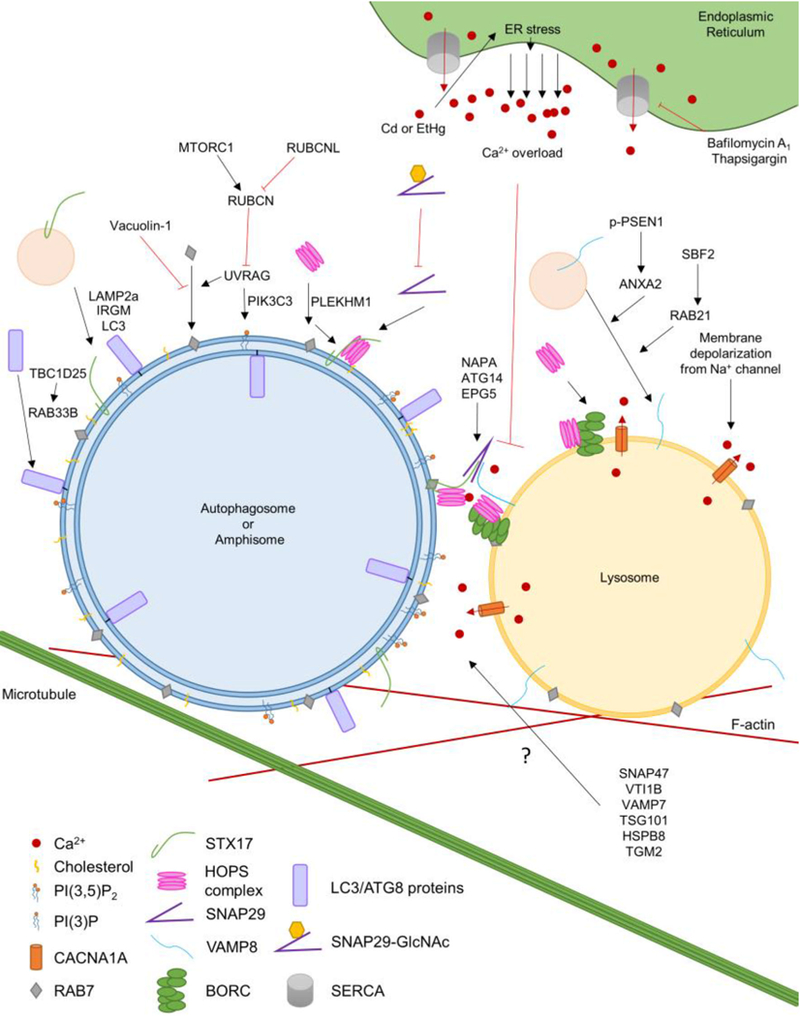
The model presented in the figure illustrates a summary of the regulation steps of autophagosome/amphisome to lysosome fusion. The core SNARE proteins STX17, SNAP29, and VAMP8 are the main proteins responsible for this membrane fusion, while the SNAREs SNAP47, VTI1B, and VAMP7 also have implicated roles. SNAP29’s GlcNAc modification is inhibitory to its role in fusion, and must be removed for efficient fusion to occur. Tethering factors, such as ATG14, the HOPS complex, the BORC complex, EPG5, PLEKHM1, and NAPA, have been shown to increase the efficiency of the fusion mediated by the core SNARE proteins. Other associated proteins have been shown to demonstrate a role in autophagosome-lysosome fusion. TBC1D25 and RAB33B help ATG8 family proteins localize to the autophagosome. LAMP2, IRGM and LC3 together help target STX17 to the autophagosome. UVRAG promotes the recruitment of RAB7 to autophagosomes, and UVRAG can be negatively regulated by RUBCN. RUBCN itself is a molecular switch with positive regulation from MTORC1 and negative regulation from RUBCNL. VAMP8’s localization to the lysosome is dependent on ANXA2, activated by phosphorylated PSEN1, and RAB21, which is activated by SBF2. A balance between PI(3)P, PI(3,5)P2 and cholesterol has been shown to be essential for fusion, as well as, trafficking of fusion-coordinating proteins. Calcium ions are necessary for fusion. Sodium mediated depolarization of the lysosome membrane can cause the release of calcium ions from the lysosome lumen. An additional source of calcium ions can come from ER storage. ER Ca2+ release can occur in either a regulated fashion, through ion pumps like SERCA, or uncontrollably due to ER stress, which can be caused by certain metals such as cadmium or ethylmercury. Uncontrolled Ca2+ can lead to a calcium ion overload which inhibits autophagosome-lysosome fusion. Cytoskeleton components, such as microtubules and actin filaments, provide the trafficking and support for autophagosome-lysosome fusion. The arrow with a question mark describes the participation of proteins that have been implicated in autophagosome-lysosome fusion, but whose specific mechanisms of actions are not yet known.
