

Abstract
Motivation
Genetic variation that disrupts gene function by altering gene splicing between individuals can substantially influence traits and disease. In those cases, accurately predicting the effects of genetic variation on splicing can be highly valuable for investigating the mechanisms underlying those traits and diseases. While methods have been developed to generate high quality computational predictions of gene structures in reference genomes, the same methods perform poorly when used to predict the potentially deleterious effects of genetic changes that alter gene splicing between individuals. Underlying that discrepancy in predictive ability are the common assumptions by reference gene finding algorithms that genes are conserved, well-formed and produce functional proteins.
Results
We describe a probabilistic approach for predicting recent changes to gene structure that may or may not conserve function. The model is applicable to both coding and non-coding genes, and can be trained on existing gene annotations without requiring curated examples of aberrant splicing. We apply this model to the problem of predicting altered splicing patterns in the genomes of individual humans, and we demonstrate that performing gene-structure prediction without relying on conserved coding features is feasible. The model predicts an unexpected abundance of variants that create de novo splice sites, an observation supported by both simulations and empirical data from RNA-seq experiments. While these de novo splice variants are commonly misinterpreted by other tools as coding or non-coding variants of little or no effect, we find that in some cases they can have large effects on splicing activity and protein products and we propose that they may commonly act as cryptic factors in disease.
Availability and implementation
The software is available from geneprediction.org/SGRF.
Supplementary information
Supplementary information is available at Bioinformatics online.
1 Introduction
Much work has gone into the development of sophisticated methods for whole-gene gene-structure prediction, as documented in the extensive literature on gene finding (e.g. Allen and Salzberg, 2005; Burge and Karlin, 1997; Guigo et al., 1992; Korf et al., 2001; Lukashin and Borodovsky, 1998; Meyer and Durbin, 2004; Pachter et al., 2002; Stanke et al., 2006; reviewed in Majoros, 2007). These methods jointly model entire sequences and their whole-gene splicing patterns, typically via hidden Markov models (HMMs) or conditional random fields (CRFs). However, traditional methods have focused exclusively on annotation of reference genomes, in which genes are assumed to be well-formed and to have conserved function. The growing importance of resequencing studies and personalized genomics has created a need for tools that can accurately annotate personal genomes and the genomes of individual animal and plant breeds. As the focus in these studies is typically on identifying genetic differences that may have functional consequences, the assumptions made by traditional gene-finding models are violated (Supplementary Fig. S1).
In eukaryotes, messenger RNAs are commonly spliced prior to nuclear export and translation to remove intronic sequences that do not encode amino acids. Failure to properly remove introns can result in large changes to the resulting polypeptide or failure to produce a functional protein, either of which can be deleterious to the organism. Similarly, splicing errors can result in loss of existing function or gain of new function, either of which may be deleterious. In humans, 95% of protein-coding genes contain introns that require splicing, and 95% of those spliced genes can be alternatively spliced to produce multiple distinct isoforms (Pan et al., 2008).
Though splicing errors commonly occur in many genes at a low rate in healthy individuals (Pickrell et al., 2010; Stepankiw et al., 2015), genetic variants that directly interrupt normal splicing signals can dramatically increase the production of aberrant splice forms [e.g. (Buratti et al., 2007; Královicová et al., 2005)]. While these can be deleterious, in many cases they result in small changes that appear to be benign. Examples include alternative splicing patterns that cause synonymous or conservative amino acid changes, or the addition or removal of a single amino acid. As an additional challenge, multiple variants in the same haplotype can act non-independently, so that methods that interpret the effect of each variant in isolation can produce incorrect predictions (Majoros et al., 2017).
Computational methods are therefore needed that can predict the joint effect of any combination of variants present together in a haplotype on resulting splicing patterns and protein structure. Previous approaches to predicting aberrant splicing have focused on each single-nucleotide polymorphism (SNP) individually and report the predicted effect on a single splice site or exon (e.g. Mort et al., 2014; Woolfe et al., 2010; Xiong et al., 2015). There is thus a need for whole-gene models that can integrate the effects of multiple variants in a haplotype and interpret the resulting splicing patterns as to their likely effect on the encoded protein as a whole (Guigo and Valcárel, 2015). Ideally, such models should be applicable to both SNPs and multi-nucleotide variants, as insertions and deletions can have large impacts on splicing signals and reading frames. It will also be ideal to develop methods that can be retrained for any species for which an annotated reference genome is available rather than requiring large training sets of confirmed aberrant splicing cases or splicing variants implicated in disease (e.g. Mort et al., 2014; Woolfe et al., 2010).
Here, we describe a novel method for annotating personal genomes that explicitly accounts for combinations of genetic variants and does not assume that genes are conserved or functional. The proposed model does not utilize translation reading frames or codon statistics, and is thus applicable to both coding and non-coding genes. Traditional ab initio gene finders focus primarily on signals within coding sequence, in particular codon biases. It has been noted that coding sequences within eukaryotic protein-coding genes contain other signals in addition to codons (Itzkovitz et al., 2010). In particular, signals that promote splicing and exon inclusion often overlap coding signals, either in-frame or out-of-frame (Woolfe et al., 2010; Zhang et al., 2008). These signals are referred to as splicing enhancers and are believed to serve primarily as binding sites for RNA-binding factors such as SR proteins. Splicing enhancer motifs can be found arbitrarily deep within exons (Woolfe et al., 2010), suggesting that SR proteins bind all along the exon. This scaffolding of splicing factors across the exon body is believed to mediate the process of exon definition (Berget, 1995; Robberson et al., 1990), whereby U1 and U2 snRNPs associated with the ends of the exon are brought into close spatial proximity and which is necessary for the exon to be included in the mature transcript (Schneider et al., 2010). Meanwhile, hnRNPs are believed to bind primarily within introns, marking them for exclusion from the mature transcript. Together, these enhancing and silencing signals allow the cell to discriminate exonic from intronic sequence (Zhang et al., 2008).
