

Abstract
Background/Aim: Genomic DNA copy number alterations (CNAs) are frequent in tumors and have been catalogued by The Cancer Genome Atlas project. Emergence of chemoresistance frequently renders drug therapies ineffective. Materials and Methods: We analyzed how CNAs recurrently found in the genomes of TCGA patients of thirty- one tumor types affect protein targets of antineoplastic (AN) agents. Results: CNA deletions more frequently affected the targets of AN agents than CNA amplifications. Interestingly, in seven tumors we observed signs of compensatory CNAs. For example, in glioblastoma multiforme, two target genes (FLT1, FLT3) of the experimental drug sorafenib were recurrently deleted, whereas another target (KDR) of sorafenib was recurrently amplified. In renal clear cell carcinoma, the target FLT1 of pazopanib, sunitinib, sorafenib, and axitinib was recurrently deleted, whereas FLT4 bound by the same drugs, was recurrently amplified. Conclusion: Deletions of AN target proteins can be compensated by amplification of alternative targets.
Keywords: Copy number variation, drug target, chemoresistance, cancer genome, The Cancer Genome Atlas
Tumor cells differ phenotypically from normal cells, for example, by showing increased levels of proliferation and evading apoptosis (1). At the genomic level, one common variation of tumor cells is DNA copy number changes that include both gene amplifications and deletions (2). When these changes occur in germline cells, they are referred to as DNA copy number variations (CNV). When they occur in somatic cells, they are termed copy number alterations (CNA) (3). It is believed that CNAs in genome sequences of cancer patients (4) may play important roles in oncogenesis and cancer therapy (5).
An important reference data set on CNAs in patients suffering from more than 30 different tumors was compiled by The Cancer Genome Atlas (TCGA) project. A pan- cancer study of these data analyzed the effect of CNAs on known oncogenic drivers and tumor suppressor genes (TSG) and identified potential new cancer drivers, TSGs and biomarkers (6). This study also analyzed the length and the distribution of somatic CNAs along the chromosomes, identified regions that recurred significantly often and compared the number of genes in amplified and deleted regions (6). Subsequent studies (7,8) of CNA data from TCGA focused either on specific genes (e.g. PD-L1, CD247, IRS4, IGF2) or on the relationship between copy number events and gene expression (7,9). From the 33 tumor types available at TCGA today, we processed the data from 31 tumors in this study (glioblastoma multiforme, kidney renal clear cell carcinoma, brain lower-grade glioma, lung squamous cell carcinoma, liver hepatocellular carcinoma, kidney renal papillary cell carcinoma, kidney chromophobe carcinoma, breast invasive carcinoma, ovarian serous cystadenocarcinoma, uterine carcinosarcoma, head and neck squamous cell carcinoma, thyroid carcinoma, prostate adenocarcinoma, colon adenocarcinoma, stomach adenocarcinoma, bladder urothelial carcinoma, cervical squamous cell carcinoma and endocervical adenocarcinoma, sarcoma, acute myeloid leukemia, esophageal carcinoma, pheochromocytoma and paraganglioma, rectum adenocarcinoma, adrenocortical carcinoma, cholangio- carcinoma, lymphoid neoplasm diffuse large B-cell lymphoma, uveal melanoma, mesothelioma, thymoma, testicular germ cell tumors, uterine corpus endometrial carcinoma, pancreatic adenocarcinoma). The original publications on the datasets collected for these thirty-one tumors focused on the rate of copy number alterations, identification of recurrently amplified/deleted CNAs, the distribution of CNAs along the chromosomes, identification of oncogenes and TSGs, and clustered the tumors into subtypes. Several follow-up studies have analyzed CNA data from TGCA and analyzed copy number changes (10-12), recurrent copy number variations/alterations (13-16), the effect of CNAs on specific genes (17-21), identified putative new druggable cancer driver genes (22), tried to predict cancer relapse (23), and studied how cancer patients may be grouped into subtypes (12,18).