A priority for the autophagic flux field of study would be to elucidate where the amphisome stage is being affected. Many studies currently describe autophagosome-lysosome fusion, but use tools that are unable to identify what percentage of the data generated include amphisome formation, the fusion between the autophagosome and endosome, or the fusion of the amphisome to the lysosome. Despite evidence of the importance of this middle step in the pathway, it is largely overlooked. The tfLC3 autophagic flux tool is often used in the literature to show autolysosome formation by measure of acidification. However, amphisomes, which are acidified compartments, cannot be distinguished from autolysosomes through use of the tfLC3 construct alone. Understanding the base requirements for autophagosome-lysosome fusion will require studies into the specific relationship of YKT6 with the other 3 core SNARE proteins, to enhance our knowledge of SNARE bundles outside of 4 coiled-coil domains. It would benefit the field’s understanding to have more data to describe the balance of microtubules that are required for fusion, as there are multiple works described here that have opposing views on what the proper balance of number or modifications is for permissive fusion.
Small cellular misregulations can alter the balance of proteins, lipids, or ions with the potential to cause an autophagosome-lysosome fusion defect that could have serious repercussions at the organism level. Lysosomal storage diseases, such as Alzheimer’s disease, Huntingtin’s disease and Parkinson’s disease, are strongly correlated to a defect in autophagy, reviewed in [85]. Additionally, redirecting autophagy away from the lysosome is a common strategy for intracellular pathogens [22,51,86–89]. Understanding how autophagosome-lysosome fusion can stall if the necessary components don’t find a “middle ground” is of high importance in the medical research community trying to clarify underlying causes of such diseases.
Figure I: The tandem fluorescent-tagged LC3 construct is a tool to study autophagy.
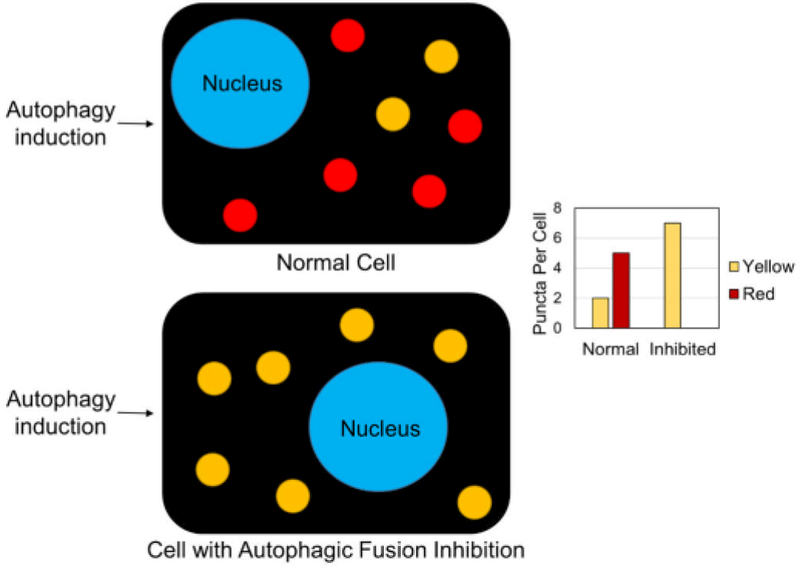
Acidification of the autophagosomes can occur by fusion between the autophagosome and an endosome or lysosome containing membrane-bound vacuolar ATPases that have the ability to acidify the lumen of the vesicle. In a cell transfected with either eGFP-RFP-LC3 or eGFP-mCherry-LC3, autophagic vesicles either will fluoresce green and red (appearing as yellow when images are overlaid) or only red for whether the vesicle is non-acidified or acidified, respectively. The ratio between red and yellow vesicles can describe the overall state of autophagy in a cell. When autophagy is high in a cell, it can be expected that there would be more red vesicles than yellow vesicles, as shown in the top cell. If autophagic flux is inhibited, it can be expected that there would be more yellow vesicles than red vesicles, as described by the bottom cell in the schematic. This important tool is now used broadly in the autophagy field to describe the state of autophagosome acidification in a cell.