A number of feature sets comprising scored nucleotide hexamers or octamers have been proposed to capture exon-definition signals (Erkelenz et al., 2014; Stadler et al., 2006; Zhang et al., 2005; Zhang and Chasin, 2004; Zhang et al., 2008). Most recently, a set of hexamer weights determined via massively parallel splicing reporter assays were used to evaluate individual SNPs in human exons for their potential to induce skipping of individual exons (Rosenberg et al., 2015). To our knowledge, such exon-definition features have not previously been incorporated into a whole-gene model of gene structure. To the extent that such features capture exon definition propensities, doing so should be informative for predicting splicing patterns of whole transcripts.
By utilizing these signals instead of codon statistics, the model we propose is applicable to predicting changes to gene structures in non-coding genes, in untranslated regions of coding genes and in coding regions that are altered by variants that disrupt the reading frame. Because the model can predict alterations to the reading frame, it can detect changes that may be deleterious. As such, the model is applicable to identifying differences in gene structures between individuals or strains of the same species, in which such differences may reflect gain or loss of function that has yet to be eliminated by natural selection but which may be of interest to breeders or clinicians. In contrast, traditional comparative gene-finding models (e.g. Majoros et al., 2005; Meyer and Durbin, 2004; Pachter et al., 2002) compare multiple reference genomes of distinct species, and assume that gene structures are conserved. Finally, because the model we propose can be trained on annotated exons and introns in a reference genome rather than relying on large numbers of curated examples of aberrant splicing, it can be retrained easily on any non-human species for which an annotated reference genome is available, a major advantage.
2 Materials and methods
2.1 Splice graph random field
Reference gene-structure prediction methods (reviewed in Majoros, 2007) are based on probabilistic graphical models such as HMMs or CRFs. Whereas HMMs model the joint probability P(ϕ, S), for sequence S and state path ϕ, CRFs directly model the posterior probability P(ϕ | S), where ϕ is considered a labeling of the sequence S:
The potential functions Φc are applied to the cliques c in a dependency graph (Sutton and McCallum, 2006). One advantage of CRFs over HMMs is that arbitrary features such as empirical hexamer weights can be incorporated into the Φ functions.
We propose a CRF for gene structures in which each vertex in the field denotes a putative splice site and each edge denotes a putative exon or intron (Fig. 1A). We refer to this model as a splice graph random field (SGRF). Labels are chosen from {0, 1}, with 0 denoting omission and 1 denoting inclusion of the splice site in the predicted gene structure. Cliques in the SGRF consist of singletons and pairs of vertices directly connected via a single edge. Clique potential functions (Fig. 1B) evaluate to non-zero values only when all labels in the clique are 1, so that splice sites not included in the prediction do not contribute to its score.
Fig. 1.
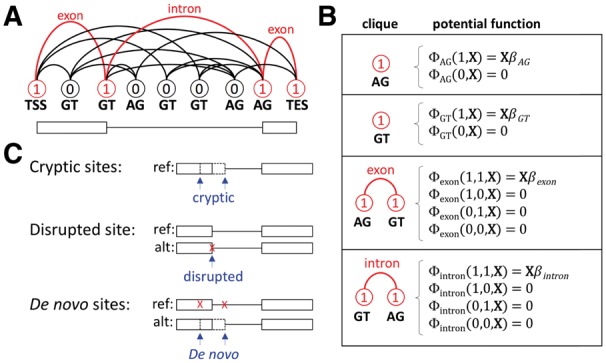
(A) A SGRF. Vertices denote splice sites, and edges denote exons and introns. A path from TSS to TES outlines a single gene structure. Labels 0 and 1 denote omission or inclusion, respectively, of a vertex on the selected path. (B) Cliques and their potential functions. SGRFs have only singleton and pair cliques. Potential functions for cliques labeled with any 0 do not contribute to the score, since they do not participate in the selected path. (C) Cryptic splice sites are unannotated splice sites near an annotated splice site. Disrupted splice sites exist in the reference but not in the alternate sequence. De novo splice sites exist in the alternate sequence but not in the reference
SGRFs are constructed in a highly constrained manner, as follows. For a given splice isoform annotated on the reference genome, we project exon co-ordinates to the genome of an individual or strain for prediction in that individual. For each projected splice site, a vertex is created and linked to the preceding vertex, resulting in a linear-chain SGRF having exactly one path that corresponds to the projected gene structure. However, if any splice site is disrupted in the alternate sequence, its vertex is removed from the SGRF and alternate splice sites of the same type (i.e. donor or acceptor) are identified via a signal sensor (Supplementary Material) in the vicinity of the disrupted site. These alternate splice sites are linked into the SGRF using edges of the appropriate type (i.e. exon or intron). In addition, any genetic variant that could potentially create a de novo splice site (Fig. 1C) that does not exist in the reference is also added to the SGRF and linked via appropriate edges to the nearest annotated vertices already in the SGRF. In this way, the SGRF represents exactly the reference annotation when that annotation can be projected perfectly onto the alternate sequence. Only when a splice site is disrupted, or when a genetic variant creates a new putative splice site, is the SGRF expanded to include more potential paths. We call this procedure constrained splice-graph construction. Decoding with an SGRF can be accomplished efficiently using dynamic programming. We use N-best decoding to find the N highest-scoring predictions, where N can be set by the user. For the experiments described here we used N = 10.