Tumor therapy often involves chemotherapy (24). The current release of Drugbank (version 5.0.11, downloaded on January 12, 2018) lists 477 drugs as antineoplastic (AN) agents that are annotated to bind to 220 different protein targets. Mapping the targets of AN agents to the KEGG database of cellular pathways using the tool KEGG mapper (25) shows that 53 target proteins from this list belong to the PI3K-Akt signaling pathway, 39 to metabolic pathways, 32 to the Rap1 signaling pathway, 30 to Th17 cell differentiation, 32 to the Ras signaling pathway, and 38 to the MAPK signaling pathway.The aim of this project was to analyze how protein targets of AN agents are affected by CNAs. To our best knowledge, no prior study addressed a related question so far. The only related work we are aware of is a study by Graham et al. who recently reported that recurrent patterns of DNA copy number alterations in tumors reflect metabolic selection pressures such as coordinated alteration of genes involved in glycolytic metabolism (26). For 31 tumor types from the TCGA dataset (see list above), we compared how recurrent CNAs affected the set of protein targets of chemotherapeutic drugs in comparison with a set of housekeeping genes and a set of cancer hallmark genes.
Materials and Methods
Figure 1 summarizes the main steps of our analysis.
Figure 1. Main steps of analysis workflow.

Data on copy number alterations. As mentioned, we analyzed genomic data from the TCGA project on CNAs observed in patients suffering from 31 different forms of tumors (listed in the introduction section). Missing from this list are the data for lung adenocarcinoma and skin cutaneous melanoma as these could not be processed with the GISTIC2.0 tool (see below). The CNA data of these patients (start and end position, chromosome, and segment mean of CNA) were downloaded from the Genomic Data Commons Portal (GDC portal) on September 29, 2017 (27).
Clinical data. From the clinical data provided at GDC, we extracted information on which drug treatment was given to specific patients. Thereby, the presence of CNAs in individual patient genomes was associated with the drug treatment applied to these patients. In our work, only data from patients that had both CNA and clinical data available were used.
Antineoplastic agents and their targets. A list of 477 ANs together with their target proteins was extracted from Drugbank (28) (version 5.0.11, downloaded on January 12, 2018). We considered only those protein targets for which pharmacological action of the respective drug molecule is reported as “yes” in Drugbank. These 477 AN agents are reported to bind to 220 different protein targets (labeled here by their Uniprot accessions numbers). After converting Uniprot accession numbers to gene symbols, we were left with 218 genes. As “tumor-specific” drugs, we considered those drugs that were applied to the patients of a particular tumor entity according to the TCGA data files. As shown in Table I and Table II, for drugs against lung cancer or breast cancer, these sets comprise a representative subset of the FDA-approved drug treatments for these tumors types (8 out of 16 and 23 out of 31), see https://www.cancer.gov/about- cancer/treatment/drugs/cancer-type. The sets for lung squamous cell carcinoma and breast cancer also included eight further drugs each that are not FDA-approved, but applied to TCGA patients possibly during ongoing clinical trials. Here, such drugs are labeled as “experimental drugs”.
Table I. Drugs applied against lung carcinoma. The first column contains FDA-approved drugs against non-small cell lung cancer (58 drugs). The second column contains a subset of the drugs from the first column after removing duplicated ones (25 drugs). The third column contains drugs that were applied to lung squamous cell carcinoma patients in TCGA (16 drugs). The drugs marked in bold are found both in the second and third column. Eight of sixteen drugs applied to lung squamous cell carcinoma patients in the TCGA panel were FDA-approved.

According to https://www.cancer.org/cancer/non-small-cell-lung-cancer/about/what-is-non-small-cell-lung-cancer.html, lung squamous cell carcinoma is a sub-type of non-small cell lung cancer.
Table II. Drugs against breast cancer. The first column contains FDA-approved drugs against breast cancer (71), the second column contains the compact list after removing duplicates (31 drugs), and the third column contains drugs that were applied to patients in the TCGA panel (38). Twentythree out of 38 drugs applied to TCGA-patients were FDA-approved drugs. The drugs marked in bold are found both in the second and third column.