Highlights:
Protein regulation of autophagosome-lysosome fusion includes 3 core SNARE proteins, YKT6, the HOPS complex, BORC complex, small GTPases such as RAB7 and RAB33, and molecular motors for vesicle transport.
Lipid regulation of autophagosome-lysosome fusion requires a balance of specific phosphoinositides and cholesterol for protein and vesicle localization.
Cationic regulation of autophagosome-lysosome fusion relies on local cytosolic levels of calcium ions, potentially regulated by sodium ion release from the lysosome.
Actin and microtubule filaments of the cytoskeleton network are required for autophagosome-lysosome fusion.
Outstanding Questions Box.
At what stage do the non-core SNARE proteins, such as SNAP47, affect autophagic flux? Does involvement of these also depend on the type of autophagy-inducing stimulus?
How does the newly described autophagosomal SNARE, YKT6, factor into the core SNARE bundle of SNAP29, STX17 and VAMP8 during autophagosome-lysosome fusion?
How is the O-GIcNAcylation modification of SNAP29 removed, and how is that process regulated by the metabolic state of the cell?
The intermediary structure known as an amphisome, formed from autophagosome-endosome fusion, is often not considered in studies relating to autophagic flux. To what degree is this structure formed and required for this process to occur?
How are cytoskeletal pathways in the cell regulated to be both sufficient and also non-inhibitory for autophagic flux?
Do the two-pore channels 1 and 2 (TPC1 and TPC2) contribute to autophagosome-lysosome fusion?
How is the web of fusion proteins and lipids regulated in the face of different types of autophagy-inducing stimuli?
Acknowledgements
This work was funded by NIAID grant AI104928 to William T. Jackson. Abigail K. Corona is supported by NIAID training grant T32AI095190 (PI: Vogel). We also thank Angel F. Corona Velazquez for discussions and manuscript edits prior to publication.
Glossary
- ATG14
Autophagy related 14, plays a role in autophagosome formation and interacts with autophagosome fusion SNAREs
- BLOC
Biogenesis of lysosome-related organelles complex 1, required for endosomal-lysosomal vesicle generation
- EPG5
Ectopic P-granules autophagy protein 5 homolog, mutations in this protein are known to be associated with Vici syndrome
- ERGIC
Endoplasmic reticulum-Golgi intermediate compartment, regulates trafficking between ER and Golgi
- ESCRT-I
Endosomal sorting complexes required for transport I, a complex of four proteins that aid in vesicle formation, vesicle abscission and recruitment of next-step ESCRT complexes
- Filipin staining
A chemical stain used to fluorescently label cholesterol in cells
- IRGM
Immunity related GTPase M, role in innate immune response hypothesized to be linked to its role in autophagy
- LAMP1
Lysosomal associated membrane protein 1, found on the endo-lysosomal pathway
- LAMP2
Lysosomal associated membrane protein 2, found on the endo-lysosomal pathway
- LBPA
Lysobisphosphatidic acid, a lipid found in late endosomes
- SBF2
SET binding factor 2, previously known as myotubularin-related protein 13 (MTMR13), phosphoinositide phosphatase with activity against PI(3)P and PI(3,5)P2
- Multivesicular bodies
Late endosomes with internal membrane-bound vesicles
- NDP52
CALCOCO2, calcium binding and coiled coil domain 2, receptor in autophagy for ubiquitinated substrates
- O-GlcNAcylation
Carbohydrate post-translational protein modification that responds to nutrient and stress conditions of the cell
- Optineurin
Autophagy receptor for ubiquitinated substrates
- Palmitoylation
The covalent addition of a palmitic acid to a protein, typically as a lipid moiety to anchor the protein to a membrane
- PI(3)P
Phosphoinositide with one phosphorylation on the 3 carbon of the inositol ring
- PLEKHM1
Pleckstrin homology domain-containing family M member 1, adaptor protein in lysosome fusion
- Prenylation
The addition of a prenyl group to a protein, typically as a lipid moiety to anchor the protein to a membrane
- RAB5
Ras-related protein Rab 5a, small GTPase
- RAB7
Ras-related protein Rab 7a, key component of endocytic pathway, small GTPase
- RAB21
Ras-related protein Rab 21, small GTPase
- RPTOR
Regulatory-associated component of the MTOR Complex 1
- SNARE priming
Partial assembly of fusion complexes, hypothesized to allow quick responses to stimuli
- SQSTM1
Sequestosome 1, also known as p62, autophagy receptor for ubiquitinated substrates
- STX6
Syntaxin 6, intracellular trafficking
- T6BP
Tax1 binding protein 1, also known as TAX1BP
- TFEB
Transcription factor EB, transcription factor associated with autophagy and the MTOR signaling pathway
- Thapsigargin
Inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA)
- Tom1
Target of Myb1 membrane trafficking protein
- UVRAG
UV Radiation resistance associated, activates BECN1-PIK3C3 complex in response to autophagy initiation signaling
- VAMP3
Vesicle-associated membrane protein 3, involved in late endosomes and Golgi traffic
- VAMP4
Vesicle-associated membrane protein 4, intracellular trafficking
- VTI1B
Vesicle-transport through interaction with t-SNAREs homolog 1B
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors declare that they have no conflicts of interest.