The SGRF model is distributed as part of our ACE/ACE+ software (Majoros et al., 2017). Inputs to the system consist of a set of FASTA files containing the reference genome, a GTF file containing reference annotations and a phased VCF file containing genetic variants for one or more individuals. The system reconstructs haplotype sequences based on the reference genome and variants in the VCF file, maps annotations onto those haplotypes, applies the SGRF to predict changes in gene structure and then interprets those changes as to possible loss of function (Supplementary Fig. S6).
2.2 SGRF features and parameter estimation
We refer to the potential functions for singleton cliques, specifically individual splice sites, as signal sensors. We refer to pair cliques, which we generate for exons and introns, as content sensors. For content sensors we use a linear combination Xβ of hexamer weights β with hexamer counts X in the interval spanned by the clique (not including splice sites at the ends of the interval), where β and X are 4096-dimensional vectors corresponding to the 4096 possible hexamers. Any collection of hexamer weights can thus be used as an SGRF content sensor. Both coding and non-coding genes can be used for training; in the case of coding genes, hexamer counts are extracted from all reading frames on the sense strand. For signal sensors, we score a fixed window spanning the putative splice site with a small number of flanking positions on both sides (Supplementary Material). Features are indicators (0 or 1) for whether each possible nucleotide (A, C, G, T) is present at each position.
Training of CRFs can be accomplished via conditional maximum likelihood (CML), which globally optimizes all model parameters jointly by maximizing P(ϕ | S). Because CML can be computationally burdensome, we instead use piecewise training (Sutton and McCallum, 2005) with the ‘factor-as-piece’ approximation (Sutton, 2008). As such, piecewise training for the SGRF consists of estimating parameters of each potential function ΦAG, ΦGT, Φexon and Φintron separately. Because each of these functions are linear combinations of hexamer counts or nucleotide indicators, and because standard logistic regression is equivalent to CML for a single-vertex CRF with a linear-combination potential function (Sutton and McCallum, 2005), we use logistic regression to separately estimate all parameters of each potential function individually. For pair cliques, we binarize the problem into classification of exons versus introns in order to apply logistic regression. The procedure is similar to that of Domke (2014), except that we employ only a single iteration of logistic regression and eliminate the belief propagation step due to time efficiency concerns. We call this simplified procedure piecewise logistic. For the experiments described here we used elastic net (regularized) logistic regression to favor sparse parameterizations (Supplementary Material). For the experiments on human sequences we trained the content sensors on 10 000 pairs of exons and introns annotated in GENCODE version 19 (Harrow et al., 2012). Signal sensors were trained on 5000 donor splice sites and 5000 acceptor splice sites annotated in GENCODE v19. Score thresholds for signal sensors were selected to admit 99% of training splice sites.
2.3 Computational validation
We tested the predictive accuracy of the SGRF on 150 human genomes from the Thousand Genomes project (The Thousand Genomes Project Consortium, 2015) for which paired RNA-seq data from lymphoblastoid cell lines were available (Lappalainen et al., 2013). For each individual and each gene in GENCODE v19, phased variants were used to construct explicit haplotype sequences for the gene, as previously described (Majoros et al., 2017). Insertion/deletion variants were used to infer an alignment between the reference sequence and the personal genome. For each annotated isoform of each gene, the isoform was projected onto the personal genomic sequence using the inferred alignment. The SGRF was then applied to produce predicted splice forms in the personal genome. We used the RNA-seq data to quantify support for predicted novel splice junctions not occurring in any annotated isoform of a gene. Transcripts not expressed in LCLs at an FPKM of at least three, or in which NMD is likely, were omitted from the analysis to avoid overestimating false positives (see Supplementary Material for additional details).
Receiver-operating characteristic (ROC) curves were constructed to enable comparison of prediction accuracy between three different content sensors: (i) piecewise logistic applied to 10 000 human exon-intron pairs; (ii) piecewise logistic applied to 10 000 exon-intron pairs from Arabidopsis thaliana, as annotated in the Araport 11 release (Cheng et al., 2017) and (iii) hexamer weights estimated previously by Rosenberg et al. (2015) by fitting a sigmoid function to exon inclusion levels resulting from a massively parallel minigene experiment in human HEK293 cells.
To investigate whether the piecewise logistic training procedure is hampering predictive accuracy by not training all parameters jointly, we incorporated an additional parameter rcontent/signal into the model, which weights the relative contributions of content sensors versus signal sensors. We performed a sensitivity analysis by applying the modified model to Thousand Genomes individual HG00096 with different values (0.1, 0.2, 0.4, 0.6, 0.8, 1, 1.5, 2, 3, 4, 5, 8, 10) for rcontent/signal. A substantial improvement in predictive accuracy for values of rcontent/signal < 1 would indicate that the content sensors are overpowering the signal sensors, and that accuracy might be improved by global training of all parameters jointly.
3 Results
3.1 Prediction accuracy on 150 human genomes
Area under the ROC curve (AUC) values for the SGRF with different content sensors indicate that on average an AUC of approximately 0.75 is achievable using either experimentally determined or bioinformatically inferred features (Fig. 2A and B). The logistic model trained on human annotations achieved the highest AUC (0.75) followed closely by the model trained on splicing minigene outputs (0.72). The logistic model trained on Arabidopsis performed worst (0.51), indicating that training for the target organism is necessary in order to learn organism-specific exon definition features. The median difference between the logistic human model and the minigene model was positive and significant (Wilcoxon signed-rank test: V = 43489, P = 2 × 10−44; Fig. 2C), indicating that training on a new organism can be done effectively using logistic regression applied to annotated exons and introns and that minigene experiments are not necessary for learning SGRF parameters on a new organism. Logistic regression training took approximately 12 h on a single CPU.