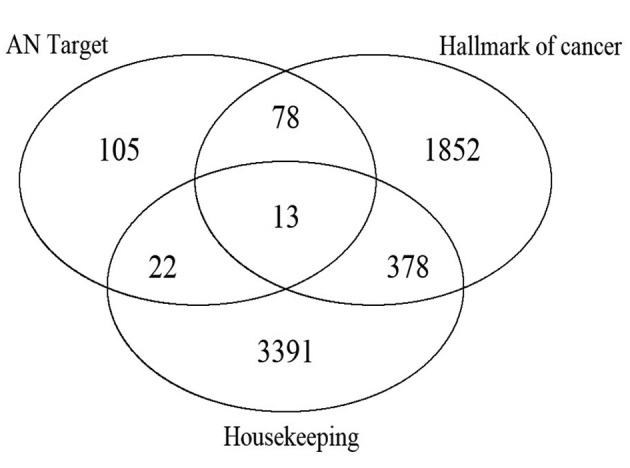
Gene Sets. Beside the set of protein targets of AN agents, we also considered a set of 3804 housekeeping genes (29) (i.e. at least one variant of these genes is expressed in all tissues uniformly; downloaded from https://www.tau.ac.il/~elieis/HKG/ on January 13, 2018) and a set of 2,338 “hallmark genes” of cancer. The latter set contains all human genes that are annotated in the Gene Ontology (30) to at least one of 37 Gene Ontology terms that were described as hallmarks of cancer (31) (downloaded from http://geneontology.org/ page/download-annotations on January 13, 2018). After converting Uniprot accession numbers to symbols, this gave 2321 gene symbols in the hallmarks of cancer gene set. Figure 2 shows the overlap of the three gene sets.
Figure 2. Overlap between the three gene sets.
Genes affected by CNAs. Genes that are recurrently affected by CNAs were identified with the GISTIC2.0 tool version 2.0.22 (32) using segmentation files and marker files created from the CNA data of the tumor samples. Following Laddha et al. (33), we used 0.2 and -0.2 as thresholds for GISTIC2.0 to identify recurrent amplification and deletion peaks and the genes contained in those peaks. Uniprot accession numbers used by Drugbank were converted to gene symbols used by GISTIC2.0 by making use of data from the HUGO Gene Nomenclature Committee (HGNC database) (34) that were downloaded in January 2017. Information on genes (chromosome, start position, and end position) was based on data from Ensembl (data downloaded from http://rest.ensembl.org on January 16, 2018).
Results
General statistics. The aims of this work were (1) to test the hypothesis that genomic CNAs observed in tumors affect the protein targets of AN agents significantly more often than expected by chance, (2) to test whether either amplifications or deletions are more common, and (3) to study the potential relevance for chemoresistance. In principle, one can expect that eventually all genes except for the essential genes will be affected by CNAs in some patients. Hence, to get more meaningful results, our analysis was focused on the set of recurrently occurring CNAs that appear statistically more often in each individual tumor entity than expected by chance. This strategy is similar to that used by Graham et al. (26).