References
- 1.Carlsson SR and Simonsen A (2015) Membrane dynamics in autophagosome biogenesis. J Cell Sci 128, 193–205 [DOI] [PubMed] [Google Scholar]
- 2.Rubinsztein DC et al. (2012) Mechanisms of Autophagosome Biogenesis. Current Biology 22, R29–R34 [DOI] [PubMed] [Google Scholar]
- 3.Kawabata T and Yoshimori T (2016) Beyond starvation: An update on the autophagic machinery and its functions. Journal of Molecular and Cellular Cardiology 95, 2–10 [DOI] [PubMed] [Google Scholar]
- 4.Berg TO et al. (1998) Isolation and Characterization of Rat Liver Amphisomes EVIDENCE FOR FUSION OF AUTOPHAGOSOMES WITH BOTH EARLY AND LATE ENDOSOMES. Journal of Biological Chemistry 273, 21883–21892 [DOI] [PubMed] [Google Scholar]
- 5.Takáts S et al. (2013) Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol 201, 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itakura E et al. (2012) The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes. Cell 151, 1256–1269 [DOI] [PubMed] [Google Scholar]
- 7.Abada A et al. (2017) SNARE priming is essential for maturation of autophagosomes but not for their formation. Proceedings of the National Academy of Sciences 114, 12749–12754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubert V et al. (2016) LAMP-2 is required for incorporating syntaxin-17 into autophagosomes and for their fusion with lysosomes. Biology Open 5, 1516–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S et al. (2018) Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. The Journal of Cell Biology DOI: 10.1083/jcb.201708039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura S et al. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 [DOI] [PubMed] [Google Scholar]
- 11.Hohenstein AC and Roche PA (2001) SNAP-29 Is a Promiscuous Syntaxin-Binding SNARE. Biochemical and Biophysical Research Communications 285, 167–171 [DOI] [PubMed] [Google Scholar]
- 12.Morelli E et al. (2014) Multiple functions of the SNARE protein Snap29 in autophagy, endocytic, and exocytic trafficking during epithelial formation in Drosophila. Autophagy 10, 2251–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo B et al. (2014) O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nature Cell Biology 16, 1215–1226 [DOI] [PubMed] [Google Scholar]
- 14.Wong SH et al. (1998) Endobrevin, a novel synaptobrevin/VAMP-like protein preferentially associated with the early endosome. Molecular biology of the cell 9, 1549–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuta N et al. (2010) Combinational soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins VAMP8 and Vti1b mediate fusion of antimicrobial and canonical autophagosomes with lysosomes. Molecular biology of the cell 21, 1001–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jean S et al. (2015) Starvation-induced MTMR13 and RAB21 activity regulates VAMP8 to promote autophagosome-lysosome fusion. EMBO reports 16, 297–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsui T et al. (2018) Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol DOI: 10.1083/jcb.201712058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takáts S et al. (2018) Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS Genet 14, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J et al. Molecular mechanism to target the endosomal Mon1-Ccz1 GEF complex to the pre-autophagosomal structure. eLife 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nozawa T et al. (2017) The STX6-VTI1B-VAMP3 complex facilitates xenophagy by regulating the fusion between recycling endosomes and autophagosomes. Autophagy 13, 57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fader CM et al. (2009) TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1793, 1901–1916 [DOI] [PubMed] [Google Scholar]
- 22.Corona AK et al. (2018) Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Reports 22, 3304–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuster A et al. (2015) The Q-soluble N-Ethylmaleimide-sensitive Factor Attachment Protein Receptor (Q-SNARE) SNAP-47 Regulates Trafficking of Selected Vesicle-associated Membrane Proteins (VAMPs). Journal of Biological Chemistry 290, 28056–28069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rieder SE and Emr SD (1997) A novel RING finger protein complex essential for a late step in protein transport to the yeast vacuole. Mol. Biol. Cell 8, 2307–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gissen P et al. (2005) Comparative evolutionary analysis of VPS33 homologues: genetic and functional insights. Hum Mol Genet 14, 1261–1270 [DOI] [PubMed] [Google Scholar]