Fig. 2.
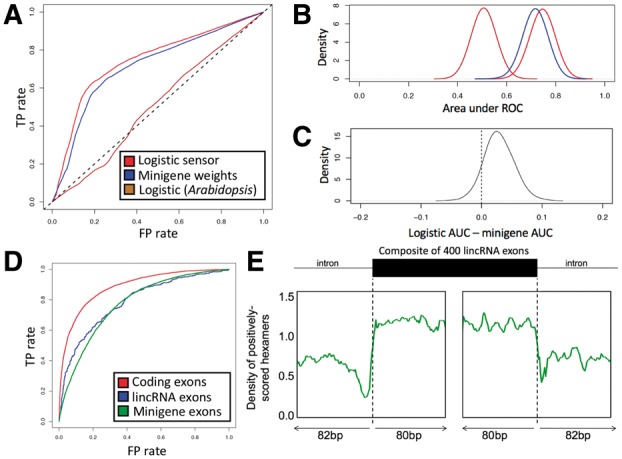
(A) ROC curves for the SGRF with three different content sensors. TP and FP rates were computed based on spliced RNA-seq reads from LCL cells. (B) Area under ROC curves shown in panel A. (C) Difference between logistic model AUC and minigene model AUC. (D) ROC for classification of coding exons versus introns (red), for lincRNA exons versus introns (blue) and for minigene exons with high versus low inclusion rates (green), using the logistic human model for classification. (E) Density of positively-scored hexamers under the human logistic model, at relative positions in non-coding lincRNA exons
Logistic weights for human data were similar across training runs (Supplementary Fig. S2). Modifying rcontent/signal away from its default value of 1 did not appreciably improve prediction accuracy on individual HG00096 (Supplementary Fig. S3), indicating that content sensor scores are not overpowering signal sensor scores to the detriment of predictive accuracy. The precipitous drop in AUC as rcontent/signal approached zero indicates that splice-site scores alone are inadequate for predicting splicing changes, consistent with previous suggestions that splice sites lack sufficient information content to allow their discrimination without genomic context information (Lim and Burge, 2001). Results of classifying 5000 annotated versus 5000 decoy human splice sites omitted from the training set indicate that both the logistic signal sensors and PWMs achieved high classification accuracy for donor splice sites (AUC = 0.984 for logistic sensor, AUC = 0.977 for PWM; Supplementary Fig. S4A) and for acceptor splice sites (AUC = 0.965 for both sensors; Supplementary Fig. S4B).
When the logistic human content sensor was used to perform direct classification of whole exons versus whole introns of matched lengths and with splice sites removed (Supplementary Material), AUC was higher when presented with coding exons than with non-coding exons (0.89 versus 0.79; Fig. 2D), indicating that while logistic regression successfully learned exon definition features that enabled the model to recognize non-coding exons with moderate accuracy, it may also be inadvertently learning some features of the coding segments present in the training exons. Classification of binarized minigene splicing results (Supplementary Material) using the human logistic model resulted in a similar AUC (0.77) to that of classifying lincRNA exons (0.79; Fig. 2D). Positively scoring hexamers under the human logistic model were enriched deep into non-coding exons relative to introns (Fig. 2E), indicating that the features learned were not due solely to sequence biases proximal to splice sites.
3.2 Logistic and splicing minigene features reflect known hnRNP but not SR protein motifs
As reported in Rosenberg et al. (2015) for the experimentally determined minigene features, G-rich hexamers in the human logistic model were enriched for negative scores (Supplementary Fig. S5), consistent with G-richness of sequences preferred by some hnRNPs (Huelga et al., 2012; Mauger et al., 2008; Rahman et al., 2015). While elastic net regularization produced a sparse model containing 1966 of the 4096 possible hexamers, all 19 hexamers containing 5 or more Gs were selected by elastic net for inclusion in the model and assigned negative scores. Consensus binding motifs for hnRNPs obtained from Huelga et al. (2012) were likewise enriched for having negative scores under the human logistic model (Wilcoxon V = 329, P = 3.9 × 10−9), as well as under the minigene model (Wilcoxon V = 1763, P = 0.0002), consistent with our expectations of depleted hnRNP binding in exons. While scrambled versions of consensus motifs are also commonly enriched for negative scores under both models (logistic model: P < 0.05 in 469/1000 scrambled motif sets; minigene model: P < 0.05 in 506/1000 scrambled motif sets), hnRNPs have been characterized as having degenerate binding motifs with low sequence specificity (Huelga et al., 2012; Singh and Valcárcel, 2005). As each consensus motif represents only the single most strongly-bound sequence for a factor, enrichment of some scrambled versions of these degenerate motifs might represent weaker binding that nevertheless supports exon definition. The most strongly negatively scoring hnRNP motif under the human logistic model was for hnRNP H, which is known to bind to poly(G) sequences (Mauger et al., 2008; Rahman et al., 2015) and has been implicated in aberrant splicing (Paul et al., 2006).
SR protein motifs obtained from Long and Caceres (2009) were not significantly biased in their scores under either the logistic model (Wilcoxon V = 6101, P = 0.08) or the minigene model (Wilcoxon V = 17713, P = 0.14). The sparse logistic model included substantially more negative features than positive features (1126 versus 840), suggesting that accurate discrimination of exons from introns relies more on features depleted from exons (e.g. hnRNP binding sites) than on features enriched in them (e.g. SR protein binding sites).
Classification of exons versus length-matched introns (with splice sites removed) using densities of known SR or hnRNP motifs produced very low AUC values (hnRNP motifs: 0.62; SR protein motifs: 0.60) that were not substantially higher than random classification (0.56), indicating that both the experimentally determined features of Rosenberg et al. (2015) and the sparse logistic features learned from human annotations represent sequence preferences not entirely explained by known SR protein or hnRNP consensus motifs.