Table III lists the number of recurrently amplified and deleted genes obtained by processing the raw CNA data for the 31 considered tumors with the GISTIC2.0 program. Specified is also how many of these amplifications/deletions affect hallmark genes, housekeeping genes, and protein targets of AN drugs. Note that, in this initial analysis, protein targets of all 477 considered AN drugs were considered irrespective of whether these drugs are actually being used to treat the particular subtype of cancer. In acute myeloid leukemia 38 of 105 cases (26.57%) received treatment prior to the time when the CNA data were taken. For glioblastoma (22 of 590 cases) and kidney renal clear cell carcinoma (18 of 530 cases), the number of such cases was around 4%. In all other tumors, the fraction of pre-treated patients was below 3%. Hence, in all tumors except for acute myeloid leukemia, the detected amplifications and deletions are unlikely to reflect resistance phenomena occurring in response to treatment (Table IV). As shown in Table III, in twenty-nine out of thirty-one studied tumors (the exceptions are thyroid carcinoma and kidney chromophobe), the number of recurrently deleted genes exceeded the number of recurrently amplified genes. However, this difference between the lower number of amplifications and the higher number of deletions was equally significant for the sets of all genes, antineoplastic targets, hallmark genes, and housekeeping genes (p-values 8.501e-09, 1.721e-08, 9.196e-09 and 8.367e-09, Wilcoxon test) and, hence, does not reflect a peculiar property of AN target genes. Table V shows that a similar behavior is observed for genes annotated to specific cancer hallmarks. For each disease, we then extracted from the GDC clinical data files the names of the drugs that were prescribed to the respective patients. The analysis was repeated with the same numbers of cases considered as in Table III but focused on the combined set of cancer-specific targets of these drugs, see Table VI. This set of target proteins was termed “specific drug targets” meaning that these are targets of the drugs that are given to patients with this specific tumor entity. By way of construction, the resulting numbers of affected genes were now far smaller. In 18 tumors, no CNA-amplifications affected the specific drug targets. In contrast, sarcoma behaved as an outlier to the other extreme with eight amplified targets. In the 12 remaining tumors, only one or two cases were observed. In contrast, in 23 tumors, CNA-deletions affected the specific drug targets of these tumor types. Among the three tumors (brain lower grade glioma, sarcoma, and mesothelioma) showing the largest number of CNA-deleted targets (10,11,14) only mesothelioma showed significantly more deletions than amplifications (adjusted p-value of 0.001, Fisher’s exact test). When taking all tumor data together, the difference between specific amplified/deleted targets for the 31 tumors was significant (p-values of 0.00016, Wilcoxon rank test).
Table III. Number of genes affected by CNAs in TCGA data for the 31 considered types of tumors.

Table IV. Treatment history of TCGA patients.

Table V. Amplifications and deletions of genes annotated with various cancer hallmarks. For this, we annotated human genes following Suzuki et al. (31) according to ten listed hallmarks of cancer. For each hallmark, we retrieved the genes that were annotated by GO terms of the hallmark. We then checked the number of genes that were affected by amplifications and deletions.

Table VI. Specific drugs and drug targets of the specified disease and the number of observed CNA-amplifications or CNA-deletions affecting the specific drug targets.

Following up on Table VI, Table VII lists the number of patient genomes where tumor-specific AN targets were affected by CNA mutations. This data show that, although the absolute number of CNA-affected AN target proteins is quite small (Table VII), the proportion of patients harboring these CNAs is in fact rather high. Respective target amplifications and deletions occur recurrently in a sizeable fraction (0 to 90%) of all patients.
Table VII. Number of cases when the specific AN targets were affected by CNAs.

To get more insight into the molecular mechanisms at place, Table VIII and Table IX list the gene symbols of the tumor- specific AN targets that were affected by CNA amplifications and deletions (Table VI) and the respective drugs that were applied to patients of these tumors. Experimental drugs were marked by labelEXP, e.g. docetaxelEXP. For acute myeloid leukemia that contains a sizeable fraction of pre-treated patients (26.57%) no information about the applied drugs is provided in the TCGA clinical data files, so we could not identify recurrent CNA amplifications or deletions of cancer- specific drug targets in this case.
Table VIII. Gene names that were recurrently amplified by CNAs. The drugs that bind to the respective AN target proteins are given in brackets. Tumors having no amplified AN targets and that are not listed in Table X are not shown.

Table IX. Names of genes that were recurrently deleted by CNAs. The drugs that bind to the respective AN target proteins are given in brackets. Tumors having no deleted AN targets and that are not listed in Table VIII are not shown.

Comparison of Table VIII and Table IX reveals that for some tumors, there exist targets of the same drugs that were both recurrently deleted and amplified in patients of the same tumor type. Table X lists all such pairs.
Table X. Drugs that bind to amplified and deleted AN targets in a single tumor type. Names of target genes are given in brackets.