- 26.Takáts S et al. (2014) Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Molecular biology of the cell 25, 1338–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutierrez MG et al. (2004) Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell. Sci 117, 2687–2697 [DOI] [PubMed] [Google Scholar]
- 28.Wang Z et al. (2016) The Vici Syndrome Protein EPG5 Is a Rab7 Effector that Determines the Fusion Specificity of Autophagosomes with Late Endosomes/Lysosomes. Molecular Cell 63, 781–795 [DOI] [PubMed] [Google Scholar]
- 29.Hegedűs K et al. (2016) The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol Biol Cell 27, 3132–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balderhaar H.J. kleine and Ungermann C (2013) CORVET and HOPS tethering complexes – coordinators of endosome and lysosome fusion. J Cell Sci 126, 1307–1316 [DOI] [PubMed] [Google Scholar]
- 31.Jiang P et al. (2014) The HOPS complex mediates autophagosome–lysosome fusion through interaction with syntaxin 17. Molecular biology of the cell 25, 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orr A et al. (2017) HOPS catalyzes the interdependent assembly of each vacuolar SNARE into a SNARE complex. Molecular biology of the cell 28, 975–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang C et al. (2008) Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature Cell Biology 10, 776–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia R et al. (2017) BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy 13, 1648–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diao J et al. (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520, 563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ebner P et al. (2018) The IAP family member BRUCE regulates autophagosome–lysosome fusion. Nature Communications 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen TN et al. (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome– lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. The Journal of Cell Biology DOI: 10.1083/jcb.201607039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuboyama K et al. (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041 [DOI] [PubMed] [Google Scholar]
- 39.Vaites LP et al. (2018) Systematic Analysis of Human Cells Lacking ATG8 Proteins Uncovers Roles for GABARAPs and the CCZ1/MON1 Regulator C18orf8/RMC1 in Macroautophagic and Selective Autophagic Flux. Molecular and cellular biology 38, e00392–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Y et al. (2014) Vacuolin-1 potently and reversibly inhibits autophagosome-lysosome fusion by activating RAB5A. Autophagy 10, 1895–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ranieri R et al. (2017) N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell death and differentiation [DOI] [PMC free article] [PubMed]
- 42.Lőrincz P et al. (2017) Rab2 promotes autophagic and endocytic lysosomal degradation. The Journal of Cell Biology 216, 1937–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujita N et al. Genetic screen in Drosophila muscle identifies autophagy-mediated T-tubule remodeling and a Rab2 role in autophagy. eLife 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Itoh T et al. (2011) OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J Cell Biol 192, 839–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y-M et al. (2015) mTORC1 Phosphorylates UVRAG to Negatively Regulate Autophagosome and Endosome Maturation. Molecular Cell 57, 207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Q et al. (2011) The RUN Domain of Rubicon Is Important for hVps34 Binding, Lipid Kinase Inhibition, and Autophagy Suppression. J Biol Chem 286, 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Q et al. (2010) Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A 107, 19338–19343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng X et al. (2017) Pacer Mediates the Function of Class III PI3K and HOPS Complexes in Autophagosome Maturation by Engaging Stx17. Molecular Cell 65, 1029–1043.e5 [DOI] [PubMed] [Google Scholar]
- 49.Bustos V et al. (2017) Phosphorylated Presenilin 1 decreases β-amyloid by facilitating autophagosome–lysosome fusion. Proceedings of the National Academy of Sciences 114, 7148–7153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McEwan DG et al. (2015) PLEKHM1 Regulates Autophagosome-Lysosome Fusion through HOPS Complex and LC3/GABARAP Proteins. Molecular Cell 57, 39–54 [DOI] [PubMed] [Google Scholar]