3.3 De novo splice sites are prevalent, have a wide range of effects and are misclassified by existing tools
Predictions of the SGRF with human logistic features were highly enriched for novel isoforms utilizing de novo splice sites present in the alternate sequence but absent from the reference genome, as compared to variants disrupting existing splice sites (predictions with posterior probability > 0.9: 3165 de novo splice sites/750 disrupted sites = enrichment of 4.2×). Predictions supported by spliced RNA-seq reads were similarly enriched relative to numbers of disrupted splice sites (Fig. 3A). De novo splice sites are distinct from cryptic splice sites, in that cryptic sites exist in the reference genome (and typically also the alternate sequence), whereas de novo splice sites exist in the alternate sequence but not in the reference (Fig. 1C). Simulation of a simple mutation process based on context-dependent substitution (Supplementary Material) also produced a large bias of de novo sites over disrupted sites (Fig. 3A). The enrichment was greatest (125.5×) when requiring only that de novo splice sites have a canonical 2 bp consensus, and least (8.8×) when requiring that de novo sites score above threshold under the logistic splice-site model and occur in a favorable exon definition context (Supplementary Material). Moreover, each individual in the Thousand Genomes sample had on average 126 predicted de novo splice sites supported by spliced RNA-seq reads in LCLs, indicating that de novo splice sites are widespread and commonly utilized by the spliceosome.
Fig. 3.
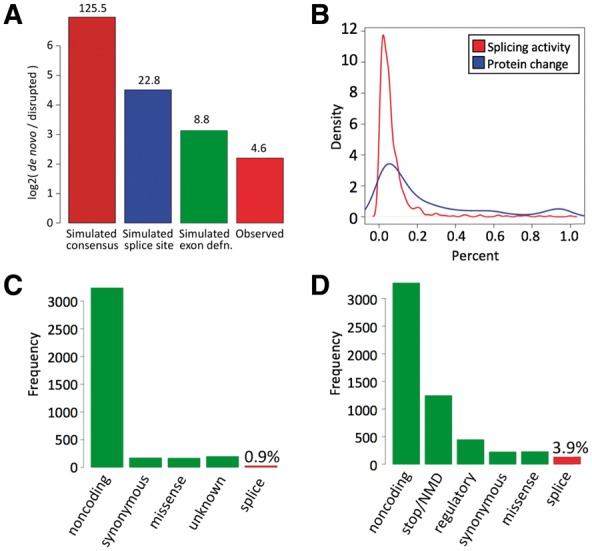
(A) Log2 (number of de novo splice sites/number of disrupted splice sites) in mutation simulations (brown: requiring only a 2 bp consensus for de novo splice sites; blue: requiring sufficiently high score under splice-site model for de novo splice sites; green: requiring sufficiently high score under the splice-site model and favorable exon definition context for de novo splice sites), and in predictions supported by RNA-seq (red). Numbers above bars are raw (non-log2) ratios. (B) Estimates of relative splicing activity (red) and protein change (blue) due to de novo splice sites supported by RNA-seq. (C) Frequencies of dbSNP classifications of variants predicted to create de novo splice sites and supported by spliced RNA-seq reads. (D) Frequencies of VEP classifications of variants creating de novo splice sites supported by RNA-seq
We found evidence that de novo splice sites are capable of having large effects on splicing ratios and on encoded proteins. As subjects in the Thousand Genomes project are reported to be healthy, we expect natural selection to result in a strong bias for these de novo splice sites to have small effects. Consistent with this expectation, most de novo splice sites appear to experience at most moderate utilization in these cells (Fig. 3B). Nevertheless, some de novo splice sites in this dataset are highly utilized (Fig. 4), consistent with examples of de novo splice sites with high utilization documented in aberrant splicing databases (Buratti et al., 2007; Královicová et al., 2005). Furthermore, while non-NMD de novo splice sites exhibited the expected bias toward having small impacts on encoded proteins, 62% of RNA-supported de novo splice sites were predicted to trigger NMD and some non-NMD de novo splice sites supported by RNA-seq are predicted to result in large protein changes (Fig. 3B).
Fig. 4.