Discussion
In this project, CNA and clinical data for 31 types of tumors from the TCGA project were combined with information on AN drugs from Drugbank. As shown in Table III, in 29 studied tumors, the number of recurrently deleted genes exceeded the number of recurrently amplified genes. This finding is generally concordant with the results of the TCGA consortium who reported in their pan-cancer study that the 70 peak amplification regions contained a median of 3 genes each, whereas 70 peak regions of CNA deletions contained a median of 4 genes (6). Earlier studies (6,9) reported that CNAs promote carcinogenesis and/or tumor progression by deleting tumor suppressor genes (TSGs). In agreement with this, in the dataset studied here the patient genomes of 29 tumors contained at least one of 71 known TSGs (13) in their list of genes recurrently deleted by CNAs. In the case of uterine corpus endometrial carcinoma and lymphoid neoplasm diffuse large B-cell lymphoma, even 22 of the 71 known TSGs were recurrently affected by CNA deletions (Table XI).
Table XI. Number of tumor suppressor genes affected by CNAs in different tumors.

The recurrently amplified/deleted genes of the 31 tumor types had no protein-coding gene in common. This is not unexpected as will be argued in the following. As shown in Table III, recurrent CNA deletions affected on average 4150 genes, which is roughly 20% of all genes. If we assume that the 31 considered tumors are unrelated, we would expect that- by chance – an overlap of (0.2)31×20.000 genes=4×10–28 genes would be affected in all tumor groups. This number is even smaller for amplified genes. This led to the expected result that all three gene sets (AN targets, housekeeping genes, and hallmark of the cancer genes) had no gene in common that is affected by CNAs in all type of tumors.
Then, we compared how CNAs affect gene subsets comprising antineoplastic (AN) target genes, housekeeping (HK) genes, hallmark of cancer (HC) genes, or tumor- specific AN target genes. Importantly, in all these gene sets, significantly more genes were affected by deletions than by amplifications. Hence, this observation is not specific to AN target genes nor to tumor-specific AN target genes.
The tumor-specific AN target genes recurrently affected by CNA amplifications are epidermal growth factor receptor (EGFR), FLT4, TYMS, TOP2A, KDR, VEGFA, BRAF, KIT, PDGFRA, HDAC2, TUBB1, PTGS2 and FGFR1. These genes belong to 13 types of tumors (Tables III and VIII). In the 18 remaining tumor types, no tumor-specific AN target gene was amplified. As an example, amplifications of EGFR gene copy numbers and overexpression of EGFR are known to be one of the most common alterations in non-small-cell lung carcinoma (NSCLC) cells (35-38) and are associated with a poor prognosis and chemoresistance. Among the histological subtypes of NSCLC, EGFR is most frequently expressed in squamous cells (39).
On the other hand, in 23 tumors, CNA-deletions affected specific drug targets of these tumor types. As shown in Table IX, CNA deletions of AN targets affected (1) the two enzymes bifunctional purine biosynthesis protein PURH (gene name ATIC) (40) and a subunit of ribonucleotide reductase (RRM1) that are both important for cell replication (41); (2) the nuclear receptor NR1I2 that regulates the metabolism and efflux of xenobiotics via CYP3A4 and MDR1 (42); (3) the mitochondrial and nuclear DNA topoisomerases TOP1MT and TOP2A; (4) the members of the vascular endothelial growth factor receptor family VEGFA, FLT1, FLT3, and (5) fibroblast grown factor FGFR2; (6) estrogen receptor ESR2; (7) the signaling MAP kinase MAPK11 and (8) the B-Raf Proto-Oncogen BRAF that regulates the MAP kinase/ERK signaling pathway (43); the inhibitory cell surface receptor PDCD1 that is involved in the regulation of T-cell function (44); and finally beta tubulin TUBB and the microtubule-associated protein MAP1A that is almost exclusively expressed in the brain (45,46) (and was CNA-deleted in glioblastoma). As all of these proteins have important roles in promoting carcinogenesis, they have likely been selected as targets of antineoplastic agents. As argued above, the CNA mutations pre-existed before the onset of the therapy.
These findings of rare CNA amplifications, but frequent CNA deletions of tumor-specific drug targets have clear consequences on drug development. In the future, considering CNA frequencies should certainly become a standard element of drug design efforts. These data also suggest that genomes of tumor patients may contain “compensating” mutations where one target protein of a drug is deleted and another target protein of the same drug is amplified. Unfortunately, due to space reasons we are restricted to discussing only a few of these cases in more detail.