- 51.Mohamud Y et al. (2018) Enteroviral Infection Inhibits Autophagic Flux via Disruption of the SNARE Complex to Enhance Viral Replication. Cell Reports 22, 3292–3303 [DOI] [PubMed] [Google Scholar]
- 52.Majumder P and Chakrabarti O (2015) Mahogunin regulates fusion between amphisomes/MVBs and lysosomes via ubiquitination of TSG101. Cell Death & Disease 6, e1970–e1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carra S et al. (2008) HspB8 and Bag3: A new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 4, 237–239 [DOI] [PubMed] [Google Scholar]
- 54.Li X-C et al. (2017) HSPB8 Promotes the Fusion of Autophagosome and Lysosome during Autophagy in Diabetic Neurons. International Journal of Medical Sciences 14, 1335–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang C et al. (2014) Thioredoxin-interacting protein mediates dysfunction of tubular autophagy in diabetic kidneys through inhibiting autophagic flux. Laboratory Investigation 94, 309–320 [DOI] [PubMed] [Google Scholar]
- 56.D’Eletto M et al. (2009) Transglutaminase 2 is involved in autophagosome maturation. Autophagy 5, 1145–1154 [DOI] [PubMed] [Google Scholar]
- 57.Brandstaetter H et al. (2014) Loss of functional MYO1C/myosin 1c, a motor protein involved in lipid raft trafficking, disrupts autophagosome-lysosome fusion. Autophagy 10, 2310–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wijdeven RH et al. (2016) Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nature Communications 7, 11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dooley HC et al. (2014) WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12–5–16L1. Molecular Cell 55, 238–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferguson CJ et al. (2009) Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet 18, 4868–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hasegawa J et al. (2016) Autophagosome–lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. The EMBO Journal 35, 1853–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang H et al. (2015) GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc Natl Acad Sci U S A 112, 7015–7020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Albanesi J et al. (2015) GABARAP-mediated targeting of PI4K2A/PI4KIIα to autophagosomes regulates PtdIns4P-dependent autophagosome-lysosome fusion. Autophagy 11, 2127–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fass E et al. (2006) Microtubules Support Production of Starvation-induced Autophagosomes but Not Their Targeting and Fusion with Lysosomes. J. Biol. Chem 281, 36303–36316 [DOI] [PubMed] [Google Scholar]
- 65.Bulinski JC (2007) Microtubule Modification: Acetylation Speeds Anterograde Traffic Flow. Current Biology 17, R18–R20 [DOI] [PubMed] [Google Scholar]
- 66.Xie R et al. (2010) Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol 11, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Köchl R et al. Microtubules Facilitate Autophagosome Formation and Fusion of Autophagosomes with Endosomes. Traffic 7, 129–145 [DOI] [PubMed] [Google Scholar]
- 68.Xu M et al. (2013) Regulation of Autophagic Flux by Dynein-mediated Autophagosomes Trafficking in Mouse Coronary Arterial Myocytes. Biochim Biophys Acta 1833, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng X-T et al. (2015) Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. The Journal of Cell Biology 209, 377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xia Q et al. (2016) TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. The EMBO Journal 35, 121–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiao Y et al. (2018) Iron promotes α-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. Journal of Neurochemistry DOI: 10.1111/jnc.14312 [DOI] [PubMed] [Google Scholar]
- 72.Ejlerskov P et al. (2013) Tubulin Polymerization-promoting Protein (TPPP/p25α) Promotes Unconventional Secretion of α-Synuclein through Exophagy by Impairing Autophagosome-Lysosome Fusion. J Biol Chem 288, 17313–17335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tumbarello DA et al. (2012) Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nature Cell Biology 14, 1024–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee J-Y et al. (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 29, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iwata A et al. (2005) HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated Huntingtin. Journal of Biological Chemistry 280, 40282–40292 [DOI] [PubMed] [Google Scholar]