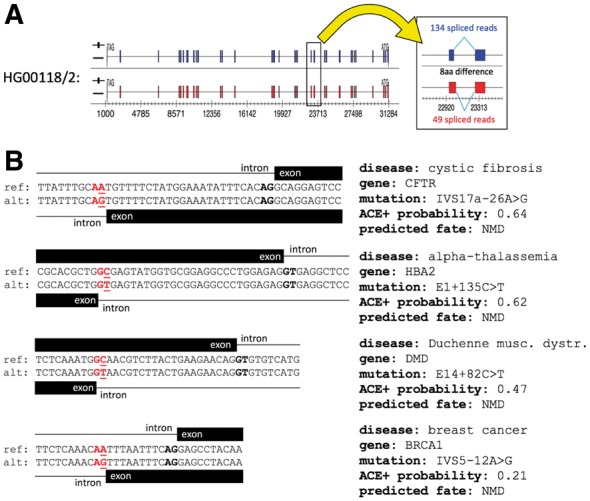
(A) Example of a de novo splice site that results in greater splicing activity than at the annotated splice site. Variant rs202069778 in haplotype 2 of Thousand Genomes individual HG00118 creates a new acceptor splice site that retains the original reading frame in the MAP4K1 gene, resulting in eight amino acids being excluded from the encoded protein; TopHat2 aligns more spliced reads to this site than to the annotated site in this haplotype. This variant has a global MAF of 0.0002 in Thousand Genomes phase three samples, indicating it is possibly deleterious. (B) ACE+ predictions on a sample of four disease mutations documented in DBASS as creating de novo splice sites. Reference and alternate sequences are labeled ref and alt, respectively. Scores are posterior probabilities under the random field model, and indicate the predicted relative usage of the site by the spliceosome. The annotated splice site is in black; the de novo site is in red. The mutation is underlined
A number of de novo splice sites have been implicated in disease, as evidenced by entries in the DBASS database (Buratti et al., 2007; Královicová et al., 2005). Examples include breast cancer, cystic fibrosis, hemophilia, muscular dystrophy, alpha- and beta-thallasemia, hypothyroidism, phenylketonuria and others. These disease-related variants are documented as having a range of effects. For example, mutation E1 + 135C > T in the HBA2 (hemoglobin alpha sub-unit 2) gene is implicated in alpha-thalassemia, and is described as having 100% splicing utilization (Harteveld et al., 2004), whereas IVS17a-26A > G in the CFTR (cystic fibrosis transmembrane conductance regulator) gene, implicated in cystic fibrosis, is described as resulting in leaky splicing and a mild form of the disease (Beck et al., 1999). De novo splice sites can thus present a wide range of effects in the clinical setting. On a sample of four disease-causing de novo splice sites listed in DBASS, the SGRF predicted substantial usage of all four sites by the spliceosome, albeit with a range of posterior probabilities (Fig. 4).
Despite their known role in a number of diseases, de novo splice sites are commonly misinterpreted as non-coding, synonymous, or missense mutations. For the 3289 Thousand Genomes variants predicted by the SGRF to result in de novo splice sites and that were supported by spliced RNA-seq reads in LCLs, dbSNP (Sherry et al., 2001) predicted only 0.9% to be involved in splicing (Fig. 3C). Similary, Ensembl’s VEP tool (McLaren et al., 2016) predicted only 3.9% to be involved in splicing (Fig. 3D).
4 Discussion
In this work, we investigated the use of a model in which features that reflect exon definition potential are used instead of coding signals, thus avoiding the assumptions imposed by previous methods. Our results on 150 human genomes indicate that exon definition features can be automatically learned via machine-learning methods applied to annotated training genes, and that those learned features can be used to discriminate splicing changes that are supported by RNA-seq. Our use of standard regularized logistic regression and reliance only on annotated exons and introns for training, as opposed to curated examples of aberrant splicing, renders this approach broadly applicable to other organisms, such as breeds of economically important animals and plants.
While most computational gene prediction in the 1980s focused on finding individual exons based on their codon usage statistics, the sequencing of large chromosome segments by the human genome project circa 2000 (Lander et al., 2001; Venter et al., 2001), together with bioinformatic advances such as the application of grammar models and dynamic programming to DNA sequence, enabled the development of methods that could efficiently and accurately predict whole gene structures in the late 1990s (Burge and Karlin, 1997; Kulp et al., 1996). These whole-gene structure annotations in turn enabled downstream analyses of genome-wide protein coding properties and the identification of gene families and functional annotation via protein similarity. As such, these bioinformatic advances had a measurable impact on our understanding of genomics.
The strong codon signals imposed on coding segments by natural selection enabled these methods to accurately chain exons together into multi-exon transcripts, by enforcing the constraint that translation reading frames must be contiguous and consistent across exons. The assumption of intact reading frames enables highly accurate prediction of genes in reference genomes, where it is natural to assume that most genes are well-formed, evolutionarily conserved and functional. For personal genomes, the assumption of contiguous reading frames matching organism-specific codon biases can result in incorrect predictions for genes that have been disrupted by genetic variants altering the reading frame, splicing patterns, or both. As the goal of personal genomics is to identify functional variants that may be implicated in disease, these biases are problematic.
Our observation that de novo splice sites appear to be readily created via simple mutation, that each individual has numerous such sites supported by spliced RNA-seq reads and that such sites have the potential to have a wide range of effects both in splicing utilization and in resulting changes to amino acid sequences, indicates that this is an important class of variants. That these variants are commonly misinterpreted by popular tools as not impacting splicing indicates that there is a pressing need for the development of new models that can accurately identify these variants, particularly as many de novo splice sites have been implicated in disease (Královicová et al., 2005; Buratti et al., 2007). Given the prevalence of de novo splice variants in individual human genomes and the difficulty that existing tools have in identifying them as such, it is conceivable that this class of cryptic variants may account for a sizeable fraction of unexplained disease cases.
The method we have proposed does not fully solve the problem of identifying modified splicing patterns in individual genomes, as there is much room for improvement above the 0.75 AUC reported here. Indeed, as our model currently relies on reference annotations, we expect that ab initio prediction of splicing patterns based on exon definition features represents an important challenge for the future, as the ability to perform such ab initio prediction may be seen as a yardstick for our current understanding of the complex biology of splicing. There are a number of possible avenues for seeking greater predictive accuracy.
First, the challenge of learning exon definition features from training examples that are enriched for coding exons, without being biased toward coding features, is an unavoidable problem given that most annotated genes available for training are coding genes. While training only on out-of-frame hexamers might seem an obvious solution, preliminary experiments indicated that the use of only out-of-frame hexamers during training did not improve prediction accuracy. Furthermore, it may be expected that di-codon patterns in reading frames impose biases even on out-of-frame hexamers, and conversely that exon definition features may occur in-frame. One possible solution is to modify the regression problem so as to simultaneously learn both coding and exon definition features using separate parameters, so that coding biases can be minimized in the learned exon definition parameters.