In renal clear cell carcinoma patients that were subsequently treated with the drug molecules pazopanib, sunitinib, sorafenib, and axitinib, the target protein FLT1 of these drugs was recurrently deleted (in 55 samples), whereas another target protein, FLT4, of the same drugs was recurrently amplified (in 337 samples). Overall, 36 samples had both deleted FLT1 and amplified FLT4. FLT4 encodes a tyrosine kinase receptor of the same protein family as vascular endothelial growth factors C and D. In agreement with what is expected from the observed CNA amplification, FLT4 was previously reported to be overexpressed in kidney clear cell carcinoma (47). Besides being a recurrent target of CNA deletions here, FLT1 was also reported to be frequently silenced through promoter hypermethylation in renal clear cell carcinoma (48).
In lung squamous cell carcinoma patients subsequently treated with the drug erlotinib, one of its targets, NR1I2, was recurrently deleted (in 20 samples) and another target, epidermal growth factor receptor (EGFR), was recurrently amplified (in 186 samples). Nine samples had NR1I2 deleted and EGFR amplified at the same time. In brain lower grade glioma, NR1I2 and EGFR were also deleted and amplified, respectively. Beside these two genes, the target KIT of sorafenib was amplified while FLT4 and FGFR1 were deleted.
There exist also cases where the same target protein can be either amplified or deleted. For example, Table X shows that, FLT4 (target of sorafenib and pazopanib), and PTGS2 (target of sulindac) were observed to be either amplified or deleted in different sarcoma samples. FLT4 was amplified in 57 samples, and was deleted in 36 samples. PTGS2 was amplified in 63 samples, and was deleted in 32 samples.
The aim of this work was to test the hypothesis whether the protein targets of AN agents in tumors are affected by genomic copy number alternations (CNAs) more strongly than expected by chance. Based on CNAs and clinical data from the TCGA repository, we found that the genome sequences of tumor patients generally contain more recurrently deleted CNAs than recurrently amplified CNAs. This is also the case for CNAs affecting target genes of the specific AN for this tumor. Interestingly, we observed certain signs of apparently compensating effects of CNAs. The data available for this study enabled us to identify CNA alterations that existed prior to therapy and that may render certain chemotherapies more or less effective. In the future, it would be desirable to also collect time-series CNA data of tumor patients at the time of diagnosis and at later time points. This would point to CNA alterations caused by application of certain chemotherapies and thus reflect chemoresistance.
Acknowledgements
Ha Vu Tran was supported by a Ph.D scholarship from DAAD.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Pollack JR, Perou CM, Alizadeh AA, Eisen MB, Pergamenschikov A, Williams CF, Jeffrey SS, Botstein D, Brown PO. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat Genet. 1999;23:41–46. doi: 10.1038/12640. [DOI] [PubMed] [Google Scholar]
- 3.Li W, Lee A, Gregersen PK. Copy-number-variation and copy-number-alteration region detection by cumulative plots. BMC Bioinformatics. 2009;10:S67. doi: 10.1186/1471-2105-10-S1-S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer. 2010;10:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]
- 6.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang C-Z, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M, Beroukhim R. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budczies J, Bockmayr M, Denkert C, Klauschen F, Gröschel S, Darb-Esfahani S, Pfarr N, Leichsenring J, Onozato ML, Lennerz JK, Dietel M, Fröhling S, Schirmacher P, Iafrate AJ, Weichert W, Stenzinger A. Pan-cancer analysis of copy number changes in programmed death-ligand 1 (PD-L1, CD274) – associations with gene expression, mutational load, and survival. Genes, Chrom Cancer. 2016;55:626–639. doi: 10.1002/gcc.22365. [DOI] [PubMed] [Google Scholar]