- 76.Ganley IG et al. (2011) Distinct Autophagosomal-Lysosomal Fusion Mechanism Revealed by Thapsigargin-Induced Autophagy Arrest. Molecular Cell 42, 731–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamamoto A et al. (1998) Bafilomycin A1 Prevents Maturation of Autophagic Vacuoles by Inhibiting Fusion between Autophagosomes and Lysosomes in Rat Hepatoma Cell Line, H-4-II-E Cells. Cell Struct. Funct 23, 33–42 [DOI] [PubMed] [Google Scholar]
- 78.Mauvezin C et al. (2015) Autophagosome–lysosome fusion is independent of V-ATPase-mediated acidification. Nature Communications 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choi J-Y et al. (2016) From the Cover: Ethylmercury-Induced Oxidative and Endoplasmic Reticulum Stress-Mediated Autophagic Cell Death: Involvement of Autophagosome–Lysosome Fusion Arrest. Toxicological Sciences 154, 27–42 [DOI] [PubMed] [Google Scholar]
- 80.Liu F et al. (2017) Cadmium disrupts autophagic flux by inhibiting cytosolic Ca 2+-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 383, 13–23 [DOI] [PubMed] [Google Scholar]
- 81.Koga H et al. (2010) Altered lipid content inhibits autophagic vesicular fusion. The FASEB Journal 24, 3052–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Miyagawa K et al. (2016) Lipid-induced endoplasmic reticulum stress impairs selective autophagy at the step of autophagosome-lysosome fusion in hepatocytes. The American journal of pathology 186, 1861–1873 [DOI] [PubMed] [Google Scholar]
- 83.Liu EA and Lieberman AP (2018) The intersection of lysosomal and endoplasmic reticulum calcium with autophagy defects in lysosomal diseases. Neuroscience Letters DOI: 10.1016/j.neulet.2018.04.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tian X et al. (2015) A Voltage-Gated Calcium Channel Regulates Lysosomal Fusion with Endosomes and Autophagosomes and Is Required for Neuronal Homeostasis. PLOS Biology 13, e1002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levine B and Kroemer G (2008) Autophagy in the Pathogenesis of Disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tian L et al. (2018) The cytotoxicity of coxsackievirus B3 is associated with a blockage of autophagic flux mediated by reduced syntaxin 17 expression. Cell Death & Disease 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ding B et al. (2014) Phosphoprotein of Human Parainfluenza Virus Type 3 Blocks Autophagosome-Lysosome Fusion to Increase Virus Production. Cell Host & Microbe 15, 564–577 [DOI] [PubMed] [Google Scholar]
- 88.Romagnoli A et al. (2012) ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8, 1357–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Siqueira M. da S. et al. (2018) Autophagy and Its Interaction With Intracellular Bacterial Pathogens. Front Immunol 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lou X and Shin Y-K (2016) SNARE zippering. Bioscience Reports 36, e00327–e00327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bombardier JP and Munson M (2015) Three steps forward, two steps back: mechanistic insights into the assembly and disassembly of the SNARE complex. Current Opinion in Chemical Biology 29, 66–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hong W (2005) SNAREs and traffic. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1744, 120–144 [DOI] [PubMed] [Google Scholar]
- 93.Sreetama SC et al. (2016) Injured astrocytes are repaired by Synaptotagmin XI-regulated lysosome exocytosis. Cell Death and Differentiation 23, 596–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo F and Südhof TC (2017) Synaptotagmin-7-Mediated Asynchronous Release Boosts High-Fidelity Synchronous Transmission at a Central Synapse. Neuron 94, 826–839.e3 [DOI] [PubMed] [Google Scholar]
- 95.Simms BA and Zamponi GW (2014) Neuronal Voltage-Gated Calcium Channels: Structure, Function, and Dysfunction. Neuron 82, 24–45 [DOI] [PubMed] [Google Scholar]
- 96.Cang C et al. (2014) The voltage-gated sodium channel TPC1 confers endolysosomal excitability. Nature Chemical Biology 10, 463–469 [DOI] [PubMed] [Google Scholar]
- 97.Cang C et al. (2013) mTor Regulates Lysosomal ATP-sensitive Two-Pore Na+ Channel to Adapt to Metabolic State. Cell 152, 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang X et al. (2012) TPC Proteins Are Phosphoinositide-activated Sodium-selective Ion Channels in Endosomes and Lysosomes. Cell 151, 372–383 [DOI] [PMC free article] [PubMed] [Google Scholar]