The use of massively parallel splicing minigene experiments to ascertain empirical hexamer weights is another solution, as the use of randomized exonic sequence mitigates biases due to natural selection on functional content. However, this solution is laborious, and other biases may remain, such as that due to NMD when the randomized exon is coding, or due to secondary structures specific to the minigene (Rosenberg et al., 2015). Nevertheless, features need to be learned anew for each new species, as our results using Arabidopsis features to predict splicing in humans indicate and as is also seen in traditional gene-finding with coding features (Korf, 2004). An important consideration for future work is the rate at which exon definition features change over evolutionary time, and the degree to which the methods outlined here are applicable to lineages with greater divergence (for example, between human and Neanderthal). While we expect that rate to be somewhat constrained by pleiotropic considerations, the Arabidopsis results demonstrate that these features are not conserved across distant timespans.
The use of collections of hexamer weights to represent exon definition potential has become popular in recent years. However, these hexamer models make strong assumptions about the independence of features residing near or far from each other in linear sequence space. It is known that SR proteins extensively interact, and that SR proteins compete with both other SR proteins and with hnRNPs for binding sites in mRNAs (Pandit et al., 2013; Rahman et al., 2015), and likewise that hnRNPs can interact in positive or negative ways (Huelga et al., 2012). Other examples of interactions between splicing regulators have been documented (reviewed in Ke and Chasin, 2011). It has also been demonstrated that splicing decisions can be influenced by epigenetic effects such as nucleosome positioning and histone modifications (reviewed in Zhou et al., 2014), and by binding of specific transcription factors at promoters and possibly even at distal enhancers (reviewed in Kornblihtt et al., 2013).
The above facts indicate that there is much room for incorporation of features beyond simple hexamer weights. Such improvements could be incorporated into the SGRF via modification of the Φ functions. Assuming the constrained graph construction process remains unchanged, modifications to the Φ functions will not negatively impact decoding efficiency, as the graph will remain sparse. Modifications to the training process may however be necessary, particularly if new features integrate information across larger intervals or consider combinatoric interactions. While piecewise training via simple logistic regression worked well for the initial model described here, for an expanded model with wider dependencies, an iterative method such as the one proposed by Domke (2014) that combines logistic regression with belief propagation may be required.
Our use of reference annotations as training examples results in a model that does not reflect splicing regulatory differences between cell types. As alternative splicing is often regulated in a cell-type specific manner, it can be expected that aberrant splicing will also exhibit cell-type specific patterns. Such cell-type-specific effects have been modeled previously, though not in a whole-gene model that can accommodate multiple variants jointly (Xiong et al., 2015). One possible means of addressing this is to train separate SGRF models on transcripts found to be expressed in individual cell types via RNA-seq data, such as that published by the GTex project (Melé et al., 2015).
Our inability to link positively-scoring hexamers in either the logistic model or the minigene model published by Rosenberg et al. (2015) to known SR protein motifs could be explained by a number of possibilities. Some SR proteins and hnRNPs have been characterized as participating in both specific and non-specific binding, and may rely on co-factors for specific binding (Singh and Valcárcel, 2005). It has also been shown that individual SR proteins can have both a positive and a negative effect on exon definition in different contexts, as can hnRNPs (Huelga et al., 2012; Pandit et al., 2013; Singh and Valcárcel, 2005). The fact that densities of known SR protein motifs or of known hnRNP motifs did not produce strong classification accuracy in the experiments described here supports the notion that while these molecules have been demonstrated to play important roles in splicing, predicting their specific effects via simple consensus motif counts may not be feasible in general. That both the logistic model and the minigene model were able to achieve much higher classification accuracy suggests that these models are detecting features relevant to exon definition, though at present it is not known with certainty what biological significance individual hexamers in these models have. Novel experimental work will likely be required to ascertain whether these features represent unknown binding motifs for known SR proteins or hnRNPs, or possibly for unknown splicing factors or co-factors.
Funding
W.H.M. and T.E.R. were supported by grants from the National Institutes of Health [R01 DK099820 and R01 DK097534]. M.S.C. was supported by a Post-doctoral Research Fellowship in Biology from the National Science Foundation [PRFB-1523793]. W.H.M., C.H. and M.Y were supported by grants from the National Science Foundation [EAGER IOS-1561337 and IOS-1126998].
Conflict of Interest: none declared.
Supplementary Material
References
- Allen J.E., Salzberg S.L. (2005) JIGSAW: integration of multiple sources of evidence for gene prediction. Bioinformatics, 21, 3596–3603. [DOI] [PubMed] [Google Scholar]
- Beck S. et al. (1999) Cystic fibrosis patients with the 3272-26A–>G mutation have mild disease, leaky alternative mRNA splicing, and CFTR protein at the cell membrane. Hum. Mutat., 14, 133–144. [DOI] [PubMed] [Google Scholar]
- Berget S.M. (1995) Exon recognition in vertebrate splicing. J. Biol. Chem., 270, 2411–2414. [DOI] [PubMed] [Google Scholar]
- Buratti E. et al. (2007) Aberrant 5’ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res., 35, 4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge C., Karlin S. (1997) Prediction of complete gene structures in human genomic DNA. J. Mol. Biol., 268, 78–94. [DOI] [PubMed] [Google Scholar]
- Cheng C.-Y. et al. (2017) Araport 11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J., 89, 789–804. [DOI] [PubMed] [Google Scholar]
- Domke J. (2014) Training structured predictors through iterated logistic regression. In: Advanced Structured Prediction, MIT Press, Cambridge, MA, USA.