- 8.Korbel JO. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2017;49:65–74. doi: 10.1038/ng.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao M, Zhao Z. Concordance of copy number loss and down-regulation of tumor suppressor genes: a pan-cancer study. BMC Genomics. 2016;17:532. doi: 10.1186/s12864-016-2904-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Xing H, Zhang NR. Estimation of parent specific DNA copy number in tumors using high-density genotyping arrays. PLOS Comput Biol. 2011;7:e1001060. doi: 10.1371/journal.pcbi.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xi R, Hadjipanayis AG, Luquette LJ, Kim T-M, Lee E, Zhang J, Johnson MD, Muzny DM, Wheeler DA, Gibbs RA, Kucherlapati R, Park PJ. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci USA. 2011;108:E1128–E1136. doi: 10.1073/pnas.1110574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jörnsten R, Abenius T, Kling T, Schmidt L, Johansson E, Nordling TEM, Nordlander B, Sander C, Gennemark P, Funa K, Nilsson B, Lindahl L, Nelander S. Network modeling of the transcriptional effects of copy number aberrations in glioblastoma. Mol Syst Biol. 2011;7:486. doi: 10.1038/msb.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Yuan Y, Lu KH, Zhang L. Identification of recurrent focal copy number variations and their putative targeted driver genes in ovarian cancer. BMC Bioinformatics. 2016;17:222. doi: 10.1186/s12859-016-1085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freire P, Vilela M, Deus H, Kim Y-W, Koul D, Colman H, Aldape KD, Bogler O, Yung WKA, Coombes K, Mills GB, Vasconcelos AT, Almeida JS. Exploratory analysis of the copy number alterations in glioblastoma multiforme. PLOS ONE. 2009;3:e4076. doi: 10.1371/journal.pone.0004076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q, Ding L, Larson DE, Koboldt DC, McLellan MD, Chen K, Shi X, Kraja A, Mardis ER, Wilson RK, Borecki IB, Province MA. CMDS: a population-based method for identifying recurrent DNA copy number aberrations in cancer from high-resolution data. Bioinformatics. 2010;26:464–469. doi: 10.1093/bioinformatics/btp708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Y, Hu X, Greshock J, Shen L, Yang X, Shao Z, Liang S, Tanyi JL, Sood AK, Zhang L. Genomic DNA copy-number alterations of the let-7 family in human cancers. PLOS ONE. 2012;7:e44399. doi: 10.1371/journal.pone.0044399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyerson M. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaelin WG. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell. 2013;24:738–750. doi: 10.1016/j.ccr.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen Y-B, Gonen M, Liu H, Takeda S, Voss MH, Tickoo SK, Reuter VE, Russo P, Cheng EH, Sander C, Motzer RJ, Hsieh JJ. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: A report by MSKCC and the KIRC TCGA Research Network. Clin Cancer Res. 2013;19:3259–3267. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerr E, Gaude E, Turrell F, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, Buchanan S, Peng SB, Staschke KA, Yadav V, Yue Y, Kouros-Mehr H. Identification of druggable cancer driver genes amplified across TCGA Datasets. PLOS ONE. 2014;9:e98293. doi: 10.1371/journal.pone.0098293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao Y, Heguy A, Huberman K, Bernstein M, Assel M, Murali R, Vickers A, Scardino PT, Sander C, Reuter V, Taylor BS, Sawyers CL. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci USA. 2014;111:11139–11144. doi: 10.1073/pnas.1411446111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sudhakar A. History of Cancer, Ancient and Modern Treatment Methods. J Cancer Sci Ther. 2009;1:1–4. doi: 10.4172/1948-5956.100000e2. [DOI] [PubMed] [Google Scholar]
- 25.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucl Ac Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Graeber TG. Recurrent patterns of DNA copy number alterations in tumors reflect metabolic selection pressures. Mol Syst Biol. 2017;13:914. doi: 10.15252/msb.20167159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM. Toward a shared vision for Cancer Genomic Data. New Engl J Med. 2016;375:1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucl Ac Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 30.Sherlock G. Gene Ontology: tool for the unification of biology. Nature Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki A, Makinoshima H, Wakaguri H, Esumi H, Sugano S, Kohno T, Tsuchihara K, Suzuki Y. Aberrant