- Erkelenz S. et al. (2014) Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res., 42, 10681–10697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigo R. et al. (1992) Prediction of gene structure. J. Mol. Biol., 226, 141–157. [DOI] [PubMed] [Google Scholar]
- Guigo R., Valcárel J. (2015) Prescribing splicing. Science, 347, 124–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J. et al. (2012) GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res., 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harteveld C.L. et al. (2004) An alpha-thalassemia phenotype in a Dutch Hindustani, caused by a new point mutation that creates an alternative splice donor site in the first exon of the alpha2-globin gene. Hemoglobin, 28, 255–259. [DOI] [PubMed] [Google Scholar]
- Huelga S.C. et al. (2012) Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep., 1, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzkovitz S. et al. (2010) Overlapping codes within protein-coding sequences. Genome Res., 20, 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S., Chasin L.A. (2011) Context-dependent splicing regulation. RNA Biol., 8, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 59.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. et al. (2001) Integrating genomic homology into gene structure prediction. Bioinformatics, 17, S140–S148. [DOI] [PubMed] [Google Scholar]
- Kornblihtt A.R. et al. (2013) Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol., 14, 153–165. [DOI] [PubMed] [Google Scholar]
- Královicová J. et al. (2005) Biased exon/intron distribution of cryptic and de novo 3’ splice sites. Nucleic Acids Res., 33, 4882–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D. et al. (1996) A generalized hidden Markov model for the recognition of human genes in DNA. Proc. Int. Conf. Intel. Syst. Mol. Biol., 4, 134–142. [PubMed] [Google Scholar]
- Lander E.S. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Lappalainen T. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L.P., Burge C.B. (2001) A computational analysis of sequence features involved in recognition of short introns. PNAS, 98, 11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J.C., Caceres J.F. (2009) The SR protein family of splicing factors: master regulators of gene expression. Biochem. J., 417, 15–27. [DOI] [PubMed] [Google Scholar]
- Lukashin A.V., Borodovsky M. (1998) GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res., 26, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros W.H. (2007) Methods for Computational Gene Prediction. Cambridge University Press, Cambridge, England. [Google Scholar]
- Majoros W.M. et al. (2005) Efficient implementation of a generalized pair hidden Markov model for comparative gene finding. Bioinformatics, 21, 1782–1788. [DOI] [PubMed] [Google Scholar]
- Majoros W.H. et al. (2017) High-throughput interpretation of gene structure changes in human and nonhuman resequencing data, using ACE. Bioinformatics, 33, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauger D.M. et al. (2008) hnRNP H and hnRNP F complex with Fox2 to silence fibroblast growth factor receptor 2 exon IIIc. Mol. Cell. Biol., 28, 5403–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W. et al. (2016) The ensembl variant effect predictor. Genome Biol., 17, 122.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melé M. et al. (2015) The human transcriptome across tissues and individuals. Science, 348, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer I.M., Durbin R. (2004) Gene structure conservation aids similarity based gene prediction. Nucleic Acids Res., 32, 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mort M. et al. (2014) MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol., 15, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachter L. et al. (2002) Applications of generalized pair hidden Markov models to alignment and gene finding problems. J. Comput. Biol., 9, 389–399. [DOI] [PubMed] [Google Scholar]
- Pandit S. et al. (2013) Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell, 50, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q. et al. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet., 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Paul S. et al. (2006) Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J., 25, 4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J.K. et al. (2010) Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet., 6, e1001236.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M.A. et al. (2015) SRSF1 and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome. Sci. Rep., 5, 13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberson B.L. et al. (1990) Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell Biol., 10, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg A.B. et al. (2015) Learning the sequence determinants of alternative splicing from millions of random sequences. Cell, 163, 698–711. [DOI] [PubMed] [Google Scholar]
- Schneider M. et al. (2010) Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol. Cell, 38, 223–235. [DOI] [PubMed] [Google Scholar]
- Sherry S.T. et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Valcárcel J. (2005) Building specificity with nonspecific RNAbinding proteins. Nat. Struct. Mol. Biol., 12, 645–653. [DOI] [PubMed] [Google Scholar]
- Stadler M.B. et al. (2006) Inference of splicing regulatory activities by sequence neighborhood analysis. PLoS Genet., 2, e191.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M. et al. (2006) Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics, 7, 62.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepankiw N. et al. (2015) Widespread alternative and aberrant splicing revealed by lariat sequencing. Nucleic Acids Res., 43, 8488–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton C. (2008) Efficient training methods for conditional random fields. PhD Thesis, University of Massachusetts, Amherst.
- Sutton C., McCallum A. (2005) Piecewise training for undirected models. In: Proceedings of the Twenty-First Conference on Uncertainty in Artificial Intelligence, pp. 568–575. AUAI Press, Arlington, Virginia, USA. [Google Scholar]
- Sutton C., McCallum A. (2006) An introduction to conditional random fields for relational learning In: Getoor L., Taskar B. (eds.) Introduction to Statistical Relational Learning. MIT Press, Cambridge, MA, USA. [Google Scholar]
- The Thousand Genomes Project Consortium. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J.C. et al. (2001) The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- Woolfe A. et al. (2010) Genomic features defining exonic variants that modulate splicing. Genome Biol., 11, R20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H.Y. et al. (2015) The human splicing code reveals new insights into the genetic determinants of disease. Science, 347, 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.H., Chasin L.A. (2004) Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev., 18, 1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. et al. (2008) RNA landscape of evolution for optimal exon and intron discrimination. PNAS, 105, 5797–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.H.F. et al. (2005) Exon inclusion is dependent on predictable exonic splicing enhancers. Mol. Cell Biol., 25, 7323–7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.L. et al. (2014) Regulation of alternative splicing by local histone modifications: potential roles for RNA-guided mechanisms. Nucleic Acids Res., 42, 701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.