transcriptional regulations in cancers: genome, transcriptome and epigenome analysis of lung adenocarcinoma cell lines. Nucl Ac Res. 2014;42:13557–13572. doi: 10.1093/nar/gku885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN in human cancers. Mol Cancer Res. 2014;12:485 LP–490. doi: 10.1158/1541-7786.MCR-13-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucl Ac Res. 2015;43:D1079–D1085. doi: 10.1093/nar/gku1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HJ, Xu X, Choe G, Chung DH, Seo JW, Lee JH, Lee CT, Jheon S, Sung SW, Chung JH. Protein overexpression and gene amplification of epidermal growth factor receptor in nonsmall cell lung carcinomas: Comparison of four commercially available antibodies by immunohistochemistry and fluorescence in situ hybridization study. Lung Cancer. 2010;68:375–382. doi: 10.1016/j.lungcan.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 36.López-Malpartida AV, Ludeña MD, Varela G, García Pichel J. Differential ErbB receptor expression and intracellular signaling activity in lung adenocarcinomas and squamous cell carcinomas. Lung Cancer. 2009;65:25–33. doi: 10.1016/j.lungcan.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Dacic S, Flanagan M, Cieply K, Ramalingam S, Luketich J, Belani C, A Yousem S. Significance of EGFR protein expression and gene amplification in non-small cell lung carcinoma. Am J Clin Pathol. 2006;125:860–865. doi: 10.1309/H5UW-6CPC-WWC9-2241. [DOI] [PubMed] [Google Scholar]
- 38.Hirsch FR, Varella-Garcia M, Bunn PA, Di Maria MV, Veve R, Bremnes RM, Barón AE, Zeng C, Franklin WA. Epidermal growth factor receptor in non–small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol. 2003;21:3798–3807. doi: 10.1200/JCO.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 39.Pancewicz-Wojtkiewicz J. Epidermal growth factor receptor and notch signaling in non-small-cell lung cancer. Cancer Medicine. 2016;5:3572–3578. doi: 10.1002/cam4.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riley JL. PD-1 signaling in primary T cells. Immun Rev. 2009;229:114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foskolou IP, Jorgensen C, Leszczynska KB, Olcina MM, Tarhonskaya H, Haisma B, D’Angiolella V, Myers WK, Domene C, Flashman E, Hammond EM. Ribonucleotide reductase requires subunit switching in hypoxia to maintain DNA replication. Mol Cell. 2017;66:206–220. doi: 10.1016/j.molcel.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang B, Xie W, Krasowski MD. PXR: a xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenom. 2008;9:1695–1709. doi: 10.2217/14622416.9.11.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hommes DW, Peppelenbosch MP, van Deventer SJH. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut. 2003;52:144–151. doi: 10.1136/gut.52.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin HT, Ahmed R, Okazaki T. Role of PD-1 in regulating Tcell immunity BT - Negative co-receptors and ligands. Ahmed R and Honjo T (eds.) Springer Berlin Heidelberg. 2011:pp. 17–37. [Google Scholar]
- 45.Leandro-García LJ, Leskelä S, Landa I, Montero-Conde C, López-Jiménez E, Letón R, Cascón A, Robledo M, Rodríguez-Antona C. Tumoral and tissue-specific expression of the major human β-tubulin isotypes. Cytoskeleton. 2010;67:214–223. doi: 10.1002/cm.20436. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Lee JW, Ackerman SL. Mutations in the Microtubule- Associated Protein 1A (Map1a) gene cause purkinje cell degeneration. J Neurosci. 2015;35:4587–4598. doi: 10.1523/JNEUROSCI.2757-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beroukhim R, Brunet J-P, Di Napoli A, Mertz KD, Seeley A, Pires MM, Linhart D, Worrell RA, Moch H, Rubin MA, Sellers WR, Meyerson M, Linehan WM, Kaelin WG, Signoretti S. Patterns of gene expression and copy-number alterations in VHL disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69:4674–4681. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim JY, Hwang J, Lee SH, Lee HJ, Jelinek J, Jeong H, Lim JS, Kim JM, Song KS, Kim BH, Lee S, Kim J. Decreased efficacy of drugs targeting the vascular endothelial growth factor pathway by the epigenetic silencing of FLT1 in renal cancer cells. Clin Epigenet. 2015;7:99. doi: 10.1186/s13148-015-0134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]